

Abstract
Purpose
To identify the magnitude and sources of variability of a generic, aseptic manufacturing process for experimental anticancer agents employed at our facility, and to estimate the effects on product quality.
Materials and Methods
In-process and quality control data of all products manufactured according to this generic process (composed of weighing, dissolution, filtration, filling, semi-stoppering and lyophilization) over a 3-year period were retrospectively analyzed using mixed-effects analysis.
Results
Variability in the filling process was shown to be marginal and of minor importance for product quality in terms of content and content uniformity. An overall content of 101% was found with batch-to-batch and vial-to-vial variability up to 4.21% and 2.57%, respectively. Estimation of the overall batch failure revealed that structural bias in content and a high batch-to-batch variability in content were the most prominent factors determining batch failure. Furthermore, content and not content uniformity was shown to be most important parameter influencing batch failure. Calculated Process Capability Indices (CpKs) calculated for each product showed that the process is capable of manufacturing products which will routinely comply with the specification of 90–110% for content. However, the CpK values decreased dramatically using the specification of 95–105% as required for approved drug products.
Conclusion
These results indicate that at the early stage of product development less tight specification limits must be applied to prevent unnecessary batch rejection of investigational agents.
Key words: investigational drugs, manufacture, mixed effects analysis, validation
INTRODUCTION
The first steps in clinical development of experimental anticancer agents are phase I and II trials. Product quality, evidently, is very significant at this stage of development as it may, apart from patient safety, affect the outcomes of clinical trials which will determine whether or not the product will be selected for further development (1). Therefore, guidelines have been established with respect to Good Manufacturing Practice (GMP) of investigational agents (1–4). Product quality starts off with drug synthesis, followed by scientific sound formulation-, compatibility- and stability studies. This also involves the development and validation of a set of analytical techniques and methods to enable characterization and quality control of both the active pharmaceutical ingredient (API) and the final pharmaceutical product. Consistent manufacturing of the pharmaceutical product of the required quality should be possible then and proven by validation studies (5).
In our institute we have a long experience in the pharmaceutical development and manufacture of experimental anticancer agents for phase I and II clinical trials (6–15).
Anticancer drug formulations for experimental use are generally intended for intravenous administration to obtain absolute bio-availability, to circumvent possible disturbance of or degradation in the gastrointestinal tract and to be able to adjust or to stop administration of the drug immediately in case of acute toxicity. Consequently, the development of a pharmaceutical formulation of a novel anticancer agent is focussed on issues associated with the design of sterile and stable injectable products.
General characteristics in this early phase of development are: often limited availability of API, precluding elaborate validation studies; small scale (both in number of units per batch as well as in number of batches); flexibility (due to relatively many changes in strength and/or composition of batches); aseptical manufacturing process as the instability of the APIs often does not allow heat sterilization.
For the manufacture of sterile anticancer agents in the early phase of development, we established a small manufacturing unit at our institute in which we apply a generic production process capable of handling intravenous investigational anticancer drugs with the characteristics inherent to this phase. The generic production process is composed of six steps: weighing, dissolution, filtration, filling, semi-stoppering, and lyophilization.
The aim of this study was to identify the magnitude and sources of variability of the production process and to estimate the effects on product quality. Therefore, we performed a retrospective validation of this generic manufacturing process using mixed effects analysis. Based on this analysis, we evaluated the specifications for investigational products.
MATERIALS AND METHODS
Manufacturing Process
A flow chart of the generic production process, including the in-process controls which are performed at the different processing steps, is depicted in Fig. 1. All manufacturing steps are performed in a class 100 (B) clean room facility. Manipulations with open product (filtration, filling, semi-stoppering, lyophilization) are performed in a class 100 (A) environment. The clean room facility is regularly validated with respect to viable (settle plates, air sampling, contact plates) and non-viable particles, both in the “at rest state” and at operating state situation. Also, the aseptic manufacturing process and personnel is regularly validated by media fills by simulating the complete manufacturing process. Suitability of sterilizing filters for the manufacture of a specific product is evaluated. This all according to current guidelines (16,17). The facility holds a manufacturer’s licence for the manufacture of experimental anticancer agents since 1999.
Fig. 1.
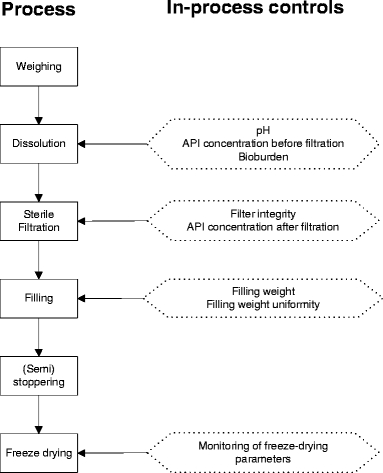
Schematic representation of the generic production process.
Data Collection
Data (i.e., batch characteristics and quality control data) were collected of all batches manufactured between January 2003 and November 2005. The following batch characteristics were used as input variables: product, batch size, dissolution vehicle, and theoretical filling volume. Table I gives an overview of the batch characteristics and their ranges. Output variables were: filling weight, content, and content uniformity.
Table I.
Batch Characteristics of all Batches Manufactured from 2003 to 2005 Used for Validation
| Batch Characteristic | Range |
|---|---|
| No. of products | 7 |
| No. of batches | 97 |
| Batch size | 75–800 vials |
| Dissolution vehicle | DMSO, WfI, TBA/WfIa |
| API concentrationb | 0.5–150 mg/ml |
| Filling volume | 1–40 ml |
aDMSO dimethyl sulfoxide, WfI water for injection, TBA/WfI 40% v/v TBA in WfI.
bConcentration of API in the formulation solution.
Data Analysis
For all batches, two types of observations were available:
In-process controls during the filling process (=filling weight)
Overall content (n = 13) of the vials calculated from both the content uniformity (n = 10) and content (n = 3) data
Both parameters were expressed as percentage of the theoretical values in order to facilitate comparison of the different batches and products.
Retrospective Data Analysis: Basic Model
Three distinct types of variability were assumed to exist:
Structural bias: The occurrence of a structural deviation from the theoretical content may be due to e.g., the production process, characteristics of the API (e.g., absorbance to materials used during manufacture) or filling of very low volumes.
Batch-to-batch variation (i.e., variation between different batches of one product)
Vial-to-vial variation (i.e., variation within one batch)
The basic model applied for data obtained from the filling process was:
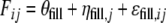 |
in which Fij is the measured filling weight in vial i of batch j, θfill is the typical value of the filling weight (if no structural bias is present θfill = 100%), ηfill,j is a random effect describing batch-to-batch variability with mean 0 and a standard deviation of ωfill, and εfill,ij is the random effect describing vial-to-vial variability with mean 0 and standard deviation σfill.
The predicted filling weight for an unknown vial in batch j equals 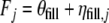
Similarly, the content data were modelled as:
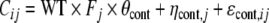 |
in which Cij is the measured content of vial i of batch j, WT is the weighted amount of API (expressed as percentage of theoretical), θcont is the typical value of content, ηcont,j is a random effect describing batch-to-batch variability with mean 0 and standard deviation ωcont and εcont,ij is the random effect describing vial-to-vial variability with mean 0 and standard deviation σcont. Because weighing is performed on a calibrated balance, it was assumed that bias and precision of the weighing could be neglected compared to the other sources of variability.
Both models were simultaneously applied to the data containing both types of observations (in-process controls during the filling process, and Overall content of the vials calculated from the content uniformity and content). Non-linear mixed effects modelling (NONMEM, version V, double precision, level 1.1, Globomax, Ellicott City, MD, USA) was used for the data analysis. NONMEM applies a maximum likelihood criterion to simultaneously estimate fixed effects (i.e., the typical values of content and the filling process) and random effects (i.e., the different variability terms). The first-order conditional estimation method with interaction between different types of variability (INTERACTION option of NONMEM) was used throughout. The following fixed effects were estimated for the basic model: θfill and θcont. The following random effects were estimated: ωfill, σfill, ωcont and σcont. Precision of parameter estimates was obtained with the COVARIANCE option of NONMEM.
Retrospective Data Analysis: Influence Of Production Parameters
For all batches the following co-variates were recorded: product (PROD), batch size (SIZE), filling volume (FILL) and, vehicle (VEH). The influence of these co-variates was tested on the different terms in the model. For instance, a product may have a systematic bias, an increased batch-to-batch variability or an increased vial-to-vial variability. The influence of these co-variates on the random effects was tested by introduction of different random effects terms for data with and without the co-variate (i.e., one product compared to the other products). The influence on the fixed effects was tested by introduction of a separate fixed effect describing the systematic bias for that co-variate. Significance was tested using the likelihood ratio test. The difference in objective function (minus twice the log likelihood of the data) between two nested models (i.e., models with and without a co-variate influence) has a chi-square distribution with one degree of freedom. Therefore, a difference of 3.84 points corresponds with a p value of 0.05.
Possible co-variates were introduced separately on the different terms of the basic model. Subsequently, all possible significant co-variates were introduced in an intermediate model. Stepwise backward elimination was used to retain only the significant co-variates in the final model.
Furthermore, the Process Capability Index (CpK) was calculated. This parameter is often used to measure the reproducibility as a function of the specification limits (18). CpK values were calculated for each product assuming a content equal to the average content for this product (optimal situation resulting in an “ideal” batch) and for each product assuming a content equal to the average content ±1 R.S.E. batch-to-batch variability, using Eqs. 1 and 2, whichever gives the lowest number.
 |
1 |
or
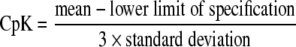 |
2 |
For the calculation of the CpK values the specification limits for content of 90–110% and 95–105% were used.
Simulation Studies
Based on the results of the retrospective data analysis, several simulations were performed in order to characterize the influence of different production parameters on batch failure (both content and content uniformity). For content uniformity the specifications according to the European Pharmacopoeia (19) were used, for content an average of 90–110% was used as specification. Several scenarios were investigated in which different sets of production parameters were defined and the influence of these parameters on batch failure was investigated. For each scenario, 1,000 batches were simulated and subsequently the batch failure based on content, content uniformity and overall batch failure was recorded. Simulations were performed using the SIMULATION option of NONMEM and the model as developed in the retrospective data analysis.
RESULTS AND DISCUSSION
Manufacturing Process
Table II gives a list of standard quality control test items for sterile, lyophilized products for parenteral use and the variables which may affect these. From these, the items and in-process controls were selected which are critical for product quality and are indicative for the general performance of the production process in relation to batch characteristics (product, batch size, dissolution vehicle, theoretical filling volume) were selected. The decision tree used to designate a variable as critical is given in Fig. 2. The test items Appearance, Reconstitution characteristics, and Residual moisture or solvent content are all output variables which are inherent to the selected freeze-drying process and are product-specific. Also, they influence more critical product quality items like content and purity only indirectly and are for this reason considered non-critical. Therefore, these items were not included in the assessment. For the same reason, pH after reconstitution was not used. Purity was not used because for all products analyzed the stability was studied well during formulation development and the compounds of interest were shown to be stable during processing time. Of all batches analyzed, no deviations in the manufacturing process occurred which may have affected purity or content. The test items content and content uniformity, however, were selected for the assessment because they are critical items for product quality. As shown from Table II, both content and content uniformity are directly influenced by filling weight and filling weight uniformity. Therefore, the in-process control filling weight was used in the analysis as well. Controls for producing an investigational new drug are primarily aimed at patient safety (4). Because it concerns the manufacture of parenteral drugs, sterility and pyrogenicity immediately affect product safety and are therefore critical items. The sterility and bacterial endotoxins content of all batches used for the assessment were according to the specifications. However, the tests performed to determine sterility and pyrogenicity are only performed on a selected number of vials and therefore, a negative result does not completely guarantee that all vials are sterile and free of endotoxins. This emphasizes that minimizing bioburden during manufacture and validation of production personnel, clean room facilities and production process is of utmost importance. No critical deviations with respect to these items occurred during the period of analysis.
Table II.
Variables Which May Influence Product Quality
| Product Quality Item | Process Variable |
|---|---|
| Appearance | Freeze-drying process |
| Reconstitution characteristics | Freeze-drying process |
| Content | Filling weight (IPC) |
| Content uniformity | Filling weight uniformity (IPC) |
| Purity | Purity API |
| Processing time | |
| pH after reconstitution | Concentration/characteristics of API and/or excipients |
| Residual moisture/solvent content | Freeze-drying process |
| Sterility | API/excipients/bioburden of formulation solution (IPC) |
| Filter integrity (IPC) | |
| Clean room/personnel performance | |
| Bacterial endotoxins | Contamination of API and/or excipients |
| Clean room/personnel performance |
IPC in-process control.
Fig. 2.
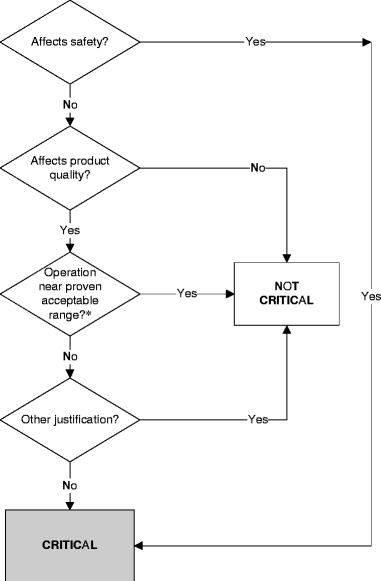
Decision tree for determination of product quality. Asterisk: generally unknown at early stage of drug development.
Data Analysis
Retrospective Data Analysis: Basic Model
In total, data of 97 batches of seven products were used for the retrospective risk assessment.
Variation in content or content uniformity found by quality control analysis is the result of an addition of errors in weighing, filling, and analytical variation. Due to the performance of extensive formulation and stability studies, it can be assumed that no degradation or loss during dissolution, filtration and freeze-drying occurs during the production process. The overall accuracy of the filling process was 99.6%, with a batch-to-batch and vial-to-vial variability of 0.796 and 0.988%, respectively (Table III). During manufacture of each batch, the pump was calibrated using the freshly prepared formulation solution which might explain the relatively small batch-to-batch variability compared to the vial-to-vial variability, although this difference is small.
Table III.
Results of the Retrospective Data Analysis: Basic Model
| Typical Value (%) (%R.S.E) | Batch-to-Batch Variability (%) (%R.S.E) | Vial-to-Vial Variability (%) (%R.S.E) | |
|---|---|---|---|
| Filling process | 99.6 (0.089) | 0.796 (20) | 0.988 (23) |
| Content | 101 (0.40) | 3.89 (19) | 1.96 (16) |
%R.S.E. Percent relative standard error of estimate
For content, an overall accuracy of 101% was found, while batch-to-batch variability and vial-to-vial variability were approximately 4 and 2%, respectively. This indicates that variability in the filling process is only marginal compared to the variability in content and therefore of minor relevance for the overall product quality.
Retrospective Data Analysis: Influence of Production Parameters
With this analysis, it was tested whether different production parameters had impact on the performance of the production process. This analysis, however, was hampered by the fact that several co-variates showed some degree of co-linearity. For instance, the dissolution vehicle dimethyl sulfoxide (DMSO) was only used as vehicle in a single product. Therefore, it was impossible to differentiate between the influence of DMSO as vehicle or other specific properties of this product (e.g., quality control methods). In these cases, it was chosen to use the product first as co-variate, except for co-variates related to the filling process where filling volume and vehicle were used preferentially. Table IV shows the results of this analysis. As can be seen, two products (3 and 5) had a systematic lower filling weight and one product (product 7) showed a lower batch-to-batch variability in the filling process than the other products. In product 3 the excipient 2-hydroxypropyl-β-cyclodextrin was used, resulting in a slight increase in viscosity of the formulation solution, probably resulting in a decrease in filling volume. For the vial-to-vial variability almost a three-fold increase (from 0.735 to 1.99%) was seen when a fill volume of 1 ml was used compared to higher filling volumes of 2–40 ml. Furthermore, the use of DMSO as vehicle increased the vial-to-vial variability with 53%, while the use of a mixture of water and tert-butyl alcohol (TBA) reduced this variability with 33.9%.
Table IV.
Results of the Retrospective Data Analysis: Influence of Production Parameters
| Typical Value (%) (%R.S.E) | Batch-to-Batch Variability (%) (%R.S.E) | Vial-to-Vial Variability (%) (%R.S.E) | |
|---|---|---|---|
| Overall filling process | 99.8 (0.063) | 0.857 (19) | 0.735 (20) |
| Product 3 | −0.552a (37) | ||
| Product 5 | −0.892a (40) | ||
| Product 7 | 0.251b (59) | ||
| FILL = 1 ml | 1.99b (49) | ||
| VEH = DMSO | +53%a (87) | ||
| VEH = TBA/WfI | −33.9%a (75) | ||
| Overall content | 100 (0.28) | 4.21 (22) | |
| Product 1 | 1.65b (11) | ||
| Product 2 | −5.04a (32) | 2.04b (64) | |
| Product 3 | 1.95b (28) | 1.51b (21) | |
| Product 4 | 2.57b (33) | ||
| Product 5 | 1.52b (16) | ||
| Product 6 | +7.14a (28) | 2.06b (21) | |
| Product 7 | 1.53b (30) | 2.12b (30) |
FILL filling volume, VEH dissolution vehicle, DMSO dimethyl sulfoxide, TBA/WfI 40% v/v tert-butyl alcohol in water for injection, %R.S.E. % relative standard error of estimate.
aRelative difference compared to typical value.
bAbsolute value.
The typical value of the overall content was exact 100%, indicating that no general structural bias was present. However, products 2 and 6 had a systematic and significant bias of –5.04 and +7.14%, respectively. This bias is not due to variation in filling weight, because no deviation in the typical value of the overall filling of these products was seen (Table IV). No specific product characteristics could be related to the structural bias of these products. Further investigation of the cause of the structural bias is required.
The batch-to-batch variability of the overall content was 4.21% while product 3 and 7 had a significant lower variability of less than 2%. Vial-to-vial variability was estimated for all products separately in order to account for differences in quality control methods. Vial-to-vial variability in content ranged from 1.51 to 2.57%. The highest vial-to-vial variability was found for product 4. Remarkable is the low vial-to-vial variability of product 3 compared to the other products, because this product is analyzed using a derivatization step, which may inherently increase variability. Apparently, the derivatization process is robust.
Furthermore, the Process Capability Index (CpK) was calculated for each product. There are generally accepted rules to relate the CpK value to the robustness of the process. A CpK value of less than or equal to 0.8 indicates that the process is not capable of meeting the specification limits routinely and therefore, further efforts have to be made in developing a more robust process. CpK values of 0.9–1.0 indicate a marginal process, of 1.0–1.25 are satisfactory, of 1.25–1.5 are good, and values higher than 1.5 are excellent. The calculated CpK values of our products are given in Table V. Within the limits of 90–110% and a deviation of one batch-to-batch variability, products 2 and 6 had a CpK value of less than 0.8, indicating that the process was not able to meet the specifications routinely for these products. The CpK values of the “ideal” batches of these products, however, were not much better. This is due to the structural bias found for these products. The presence of a structural bias can be a pitfall when using CpK values, because it can result in a low CpK value while the manufacturing process is acceptable and vice versa (18). The CpK values were satisfactory for products 1 and 4, good for product 7 and excellent for products 3 and 5.
Table V.
Calculated CpK Values for Content
| Product | Limit 90–110% | Limit 95–105% | ||
|---|---|---|---|---|
| Meana | Mean±1 bbvb | Meana | Mean±1 bbvb | |
| 1 | 2.02 | 1.17 | 1.01 | 0.16 |
| 2 | 0.81 | 0.12 | −0.01 | −0.69 |
| 3 | 2.21 | 1.78 | 1.10 | 0.67 |
| 4 | 1.30 | 1.04 | 0.65 | 0.40 |
| 5 | 2.19 | 1.77 | 1.10 | 0.67 |
| 6 | 0.46 | 0.15 | −0.35 | −0.66 |
| 7 | 1.57 | 1.33 | 0.79 | 0.55 |
aCpK value calculated for the most ideal batch with a batch-to-batch variability of 0.
bCpK value calculated for a batch with a deviation of 1 batch-to-batch variability (bbv).
As expected, using the specification limits of 95–105% according to the European guidelines for approved drugs, the CpK values were lower (20). The CpK values were less than 0.8 for all products with a deviation of one batch-to-batch variability. For the “ideal” batches, the CpK values of four of the seven products were still less than 0.8. It should be noted that the estimates for batch-to-batch variability were based on a limited number of batches. These values may therefore be highly dependent on single outlying batches. Further process optimization may be required before the products can fulfil the specifications for approved drug products routinely, especially when no outliers were found.
Simulation Studies
From the retrospective data analysis, it was established between which limits the different terms of the model varied according to the different production parameters. The ranges for the different terms were used in simulation studies to assess the influence of the different parameters on batch failure. The ranges used in the simulation study are shown in Table VI. The lower limits were defined as the best case scenario, the upper limits were defined as the worst case scenario.
Table VI.
Parameter Ranges for Simulation Study
| Filling Process (%) | Content (%) | |
|---|---|---|
| Absolute structural bias | 0–1 | 0–7 |
| Batch-to-batch variability | 0.25–1 | 1.5–5 |
| Vial-to-vial variability | 0.75–3 | 1.5–2.5 |
In a first series of simulations, it was investigated to what extent parameters derived from the filling process influenced the results. In the best case scenario as well as the worst case scenario no batches (out of the 1,000 simulated batches) were out of specifications for content and/or content uniformity. Therefore, it was concluded that the filling process was not a critical step in the manufacturing process within our current assessment.
The results of the same series of simulation for content are shown in Fig. 3. As can be seen, an increased batch-to-batch variability in content (1.5 to 5%, Table VI) was the most prominent factor determining batch failure in the absence of a structural bias. Furthermore, it was observed that the batch-to-batch variability had more impact on content than on content uniformity. It should be noted that overall batch failure was only determined by failure on content and not on content uniformity.
Fig. 3.
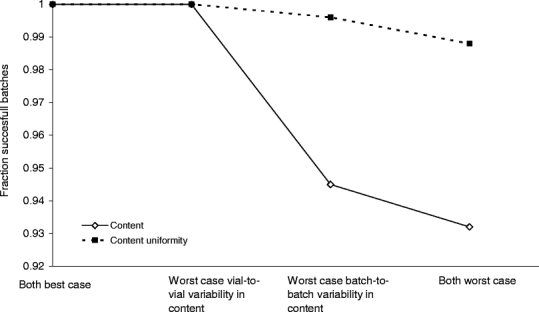
Calculated effect of vial-to-vial and batch-to-batch variability on the risk of batch failure based on the content and content uniformity in absence of structural bias.
In a next step, the influence of a structural bias in content and content uniformity on the chance of batch failure was tested. This was performed using a best and a worst case scenario (indicated in Table VI). The results of these simulations are shown in Figs. 4 and 5. Again, overall batch failure was only determined by batch failure due to content being out of specification. In the worst case scenario defined in our retrospective analysis (for all parameters involved) a structural bias of 7% in content could be found (Table VI), resulting in an overall content of 93 or 107%. In this situation approximately 30 and 15% of the batches are out of specification for content and content uniformity, respectively (Fig. 5). Caution should be taken in interpretation of these results as it is a simulation of the worst case scenario for all parameters simultaneously, which has not been encountered in practice thus far. Nonetheless, it indicates that structural bias has an important influence on batch failure.
Fig. 4.
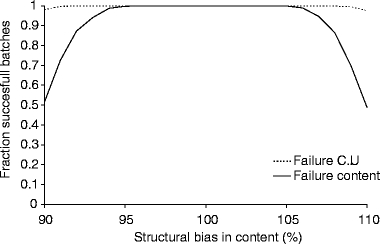
Estimated fraction of successful batches as function of the overall bias in the best case scenario. CU Content Uniformity.
Fig. 5.
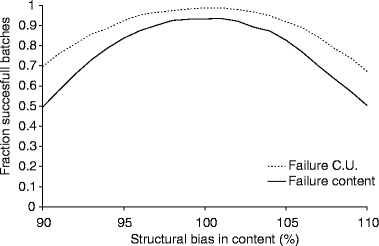
Estimated fraction of successful batches as function of the overall bias in the worst case scenario. CU Content Uniformity.
Overall it was found that within the limits as found in our retrospective data analysis, a structural bias in content and a high batch-to-batch variability in content are the most prominent factors determining batch failure. A variation in structural bias from –5 to +7% was seen. The presence of such a bias would result in a dramatic increase in batch failure if the limits 95–105% for content, according to the European guidelines for approved drugs, were used (20).
These results indicate that at the early stages of product development, less tight specification limits must be applied to prevent unnecessary batch rejection of investigational agents. It is generally accepted that product specifications will evolve during development, starting off with relative wide limits followed by tightening of these specifications in the course of product development as more information is gained and manufacture and analysis becomes more qualified (18,21–24). It is shown that the 90–110% specification limits we apply for content is feasible in this respect.
CONCLUSION
This analysis showed that for our generic manufacturing process, variability in the filling process was marginal and of minor importance for product quality in terms of content and content uniformity although it can be influenced by production parameters like dissolution vehicle and filling volume. An overall content of 101% was found with batch-to-batch and vial-to-vial variability up to 4.21 and 2.57%, respectively. Estimation of the overall batch failure revealed that structural bias in content and a high batch-to-batch variability in content were the most prominent factors determining batch failure. Furthermore, content and not content uniformity was shown to be most important parameter influencing batch failure. Calculated Process Capability Indices (CpKs) were calculated for each product, which showed that our manufacturing process is capable of manufacturing products which will routinely comply with the specification of 90–110% for content. However, the CpK values decreased dramatically using the specification of 95–105% as required for approved drug products. These results indicate that at the early stages of product development, less tight specification limits must be applied to prevent unnecessary batch rejection of investigational agents. Mixed effect analysis was shown to be a valuable tool in the assessment of the performance of our manufacturing procedures, and is implemented as an adjuvant to the application of GMP at our facility.
References
- 1.Food and Drug Administration. Guideline on the preparation of investigational new drug products (human and animal), FDA guidelines (1991).
- 2.European Commission. Eudralex volume 4—medicinal products for human and veterinary use: Good Manufacturing Practices Annex 13: Manufacture of investigational medicinal products (2003).
- 3.J. H. Beijnen, K. P. Flora, G. W. Halbert, R. E. Henrar, and J. A. Slack. CRC/EORTC/NCI Joint Formulation Working Party: experiences in the formulation of investigational cytotoxic drugs. Br. J. Cancer72:210–218 (1995). [DOI] [PMC free article] [PubMed]
- 4.Food and Drug Administration. INDs—Approaches to Complying with CGMP During Phase 1, Guidance for Industry, Draft (2006).
- 5.European Commission. Eudralex volume 4—medicinal products for human and veterinary use: Good Manufacturing Practices Annex 15: Qualification and validation (2001).
- 6.M. Bouma, B. Nuijen, G. Sava, A. Perbellini, A. Flaibani, M. J. van Steenbergen, H. Talsma, J. J. Kettenes-Van Den Bosch, A. Bult, and J. H. Beijnen. Pharmaceutical development of a parenteral lyophilized formulation of the antimetastatic ruthenium complex NAMI-A. Int. J. Pharm.248:247–259 (2002). [DOI] [PubMed]
- 7.M. Bouma, B. Nuijen, R. Harms, J. R. Rice, D. P. Nowotnik, D. R. Stewart, B. A. Jansen, S. van Zutphen, J. Reedijk, M. J. van Steenbergen, H. Talsma, A. Bult, and J. H. Beijnen. Pharmaceutical development of a parenteral lyophilized formulation of the investigational polymer-conjugated platinum anticancer agent AP 5280. Drug Dev. Ind. Pharm.29:981–995 (2003). [DOI] [PubMed]
- 8.M. W. Den Brok, B. Nuijen, J. J. Kettenes-Van Den Bosch, M. J. van Steenbergen, J. N. Buluran, M. D. Harvey, C. K. Grieshaber, and J. H. Beijnen. Pharmaceutical development of a parenteral lyophilised dosage form for the novel anticancer agent C1311, PDA. J. Pharm. Sci. Technol.59:285–297 (2005). [PubMed]
- 9.M. W. Den Brok, B. Nuijen, D. M. Meijer, E. Millan, C. Manada, and J. H. Beijnen. Pharmaceutical development of a parenteral lyophilised formulation of the investigational anticancer agent ES-285.HCl, PDA. J. Pharm. Sci. Technol.59:246–257 (2005). [PubMed]
- 10.M. W. J. den Brok, B. Nuijen, C. Lutz, H. G. Opitz, J. H. Beijnen. Pharmaceutical development of a lyophilised dosage form for the investigational anticancer agent Imexon using dimethyl sulfoxide as solubilising and stabilising agent. J. Pharm. Sci.94(5):1101–1114 (2005). [DOI] [PubMed]
- 11.J. D. Jonkman-de Vries, H. Talsma, R. E. C. Henrar, J. J. Kettenes-van den Bosch, A. Bult, and J. H. Beijnen. Pharmaceutical Formulation Development of a Parental Lyophilized Formulation of the Novel Indoloquinone Antitumor Agent EO9. Cancer Chemother. Pharmacol.34:416–422 (1994). [DOI] [PubMed]
- 12.B. Nuijen, M. Bouma, R. E. C. Henrar, P. Floriano, J. M. Jimeno, H. Talsma, J. J. Kettenes-van den Bosch, A. J. R. Heck, A. Bult, and J. H. Beijnen. Pharmaceutical development of a parenteral lyophilized formulation of the novel antitumor agent aplidine, PDA. J. Pharm. Sci. Technol.54:193–208 (2000). [PubMed]
- 13.B. Nuijen, M. Bouma, C. Manada, J. M. Jimeno, J. H. Schellens, A. Bult, and J. H. Beijnen. Pharmaceutical development of anticancer agents derived from marine sources. Anti-cancer Drugs11:793–811 (2000). [DOI] [PubMed]
- 14.B. Nuijen, M. Bouma, H. Talsma, C. Manada, J. M. Jimeno, L. Lopez-Lazaro, A. Bult, and J. H. Beijnen. Development of a lyophilized parenteral pharmaceutical formulation of the investigational polypeptide marine anticancer agent kahalalide F. Drug Dev. Ind. Pharm.27:767–780 (2001). [DOI] [PubMed]
- 15.B. Nuijen. Pharmaceutical formulation of novel anticancer agents. In D. R. Budman, A. H. Calvert, and E. K. Rowinsky (eds.), Handbook of Anticancer drug Development, Williams & Wilkins, Baltimore, 2003, pp. 205–223.
- 16.International Organization for Standardization. Aseptic processing of health care products, ISO 13408 (1998).
- 17.European Commission. Eudralex volume 4—medicinal products for human and veterinary use: Good Manufacturing Practices Annex 1: manufacture of sterile medicinal products (2003).
- 18.M. Gibson. Product optimization. In M. Gibson (ed.), Pharmaceutical Preformulation and Formulation, Interpharm/CRC, Boca Raton, 2004, pp. 295–329.
- 19.Directorate for the Quality of Medicines of the Council of Europe. European Pharmacopoeia, 2005.
- 20.European Commission. Eudralex volume 3—medicinal products for human and veterinary use: Guidelines 3AQ11A: Specifications and control tests on the finished product (1992).
- 21.J. P. Davignon, A. J. Slack, J. H. Beijnen, W. R. Vezin, and T. J. Schoemaker. EORTC/CRC/NCI Guidelines for the Formulation of Investigational Cytotoxic Drugs. Eur. J. Cancer Clin. Oncol.24:1535–1538 (1988). [DOI] [PubMed]
- 22.J. Stafford. Calculating the Risk of Batch Failure in the Manufacture of Drug Products. Drug Dev. Ind. Pharm.25:1083–1091 (1999). [DOI] [PubMed]
- 23.J. Geigert. Appropriate specifications at the IND stage, PDA. J. Pharm. Sci. Technol.51:78–80 (1997). [PubMed]
- 24.Food and Drug Administration. Content and Format of Investigational New Drug Applications (INDs) for Phase 1 Studies of Drugs, Including Well-characterized, Therapeutic, Biotechnology-derived Products, Guidance for Industry (1995).