

Abstract
Nitric oxide (NO) is an important regulator of renal transport processes. In the present study, we investigated the role of NO, produced by inducible NO synthase (iNOS), in the regulation of renal ATP-binding cassette (ABC) transporters in vivo during endotoxemia. Wistar–Hannover rats were injected with lipopolysaccharide (LPS+) alone or in combination with the iNOS inhibitor, aminoguanidine. Controls received detoxified LPS (LPS−). After LPS+, proximal tubular damage and a reduction in renal function were observed. Furthermore, iNOS mRNA and protein, and the amount of NO metabolites in plasma and urine, increased compared to the LPS− group. Coadministration with aminoguanidine resulted in an attenuation of iNOS induction and reduction of renal damage. Gene expression of 20 ABC transporters was determined. After LPS+, a clear up-regulation in Abca1, Abcb1/P-glycoprotein (P-gp), Abcb11/bile salt export pump (Bsep), and Abcc2/multidrug resistance protein (Mrp2) was found, whereas Abcc8 was down-regulated. Up-regulation of Abcc2/Mrp2 was accompanied by enhanced calcein excretion. Aminoguanidine attenuated the effects on transporter expression. Our data indicate that NO, produced locally by renal iNOS, regulates the expression of ABC transporters in vivo. Furthermore, we showed, for the first time, expression and subcellular localization of Abcb11/Bsep in rat kidney.
Electronic supplementary material
The online version of this article (doi: 10.1007/s00424-007-0210-x) contains supplementary material, which is available to authorized users.
Keywords: Renal drug transport, iNOS, Lipopolysaccharide, Aminoguanidine, Nephrotoxicity
Introduction
The free radical nitric oxide (NO) has been shown to play an important role in various physiological processes in the kidney, including salt and fluid reabsorption, renal hemodynamics, renin secretion, and tubuloglomerular feedback [1]. Endogenous NO is enzymatically produced from conversion of the amino acid l-arginine to l-citrulline, a reaction that is catalyzed by the enzyme NO synthase (NOS). This enzyme exists in three isoforms; the neuronal (nNOS) and endothelial, both of which are Ca2+- and calmodulin-dependent constitutive isoforms, and an inducible (iNOS) isoform [2]. Mediators associated with sepsis, such as endotoxin and the proinflammatory cytokines, interleukin-1β (IL-1β), IL-2, and tumor necrosis factor-α (TNF-α), have been shown to induce iNOS, thereby increasing production of NO. Sustained, high-output generation of NO by iNOS can cause cell death and tissue damage through lipid peroxidation, DNA damage, and proapoptotic effects [3]. This is the result of NO itself and the oxidant peroxynitrite, which is generated after reaction of NO with superoxide anions [4].
In contrast to many other organs, iNOS is constitutively expressed in the kidney, in particular in the medulla and in proximal tubules [5]. Many in vivo animal studies showed the importance of iNOS as a contributor to the pathophysiology of acute kidney injury (AKI). For example, it has been shown that selective chemical inhibitors of iNOS [6, 7], NO scavengers [8], iNOS gene knockout mice [9], or antisense oligodeoxynucleotides to iNOS [10] resulted in less renal tubular injury and improved structural and functional outcome. This renoprotective effect was partly due to the rescue of tubular epithelial cells from injury by NO [3].
Using killifish renal proximal tubules, we found previously that NO has a regulatory role in the transport activity of the ATP-binding cassette (ABC) transporter multidrug resistance protein 2 (Abcc2/Mrp2) via an intracellular signaling pathway in response to the action of several nephrotoxic chemicals in vitro [11]. This pathway involved at least endothelin (ET) release, binding to the basolateral ETB receptor, and activation of NOS, soluble guanylyl cyclase, and protein kinase C [12]. A similar regulatory pathway was found for the ABC transporter P-glycoprotein (Abcb1/P-gp) in rat brain capillaries [13]. Both studies indicate that the activation of NOS is most likely Ca2+-independent and we concluded that the production of NO is a result of the activation of iNOS.
The proximal tubule is responsible for the excretory transport of xenobiotics, xenobiotic metabolites, and waste products of metabolism from blood into urine. Consequently, the proximal tubule is also an important target for toxic effects and is often the first site of damage in AKI [14]. Luminal transport proteins, like the ABC transporters ABCB1/P-gp, ABCC2/MRP2, and ABCC4/MRP4, are important efflux transporters in the kidney [15] and, therefore, play a role in the detoxification of tubule cells. The observed effects of NO on Abcc2/Mrp2 in killifish proximal tubule may be part of the normal progression of cellular events that occur during AKI.
In the present study, we hypothesized that iNOS has a role in the regulation of renal ABC transporters during nephrotoxicity in vivo. An endotoxemia model was used in which rats were exposed to lipopolysaccharide (LPS), and the gene expression of 20 ABC transporters was evaluated. In addition, we examined the effect of the iNOS inhibitor, aminoguanidine, to determine whether the change in expression of the renal efflux transporters is caused directly by NO generated by iNOS.
Materials and methods
Animals and experimental design
Specified pathogen-free male Wistar–Hannover rats (240–295 g; purchased from Harlan, Zeist, The Netherlands) were kept under routine laboratory conditions at the Central Animal Laboratory of the Radboud University Nijmegen Medical Centre. This strain was used because they do not develop hydronephrosis spontaneously like normal Wistar rats, which in turn could have a down-regulating effect on iNOS expression [16]. The local Animal Care Committee approved all experimental procedures.
Different pilot studies have been conducted to determine the route of administration, intravenously or intraperitoneally (IP), concentration of the endotoxin LPS (LPS+, Escherichia coli 0127:B8, Sigma-Aldrich, Zwijndrecht, The Netherlands) and aminoguanidine (Sigma-Aldrich), and the time point of coadministration in this strain. LPS and aminoguanidine were dissolved in sterile Hank’s balanced salt solution (HBSS, Gibco, Paisly, UK) supplemented with 10 mM 4-(2-hydroxyethyl)-1-piperazine ethanesulfonic acid (HEPES, Gibco), and set to pH 7.4 with NaOH.
Thirty rats were, stratified to body weight, injected IP with 5 mg/kg LPS+ and were killed after 3, 6, 12, 24, or 48 h (n = 6 for each experimental group). Fifteen rats served as controls and received 10 mg/kg detoxified LPS (LPS−, n = 3 per group), in which the biologically active component, lipid A, essential for the activation of NOS, is lacking [17]. Although the control group is small, standard errors appeared to be satisfactory to allow a reduction and refinement of animals in this group. Twelve rats were injected IP with both 5 mg/kg LPS+ and, after 1 h, 100 mg/kg of the iNOS inhibitor aminoguanidine (Sigma-Aldrich), and were killed 6 and 12 h after LPS+ (n = 6 per group). For the isolated perfused kidney experiments, six rats received LPS+, four received LPS−, and four were untreated and served as controls, and were killed after 12 h as described under the “Isolated kidney perfusion” section.
To collect urine before and after the injection, rats were housed in metabolic cages, washed with 2% chlorohexidine (Genfarma, Zaandam, The Netherlands) to prevent bacterial growth. Urine volumes were recorded and samples were snap-frozen in liquid nitrogen and stored at −80°C until assayed. During the experiment, body weight and body temperature were determined. At indicated time points after LPS injection, the rats were anesthetized with 5% isoflurane and heparin (125 IU/100 g) was injected via the penile vein to minimize blood clotting. The left kidney was removed, divided in four pieces, and snap-frozen in liquid nitrogen for immunohistology or fixed in Bouin’s solution for routine histology. Arterial blood was obtained by aortic puncture and the rats were killed by exsanguination. The right renal vein was punctured and, subsequently, the kidney was perfused with HBSS–HEPES for 2 min at 7.5 ml/min (Minipuls 2, Gilson, Villiers-le-Bel, France). After perfusion, the kidney was removed, divided, and snap-frozen in liquid nitrogen for quantitative real-time polymerase chain reaction (RQ-PCR) and Western blotting. Thrombocyte and leukocyte counts were determined in blood as inflammatory parameters, and the concentrations of creatinine, urea, protein, glucose, and sodium were analyzed in blood and urine by routine clinical chemistry. The activity of alkaline phosphatase, a proximal tubule injury marker, was determined in 26-fold diluted urine samples, as described before [18].
Determination of mRNA expression
RNA was isolated using a Mikro-dismembrator U (Sartorius B. Braun Biotech, Melsungen, Germany). To prevent RNA degradation, we washed the metal cylinders with 0.5 M NaOH. Perfused kidneys were pulverized (2,000 rpm, 30 s) and transferred in ice-cold TRIzol reagent (Invitrogen, Breda, The Netherlands), and then RNA was isolated as described previously [19]. RQ-PCR on cDNA was performed according to the TaqMan® protocol in optical tubes using either the ABI PRISM 7700 single reporter Sequence Detection System (n = 6, Applied Biosystems, Zwijndrecht, The Netherlands) or the ABI/PRISM 7900HT Gene Expression Micro Fluidic Card Sequence Detection System (three pooled samples from nine different rats, Applied Biosystems) according to the manufacturer’s instructions. All experiments were performed in triplicate. Different rat genes were amplified with a predeveloped Gene Expression Assay, provided by Applied Biosystems.
Western blotting
Total membrane and cytosolic fractions from perfused kidneys were isolated using a Mikro-dismembrator U (Sartorius BBI Systems GmbH, Melsungen, Germany; 2,000 rpm for 30 s) and subjected to Western blotting as described previously [20]. Protein concentration was determined with the Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA) using bovine serum albumin (BSA) as standard. Samples were separated on a 6 or 12% sodium dodecylsulfate polyacrylamide gel and transferred to Hybond-C pure nitrocellulose membrane (Amersham, Buckinghamshire, UK). The membrane was incubated overnight at 4°C with antibodies against NOSII/iNOS (1:1,000, according to Vos et al. [21]), ABCB1/P-gp (1:200, C219, DakoCytomation, Dako, Denmark), Abcc2/Mrp2 (1:250, M2III-6, Alexis Biochemicals, San Diego, CA, USA), ABCC4/MRP4 (1:1,000, as described by van Aubel et al. [22]), Abcb11/bile salt export pump (Bsep) (1:1,000, kindly provided by Dr. Bruno Stieger, University Hospital Zürich, Switzerland), or β-actin (1:10,000, Sigma) in Tris-buffered saline supplemented with 0.1% Tween-20 and 1% nonfat dried milk.
Nitrate and nitrite determination
The total amount of the stable NO metabolites (NOx), nitrate and nitrite, were determined as a measure of NO radicals production, using the Griess reaction, according to Moshage et al. [23]. Heparinized plasma was diluted 1:4 and urine samples were diluted 1:40 or 1:80 with distilled water. Each sample was determined in triplicate. Calibration curves were made with sodium nitrite and sodium nitrate in distilled water. The nitrate recovery was 90–100% over the entire range tested (0–50 μmol/l). The coefficient of variation between the different experiments was less than 5%.
Histology and immunohistology
Kidneys fixed in Bouin’s solution were dehydrated, embedded in paraplast (Amsterstad, Amsterdam, The Netherlands), and cut into 3-μm sections, which were stained with periodic acid-Schiff for routine histology. For immunohistologic studies, serial tissue sections of 1.5 μm were cut from frozen kidneys using a cryostat as described previously [24]. The slices were incubated for 60 min at room temperature with antibodies against either NOSII/iNOS (1:200, according to Vos et al. [21]) or ABCB1/P-gp (1:50, C219, DakoCytomation) in PBS supplemented with 1% BSA. For the antibodies peroxynitrite (1:500, Sigma) and Abcb11/Bsep (1:1,000, as described by Huls et al. [24]), cryostat slices of 4 μm were used, fixed in acetone for 5 min. Endogenous peroxidase was blocked by incubation with 5% H2O2 for 5 min, and nonspecific protein binding was blocked with 10% normal goat serum (Vector, Burlingame, CA, USA) in PBS for 10 min at room temperature. Slices were incubated with the primary antibody in PBS 1% BSA overnight at 4°C. Primary antibody was detected using the Vectastain Elite peroxidase ABC kit (Vector) with 3,3′-diaminobenzidine as substrate. Finally, slices were counterstained with hematoxylin, dehydrated, and mounted in mounting medium. Sections were examined with a confocal laser-scanning microscope (Leica lasertechnik GmbH, Heidelberg, Germany).
Isolated kidney perfusion
Twelve hours after the administration of LPS+ (n = 6) or LPS− (n = 4), the transport activity of Abcc2/Mrp2 was determined by monitoring the urinary excretion of calcein in an isolated perfused rat kidney, as described in detail previously [25]. Briefly, rats were anesthetized IP with pentobarbital (6 mg/100 g), and furosemide was injected IP (1 mg/100 g) to prevent deterioration of the distal nephron. Heparin (125 IU/100 g) was injected in the spleen. The kidney was excised and placed in a fluid bath with a constant temperature of 37.5°C and rats were killed by exsanguination. Vitamin B12 was used for the determination of the glomerular filtration rate (GFR) and calcein-acetoxymethylester was used as a source for the fluorescent substrate calcein [25].
Statistical analysis
Values are given as mean ± SE. Differences between the experimental groups were tested using one-way ANOVA with Bonferroni’s correction or two-way ANOVA corrected for repeated measurements. A two-sided P value < 0.05 was considered significant.
Results
Signs of inflammation
A few hours after LPS+ administration, inflammatory symptoms appeared, like diarrhea and piloerection. Body temperature was elevated after 24 h and the loss in body weight increased from −3% (t = 3 h), due to normal circadian rhythm, to 12% (t = 48 h), compared to the controls (Table a in the supplementary material 1). Furthermore, administration of LPS+ resulted in the expected leucopenia (t = 3 h) followed by leukocytosis (t = 48 h) and thrombocytopenia, with a maximum at 48 h. Coadministration of LPS+ with aminoguanidine did not affect the inflammatory parameters, except for a greater loss in body weight 12 h after LPS administration.
iNOS induction after LPS treatment
RQ-PCR was used to determine the iNOS mRNA levels in kidney samples. The relative expression of iNOS in LPS− rats was normalized for the average cycle threshold (CT) value for the housekeeping gene, GAPDH (CT = 16.06 ± 0.04, n = 15), and set to 1. GAPDH expression remained stable during endotoxemia (CT = 16.22 ± 0.09, n = 42). Furthermore, no circadian rhythm for the different genes was detected in control animals.
Analysis of three separate experiments revealed a more than 40,000 times induction in iNOS mRNA 3 h after LPS+ compared to controls (Fig. 1a), which decreased with time, but was still 100 times higher than control values 48 h after LPS+. Treatment with both LPS+ and aminoguanidine slightly, but not significantly, reversed iNOS mRNA induction compared to LPS+ alone (Fig. 1a). Maximal iNOS protein expression was observed in cytosolic fractions 6 h after LPS+, and returned to LPS− level at 48 h (Fig. 1b). In contrast to its effect on iNOS mRNA expression, aminoguanidine reversed the induction in protein expression (Fig. 1c).
Fig. 1.
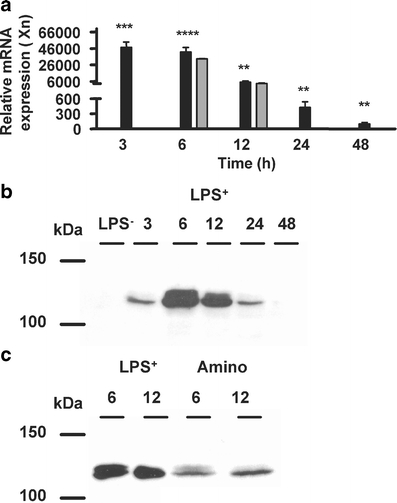
iNOS mRNA and protein. a iNOS mRNA expression in rat kidney at different time points after LPS+ was determined with RQ-PCR without (closed bars, n = 6) or with coadministration of aminoguanidine (gray bars, n = 6). The iNOS mRNA expression was normalized for the GAPDH CT value (16.06 ± 0.04, n = 15), and ΔCT values for the LPS− were set to 1. Data are expressed as mean±SE. Significantly different compared to the LPS− (double asterisksP < 0.01, triple asterisksP < 0.001, quadruple asterisksP < 0.0001). Cytosolic fractions of rat kidney were isolated and expression of iNOS was determined by Western blotting. Representative images (n = 4) show maximal iNOS protein expression 6 h after LPS+ administration (b), and coadministration with aminoguanidine reversed this induction (c)
NOx levels in plasma and urine were measured using the Griess assay. An increase in NOx concentration in plasma was already visible 3 h after LPS+, with a peak concentration at 12 h. Coadministration with aminoguanidine reversed the increase in NOx significantly at both 6 and 12 h (Fig. 2a). The urinary excretion of NOx increased 12 h after LPS+, and was still elevated 48 h later. Aminoguanidine returned this increase to baseline levels at both time points (Fig. 2b).
Fig. 2.
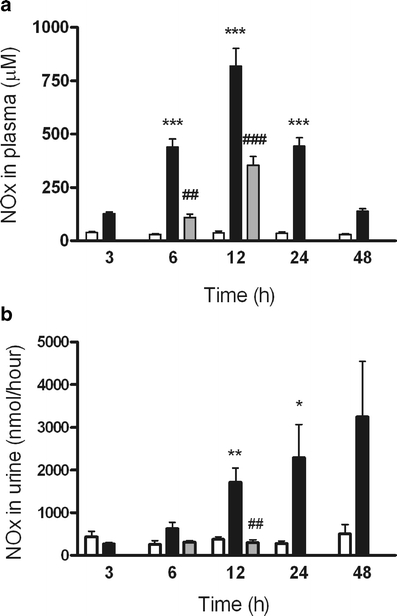
NOx in plasma and urine. NOx levels in plasma and urine were measured at various times after LPS− (open bars, n = 3) or LPS+ (closed bars, n = 6) treatment or LPS+ together with aminoguanidine (gray bars, n = 6). a Blood samples show a peak concentration at 12 h and coadministration with aminoguanidine reversed this increase at both 6 and 12 h. b The urinary excretion of NOx increased 12 h after LPS+ treatment, and aminoguanidine returned this increase to baseline levels. Data are expressed as mean±SE. Significantly different compared to the LPS− (single asterisksP < 0.05, double asterisksP < 0.01, triple asterisksP < 0.001) or LPS+ (double sharp signsP < 0.01, triple sharp signsP < 0.001)
Renal localization of iNOS and formation of 3-nitrotyrosine-protein adducts, a reliable biomarker of peroxynitrite generation [26], were determined by immunohistology. There was no iNOS staining visible in kidney sections of rats treated with LPS−. Positive iNOS staining was seen in both experimental groups after LPS+, in inflammatory cells, and in proximal tubule cells. Forty-eight hours after LPS+, iNOS staining was no longer detectable. Figure 3 shows representative immunohistochemical images with iNOS expression in all segments of the proximal tubule (3A+C). After colocalization with Abcb1/P-gp (3B), iNOS expression in the vicinity of the apical membrane was visible (3A+C). 3-Nitrotyrosine-protein adducts were predominantly present in the cortex in peritubulary capillaries and proximal tubules after LPS+ (Fig. 3d,e). Coadministration with aminoguanidine attenuated the formation of adducts in the cortex (Fig. 3f,g).
Fig. 3.
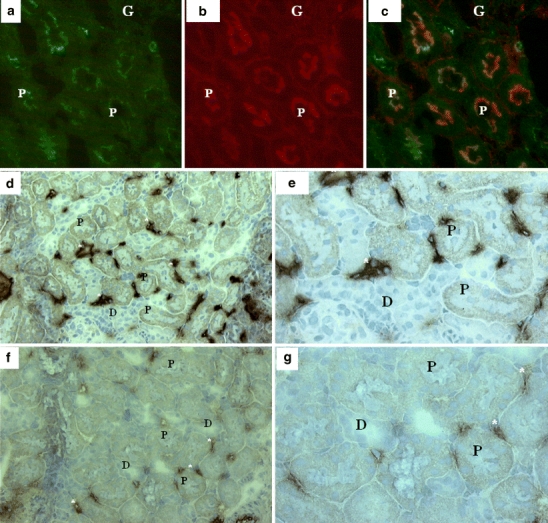
Immunofluorescence microscopic analysis of iNOS and peroxynitrite in the kidney. a–c Representative immunohistochemical images show that iNOS (green staining) is expressed in the vicinity of the apical membrane (P-gp counterstaining, red) in all segments of the proximal tubule 12 h after LPS+ treatment. d, e Formation of 3-nitrotyrosine-protein adducts, a reliable biomarker of peroxynitrite generation, were visible in the cortex in peritubulary capillaries and proximal tubules 12 h after LPS+ treatment. f, g Coadministration with aminoguanidine resulted in attenuated formation of 3-nitrotyrosine-protein adducts at 12 h. G glomerulus, asterisks peritubular staining, P proximal tubule, D distal tubule. Original magnification d, f ×200 and a–c, e, g ×400
Aminoguanidine restores the slightly impaired kidney function and loss of brush border membrane
Kidney function was slightly impaired after LPS+ as indicated by increased blood urea nitrogen (BUN), an increase in plasma creatinine, and an increased urinary glucose excretion (Table 1). Furthermore, the urine/plasma creatinine ratio was decreased (data not shown) as was the creatinine clearance, although not significantly.
Table 1.
Renal functional parameters
| Kidney function | Time (h) | Controls (LPS−) | LPS+ | LPS+ and aminoguanidine |
|---|---|---|---|---|
| Total urine volume (ml) | 3 | 1.6 ± 0.24 | 1.7 ± 0.27 | |
| 6 | 2.4 ± 0.65 | 4.45 ± 0.98 | 7.3 ± 0.76 | |
| 12 | 3.2 ± 0.57 | 10.0 ± 0.70a | 9.4 ± 0.65a | |
| 24 | 11.5 ± 0.25 | 13.7 ± 2.11 | ||
| 48 | 23.5 ± 3.47 | 18.0 ± 1.57 | ||
| BUN (mmol/l) | 3 | 6.6 ± 0.20 | 9.9 ± 0.74 | |
| 6 | 4.6 ± 0.72 | 14.0 ± 1.48b | 12.1 ± 0.82b | |
| 12 | 6.3 ± 0.24 | 12.3 ± 1.38a | 15.4 ± 1.27b | |
| 24 | 7.0 ± 0.58 | 10.6 ± 0.58 | ||
| 48 | 6.5 ± 0.10 | 9.1 ± 0.38 | ||
| Blood creatinine (mmol/l) | 3 | 0.035 ± 0.002 | 0.049 ± 0.002b | |
| 6 | 0.039 ± 0.002 | 0.053 ± 0.002b | 0.043 ± 0.019d | |
| 12 | 0.037 ± 0.002 | 0.048 ± 0.002c | 0.036 ± 0.032e | |
| 24 | 0.038 ± 0.001 | 0.051 ± 0.001a | ||
| 48 | 0.042 ± 0.001 | 0.052 ± 0.001 | ||
| Glucose excretion (μmol/h) | 3 | 0.290 ± 0.045 | 0.481 ± 0.341 | |
| 6 | 0.333 ± 0.086 | 0.420 ± 0.100 | 0.478 ± 0.051 | |
| 12 | 0.425 ± 0.106 | 0.537 ± 0.054 | 0.332 ± 0.080 | |
| 24 | 0.254 ± 0.006 | 0.984 ± 0.039c | ||
| 48 | 0.325 ± 0.037 | 0.606 ± 0.128 | ||
| Creatinine clearance (ml/min) | 3 | 1.33 ± 0.12 | 0.78 ± 0.19 | |
| 6 | 1.06 ± 0.15 | 0.87 ± 0.09 | 0.93 ± 0.35 | |
| 12 | 1.01 ± 0.05 | 0.90 ± 0.03 | 1.01 ± 0.13 | |
| 24 | 1.19 ± 0.02 | 0.91 ± 0.20 | ||
| 48 | 1.00 ± 0.07 | 1.08 ± 0.37 | ||
| FENa (%) | 3 | 0.816 ± 0.004 | 0.365 ± 0.073 | |
| 6 | 1.046 ± 0.145 | 0.358 ± 0.094b | 1.423 ± 0.310e | |
| 12 | 1.094 ± 0.097 | 0.458 ± 0.075a | 1.610 ± 0.282e | |
| 24 | 1.070 ± 0.024 | 0.429 ± 0.065c | ||
| 48 | 1.033 ± 0.077 | 0.121 ± 0.024a |
Data are expressed as mean±SE, n = 6 for each time point for both LPS+ and LPS+ +aminoguanidine and n = 3 for LPS−
FENa fractional sodium excretion
aSignificantly different compared to the control group; P < 0.01
bSignificantly different compared to the control group; P < 0.001
cSignificantly different compared to the control group; P < 0.05
dSignificantly different compared to the LPS+ group; P < 0.01
eSignificantly different compared to the LPS+ group; P < 0.001
Histological examination after LPS+ revealed damage to the proximal tubules, compared to LPS− (Fig. 4a,b). Loss of brush border membranes and formation of vacuoles were observed. Aminoguanidine reduced LPS+-induced damage to the proximal tubule (Fig. 4c). The activity of alkaline phosphatase in urine samples was assayed to get an indication of proximal tubule injury. Twelve hours after LPS+, the activity of alkaline phosphatase was increased in urine samples and was still significantly increased after 48 h (Fig. 4d). Furthermore, the expression of kidney injury molecule 1 (Kim-1 [27]) was strongly up-regulated, 400 times more than control values, 6 and 12 h after LPS+ (Table b in the supplementary material 1). Except for BUN, aminoguanidine improved all parameters of renal function, as shown in Table 1 and Fig. 4. No differences in urinary protein excretion were found and, remarkably, the fractional excretion of filtered sodium was decreased (Table 1).
Fig. 4.
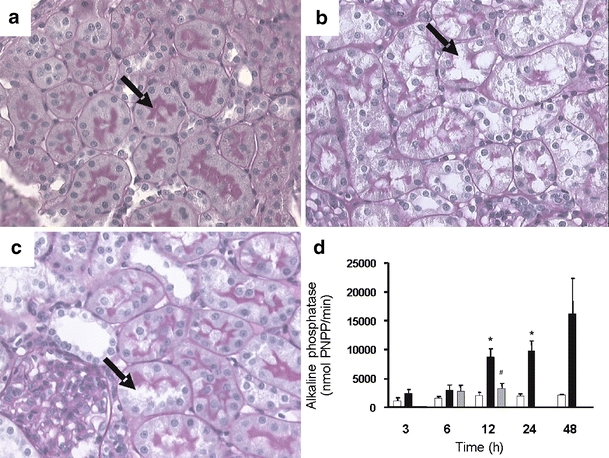
Proximal tubular damage during endotoxemia. Histological examination after LPS+ revealed damage to the proximal tubules compared to LPS−. a LPS−-treated rat with intact brush border membrane in proximal tubules. b Twelve hours after LPS+, damage to the proximal tubules is observed with loss of brush border membranes and formation of vacuoles. c Treatment with both LPS+ and aminoguanidine resulted in less damage to the proximal tubule. Kidneys of LPS+-treated rats killed after 24 or 48 h also showed signs of proximal tubule damage (data not shown). Proximal tubules are indicated by arrows. Original magnifications (a–c) ×400. d Histological data are supported by an increase in the activity of alkaline phosphatase, marker for proximal tubule damage, in urine samples 12 h after LPS+ administration (closed bars, n = 6), compared to controls (open bars, n = 3). Coadministration with aminoguanidine (gray bars, n = 6) reduced this proximal tubule damage. Data are expressed as mean±SE. Significantly different compared to the LPS− (double asterisksP < 0.01) or LPS+ (sharp signP < 0.05)
Furthermore, the mRNA expression of different biotransformation enzymes [28], enzymes involved in the NO pathway [2] or known to be activated during oxidative stress [29, 30] was determined. Twelve and 24 h after LPS+ treatment, expression of the metabolizing enzymes glutathione S-transferases (GST-α2 and GST-α5) and uridine diphosphate-glucuronosyltransferases (UDPGT1a6) was down-regulated (Table b in the supplementary material 1, at least a 2 times lower expression). The arginine metabolic enzymes, argininosuccinate synthetase and arginase 2, were not changed after LPS+. In contrast, arginase 1 mRNA expression was reduced. The gene expression of the ETB receptor, which gets activated as part of the NO-dependent signaling pathway regulating Abcb1/P-gp and Abcc2/Mrp2 [13, 31], did not change during endotoxemia. Three and 6 h after LPS+ the mRNA expressions of super oxide dismutase 2, heam-oxygenase 1, and nuclear factor κB (NF-κB) were up-regulated (i.e., at least a 2 times higher expression). Coadministration of aminoguanidine had no effect on the gene expression of all these proteins. In contrast, aminoguanidine reversed the down-regulation of nNOS gene expression during endotoxemia (Table b in the supplementary material 1).
LPS+ treatment affects ABC drug transporter expression in the kidney
The mRNA expression of 20 renal ABC transporters was profiled. Abcb1a/P-gp, Abcb1b/P-gp, Abcc2/Mrp2 and Abcc4/Mrp4 expression was determined with the ABI PRISM 7700 single reporter sequence detection system. In rodents, Abcb1/P-gp exists as two functional isoforms, which are encoded by two different genes: Abcb1a and Abcb1b [32]. The other ABC transporters were assessed with the ABI/PRISM 7900HT Gene Expression Micro Fluidic Card (Table 2). Their expression was normalized for the average CT value for GAPDH, as described for iNOS. Other housekeeping genes, like glucuronidase and hypoxanthine guanine phosphoribosyl transferase 1, gave comparable values to GAPDH (data not shown). Abcb1b/Pg-p and Abcc2/Mrp2 were clearly up-regulated 3 h after LPS+ and were still elevated 12 and 24 h later (Fig. 5b,c). Furthermore, Abcb1a/P-gp and Abcc4/Mrp4 were slightly, but not significantly, increased 12 h after LPS+ treatment (Fig. 5a,d). Table 2 presents a list of the expression of other transporter genes during endotoxemia. Abca1 showed a slight up-regulation 3 h after LPS+, but returned to basal levels within 12 h. Furthermore, Abcb11 was up-regulated, but only significantly after 48 h. A down-regulation was observed for Abcb4 and Abcc8 (at least a 2 times lower expression). The expression of the Abcg genes was not affected by endotoxemia.
Table 2.
Gene expression of efflux transporters during endotoxemia
| Gene symbol | Protein synonym | Assay IDs | Delta CT valuesa | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| LPS− (controls) | LPS+t = 3 h | LPS+t = 6 h | LPS+t = 12 h | LPS+t = 24 h | LPS+t = 48 h | LPS+ + amino t = 6 h | LPS+ + amino t = 12 h | |||
| Abca1 | Abca 1 | Rn00710172_m1 | 7.1 ± 0.31 | 5.1 ± 0.16 | 5.8 ± 0.22 | 7.3 ± 0.55 | 7.3 ± 0.35 | 6.8 ± 0.23 | 6.1 ± 0.23 | 8.0 ± 0.12 |
| Abca2 | Abca 2 | Rn00577821_m1 | 3.5 ± 0.40 | 3.5 ± 0.35 | 4.1 ± 0.15 | 4.5 ± 0.38 | 4.3 ± 0.35 | 3.6 ± 0.06 | 4.4 ± 0.38 | 4.4 ± 0.12 |
| Abca5 | Abca 5 | Rn00597824_m1 | 9.8 ± 0.06 | 9.8 ± 0.35 | 9.8 ± 0.10 | 9.5 ± 0.12 | 10.0 ± 0.23 | 10.2 ± 0.21 | 9.5 ± 0.25 | 9.7 ± 0.35 |
| Abcb1a | P_gp | Rn00591394_m1 | 8.4 ± 0.01 | Fig. 5a | ||||||
| Abcb1b | P_gp | Rn00561753_m1 | 10.8 ± 0.04 | Fig. 5b | ||||||
| Abcb4 | Abcb 4 | Rn00562185_m1 | 10.7 ± 0.62 | 12.3 ± 0.16 | 12.5 ± 0.21 | 12.8 ± 0.15 | 10.9 ± 0.15 | 7.3 ± 0.42 | 13.2 ± 0.46 | 12.4 ± 0.24 |
| Abcb6 | Abcb 6 | Rn00589801_m1 | 5.5 ± 0.35 | 5.9 ± 0.26 | 6.4 ± 0.25 | 6.5 ± 0.52 | 5.9 ± 0.38 | 5.4 ± 0.14 | 6.4 ± 0.25 | 6.2 ± 0.32 |
| Abcb9 | Abcb 9 | Rn00573757_m1 | 8.1 ± 0.20 | 8.5 ± 0.38 | 9.3 ± 0.29 | 8.8 ± 0.17 | 8.6 ± 0.10 | 8.7 ± 0.17 | 9.2 ± 0.42 | 8.9 ± 0.15 |
| Abcb11 | Bsep | Rn00582179_m1 | 10.5 ± 0.40 | 8.6 ± 0.37 | 10.3 ± 0.24 | 11.9 ± 0.21 | 8.7 ± 0.17 | 5.4 ± 0.49 | 9.9 ± 0.21 | 9.7 ± 0.21 |
| Abcc1 | Mrp1 | Rn00574093_m1 | 5.9 ± 0.25 | 6.2 ± 0.26 | 6.1 ± 0.25 | 6.1 ± 0.25 | 6.3 ± 0.21 | 5.9 ± 0.23 | 6.4 ± 0.40 | 6.2 ± 0.15 |
| Abcc2 | Mrp2 | Rn00563231_m1 | 6.7 ± 0.31 | Fig. 5c | ||||||
| Abcc3 | Mrp3 | Rn00589786_m1 | 8.4 ± 0.15 | 8.7 ± 0.12 | 8.8 ± 0.508 | 8.6 ± 0.06 | 8.0 ± 0.35 | 8.2 ± 0.12 | 8.8 ± 0.32 | 8.7 ± 0.23 |
| Abcc4 | Mrp4 | Rn01465699_q1 | 6.6 ± 0.06 | Fig. 5d | ||||||
| Abcc5 | Mrp5 | Rn00588341_m1 | 7.1 ± 0.01 | 7.9 ± 0.03 | 7.6 ± 0.15 | 6.4 ± 0.06 | 6.5 ± 0.50 | 6.5 ± 0.38 | 7.6 ± 0.45 | 6.4 ± 0.36 |
| Abcc6 | Mrp6 | Rn00578778_m1 | 7.0 ± 0.14 | 7.0 ± 0.01 | 8.4 ± 0.21 | 8.4 ± 0.12 | 7.8 ± 0.05 | 6.8 ± 0.35 | 8.6 ± 0.07 | 8.3 ± 0.14 |
| Abcc8 | Sur1 | Rn00564778_m1 | 11.5 ± 0.40 | 11.4 ± 0.21 | 11.4 ± 0.26 | 13.4 ± 0.15 | 14.8 ± 0.15 | 13.2 ± 0.25 | 11.7 ± 0.23 | 13.7 ± 0.31 |
| Abcc9 | Sur2 | Rn00564842_m1 | 6.8 ± 0.01 | 7.7 ± 0.26 | 8.4 ± 0.31 | 8.1 ± 0.15 | 7.2 ± 0.32 | 6.8 ± 0.40 | 8.0 ± 0.35 | 8.2 ± 0.32 |
| Abcg1 | Abcg1 | Rn00585262_m1 | 6.8 ± 0.15 | 7.8 ± 0.25 | 6.8 ± 0.25 | 7.1 ± 0.40 | 6.8 ± 0.31 | 6.7 ± 0.21 | 6.4 ± 0.31 | 7.3 ± 0.36 |
| Abcg2 | Abcg2 | Rn00710585_m1 | 4.1 ± 0.24 | 4.5 ± 0.07 | 4.6 ± 0.21 | 5.0 ± 0.06 | 4.5 ± 0.14 | 4.5 ± 0.07 | 4.9 ± 0.42 | 5.2 ± 0.07 |
| Abcg5 | Abcg5 | Rn00587092_m1 | 18.8 ± 0.29 | b | ||||||
aData of nine pooled samples, measured in triplicate, are expressed as mean ± SD
bLow expressed transport protein: CT value is not considered as valid
Fig. 5.
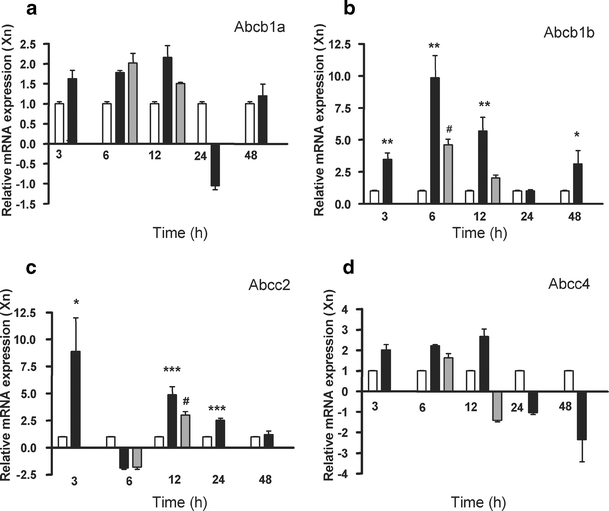
mRNA expression of different renal ABC transporters. Gene expression of Abcb1a/P-gp (a) and Abcb1b/P-gp (b), Abcc2/Mrp2 (c), and Abcc4/Mrp4 (d) were determined in rat kidney at different time points after LPS− (open bars, n = 3) or LPS+ (closed bars, n = 6) treatment or LPS+ together with aminoguanidine (gray bars, n = 6). ΔCT values for the LPS−, given in Table 2, were set to 1. Data are expressed as mean±SE. Significantly different compared to the LPS− (single asterisks P < 0.05, double asterisks P < 0.01, triple asterisks P < 0.001) or LPS+ (sharp signs P < 0.05)
Coadministration with aminoguanidine blocked the induction in mRNA expression of both Abcb1b/P-gp and Abcc2/Mrp2 at 6 and 12 h, respectively (Fig. 5b,c). Furthermore, aminoguanidine reduced the increased protein expression of both transporters 12 h after LPS+ treatment (Fig. 6b,d), indicating a regulatory role for NO in the expression of these efflux transporters. Abcc4/Mrp4 protein levels, on the other hand, decreased even more after treatment with both LPS+ and aminoguanidine. Gene expression levels of other transporters were not affected by aminoguanidine (Table 2).
Fig. 6.
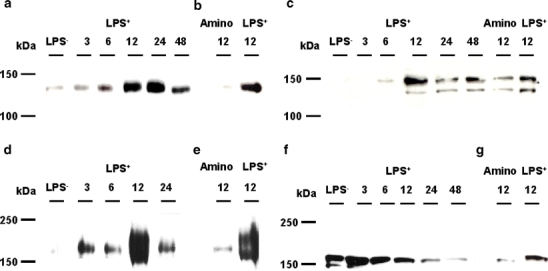
Protein expression of different renal ABC transporters. Total membrane fractions of rat kidney were isolated and expression of Abcb1/P-gp (a, b), Abcb11/Bsep (c), Abcc2/Mrp2 (d, e), and Abcc4/Mrp4 (f, g) was determined by Western blotting. Representative images (n = 4) show maximal Abcb1/P-gp protein expression 12 and 24 h after LPS+ (a), and coadministration with aminoguanidine reversed this induction (b). The protein expression of Abcb11/Bsep was up-regulated 12 h after LPS+ treatment, which was reversed by aminoguanidine (c). Maximal Abcc2/Mrp2 protein expression was seen 12 h after LPS+ (d) and aminoguanidine reversed this induction (e). Abcc4/Mrp4 protein expression was down-regulated during endotoxemia (f), which was intensified by aminoguanidine (g)
The expression patterns of Abcb1/P-gp, Abcb11, Abcc2/Mrp2, and Abcc4/Mrp4 were investigated further. Using total membrane fractions, we found an up-regulation of Abcb1/P-gp 6 h after LPS+, with maximal protein expression 12 and 24 h after LPS+ (Fig. 6a). Abcb11 is the major liver canalicular bile salt export pump (Bsep), but surprisingly, this transporter is also expressed at the apical membrane of mice renal proximal tubule, as we discovered recently [24]. In agreement with an increased mRNA expression, Abcb11/Bsep protein was up-regulated after LPS+ (Fig. 6c). Although we are not completely certain whether the C219 antibody against Abcb1/P-gp also detects Abcb11/Bsep [33], our Western blot results showed expression of Abcb1/P-gp, with a molecular weight of 130 kDa, and it does not seem to cross-react with Bsep at 160 kDa.
By using immunohistochemistry, the expression and localization of Abcb11/Bsep was investigated in endotoxemic rat kidneys. In agreement with mouse kidney, positive staining was observed in the apical membranes of the proximal tubule 12 h after LPS+ treatment (Fig. 7). In addition, we observed staining in rat glomerular endothelium and peritubular capillaries.
Fig. 7.
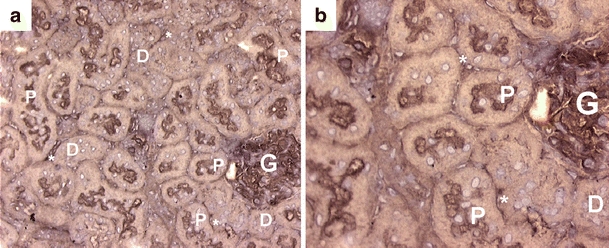
Distribution of Abcb11/Bsep in rat kidney cortex. a, b Representative immunohistochemical images show apical staining in proximal tubules (indicated by arrows). In addition, glomerular and peritubular staining of Bsep in the cortex was found. G glomerulus, asterisks peritubular staining, P proximal tubule, D distal tubule. Original magnification in a ×200, and in b ×400
Furthermore, a clear increase in Abcc2/Mrp2 protein was observed after LPS+ (Fig. 6d), but a down-regulation in Abcc4/Mrp4 was found (Fig. 6f). We measured the function of Abcc2/Mrp2 by monitoring the urinary excretion of calcein in an isolated perfused rat kidney 12 h after LPS+ or LPS−. A 16 ± 3% increase in calcein excretion was observed after LPS+, as shown in Fig. 8. We do not expect calcein to be a Bsep substrate because Mrp2-deficient (TR−) rats clearly showed that calcein excretion was highly reduced [25], despite the presence of Bsep in these strains (unpublished data). Twenty-four hours after LPS+, Abcc2/Mrp2 activity was still elevated, although not significantly. In accordance, endotoxemia affected renal function as observed by diminished GFR and fractional reabsorption of water in isolated perfused rat kidneys (Table 3).
Fig. 8.
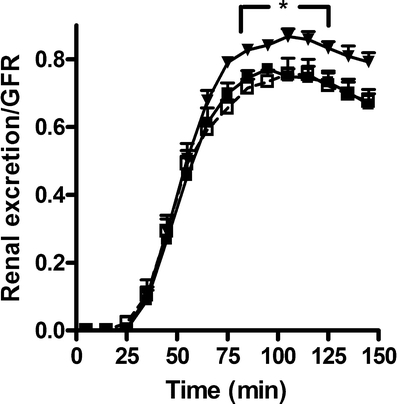
Renal calcein excretion in isolated perfused rat kidneys. Secretion of the fluorescent calcein into the urine was measured after 12 h in untreated (closed squares control; n = 4), LPS− (open squaresn = 4), or LPS+ (trianglesn = 6) rats. Urinary excretion was corrected for GFR. Significantly different compared to the LPS− (asterisk P < 0.05)
Table 3.
Functional parameters of isolated perfused rat kidneys in control rats (no additional treatment) and after exposure to LPS− or LPS+ for 12 h
| Parameter | Controls (n = 4) | LPS− (n = 4) | LPS+ (n = 6) |
|---|---|---|---|
| GFR (μl/min) | 350 ± 25 | 380 ± 17 | 290 ± 20a |
| Diuresis (μl/min) | 18.6 ± 1.8 | 21.1 ± 1.3 | 17.4 ± 1.0 |
| FRwater (%) | 94.7 ± 0.2 | 94.5 ± 0.3 | 92.7 ± 0.6a |
| RPP (mmHg)b | 88.4 ± 1.5 | 84.5 ± 1.7 | 88.7 ± 1.0 |
Data are expressed as mean ± SE of four to six perfused kidney experiments over the period 25-115 min. Untreated rats served as controls for this model. Kidneys were perfused for 150 min as described in the “Materials and methods” section
aSignificantly different from perfused control kidney (P < 0.05)
bConstant flow during the experiments was 18 ± 0.5 ml/min
FR fractional reabsorption, RPP renal perfusion pressure
Discussion
In the present study, we investigated the role of NO, produced by iNOS, in the differential expression of renal ABC transporters in rats in vivo. A clear up-regulation of Abca1, Abcb1/P-gp, Abcb11/Bsep, and Abcc2/Mrp2 expression was found after LPS+. In addition, we observed a functional up-regulation of Abcc2/Mrp2 in perfused kidneys. This supports a detoxifying role of the efflux pump, which is in accordance with the up-regulation of Abcc2/Mrp2 protein in killifish renal tubules [34, 35], in rat kidney, and in MDCKII cells overexpressing human ABCC2/MRP2 after long-term exposure to the nephrotoxicant gentamicin [20], and of abcb1/P-gp in mouse kidneys after ischemic reperfusion injury [24]. Indeed, blocking Abcb1/P-gp during endotoxemia leads to further renal damage (Heemskerk, Wouterse, Russel, and Masereeuw, preliminary findings). The renal drug transporters are likely to be under direct influence of NO produced by iNOS, as coadministration of the iNOS inhibitor, aminoguanidine, attenuated the LPS-induced effects on transporter expression. Aminoguanidine treatment results in decreased NO production and, subsequently, reduced oxidative stress by decelerating the formation of peroxynitrite. Under these conditions, less toxic metabolites may be produced, explaining the restoration in transporter expression and reversal of proximal tubular damage.
In contrast to our findings, Cherrington et al. [36] reported that coadministration of aminoguanidine failed to change the expression of different hepatic drug transporters during endotoxemia. This discrepancy might be due to organ differences and the fact that Cherrington et al. pretreated the animals with the NOS inhibitor, whereas we gave aminoguanidine 1 h after LPS+. The lack of an effect of aminoguanidine on the expression of different biotransformation enzymes, enzymes involved in the iNOS pathway and during oxidative stress, supports a direct function of NO itself in ABC transporter regulation.
The role of two up-regulated efflux pumps, Abca1 and Abcb11/Bsep, in the kidney is unknown. In humans, ABCA1 is widely expressed and found to be the causative gene in Tangier disease, a disorder of cholesterol transport between tissues and the liver, mediated by binding of the cholesterol onto high-density lipoprotein particles [37]. ABCA1 controls the extrusion of membrane phospholipid and cholesterol toward specific extracellular acceptors; however, the exact role of the protein in this process is not known. Moreover, its expression and function in the kidney requires further investigation. The presence of Abcb11/Bsep, the major canalicular bile salt export pump, was recently demonstrated by us in mouse kidney [24], and here we show a clear apical expression in rat kidney proximal tubules for the first time as well. Hence, the function of Abcb11/Bsep in the kidney still needs to be determined. In addition to their up-regulation in the kidney, we found a down-regulation of Abcb1/P-gp, Abcc2/Mrp2, and Abcb11/Bsep in the liver (data not shown). This is in agreement with previous findings for the hepatic expression of the transporters during inflammation [36, 38–40], obstructive jaundice [41], and cholestasis [42]. An induction of the transporters in the kidney during liver failure may function as a compensatory elimination mechanism to reduce enhanced levels of circulating bile salts caused by endotoxin-induced cholestasis [39] and/or to protect proximal tubular cells against the higher exposure via primary urine.
A differential regulation was found for Abcc4. Although this transporter is expressed in the same nephron segments as Abcb1/P-gp and Abcc2/Mrp2, the apically expressed efflux pump clearly has a distinct regulation. In killifish, we previously found renal proximal tubules exposed to nephrotoxicants or ET-1 that Abcc2/Mrp2-mediated transport and Mrp2 expression in the luminal membrane were increased after 24 h [34], whereas the expression of Abcc4/Mrp4 remained unchanged [35]. Furthermore, Abcc4/Mrp4 protein levels in rat kidney were decreased during cholestasis in contrast to the Abcc2/Mrp2 levels [43]. No changes in Abcc4/Mrp4 mRNA were found, suggesting a posttranscriptional regulation of transporter expression, including phosphorylation and protein routing. A down-regulation of ABC transporters, as observed for Abcc8/Sur1 and initially also for Abcb4, may be explained by the different functions of these ABC members in the kidney.
LPS itself may affect transporter expression after binding to the Toll-like receptor 4 and a second, not yet identified, factor through activation of NOS-dependent pathways, as was found for Abcb1/P-gp in rat brain capillaries [44]. Furthermore, systemic TNF-α, released during endotoxemia, may regulate efflux pumps, including ABC transporters [45, 46]. This cytokine might act through NOS activation [44], but also through a pathway that does not involve iNOS-derived NO during endotoxin-associated renal failure [47]. Furthermore, regulation of iNOS after exposure to LPS occurs at the level of gene transcription and may require activation of the NF-κB pathway [48]. It has been shown previously that an up-regulation of Abcb1/P-gp in renal failure, i.e., after ischemic–reperfusion damage [24] or exposure to cadmium [49], might be signaled through an NF-κB-dependent pathway. Consistent with these findings, we observed a clear induction (sevenfold) of NF-κB expression 3 h after LPS+ as well.
Selective inhibition of renal iNOS has important implications for the treatment of septic acute kidney disease. Changes in the expression levels of renal drug transporters alters the pharmacokinetics of many clinically important drugs [50] and has implications for the prediction of overall drug disposition during sepsis. For example, glucocorticoids are substrates for Abcb1/Pg-p and are often prescribed for their anti-inflammatory properties in septic patients. An up-regulation of Abcb1/Pg-p, as observed in our study, might have consequences for therapeutic levels of these substrates [51].
In conclusion, after LPS+, a clear up-regulation in Abca1, Abcb1/P-gp, Abcb11/Bsep, and Abcc2/Mrp2 was found, whereas Abcc8/Sur1 was down-regulated. Abcb4 and Abcc4/Mrp4 show a differential pattern during the course of endotoxemia. The renal drug transporters are likely to be under direct influence of NO produced by iNOS, as coadministration of the iNOS inhibitor, aminoguanidine, attenuated the LPS-induced effects on their expression. Apparently, an up-regulation of important efflux pumps diminishes the renal accumulation of toxic compounds and attenuates further proximal tubular damage during endotoxemia.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Jeroen J.M.W. van den Heuvel for his help with the RQ-PCR experiments. Furthermore, we thank Dr. Han Moshage (University Hospital Groningen, The Netherlands) for providing us with the antibody against iNOS and Dr. Bruno Stieger (University Hospital Zürich, Switzerland) for the antibody against Bsep. S. Heemskerk was supported by a grant from The Netherlands Organisation for Scientific Research (NWO).
Abbreviations
- ABC
ATP-binding cassette
- AKI
acute kidney injury
- BSA
bovine serum albumin
- Bsep
bile salt export pump
- BUN
blood urea nitrogen
- calcein-AM
calcein-acetoxymethylester
- ET
endothelin
- FENa
fractional sodium excretion
- FR
fractional reabsorption
- GFR
glomerular filtration rate
- HBSS
Hank’s balanced salt solution
- IL
interleukin
- LPS
lipopolysaccharide
- M-MLV
Moloney murine leukemia virus
- Mrp
multidrug resistance protein
- NF-κB
nuclear factor kappa B
- NOx
NO metabolites
- NOS
nitric oxide synthase
- P-gp
P-glycoprotein
- RPP
renal perfusion pressure
- RQ-PCR
quantitative real-time polymerase chain reaction
- TBS
Tris-buffered saline
- TNF-α
tumor necrosis factor-α
Footnotes
Electronic supplementary material
The online version of this article (doi: 10.1007/s00424-007-0210-x) contains supplementary material, which is available to authorized users.
References
- 1.Ortiz PA, Garvin JL (2002) Role of nitric oxide in the regulation of nephron transport. Am J Physiol Renal Physiol 282:F777–F784 [DOI] [PubMed]
- 2.Xie Q, Nathan C (1994) The high-output nitric oxide pathway: role and regulation. J Leukoc Biol 56:576–582 [DOI] [PubMed]
- 3.Goligorsky MS, Brodsky SV, Noiri E (2004) NO bioavailability, endothelial dysfunction, and acute renal failure: new insights into pathophysiology. Semin Nephrol 24:316–323 [DOI] [PubMed]
- 4.Noiri E, Nakao A, Uchida K, Tsukahara H, Ohno M, Fujita T, Brodsky S, Goligorsky MS (2001) Oxidative and nitrosative stress in acute renal ischemia. Am J Physiol Renal Physiol 281:F948–F957 [DOI] [PubMed]
- 5.Ortiz PA, Garvin JL (2003) Cardiovascular and renal control in NOS-deficient mouse models. Am J Physiol Regul Integr Comp Physiol 284:R628–R638 [DOI] [PubMed]
- 6.Schwartz D, Mendonca M, Schwartz I, Xia Y, Satriano J, Wilson CB, Blantz RC (1997) Inhibition of constitutive nitric oxide synthase (NOS) by nitric oxide generated by inducible NOS after lipopolysaccharide administration provokes renal dysfunction in rats. J Clin Invest 100:439–448 [DOI] [PMC free article] [PubMed]
- 7.Szabo C, Southan GJ, Thiemermann C (1994) Beneficial effects and improved survival in rodent models of septic shock with S-methylisothiourea sulfate, a potent and selective inhibitor of inducible nitric oxide synthase. Proc Natl Acad Sci USA 91:12472–12476 [DOI] [PMC free article] [PubMed]
- 8.Yoshida M, Akaike T, Wada Y, Sato K, Ikeda K, Ueda S, Maeda H (1994) Therapeutic effects of imidazolineoxyl N-oxide against endotoxin shock through its direct nitric oxide-scavenging activity. Biochem Biophys Res Commun 202:923–930 [DOI] [PubMed]
- 9.Ling H, Edelstein C, Gengaro P, Meng X, Lucia S, Knotek M, Wangsiripaisan A, Shi Y, Schrier R (1999) Attenuation of renal ischemia–reperfusion injury in inducible nitric oxide synthase knockout mice. Am J Physiol 277:F383–F390 [DOI] [PubMed]
- 10.Noiri E, Peresleni T, Miller F, Goligorsky MS (1996) In vivo targeting of inducible NO synthase with oligodeoxynucleotides protects rat kidney against ischemia. J Clin Invest 97:2377–2383 [DOI] [PMC free article] [PubMed]
- 11.Notenboom S, Miller DS, Smits P, Russel FG, Masereeuw R (2002) Role of NO in endothelin-regulated drug transport in the renal proximal tubule. Am J Physiol Renal Physiol 282:F458–F464 [DOI] [PubMed]
- 12.Notenboom S, Miller DS, Smits P, Russel FG, Masereeuw R (2004) Involvement of guanylyl cyclase and cGMP in the regulation of Mrp2-mediated transport in the proximal tubule. Am J Physiol Renal Physiol 287:F33–F38 [DOI] [PubMed]
- 13.Hartz AM, Bauer B, Fricker G, Miller DS (2004) Rapid regulation of P-glycoprotein at the blood–brain barrier by endothelin-1. Mol Pharmacol 66:387–394 [DOI] [PubMed]
- 14.van de Water FM, Masereeuw R, Russel FG (2005) Function and regulation of multidrug resistance proteins (MRPs) in the renal elimination of organic anions. Drug Metab Rev 37:443–471 [DOI] [PubMed]
- 15.Russel FG, Masereeuw R, van Aubel RA (2002) Molecular aspects of renal anionic drug transport. Annu Rev Physiol 64:563–594 [DOI] [PubMed]
- 16.Zhou Y, Takahashi G, Yonamine K (2003) Down-regulation of inducible nitric oxide synthase (iNOS) in rat with congenital hydronephrosis. Int J Urol 10:536–543 [DOI] [PubMed]
- 17.Traylor LA, Mayeux PR (1997) Nitric oxide generation mediates lipid A-induced oxidant injury in renal proximal tubules. Arch Biochem Biophys 338:129–135 [DOI] [PubMed]
- 18.Mircheff AK, Wright EM (1976) Analytical isolation of plasma membranes of intestinal epithelial cells: identification of Na, K-ATPase rich membranes and the distribution of enzyme activities. J Membr Biol 28:309–333 [DOI] [PubMed]
- 19.Heemskerk S, Pickkers P, Bouw MP, Draisma A, van der Hoeven JG, Peters WH, Smits P, Russel FG, Masereeuw R (2006) Up-regulation of renal iNOS during human endotoxemia and sepsis is associated with proximal tubule injury. Clin J Am Soc Nephrol 1:853–862 [DOI] [PubMed]
- 20.Notenboom S, Wouterse AC, Peters B, Kuik LH, Heemskerk S, Russel FG, Masereeuw R (2006) Increased apical insertion of the multidrug resistance protein 2 (MRP2/ABCC2) in renal proximal tubules following gentamicin exposure. J Pharmacol Exp Ther 318:1194–1202 [DOI] [PubMed]
- 21.Vos TA, Gouw AS, Klok PA, Havinga R, van Goor H, Huitema S, Roelofsen H, Kuipers F, Jansen PL, Moshage H (1997) Differential effects of nitric oxide synthase inhibitors on endotoxin-induced liver damage in rats. Gastroenterology 113:1323–1333 [DOI] [PubMed]
- 22.van Aubel RA, Smeets PH, Peters JG, Bindels RJ, Russel FG (2002) The MRP4/ABCC4 gene encodes a novel apical organic anion transporter in human kidney proximal tubules: putative efflux pump for urinary cAMP and cGMP. J Am Soc Nephrol 13:595–603 [DOI] [PubMed]
- 23.Moshage H, Kok B, Huizenga JR, Jansen PL (1995) Nitrite and nitrate determinations in plasma: a critical evaluation. Clin Chem 41:892–896 [PubMed]
- 24.Huls M, van den Heuvel JJ, Dijkman HB, Russel FG, Masereeuw R (2006) ABC transporter expression profiling after ischemic reperfusion injury in mouse kidney. Kidney Int 69:2186–2193 [DOI] [PubMed]
- 25.Masereeuw R, Notenboom S, Smeets PH, Wouterse AC, Russel FG (2003) Impaired renal secretion of substrates for the multidrug resistance protein 2 in mutant transport-deficient (TR−) rats. J Am Soc Nephrol 14:2741–2749 [DOI] [PubMed]
- 26.Beckman JS, Koppenol WH (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am J Physiol 271:C1424–C1437 [DOI] [PubMed]
- 27.Vaidya VS, Ramirez V, Ichimura T, Bobadilla NA, Bonventre JV (2006) Urinary kidney injury molecule-1: a sensitive quantitative biomarker for early detection of kidney tubular injury. Am J Physiol Renal Physiol 290:F517–F529 [DOI] [PubMed]
- 28.Shelby MK, Cherrington NJ, Vansell NR, Klaassen CD (2003) Tissue mRNA expression of the rat UDP-glucuronosyltransferase gene family. Drug Metab Dispos 31:326–333 [DOI] [PubMed]
- 29.Wang W, Jittikanont S, Falk SA, Li P, Feng L, Gengaro PE, Poole BD, Bowler RP, Day BJ, Crapo JD, Schrier RW (2003) Interaction among nitric oxide, reactive oxygen species, and antioxidants during endotoxemia-related acute renal failure. Am J Physiol Renal Physiol 284:F532–F537 [DOI] [PubMed]
- 30.Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A (2004) The story so far: molecular regulation of the heme oxygenase-1 gene in renal injury. Am J Physiol Renal Physiol 286:F425–F441 [DOI] [PubMed]
- 31.Masereeuw R, Terlouw SA, van Aubel RA, Russel FG, Miller DS (2000) Endothelin B receptor-mediated regulation of ATP-driven drug secretion in renal proximal tubule. Mol Pharmacol 57:59–67 [PubMed]
- 32.Croop JM, Raymond M, Haber D, Devault A, Arceci RJ, Gros P, Housman DE (1989) The three mouse multidrug resistance (mdr) genes are expressed in a tissue-specific manner in normal mouse tissues. Mol Cell Biol 9:1346–1350 [DOI] [PMC free article] [PubMed]
- 33.Vos TA, Ros JE, Havinga R, Moshage H, Kuipers F, Jansen PL, Muller M (1999) Regulation of hepatic transport systems involved in bile secretion during liver regeneration in rats. Hepatology 29:1833–1839 [DOI] [PubMed]
- 34.Terlouw SA, Graeff C, Smeets PH, Fricker G, Russel FG, Masereeuw R, Miller DS (2002) Short- and long-term influences of heavy metals on anionic drug efflux from renal proximal tubule. J Pharmacol Exp Ther 301:578–585 [DOI] [PubMed]
- 35.Notenboom S, Miller DS, Kuik LH, Smits P, Russel FG, Masereeuw R (2005) Short-term exposure of renal proximal tubules to gentamicin increases long-term multidrug resistance protein 2 (Abcc2) transport function and reduces nephrotoxicant sensitivity. J Pharmacol Exp Ther 315:912–920 [DOI] [PubMed]
- 36.Cherrington NJ, Slitt AL, Li N, Klaassen CD (2004) Lipopolysaccharide-mediated regulation of hepatic transporter mRNA levels in rats. Drug Metab Dispos 32:734–741 [DOI] [PubMed]
- 37.Brunham LR, Singaraja RR, Hayden MR (2006) Variations on a gene: rare and common variants in ABCA1 and their impact on HDL cholesterol levels and atherosclerosis. Annu Rev Nutr 26:105–129 [DOI] [PubMed]
- 38.Vos TA, Hooiveld GJ, Koning H, Childs S, Meijer DK, Moshage H, Jansen PL, Muller M (1998) Up-regulation of the multidrug resistance genes, Mrp1 and Mdr1b, and down-regulation of the organic anion transporter, Mrp2, and the bile salt transporter, Spgp, in endotoxemic rat liver. Hepatology 28:1637–1644 [DOI] [PubMed]
- 39.Hartmann G, Vassileva V, Piquette-Miller M (2005) Impact of endotoxin-induced changes in P-glycoprotein expression on disposition of doxorubicin in mice. Drug Metab Dispos 33:820–828 [DOI] [PubMed]
- 40.Teng S, Piquette-Miller M (2005) The involvement of the pregnane X receptor in hepatic gene regulation during inflammation in mice. J Pharmacol Exp Ther 312:841–848 [DOI] [PubMed]
- 41.Tanaka Y, Kobayashi Y, Gabazza EC, Higuchi K, Kamisako T, Kuroda M, Takeuchi K, Iwasa M, Kaito M, Adachi Y (2002) Increased renal expression of bilirubin glucuronide transporters in a rat model of obstructive jaundice. Am J Physiol Gastrointest Liver Physiol 282:G656–G662 [DOI] [PubMed]
- 42.Lee J, Azzaroli F, Wang L, Soroka CJ, Gigliozzi A, Setchell KD, Kramer W, Boyer JL (2001) Adaptive regulation of bile salt transporters in kidney and liver in obstructive cholestasis in the rat. Gastroenterology 121:1473–1484 [DOI] [PubMed]
- 43.Denk GU, Soroka CJ, Takeyama Y, Chen WS, Schuetz JD, Boyer JL (2004) Multidrug resistance-associated protein 4 is up-regulated in liver but down-regulated in kidney in obstructive cholestasis in the rat. J Hepatol 40:585–591 [DOI] [PubMed]
- 44.Hartz AM, Bauer B, Fricker G, Miller DS (2006) Rapid modulation of P-glycoprotein-mediated transport at the blood–brain barrier by tumor necrosis factor-alpha and lipopolysaccharide. Mol Pharmacol 69:462–470 [DOI] [PubMed]
- 45.Jiang Q, Matsuzaki Y, Li K, Uitto J (2006) Transcriptional regulation and characterization of the promoter region of the human ABCC6 gene. J Invest Dermatol 126:325–335 [DOI] [PubMed]
- 46.Lee G, Piquette-Miller M (2003) Cytokines alter the expression and activity of the multidrug resistance transporters in human hepatoma cell lines; analysis using RT-PCR and cDNA microarrays. J Pharm Sci 92:2152–2163 [DOI] [PubMed]
- 47.Knotek M, Rogachev B, Wang W, Ecder T, Melnikov V, Gengaro PE, Esson M, Edelstein CL, Dinarello CA, Schrier RW (2001) Endotoxemic renal failure in mice: role of tumor necrosis factor independent of inducible nitric oxide synthase. Kidney Int 59:2243–2249 [DOI] [PubMed]
- 48.Xie QW, Kashiwabara Y, Nathan C (1994) Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J Biol Chem 269:4705–4708 [PubMed]
- 49.Thevenod F, Friedmann JM, Katsen AD, Hauser IA (2000) Up-regulation of multidrug resistance P-glycoprotein via nuclear factor-kappaB activation protects kidney proximal tubule cells from cadmium- and reactive oxygen species-induced apoptosis. J Biol Chem 275:1887–1896 [DOI] [PubMed]
- 50.Sun H, Frassetto L, Benet LZ (2006) Effects of renal failure on drug transport and metabolism. Pharmacol Ther 109:1–11 [DOI] [PubMed]
- 51.Huang ZH, Murakami T, Okochi A, Yumoyo R, Nagai J, Takano M (2001) Expression and function of P-glycoprotein in rats with carbon tetrachloride-induced acute hepatic failure. J Pharm Pharmacol 53:873–881 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Below is the link to the electronic supplementary material.