

Abstract
In patients with malignancies and immune disorders expressing Tac (α chain of the interleukin 2 receptor; CD25), physiologic shedding of this receptor may lead to high blood levels of the soluble form (sTac). This system was used to model the interaction of soluble antigen with antibody in therapeutic settings and to develop rational principles to optimize the delivery of antibody to tumor target cells. First, we confirmed that sTac in vivo can block anti-Tac binding sites and diminish antibody binding to Tac+ cells. Second, the bioactivity of antibody in vivo correlated directly with the amount of antibody infused and inversely with the sTac concentration. Third, bindability of antibody declined in the hours and days after anti-Tac infusion in patients. Finally, tumor targeting was achieved even in the presence of excess sTac, demonstrating a partition of antibody between soluble and cell-bound antigen. A role is proposed for the Brambell receptor (FcRB) to delay saturation of human or chimeric antibodies via differential catabolism of antigen–antibody complexes. Principles are developed for predicting activity of administered antibody in the presence of soluble antigen to assist in dose selection in passive, radioimmuno and immunotoxin therapies.
Antigenemia occurs with several membrane-bound, tumor-associated antigens: e.g., CEA (1) and TAG-72 (2) in gastrointestinal cancers, Her2/neu in breast carcinoma (3), CA125 in ovarian carcinoma (4), ganglioside in neuroblastoma (5), antibody idiotypes in B cell lymphomas (6), and others. The prototypical example of such shed molecules is the α chain of the interleukin 2 receptor (IL-2Rα; Tac; ref. 7). Tac is up-regulated markedly during normal T cell activation and in several immune disorders and malignant states (8). Accompanying this up-regulation is a concomitant active shedding of Tac from the membrane surface (7, 9, 10). In normal physiology, this shedding of receptors is thought to mediate rapid down-regulation of activated T cells when the activating stimulus is removed. In Tac+ malignancies, however, the constitutive high expression of antigen and shedding leads to accumulations of soluble antigen (sTac) in plasma up to 1,000 times the normal levels (7).
Since 1985, we have treated >100 individuals with antibody to Tac (anti-Tac), both as unmodified antibody and as a carrier for isotopes or toxins. As a consequence of associated studies, we became aware that surface Tac antigen occasionally was unsaturated in some patients despite adequate plasma antibody concentrations, and this failure to saturate cellular Tac receptors appeared to correlate with high plasma levels of sTac. This system thus appeared to be a suitable model for a more quantitative study of the interaction of soluble antigen with antibody in therapeutic settings and with which to develop rational principles to optimize the delivery of antibody to tumor targets in vivo.
MATERIALS AND METHODS
Patients and Treatment.
Patients were enrolled according to eligibility criteria that included Tac-expressing malignancy (11, 12). Anti-Tac, a murine IgG2a,κ mAb, was administered by 2-hr infusion. 1B4M-DTPA-anti-Tac (13) (≈1 mg) was labeled with 111In and/or 90Y, to which unmodified anti-Tac was added for doses >1 mg.
Antibody Bindability (Bioactivity).
Radiolabeled anti-Tac (500–2,000 cpm) was added to 10–20 × 106 paraformaldehyde-fixed Tac+ HuT102 cells (antigen excess) at 4°C for 30 min as described (13). The fraction of radioactivity in the cell pellet was taken as the antibody “bindability.”
Antibody Bindability Analysis.
(The derivations are available from the authors.) Four chemical forms are recognized based on a bivalent antibody: A, AB, AB2, and B, in which A is antibody and B is antigen. The body is treated as a single, unified compartment for antigen–antibody binding. See ref. 10 for elaboration of the joint catabolic model.
(i) The bindability β implies, for a given total moles of antibody, Atot, a corresponding total moles of antigen, Btot, assuming A and AB are bindable and AB2 is not:
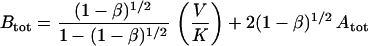 |
1 |
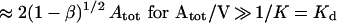 |
1a |
V is the distribution volume, and K is the monovalent Fab affinity, 5 × 109/M for anti-Tac (14). In text, Aβ (e.g., A50) indicates the dose in moles of Atot that yields β% bindability.
(ii) The kinetics of loss of bindability after infusion is modeled as β vs. t, the time to achieve the ratio α = Btot/Atot corresponding to a given bindability fraction. For the typical antibody dose where A0 ≫ Kd, α ≈ 1.41 for β = 50% and α ≈ 1.90 for β = 10% (Eq. 1), and the duration of bindability is specified by:
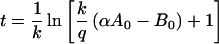 |
2 |
k = ln2/t1/2 is the antibody beta phase rate constant for IgG, which is undisturbed by antigen binding (10) and provisionally is assigned also as the clearance rate constant for antigen bound to antibody. A0 and B0 are moles of A and B at time = 0. q, the sTac production rate, is selected to provide the best fit of the data. Eq. 2 may be simplified via the relation q = cB0:
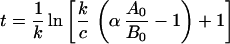 |
3 |
and for c ≫ k (t1/2antigen ≪ t1/2antibody)
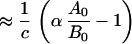 |
3a |
The antibody dose providing the bindability duration t is obtained by rearrangement of Eq. 3:
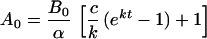 |
4 |
(iii) The rate of sTac production q can also be estimated from the descending slope of the bindability curve, dβ/dt, in the postinfusion period, which has the advantage of not depending on prior measurement of B0.
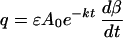 |
5 |
where ɛ = dα/dβ = d(Btot/Atot)/dβ is obtained by numerical differention of a rearrangement of Eq. 1. ɛ = −1.5 to −2.5 for β ≈ 20–80% for the range of doses in this application.
RESULTS
Clinical Intervention.
Thirteen patients were injected with a total of 28 doses of anti-Tac antibody labeled with 111In and/or 90Y. Eleven patients had HTLV-1-positive adult T cell leukemia, one had T prolymphocytic leukemia, and one had mycosis fungoides. All patients had circulating Tac+ malignant cells (595–60,843 per μl), and all patients had elevated sTac levels (>502 units/ml; range 2,113–230,391), excepting the mycosis fungoides patient (251 units/ml). Six patients had infusions of 1 mg and 50 mg at times 1 week apart, whereas all others had 10-mg infusions with a minimum of 6 weeks between doses. The details of the therapeutic interventions and responses have been reported in part elsewhere (11, 12).
Plasma beta phase half-lives ranged from 15 to 80 hr (mean 45 ± 17), a range that is typical for murine mAbs in humans. The fraction of circulating radioactivity associated with cell pellets was occasionally as high as 20–60% with 1-mg doses and high leukemic cell counts but was generally <5% for all higher doses or low cell counts. Overall, antibody binding to circulating Tac+ cells was a significant feature of the kinetics in only a minority of doses.
Antigenemia Reduces the Bioactivity of Infused Anti-Tac.
Antibody bioactivity is defined operationally as that portion of antibody that can bind to cell-associated antigen ex vivo (“bindability,” β) (see Materials and Methods). All preinfusion bindabilities were 70–95%, but marked in vivo bindability losses were encountered in several patients. A study was performed to determine whether bioactivity loss could result from binding by circulating antigen. Patient 1 had a high sTac level (230,391 units/ml). After infusion of a 1-mg dose of anti-Tac, circulating antibody persisted in vivo with a survival t1/2 of 30 hr (Fig. 1A). However, none of this antibody was bindable (Fig. 1B) in our standard 30-min assay at 4°C that minimizes dissociation of preformed antigen–antibody complexes while permitting binding of all nonsaturated antibody with the excess of coincubated Tac+ cells, as confirmed with control noninfused antibody. A parallel sample, incubated for 28 hr at 22°C, recovered 90% of the preinfusion bindability (Fig. 1B). This result is compatible with antigen-saturated but biologically intact antibody in circulation at the time of blood sampling that releases bound soluble antigen and subsequently binds Tac+ cells.
Figure 1.

Reversibility of sTac binding. (A) Pharmacokinetics of 111In anti-Tac (1 mg) in patient 1. Plasma radioactivity is shown for blood sampling during (−2 to 0 hr) and after infusion. (B) Bindability of 111In anti-Tac in patient plasma at time = 0 (solid bar) and of a control sample without injection (open bar). Conditions were designed to minimize (30′ 4°C) or favor (28HR 22°C) antigen–antibody exchange.
Next, we examined the relationship of antibody dose to bindability. The patient of Fig. 1 received a 50-mg dose 1 week after the 1-mg dose. Serial plasma samples obtained during the infusion showed a dose-dependent increase of antibody bioactivity, with the A50 level at ≈15 mg (antibody dose yielding 50% bioactivity). The bindable fraction began to plateau when ≈30 mg had been infused (Fig. 2A). [Note: This curve and following curves do not represent the mass of antibody in circulation but show instead the portion of that antibody in circulation that can bind to antigen-expressing tumor cells.] All antibody having one or two “arms” free is defined as 100% bioactive (A or AB in Materials and Methods); 0% bioactive means all antibody has both sites saturated (AB2); and 50% bioactive means one-half of all antibody has both sites saturated, in which we refer to antibody as “half neutralized,” and the remainder has one or two sites free for antigen binding. Full profile modeling showed good concordance between experimental data and theoretical curves (Fig. 2A, Left). This modeling also is instructive in revealing the narrowness of the transition between bindability and saturation.
Figure 2.

Bioactivity of anti-Tac in vivo. The patient of Fig. 1 received 50 mg of 111In anti-Tac over 2 hr. Bindability was assessed in blood samples at various times during (A) and after (B) the infusion. Data points are means of duplicate or triplicate samples as fraction of preinfusion bindability (0.91). Best fit of the theoretical curve in A (Eq. 1) was obtained with sTac (B0) = 130 nmol. In B, a second 50-mg dose of unlabeled anti-Tac was administered at 48 hr. The theoretical curves for bindability kinetics (Eq. 2) are based on a measured antibody beta t1/2 of 47 hr, B0 = 130 nmol, and an sTac production rate (q) of 13 nmol/hr. The following curve is fit with a total antibody mass of 75 mg after infusion 2; a new B0 (450 nmol) equal to the sTac bound to antibody before the second infusion (320 nmol) plus the same steady-state of free sTac that preceded the first dose (130 nmol); and q = 11 nmol/hr. Size exclusion chromatography profiles of patient sera: “Pre-” is 111In anti-Tac before infusion, eluting at ≈37 min; antibody saturated with sTac elutes at 33–35 min.
Antibody bioactivity was correlated with the HPLC profile of 111In anti-Tac during infusion (Fig. 2A, Right). In samples with low bindability, there was a shift in the size of antibody to a higher molecular weight position (15). The HPLC pattern returned to native position as the bindability increased with more antibody administered. This pattern (high molecular weight and low bindability) is once again compatible with the formation of complexes of antibody with antigen as the cause of low bioactivity.
An analysis of all patient doses showed a general, direct correlation of antibody bioactivity with quantity of antibody administered and an inverse correlation with sTac in the circulation. Fig. 3 relates the doses of anti-Tac to achieve 50% of initial bindability (A50) vs. sTac levels, determined as in Fig. 2A. A theoretical curve models the A50 based on the affinity of Tac for anti-Tac, the unit definition of sTac (3 pg or 0.13 fmol/unit), and an assumption of a whole body distribution volume for sTac of twice plasma volume (6 liters). Twice plasma volume roughly approximates distribution estimates of Tac and anti-Tac (10) and IgG and albumin (16) in murine models and of IgG (17) and albumin (18) in humans.¶
Figure 3.

Mass of antibody (A50) to achieve 50% bindability as a function of sTac level. ▪, A50 derived from patient antibody infusions, as in Fig. 2A. □, A50 estimated from Eq. 1 in which bindability at the end of infusion was <50% (from the left, 44%, 39%, and 23%). Curves predict antibody dose to achieve 50%, 90%, and 99% bindable antibody by Eq. 1 (e.g., for A50, Btot = 2.41V/K + 1.41A50). The dotted line is the predicted linear stoichiometric relation of A50 for Kd ≪ A0 (Btot = 1.41A50). Thirteen doses in eight patients allowed estimation of A50. The weight range for the study group was narrow [63 ± 7 kg (SD)], and corrections for weight and plasma volume improved the fit only slightly; hence, the uncorrected dose data are presented in the figure. Equivalent doses for patients significantly larger or smaller than 63 kg would be proportionately higher or lower.
For sTac > 9,000 units/ml (Fig. 3), there was a strong correlation between sTac and the dose of antibody required to achieve 50% bioactivity (A50) (r = 0.97), with good correspondence to theory. The log–log regression slope is ≈1, as expected for a linear stoichiometric relation in which total antibody significantly exceeds the Kd (Eq. 1a). (At the 1-mg dose, plasma antibody concentration is 1–2 nM, exceeding the monovalent Kd (14) by a factor of 5–10.) The curve of Fig. 3 deviates from linearity as the concentration of sTac falls to approach the monovalent Kd (0.2 nM, 1,500 units/ml) (Eq. 1). For 12 of 14 of the patient doses with sTac < 7,000 units/ml, bindability was >50% for all minimum evaluated quantities of administered antibody (0.5–2 mg), and thus were not graphable. Two values from one patient (patient 3) violated this pattern (Fig. 3); for these values, we suspect technical errors in estimating bindability.
In general, for each 10,000 units/ml sTac, ≈1 mg of anti-Tac will be half-neutralized (i.e., one-half is AB2), corresponding to a whole body molar ratio of sTac to anti-Tac of 1.41 (Eq. 1). When β = 50%, it can be shown that the fraction of anti-Tac binding to two sTac molecules, AB2 = 0.5; anti-Tac binding to one sTac molecule, AB = 0.41; and unbound anti-Tac, A = 0.09. To ensure that 90% of antibody is bindable after infusion (A90; Fig. 3), this ratio of sTac-to-anti-Tac must be <0.63 (Eq. 1), requiring 2.5 mg of anti-Tac per 10,000 units/ml sTac, whereas 99% bindability (A99) requires 10 mg and 99.9% bindability (A99.9) requires 20 mg of anti-Tac per 10,000 units/ml sTac.
Continued Antigen Production Progressively Reduces Anti-Tac Bioactivity.
Concurrent with measurement of antibody survival postinfusion, we assessed the kinetics of bioactivity change. For the patient of Fig. 2B, anti-Tac survival was in the normal range (t1/2 = 47 hr), but the bindability declined from its maximum at time 0 to virtually no bindable material at 40 hr. This activity loss again correlates with HPLC profile changes that reflect the binding of newly synthesized antigen to antibody. At 48 hr when one half of dose 1 of antibody was still surviving, an additional 50 mg of unlabeled antibody was administered, for a total body load of 75-mg anti-Tac. After a period of re-equilibration of bound antigen between labeled and new, unlabeled antibody, the bindability rose to 60%, compatible with the increased body load of anti-Tac, but fell subsequently with kinetics similar to that of the 0- to 40-hr phase.
Our work with animal models showed that Tac is catabolized rapidly in vivo by mainly renal mechanisms, and this catabolism is delayed markedly by antibody binding with no impact on survival of the antibody itself (10). Therefore, the progressive decline of antibody bioactivity in these patients implies a continued production of antigen during therapy that can be judged by the time course of the bindability change.
The rate (q) of sTac production can be derived from the bindability kinetics by (i) full profile modeling (Eq. 2) or from (ii) the downward slope of the curve (Eq. 5). For the patient in Fig. 2B, two consecutive doses of antibody were modeled, yielding q = 13 and 11 nmol/hr by full profile modeling and 11 and 8 nmol/hr by the slope of decline of antibody bindability. Loss of bioactivity in the period postinfusion was duplicated in one or more doses in most of our patients. For all patient doses with measurable negative slopes of bioactivity after infusion, values for q were derived by Eq. 5. q ranged over two orders of magnitude, from 0.1 to 10 nmol/hr, and correlated positively with estimated tumor burdens (not shown) and with pretherapy (steady-state) sTac levels in vivo (Fig. 4). The slope of 1 on a log–log plot in Fig. 4 implies a linear relation between production rate and sTac level, as expected by most pharmacokinetic models; i.e., q = cB0 in which c = 0.1 ± 0.06 hr−1.
Figure 4.

Relation of plasma sTac to sTac production in vivo. Tac production (q) was estimated by the bindability loss kinetics postinfusion (Eq. 5) and plotted against sTac. The line indicates slope = 1, predicted for a linear relation, from which q = (6 × 10−5) × [sTac], with sTac in units/ml and q in nmol/hr, corresponding to a value of c ≈ 0.1 (Eq. 3). Regression of the log-transformed data yields a slope of 0.85, which is not significantly different from 1 (P > 0.2 by a two-tailed t test). r = 0.91 on the log-transformed data and 0.99 on the nontransformed data. Eleven doses in eight patients allowed estimation of q.
Inasmuch as bindability is predictable by the sTac level and antibody dose and production of sTac is predictable by baseline sTac levels, the duration of bindability also should be predictable. Fig. 5 estimates the duration of antibody bindability in the presence of continued antigen production as a function of antibody t1/2, up to whole human IgG (23 days; ≈ 600 hr). For this, we chose three dosages, A90, A99, and A99.9, and predicted the time to 50 or 10% bindability (50 or 90% antibody neutralization). In general, 90% neutralization follows fairly closely after 50% neutralization: α (= Btot/Atot) is 1.90 for β = 10% vs. 1.41 for β = 50% (Eq. 1a), requiring only ≈one-third longer antigen synthesis to achieve. It is apparent that it will take less time for antigen to saturate antibody if the antibody concentration also decreases with time because of catabolism. For all curves, therefore, increased t1/2 predicts the increased bindability duration, but this effect plateaus as antibody t1/2 greatly exceeds antigen t1/2. This plateau occurs earliest for A90 and predicts ≈20 hr of bindability (90–10%) for all t1/2 ≥ 20 hr. For A99, the bindability duration (99–10%) increases steadily until t1/2 ≈ 100 hr, after which bindability durations of 70–90 hr are predicted. Of the curves shown, only the A99.9 doses can generate bioactivities of >100 hr. On the other hand, when antibody survival is longer, the cumulative impact of soluble antigen production is more important over the life of the antibody. That is, to obtain one half-life of antibody bioactivity, A90 will be adequate for antibody t1/2 of 10–20 hr, whereas A99 is required for t1/2 of 50 hr, and still higher doses (e.g., A99.9) are required for longer t1/2 values. The Inset of Fig. 5 provides some validation of these curves by comparing predicted with observed durations to 10% bindability. The correspondence is slightly less good with an assumption of a uniform value of c = 0.1.
Figure 5.

Predicted bindability duration with continued sTac production. Six curves based on Eq. 3 and c = 0.1 (q = cB0) are indicated as a function of antibody t1/2: 90 → 50 and 90 → 10, time for A90 dose to reduce to 50 and 10% bindability; and similarly for A99 and A99.9 doses. Dashed line separates curve segments for bindability durations exceeding (above) or less than (below) one antibody half-life. Inset: Observed vs. predicted duration to 10% bindability postinfusion. □, based on individual measured rates of q by Eq. 5; ▪, based on assumption of a uniform value of c = 0.1 for all subjects. Seven doses in six patients allowed estimation of hours to 10% bindability.
Antigenemia Reduces but Does Not Prevent Anti-Tac Binding to Cellular Antigen in Vivo.
The importance of soluble antigen derives from its impact on binding of antibody to antigen-expressing target cells. This was examined by flow cytometry and tumor radioimaging. Normal donors have low Tac expression on their peripheral T cells. In contrast, leukemic cells from patients were typically 90% strongly Tac+ (Fig. 6A), corresponding to a mean number of 8,000 Tac per cell for this degree of staining (not shown). The patient of Fig. 6 received 10 mg of anti-Tac, a dose estimated near the A50 for his high sTac level (115,334 units/ml) (Fig. 3). At 22-hr postinfusion, circulating anti-Tac showed 0% bindability but binding to tumor cells was still apparent (Fig. 6B). However, the signal was less than that obtained pretherapy (Fig. 6A), implying a fraction of unoccupied cellular sites. This was confirmed by the increment in mean fluorescence intensity with exogenously added unlabeled anti-Tac (288 vs. 100), by which we estimated that cellular Tac was only 35% saturated. On the other hand, the picture was different some months later during a disease relapse when the sTac level was fivefold lower (23,767 units/ml). The patient received the same 10-mg dose as previously, now equivalent to A90–A99 (Fig. 3). When assessed at 18 hr, anti-Tac bindability was still >50%, and all sites on the tumor cells were saturated as shown by the lack of increment of signal with in vitro anti-Tac supplementation.
Figure 6.

Flow cytometry of leukemic cells of an adult T cell leukemia patient. (A) Pre-therapy. (B) Twenty-two hours after first infusion of 10 mg of anti-Tac labeled with 90Y. The patient (patient 9) had a partial remission after this therapy. (C) Eighteen hours after antibody infusion during disease relapse. Cells are (1) unstained, (2) stained with goat anti-mouse-FITC, or (3) stained with added anti-Tac followed by goat anti-mouse-FITC. The difference between 2 and 3 indicates the degree of unsaturation of Tac antigen on the tumor cells in vivo.
Finally, we reviewed 111In imaging studies on six patients who received consecutive 1- and 50-mg doses of anti-Tac 1 week apart. The 1-mg dose was saturated rapidly with soluble Tac in all patients, whereas the 50-mg dose typically maintained bindability for a prolonged period; yet, tumor imaging was apparent in both settings (Fig. 7). Together, the experience in vitro in Fig. 1 and in vivo with circulating leukemic cells and solid tumors in Figs. 6 and 7 imply that antibody successfully exchanges between soluble antigen and tumor-bound antigen.
Figure 7.

Scanning images of 111In anti-Tac in vivo. An adult T cell leukemia patient (patient 3) was treated with low (1 mg) and, 7 days later, with high (50 mg) doses of 111In-anti-Tac. Images obtained by γ camera at 48 hr postinfusion; tumor sites are indicated by arrows. sTac was 13,832 units/ml.
DISCUSSION
Targeting of tumor by radioantibody (or immunotoxin) may be viewed as a problem of transferring the largest proportion of administered radioactivity (or toxin) to tumor. Neglecting for the moment mass action and kinetic considerations, an optimal strategy would be to place the maximum tolerated dose of radioactivity or toxin on a mass of antibody that just equals that required to saturate all tumor sites. Once tumor is saturated, further unlabeled antibody only reduces the radioactivity per molecule with a net loss in dose to tumor because a portion of the radioactivity cannot be bound. With soluble antigen, the optimal dose similarly is conceived as that which does not exceed the amount necessary to bind total antigen (tumor + soluble). Although this first construction concentrates 100% of the dose in tumor, the tumor dose in the presence of soluble antigen can be only that fraction of total antigen that is tumor-associated—e.g., 50 or 10% of radioactivity when tumor associated antigen is 50 or 10% of total antigen. Similarly, any mass of antibody lower than that which saturates antigen will be just as effective if it carries the same total dose of radioactivity: In the absence of soluble antigen, 100% of the radioactivity still binds to tumor, and in the presence of soluble antigen, 50 or 10% of all radioactivity will be apportioned to tumor when tumor accounts for 50 or 10% of total antigen.
Ultimately, these simplistic models must be modified to incorporate mass action, distribution kinetics, antigen production rates, on and off rates for antigen–antibody interaction, affinities for soluble vs. cell-bound antigen, and antibody and radioisotope half-lives. Mass action specifies minimum free (unbound) antibody concentrations that must accompany a given degree of antigen saturation. Kinetics of tissue distribution means that substantial concentration gradients must be applied to achieve tumor penetration in reasonable time frames, which further diminishes the dose concentration in tumor by the antibody excesses and reduced specific activities implied. On and off rates determine the rapidity of antigen–antibody exchange, and the relative affinities determine the equilibrium binding ratios between soluble and cellular antigen. Finally, the t1/2 of antibody survival and radioisotope decay both provide dimensions against which all other kinetic processes are measured.
Our results address several of these principles. First, we showed that soluble antigen binds antibody in a predictable and quantitative fashion and in a stoichiometry that accords with simple models of antibody bioactivity. Second, continued soluble antigen production in vivo led to progressive saturation of antibody and decline in “bindability.” Third, the duration of antibody bindability may be predicted based on formulations derived in this study. Fourth, excess soluble antigen competed with tumor for antibody delivery as demonstrated by reduced tumor cell saturation by flow cytometery. Finally, the binding of antibody to leukemic cells and solid tumor occurred in the presence of soluble antigen even in the absence of free (“bindable”) antibody, thus confirming antigen–antibody exchange and partitioning of antibody between soluble and tumor-bound antigen.
The actual application of these principles in dose selection must be tempered by the goals of the therapy. When the goal is to deliver a certain number of antibody molecules to each tumor cell for effective toxin or radioisotope therapy or to attract cytolytic host mechanisms in passive therapies, then saturation of cellular antigen may not strictly be required and antibody excess over soluble antigen may be less critical. Specifically, the final point of soluble and cellular antigen exchange by antibody means that a low total dose strategy (e.g., 1 mg of antibody) is generally rational for treatments in which a maximum tolerated dose of radioisotope or toxin is to be administered, regardless of the presence or absence of soluble antigen.
In contrast, when a certain fraction of surface antigen must be bound, as in targeting growth factor receptors that may require >90% blockade for suppression (e.g., 14, 19), then free antibody (A + AB) must be high enough to ensure this saturation (e.g., 10 × Kd), and excess over soluble antigen must be maintained continuously. This setting mandates a more complex strategy that exploits principles developed in this study. As an example for dose calculation, we specify an antibody t1/2 of 50 hr and an sTac level of 30,000 units/ml. By using Fig. 5, A99 for this t1/2 has 45 hr to 50% bindable, and Fig. 3 shows that A99 is ≈20 mg for this sTac level, thus indicating a dose of 20 mg to obtain 45 hr of bindability ≥50%. Repeat dosing at this interval (45 hr) will ensure bindability ≥50% throughout therapy. Alternatively, one may directly apply Eq. 4, which yields 19 mg as the dose for 45 hr, or 21 mg for a full half-life (50 hr) of bindability.‖
If antibody promptly inhibits antigen production by cytotoxicity or other mechanisms, these strategies will overestimate the necessary doses, and antibody saturation rates will need to be obtained directly as in the present study. Furthermore, whereas anti-Tac binds cellular Tac monovalently (20), other antibodies may bind cellular antigen bivalently with affinity enhancement, and the impact of soluble antigen (necessarily monovalent binding) will be less; this may be modeled by extension of the equations in Materials and Methods.
The factor c, the whole body fractional rate constant for Tac production, was constant within a factor of 2–3 in this application. For antigen t1/2 ≪ antibody t1/2, there is an inverse proportionality in the duration of antibody bindability for change in c (Eq. 3a): that is, halving c will double the bindability duration. If antigen survival approximates or exceeds that of antibody, the impact of changes in c is reduced (Eq. 3).
Because synthesis and catabolism of plasma proteins are in balance at steady-state, c is also the fractional whole body rate constant for Tac catabolism. In usual pharmacokinetic nomenclature, the catabolic rate constant for sTac, k10, is based solely on the plasma pool of antigen from which Tac is catabolized mainly by renal filtration (10). k10 is related to c by the ratio of steady-state to initial (plasma) volumes: k10 ≈ Vss/Vi × c ≈ 0.2 hr−1.** This implies an sTac catabolic t1/2 of ≈3 hr in humans, which is somewhat slower than the 1–1.4 hr value measured in mice (10). Binding to antibody blocks renal filtration of Tac and suppresses its catabolism, thereby accumulating sTac with continued synthesis to mediate the decline in anti-Tac bindability.
A remarkable result from our prior examination of antigen–antibody interactions in mouse models is of potentially great importance to antibody therapies in humans. In these studies, the survival of bound antigen, albeit prolonged, was still substantially less than that of the associated antibody. This phenomenon, termed “differential catabolism,” was explained by passive internalization of complexes by vascular endothelium via ongoing fluid phase pinocytosis, antigen–antibody dissociation in the acidic endosome, and then continuation down separate pathways: passage of antigen to lysosomes for catabolism but selective binding and return of antibody to circulation by the endosomal IgG protection receptor (FcRp; generically termed the “Brambell receptor”, FcRB) (10, 21, 22).
By using our methods, Tac production is derived indirectly from the rate of antibody saturation in which it was assumed provisionally that bound antigen clears at the same rate as associated antibody. This is probably correct for anti-Tac, a mouse mAb, because there is good reason to doubt that human FcRB binds to, thus “protects,” any murine IgG, which is the sine qua non of differential catabolism (21). The short survival of murine IgGs in humans approximates that of other “nonprotected” Igs such as IgA and IgM (17). In contrast, human IgG is protected with a net mean survival t1/2 of 23 days in humans (17); accordingly, molecules containing human IgG Fc domains will gain access to the IgG protection system and to differential catabolism in human therapies. Catabolic “stripping” of antigen that “cleanses” the antibody may significantly prolong its in vivo effectiveness; in mice, anti-Tac bindability with sTac production is prolonged 2.5-fold by differential catabolism (10, 21). The degree of this effect in human therapies ultimately depends on the sensitivity of particular antigen–antibody pairs to dissociation at endosomal pHs. Differential catabolism effectively conceals a portion of the additional binding by newly synthesized antigen, underrepresenting true antigen production. Nevertheless, in the context of these clinical studies, our measured apparent production rates (q and c), with or without antibody “cleansing,” are those that are most relevant to the prediction of antibody bindability kinetics postinfusion.
One final point of potential confusion merits attention. It may be thought that continuous soluble antigen production always will lead to saturation of any mass of antibody if one simply waits long enough. This is false. The new body burden of antigen (B′) will in fact be limited by the ratio of the new (bound to antibody) to the old (unbound) catabolic t1/2 values for antigen (10):
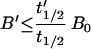 |
6 |
With A > B/1.41, A is always >50% bindable (Eq. 1; Fig. 3). Thus, maintenance of A ≥ B′/1.41 will ensure adequate bindability regardless of the duration of the treatment. Using the example cited above, Eq. 6 is seen to provide the same dosing information, as expected. A mass of antibody of B′/1.41 = 21 mg,‡‡ if maintained, always would be ≥50% bindable. Dosing at 21 mg once per antibody half-life (50 hr) provides adequate bindability for the initial doses‖ (Eq. 4) and at steady-state achieves trough levels of 21 mg as required by Eq. 6. In settings where differential catabolism applies (e.g., with human or chimeric antibodies), the t1/2′ for antigen bound to antibody (Eq. 6) is now lower than that of the antibody, thereby reducing B′ and the mass of A required to maintain bindability. Analogous modifications to Eqs. 3 and 4 predict longer bindabilities for any dose of antibody in the presence of differential catabolism. In the absence of specific knowledge, an assumption of no differential catabolism provides an upper bound on dosing needs.
In general, complete specification of the bindability profile postinfusion requires only antibody t1/2, monovalent affinity constant (K), baseline soluble antigen load (B0), and apparent fractional antigen production rate (c), in which the latter, once determined, is likely to apply broadly to patients treated for a particular antigen specificity. With human or chimeric antibodies, differential catabolism that “cleanses” antibody in vivo may reduce dosing needs to overcome new soluble antigen synthesis during therapy. Measurement of c by change in antibody bindability as we do here bypasses any need for the separate study of differential catabolism and permits an accurate modeling of bioactivity for a given therapeutic antibody in the presence of soluble antigen.
Acknowledgments
We gratefully acknowledge Dr. C. Kasten-Sportes for contributing bindability data on 5 of the 28 patient infusions, L. Top, R.N., for compiling diverse clinical data, C. Goldman, P. Perentesis, and S. Mhatre, M.S., for excellent technical assistance, and Drs. D. Nelson and J. Reynolds for contributing to patient management and radioimaging assessments during the course of these clinical studies, respectively. This work was supported in part by grants to R.P.J. from the Milheim Foundation and the Food and Drug Administration and by Career Development Awards to R.P.J. from the American Cancer Society and the National Cancer Institute.
ABBREVIATIONS
- IL-2Rα
α chain of the interleukin 2 receptor
- sTac
soluble Tac
- A
antibody
- B
soluble antigen
- AB and AB2
antibody with one or two antigens bound
- A0 and B0
moles of A and B at time 0
- β
bindability
- Aβ (β≠0)
dose of A yielding β% bindability
- k and t1/2
beta phase rate constant and t1/2 for antibody survival
- q and c
total and fractional in vivo production rates for soluble antigen. To reduce confusion with β for bindability, “beta” is spelled out when referring to the “beta phase” of pharmacokinetics
Footnotes
Although the initial dilution space is only one plasma volume, the bindability estimates during the 2-hr infusion are compatible with an antigen contact equivalent to the two volumes of the predicted approximate equilibrium space. This result may be explained by a coincidence of rapid distribution rate constants for sTac and concurrent suppression of sTac catabolism over this interval by antibody binding (10).
Specifying desired duration, t = 45 hr; k = ln2/50 hr = 0.014 hr−1; c = 0.1 hr−1; α = 1.41 for final β = 50% (or α = 1.90 for final β = 10%); and B0 = 30,000 units/ml × 0.13 fmol/unit × 6,000 ml = 23 nmol, solving for A0 in moles (Eq. 4), converting to mass = 19 mg. For t = 50 hr, A0 = 21 mg.
This relation holds only when distribution is rapid relative to catabolism, as is generally true for complete antibodies and the sTac–anti-Tac complexes. The catabolic t1/2 of IgG is typically 0.5 × the beta t1/2 (10, 17).
By using Eq. 6, B′ = 192 nmol, using an sTac catabolic t1/2 ≈3 hr (above), a Tac-bound-to-antibody catabolic t1/2′ = 25 hr [using that of anti-Tac, 0.5 × the beta t1/2 of 50 hr**], and a B0 of 23 nmol (30,000 units/ml).
References
- 1.Hansen H J, Snyder J J, Miller E, Vandevoorde J P, Miller O N, Hines L R, Burns J J. Hum Pathol. 1974;5:139–147. doi: 10.1016/s0046-8177(74)80061-4. [DOI] [PubMed] [Google Scholar]
- 2.Gero E J, Colcher D, Ferroni P, Melsheimer R, Giani S, Schlom J, Kaplan P. J Clin Lab Anal. 1989;3:360–369. doi: 10.1002/jcla.1860030609. [DOI] [PubMed] [Google Scholar]
- 3.Kath R, Hoffken K, Otte C, Metz K, Scheulen M E, Hulskamp F, Seeber S. Ann Oncol. 1993;4:585–590. doi: 10.1093/oxfordjournals.annonc.a058593. [DOI] [PubMed] [Google Scholar]
- 4.Bast R C, Jr, Klug T L, St. John E, Jenison E, Niloff J M, Lazarus H, Berkowitz R S, Leavitt T, Griffiths C T, Parker L, et al. N Engl J Med. 1983;309:883–887. doi: 10.1056/NEJM198310133091503. [DOI] [PubMed] [Google Scholar]
- 5.Ladisch S, Wu Z L, Feig S, Ulsh L, Schwartz E, Floutsis G, Wiley F, Lenarsky C, Seeger R. Int J Cancer. 1987;39:73–76. doi: 10.1002/ijc.2910390113. [DOI] [PubMed] [Google Scholar]
- 6.Meeker T C, Maloney D G, Miller R A, Thielmans K, Warnke R, Levy R. Blood. 1985;65:1349–1363. [PubMed] [Google Scholar]
- 7.Greene W C, Leonard W J, Depper J M, Nelson D L, Waldmann T A. Ann Intern Med. 1986;105:560–572. doi: 10.7326/0003-4819-105-4-560. [DOI] [PubMed] [Google Scholar]
- 8.Waldmann T A, Pastan I, Gansow O A, Junghans R P. Ann Intern Med. 1992;116:148–160. doi: 10.7326/0003-4819-116-2-148. [DOI] [PubMed] [Google Scholar]
- 9.Jacques Y, LeMauff B, Boeffard F, Godard A, Soulillou J P. J Immunol. 1987;139:2308–2316. [PubMed] [Google Scholar]
- 10.Junghans R P, Waldmann T A. J Exp Med. 1996;183:1587–1602. doi: 10.1084/jem.183.4.1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Waldmann T A, White J, Goldman C K, Top L, Grant A, Bamford R, Roessler E, Horak I D, Zaknoen S, Kasten-Sportes C, et al. Blood. 1993;82:1701–1712. [PubMed] [Google Scholar]
- 12.Waldmann T A, White J D, Carrasquillo J A, Reynolds J C, Paik C H, Gansow O A, Brechbiel M W, Jaffe E S, Fleisher T A, Goldman C K, et al. Blood. 1995;86:4063–4075. [PubMed] [Google Scholar]
- 13.Kozak R W, Raubitschek A, Mirzadeh S, Brechbiel M W, Junghans R P, Gansow O A, Waldmann T A. Cancer Res. 1989;49:2639–2644. [PubMed] [Google Scholar]
- 14.Junghans R P, Waldmann T A, Landolfi N F, Avdalovic N M, Schneider W P, Queen C. Cancer Res. 1990;50:1495–1502. [PubMed] [Google Scholar]
- 15.Junghans R P, Stone A, Lewis M S. J Biol Chem. 1996;271:10453–10460. doi: 10.1074/jbc.271.18.10453. [DOI] [PubMed] [Google Scholar]
- 16.Humphrey J H, Fahey J L. J Clin Invest. 1961;40:1696–1705. doi: 10.1172/JCI104392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Waldmann T A, Strober W. Prog Allergy. 1969;13:1–110. doi: 10.1159/000385919. [DOI] [PubMed] [Google Scholar]
- 18.Cohen S, Freeman T, McFarlane A S. Clin Sci (Lond) 1961;20:161–170. [PubMed] [Google Scholar]
- 19.Begley C G, Metcalf D, Nicola N A. Blood. 1988;71:640–645. [PubMed] [Google Scholar]
- 20.Robb J R, Greene W C, Rusk C M. J Exp Med. 1984;160:1126–1146. doi: 10.1084/jem.160.4.1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Junghans R P, Anderson C L. Proc Natl Acad Sci USA. 1996;93:5512–5516. doi: 10.1073/pnas.93.11.5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Junghans R P. Immunol Res. 1997;16:29–57. doi: 10.1007/BF02786322. [DOI] [PubMed] [Google Scholar]