

Abstract
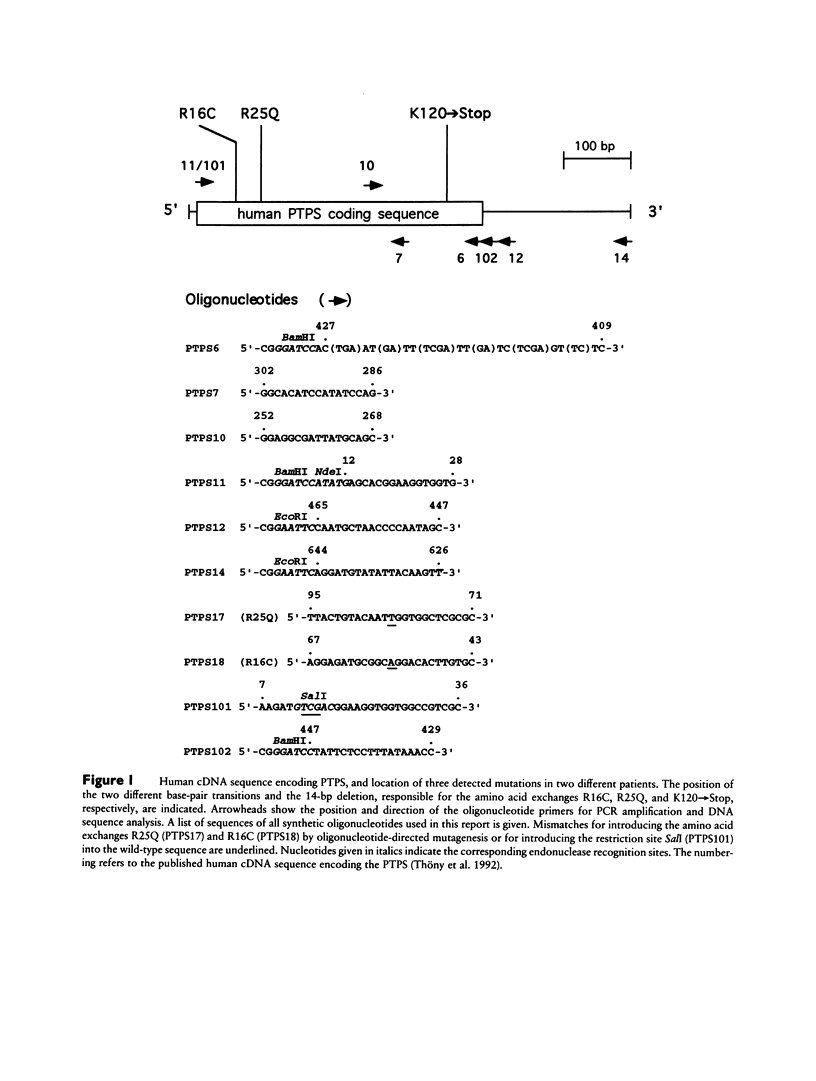
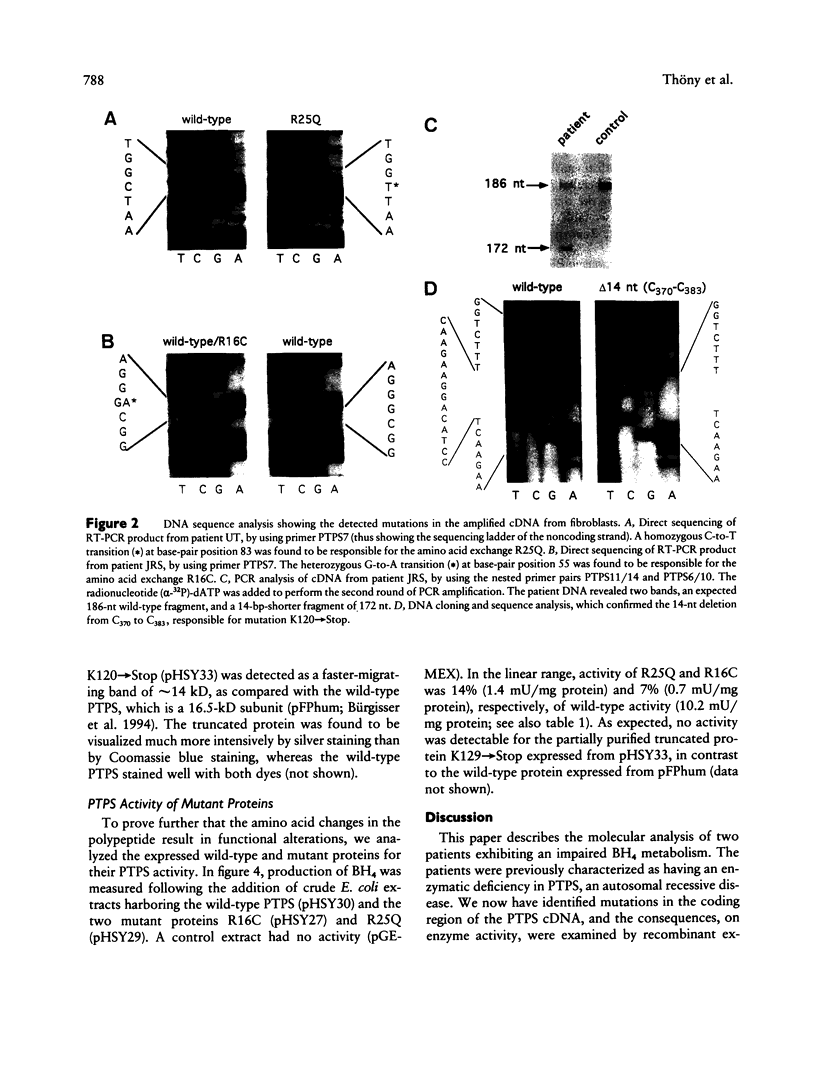
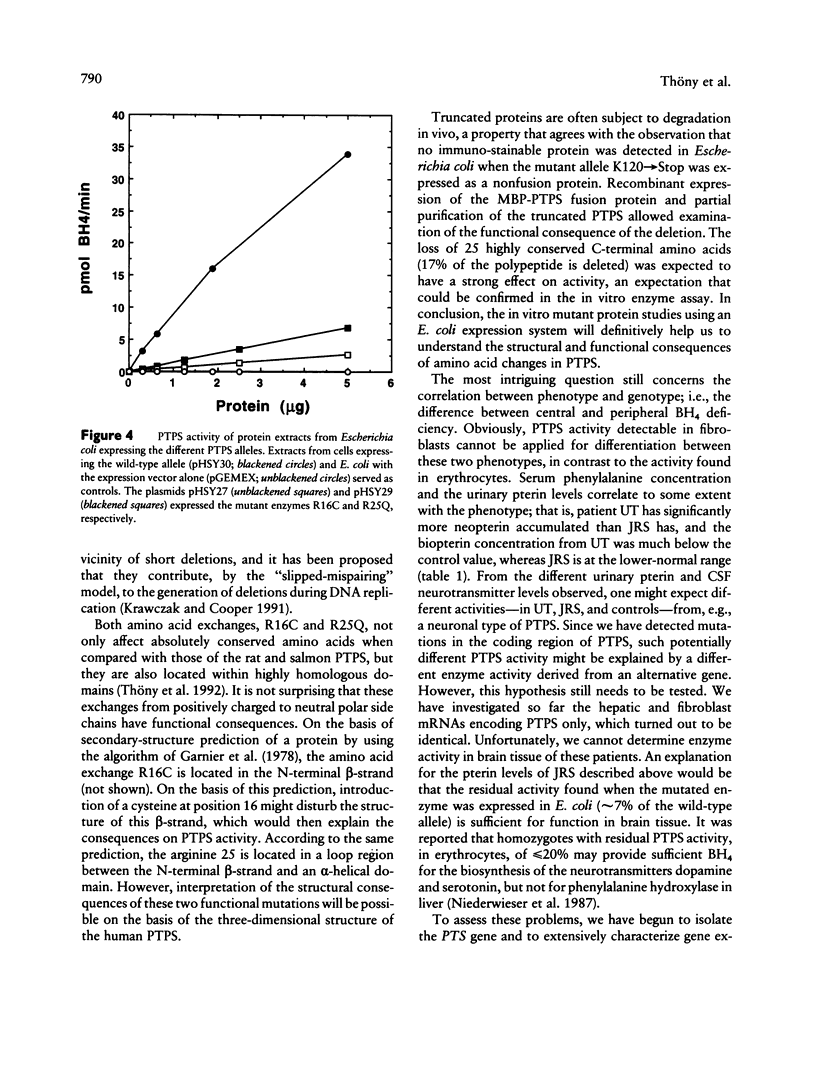
A variant type of hyperphenylalaninemia is caused by a deficiency of tetrahydrobiopterin (BH4), the obligatory cofactor for phenylalanine hydroxylase. The most frequent form of this cofactor deficiency is due to lack of 6-pyruvoyl-tetrahydropterin synthase (PTPS) activity, the second enzyme in the biosynthetic pathway for BH4. The human liver cDNA for PTPS was previously isolated, and the recombinant protein was found to be active when expressed in Escherichia coli. We now have investigated two patients for their molecular nature of this autosomal recessive disorder. Both patients were diagnosed as PTPS deficient, one with the central and one with the peripheral form, on the basis of an elevated serum phenylalanine concentration concomitant with lowered levels of urinary biopterin and PTPS activity in erythrocytes. Molecular analysis was performed on the patients' cultured primary skin fibroblasts. PTPS activities were found in vitro to be reduced to background activity. Direct cDNA sequence analysis using reverse transcriptase-PCR technology showed for the patient with the central from a homozygous G-to-A transition at codon 25, causing the replacement of an arginine by glutamine (R25Q). Expression of this mutant allele in E. coli revealed 14% activity when compared with the wild-type enzyme. The patient with the peripheral form exhibited compound heterozygosity, having on one allele a C-to-T transition resulting in the substitution of arginine 16 for cysteine (R16C) in the enzyme and having on the second allele a 14-bp deletion (delta 14bp), leading to a frameshift at lysine 120 and a premature stop codon (K120-->Stop). Heterologous expression of the enzyme with the single-amino-acid exchange R16C revealed only 7% enzyme activity, whereas expression of the deletion allele delta 14bp exhibited no detectable activity. All three mutations, R25Q, R16C, and K120-->Stop, affect evolutionarily conserved residues in PTPS, result in reduced enzymatic activity when reconstituted in E. coli, and are thus believed to be the molecular cause for the BH4 deficiency. This is the first report describing mutations in PTPS that lead to BH4 deficiency.
Full text
PDF







Images in this article


Selected References
These references are in PubMed. This may not be the complete list of references from this article.
- Abadie V., Lyonnet S., Maurin N., Berthelon M., Caillaud C., Giraud F., Mattei J. F., Rey J., Rey F., Munnich A. CpG dinucleotides are mutation hot spots in phenylketonuria. Genomics. 1989 Nov;5(4):936–939. doi: 10.1016/0888-7543(89)90137-7. [DOI] [PubMed] [Google Scholar]
- Bürgisser D. M., Thöny B., Redweik U., Hunziker P., Heizmann C. W., Blau N. Expression and characterization of recombinant human and rat liver 6-pyruvoyl tetrahydropterin synthase. Modified cysteine residues inhibit the enzyme activity. Eur J Biochem. 1994 Jan 15;219(1-2):497–502. doi: 10.1111/j.1432-1033.1994.tb19964.x. [DOI] [PubMed] [Google Scholar]
- Citron B. A., Davis M. D., Milstien S., Gutierrez J., Mendel D. B., Crabtree G. R., Kaufman S. Identity of 4a-carbinolamine dehydratase, a component of the phenylalanine hydroxylation system, and DCoH, a transregulator of homeodomain proteins. Proc Natl Acad Sci U S A. 1992 Dec 15;89(24):11891–11894. doi: 10.1073/pnas.89.24.11891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron B. A., Kaufman S., Milstien S., Naylor E. W., Greene C. L., Davis M. D. Mutation in the 4a-carbinolamine dehydratase gene leads to mild hyperphenylalaninemia with defective cofactor metabolism. Am J Hum Genet. 1993 Sep;53(3):768–774. [PMC free article] [PubMed] [Google Scholar]
- Cooper D. N., Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988 Feb;78(2):151–155. doi: 10.1007/BF00278187. [DOI] [PubMed] [Google Scholar]
- Dahl H. H., Hutchison W., McAdam W., Wake S., Morgan F. J., Cotton R. G. Human dihydropteridine reductase: characterisation of a cDNA clone and its use in analysis of patients with dihydropteridine reductase deficiency. Nucleic Acids Res. 1987 Mar 11;15(5):1921–1932. doi: 10.1093/nar/15.5.1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhondt J. L. Tetrahydrobiopterin deficiencies: preliminary analysis from an international survey. J Pediatr. 1984 Apr;104(4):501–508. doi: 10.1016/s0022-3476(84)80537-5. [DOI] [PubMed] [Google Scholar]
- Garnier J., Osguthorpe D. J., Robson B. Analysis of the accuracy and implications of simple methods for predicting the secondary structure of globular proteins. J Mol Biol. 1978 Mar 25;120(1):97–120. doi: 10.1016/0022-2836(78)90297-8. [DOI] [PubMed] [Google Scholar]
- Hauer C. R., Rebrin I., Thöny B., Neuheiser F., Curtius H. C., Hunziker P., Blau N., Ghisla S., Heizmann C. W. Phenylalanine hydroxylase-stimulating protein/pterin-4 alpha-carbinolamine dehydratase from rat and human liver. Purification, characterization, and complete amino acid sequence. J Biol Chem. 1993 Mar 5;268(7):4828–4831. [PubMed] [Google Scholar]
- Howells D. W., Forrest S. M., Dahl H. H., Cotton R. G. Insertion of an extra codon for threonine is a cause of dihydropteridine reductase deficiency. Am J Hum Genet. 1990 Aug;47(2):279–285. [PMC free article] [PubMed] [Google Scholar]
- Krawczak M., Cooper D. N. Gene deletions causing human genetic disease: mechanisms of mutagenesis and the role of the local DNA sequence environment. Hum Genet. 1991 Mar;86(5):425–441. doi: 10.1007/BF00194629. [DOI] [PubMed] [Google Scholar]
- Lockyer J., Cook R. G., Milstien S., Kaufman S., Woo S. L., Ledley F. D. Structure and expression of human dihydropteridine reductase. Proc Natl Acad Sci U S A. 1987 May;84(10):3329–3333. doi: 10.1073/pnas.84.10.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederwieser A., Shintaku H., Leimbacher W., Curtius H. C., Hyànek J., Zeman J., Endres W. "Peripheral" tetrahydrobiopterin deficiency with hyperphenylalaninaemia due to incomplete 6-pyruvoyl tetrahydropterin synthase deficiency or heterozygosity. Eur J Pediatr. 1987 May;146(3):228–232. doi: 10.1007/BF00716465. [DOI] [PubMed] [Google Scholar]
- Niederwieser A., Staudenmann W., Wetzel E. High-performance liquid chromatography with column switching for the analysis of biogenic amine metabolites and pterins. J Chromatogr. 1984 May 4;290:237–246. doi: 10.1016/s0021-9673(01)93579-4. [DOI] [PubMed] [Google Scholar]
- Shintaku H., Niederwieser A., Leimbacher W., Curtius H. C. Tetrahydrobiopterin deficiency: assay for 6-pyruvoyl-tetrahydropterin synthase activity in erythrocytes, and detection of patients and heterozygous carriers. Eur J Pediatr. 1988 Jan;147(1):15–19. doi: 10.1007/BF00442604. [DOI] [PubMed] [Google Scholar]
- Smooker P. M., Howells D. W., Cotton R. G. Identification and in vitro expression of mutations causing dihydropteridine reductase deficiency. Biochemistry. 1993 Jun 29;32(25):6443–6449. doi: 10.1021/bi00076a018. [DOI] [PubMed] [Google Scholar]
- Takikawa S., Curtius H. C., Redweik U., Leimbacher W., Ghisla S. Biosynthesis of tetrahydrobiopterin. Purification and characterization of 6-pyruvoyl-tetrahydropterin synthase from human liver. Eur J Biochem. 1986 Dec 1;161(2):295–302. doi: 10.1111/j.1432-1033.1986.tb10446.x. [DOI] [PubMed] [Google Scholar]
- Thöny B., Heizmann C. W., Mattei M. G. Chromosomal location of two human genes encoding tetrahydrobiopterin-metabolizing enzymes: 6-pyruvoyl-tetrahydropterin synthase maps to 11q22.3-q23.3, and pterin-4 alpha-carbinolamine dehydratase maps to 10q22. Genomics. 1994 Jan 15;19(2):365–368. doi: 10.1006/geno.1994.1071. [DOI] [PubMed] [Google Scholar]
- Thöny B., Leimbacher W., Bürgisser D., Heizmann C. W. Human 6-pyruvoyltetrahydropterin synthase: cDNA cloning and heterologous expression of the recombinant enzyme. Biochem Biophys Res Commun. 1992 Dec 30;189(3):1437–1443. doi: 10.1016/0006-291x(92)90235-d. [DOI] [PubMed] [Google Scholar]
- Thöny B., Neuheiser F., Hauer C. R., Heizmann C. W. Molecular cloning and recombinant expression of the human liver phenylalanine hydroxylase stimulating factor revealed structural and functional identity to the dimerization cofactor for the nuclear transcription factor HNF-1 alpha. Adv Exp Med Biol. 1993;338:103–106. doi: 10.1007/978-1-4615-2960-6_20. [DOI] [PubMed] [Google Scholar]
- Togari A., Ichinose H., Matsumoto S., Fujita K., Nagatsu T. Multiple mRNA forms of human GTP cyclohydrolase I. Biochem Biophys Res Commun. 1992 Aug 31;187(1):359–365. doi: 10.1016/s0006-291x(05)81501-3. [DOI] [PubMed] [Google Scholar]
- de Boer M., de Klein A., Hossle J. P., Seger R., Corbeel L., Weening R. S., Roos D. Cytochrome b558-negative, autosomal recessive chronic granulomatous disease: two new mutations in the cytochrome b558 light chain of the NADPH oxidase (p22-phox). Am J Hum Genet. 1992 Nov;51(5):1127–1135. [PMC free article] [PubMed] [Google Scholar]