

Abstract
Papillomavirus-like particles (VLPs) are a promising prophylactic vaccine candidate to prevent human papillomavirus (HPV) infections and associated epithelial neoplasia. However, they are unlikely to have therapeutic effects because the virion capsid proteins are not detected in the proliferating cells of the infected epithelia or in cervical carcinomas. To increase the number of viral antigen targets for cell-mediated immune responses in a VLP-based vaccine, we have generated stable chimeric VLPs consisting of the L1 major capsid protein plus the entire E7 (11 kDa) or E2 (43 kDa) nonstructural papillomavirus protein fused to the L2 minor capsid protein. The chimeric VLPs are indistinguishable from the parental VLPs in their morphology and in their ability to agglutinate erythrocytes and elicit high titers of neutralizing antibodies. Protection from tumor challenge was tested in C57BL/6 mice by using the tumor cell line TC-1, which expresses HPV16 E7, but not the virion structural proteins. Injection of HPV16 L1/L2-HPV16 E7 chimeric VLPs, but not HPV16 L1/L2 VLPs, protected the mice from tumor challenge, even in the absence of adjuvant. The chimeric VLPs also induced protection against tumor challenge in major histocompatibility class II-deficient mice, but not in β2-microglobulin or perforin knockout mice implying that protection was mediated by class I-restricted cytotoxic lymphocytes. These findings raise the possibility that VLPs may generally be efficient vehicles for generating cell-mediated immune responses and that, specifically, chimeric VLPs containing papillomavirus nonstructural proteins may increase the therapeutic potential of VLP-based prophylactic vaccines in humans.
Human papillomaviruses (HPVs) that infect the genital tract are associated with human anogenital tract cancer, particularly cervical cancer (reviewed in ref. 1). HPVs are thought to be the primary causative agent in >90% of cervical cancers (2), with HPV16 being the type most frequently found in these tumors. Approximately 500,000 women develop cervical cancer each year, and 200,000 women die from it, making this disease the second-most common cause of cancer deaths in women worldwide (3).
Significant advances have been made recently in the development of a candidate prophylactic vaccine against papillomavirus infections (reviewed in ref. 4). Expression of the papillomavirus major capsid protein, L1, in eukaryotic cells leads to self-assembly into virus-like particles (VLPs) that are morphologically indistinguishable from native virions and present the conformational epitopes required for the induction of high titer neutralizing antisera (5). L2, the minor capsid protein, coassembles with L1 at a ratio of ≈1 L2 molecule to 30 L1 molecules (6). Although L2 presents some epitopes that induce the production of neutralizing antiserum (7), most neutralizing antibodies induced by L1/L2 VLPs recognize L1 determinants (8). Several studies have shown that L1 and L1/L2 VLP-based vaccines protect animals against high dose experimental papillomavirus infection (9–11). Protection was passively transferred by the sera of immunized animals, indicating that neutralizing antibodies were sufficient to confer protection (9, 11). The initial clinical trials of HPV VLP-based vaccines in humans are now under way (12).
It is unlikely that cell-mediated responses to L1 or L1/L2 VLPs would have a significant therapeutic effect against established papillomaviral infections. This speculation is based on the observation that L1 and L2 proteins are undetectable in the most likely targets of immune regression: the basal epithelial cells of benign productive lesions and the abnormal proliferative cells in premalignant and malignant lesions (13). In contrast, other viral genes, such as E7 or E2, are likely to be expressed in these cells. Therefore these proteins are potential targets for cell-mediated immune regression.
In an effort to generate an HPV vaccine candidate with both improved prophylactic and, in addition, therapeutic potential, we have generated chimeric VLPs containing L1 and fusion proteins consisting of L2 linked to another papillomavirus protein. We chose L2 as the fusion partner because it is not required for capsid assembly or cell surface interactions (14, 15). Therefore larger and more varied insertions might be compatible with normal assembly and cell binding, in comparison with L1 chimeras. In addition, we did not wish to adversely affect the immunodominant conformational neutralizing epitopes on L1. We have generated HPV16 E7 chimeras because the 11-kDa E7 protein is selectively expressed at relatively high levels in high grade cervical dysplasias and cancers (13), and mouse tumor vaccine models have been developed for this protein (16–18). We also show that chimeric VLPs can be made with L2 fused to a 43-kDa full-length viral E2 protein.
MATERIALS AND METHODS
Generation of HPV16 L2 Chimeric Recombinant Baculoviruses.
Two HPV16 E7 chimeras were generated: 16L2-16E7 and BL2-16E7. They each contain the full-length HPV16 E7 gene (coding for 98 aa) fused to the 3′ end of the full-length HPV16 L2 (473 aa) or BPV1 L2 (469 aa) gene, respectively.
The chimeras were generated via recombinant PCR (19) whereby L2 was amplified with a 5′ oligonucleotide containing a restriction enzyme site, and a 3′ oligonucleotide that was complementary to the E7 5′ oligonucleotide. E7 was then amplified with a 5′ oligonucleotide complementary to the L2 3′ oligonucleotide and a 3′ oligonucleotide containing a restriction enzyme site. The L2 and E7 genes were then fused in a second primer extension reaction by using only the outside (L2 5′, and E7 3′) oligonucleotides. The fused L2-E7 genes were then cloned immediately downstream of the pSyn promoter into the baculovirus double expression vector pSynwtVI−, which already contained the respective L1 genes cloned under the polyhedrin promoter (6). BL2–16E7 was cloned as a 5′ BglII to 3′ BglII fragment, and primers used were as follows (restriction sites are underlined): BL2, sense, 5′-GCGGTAGATCTACCTATAAATATGAGTGCACGAAAAAGAGTAAAACGT-3′, and antisense, 5′-GCAATGTAGGTGTATCTCCATGCATGGCATGTTTCCGTTTTTTTCGTTTCCTCAACAAGGAGGG-3′; HPV16E7, sense, 5′-CCCTCCTTGTTGAGGAAACGAAAAAAACGGAAACATGCCATGCATGGAGATACACCTACATTGC-3′ and antisense 5′-CCGCTAGATCTGGTACCTGCAGGATCAGCCATGG-3′. 16L2-16E7 was cloned as a 5′ SstII to 3′ SstII fragment, and the primers used were as follows (restriction sites are underlined): HPV16L2, sense, 5′-GCGGTCCGCGGAATATGCGACACAAACGTTCTGCAAAACGCACAAAACGT-3′ and antisense, 5′-ATCTCCATGCATGGCAGCCAAAGAGAC-3′; HPV16E7, sense, 5′-GTCTCTTTGGCTGCCATGCATGGAGAT-3′ and antisense, 5′-GCTCCGCGGGGTACCTGCAGGATCAGCC-3′.
Recombinant baculovirus stocks were generated by cotransfection with baculovirus DNA (Baculo-Gold; PharMingen) by using lipofectin (GIBCO/BRL). Plaque purification was performed by using published techniques (5).
An L2-E2 chimera was also generated by fusing the coding sequence of the full-length cottontail rabbit papillomavirus (CRPV) E2 (391 aa) to the C-terminal amino acid of HPV16 L2. This chimera (16L2-CE2) was generated by simultaneously ligating L2 and E2 into the baculovirus expression vector, pFastBac1 (GIBCO/BRL). An XhoI site was inserted into the sense L2 primer, and a KpnI site was used in the antisense E2 primer for cloning into pFastBac1. An SstII site was used to fuse the two genes. The primers used were as follows (restriction sites are underlined): 16L2 sense, 5′-CCGCTCGAGAATATGCGACACAAACGTTCTG-3′; 16L2 antisense, 5′-TCCCCGCGGGGCAGCCAAAGAGACATC-3′ and CE2 sense, 5′-GGCGCCGCGGATGGAGGCTCTCAGCCAGCG-3′; CE2 antisense, 5′-GGCGGGTACCGCTGCTGATGGGAATGGG-3′.
Recombinant 16L2-CE2 baculoviruses were generated by using the Bac-to-Bac system according to the manufacturer’s instructions (GIBCO/BRL).
Coexpression of Chimeric L2 Proteins with L1 in Insect Cells.
Sf9 cells were mock infected or infected at a multiplicity of infection (moi) of ≈10 with either wild-type L1/L2 or L1/L2-E7 chimeric recombinant baculoviruses. For generation of 16L1/L2-CE2, Sf9 cells were coinfected with the 16L2-CE2 baculoviruses and baculoviruses containing the gene for HPV16 L1 (6) at an moi of ≈5 for each. After 72 h, cells were lysed by boiling in SDS sample buffer and analyzed by SDS/PAGE in 10% gels. Proteins were stained with 0.25% Coomassie blue or analyzed by Western blotting with HPV16 L1-VLP (6), GST-HPV16 L2 (6), BPV1 L1-VLP (5), GST-BPV1 L2 (7), trp-HPV16 E7 (20) rabbit antisera, or an anti-HPV16 E7 mAb (CIBA-Corning, Alameda, CA) (GST, glutathione S-transferase).
Purification of Chimeric VLPs and Transmission Electron Microscopy (TEM).
After purification by CsCl gradient centrifugation as described (6), particles were adsorbed to carbon-coated grids, stained with 1% uranyl acetate, and examined with a Philips electron microscope model EM 400RT at ×36,000 magnification.
Coimmunoprecipitation of L1/L2-E7 Complexes.
CsCl gradient-purified chimeric VLPs were immunoprecipitated in PBS, 1% Triton X-100 with anti-L1 mAb 5B6 for the bovine papillomavirus (BPV) VLPs (7), mAb H16.V5 for the HPV16 VLPs (kindly provided by N. Christensen) (21), or an irrelevant antibody (anti-E1A mAb) and protein A-Sepharose and subjected to SDS/PAGE. Proteins were immunoblotted and the blots were probed with rabbit antisera to trp-HPV16 E7 and GST-BPVL2 or GST-HPV16L2 for the BPV and HPV16 VLPs containing the L2-E7 chimeras, respectively. The blots of the 16L2-CE2 chimeric VLPs were probed with anti-GST-HPV16L2. The blots were probed with goat, anti-rabbit horseradish peroxidase-conjugated antiserum and developed via chemiluminescence (Kirkegaard & Perry Laboratories).
Chimeric VLP Antisera.
Rabbits were immunized by s.c. injection with three doses of 300 μg of CsCl gradient-purified VLPs in PBS (native) or VLPs boiled for 5 min in PBS/1% SDS (denatured) as described (6).
To determine the L1 or E7-specific antibody titers of VLP-immunized mice, 0.5 μg of purified papillomavirus VLPs, or 6-histidine tagged (6-His) E7 (R. M. Melillo, personal communication), was bound to each well of a 96-well plate, and ELISAs were performed by using a mouse-hybridoma subtyping kit (Boehringer Mannheim). Polyomavirus VP1 VLPs (VP1-baculovirus kindly provided by R. Garcea) (22), or 6-His chloramphenicol acetyltransferase (GIBCO/BRL) were bound to plates and served as negative controls for the papillomavirus VLPs or 6-His E7, respectively. The sera were tested at dilutions from 1:100 to 1:100,000. Titers were determined by subtracting averaged readings of sera from PBS-injected (naive) mice at each dilution from the mean readings of sera from immunized mice (n = 5) at the same dilution. Reported titers were those found to be >0.2 OD405nm unit over that of titers obtained from the plates bound with the negative control proteins.
Cell Lines and Mice.
The TC-1 cell line was generated by transduction of C57BL/6 (B6) primary lung epithelial cells with a retroviral vector expressing HPV16 E6/E7 plus a retrovirus expressing activated c-Ha-ras (16). B6 mice were obtained from The Jackson Laboratory and were used at 7–10 weeks of age. B6 mice with germ-line disruption of β2-microglobulin, perforin, or major histocompatibility complex (MHC) class II expression have been described (23–25).
Tumor Challenge.
Mice were immunized by s.c. injection with 140 μg VLPs in PBS and boosted once 3 weeks later with 10 μg VLPs or by a single inoculation of 10 μg VLPs. The mice were challenged 2 weeks later with 2 × 104 TC-1 tumor cells administered s.c. Tumor take was assessed on day 45. Natural killer (NK) cell depletion was accomplished by injection of monoclonal anti-NK1.1 isolated from supernatants of the PK136 hybridoma (American Type Culture Collection) as described (26). Differences in the results of the tumor challenge assays were evaluated by the two-tailed Fisher’s exact test by using SAS software release 6.12.
RESULTS
To generate a candidate vaccine that contained as many HPV16 E7 epitopes as possible, we attempted to incorporate the entire HPV16 E7 protein into papillomavirus VLPs. We constructed L2-E7 fusions by using the L2 of HPV16 or BPV1, and coexpressed the chimeras with the homologous L1 genes in Sf9 insect cells via double recombinant baculoviruses. E7 was fused to the C terminus of L2 because, except for several basic amino acids, this portion of L2 is poorly conserved among papillomaviruses. The 16L1/L2–16E7 and BL1/L2–16E7 chimeric VLPs were predicted to consist of the L1 protein (506 aa for HPV16 and 496 aa for BPV1) plus a full-length HPV16E7 (98 aa) fused to the C-terminal end of the entire L2 protein (473 aa and 469 aa for HPV16 and BPV, respectively). The L2 fusions and the corresponding L1 proteins were coexpressed in Sf9 insect cells, leading to the appearance of L2, E7 and L1 immunoreactive species of the predicted molecular weight on immunoblots of extracts of the infected cells (Fig. 1 and data not shown).
Figure 1.

Coimmunoprecipitation of 16L2–16E7 or 16L2-CE2 with 16L1 when coexpressed in insect cells. (A) Purified VLPs consisting of 16L1 alone (lanes a), 16L1 and 16L2 (lanes b), or 16L1 and 16L2–16E7 (lanes c) were immunoprecipitated with anti-16L1 mAb V.5 and analyzed by Western blotting with anti-GST 16L2 (Left) or anti-trp-16E7 (Right). (B) Crude lysates containing 16L1 and 16L2 (lane a), 16L2-CE2 (lane b), or 16L1 and 16L2-CE2 (lane c) were immunoprecipitated with anti-16L1 mAb V.5 and analyzed by Western blotting with anti-GST 16L2.
Coassembly of L2-E7 into VLPs.
To obtain evidence for the formation of a stable complex between L1 and the L2-E7 chimera, we immunoprecipitated chimeric BPV and HPV16 VLPs with the appropriate polyclonal or mAb to L1 and analyzed the precipitated complexes by SDS/PAGE and immunoblotting with L2 and E7 antibodies. L1 antibodies, but not the preimmune sera or a control mAb, were able to precipitate a complex that contained the expected molecular weight immunoreactive L2 and HPV16 E7 species from the chimeric VLP preparations, but not from control L1 alone or L1/L2 VLPs (Fig. 1A, data shown for HPV VLPs). These results established that the L2-E7 fusion proteins form a stable complex with L1 when coexpressed in insect cells. By using equivalent amounts of L1 protein, similar levels of L2 and the L2-E7 fusion protein were coimmunoprecipitated from parental and chimeric VLPs, respectively, with an L1 to L2 ratio of approximately 30:1 (Fig. 1 and data not shown).
To examine VLP morphology, VLP preparations were purified by centrifugation through a 40% sucrose cushion and banded in a CsCl density gradient. By TEM, negatively stained samples of both the chimeric HPV16 and BPV VLPs were found to contain similarly large numbers of particles that appeared morphologically indistinguishable from the parental L1/L2 VLPs (Fig. 2; data shown for HPV VLPs).
Figure 2.
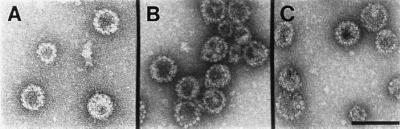
Chimeric VLPs containing the L2-E7 or L2-E2 fusion proteins retain the ability to self-assemble into well-formed particles. VLP preparations were stained with uranyl acetate and examined by TEM. (A) 16L1/L2. (B) 16L1/L2-16E7. (C) 16L1/L2-CE2. (×36,000; bar = 100 nm.)
Cosedimentation analysis was performed to obtain additional evidence that the chimeric L2s were coassembling with L1 into the VLPs. After purification over two sequential CsCl density gradients, VLP preparations were analyzed by analytical sucrose gradient centrifugation. The composition of the resulting gradient fractions was determined by Coomassie-stained SDS/PAGE and Western blot analysis with antibodies specific for L1, L2, or E7, as well as by electron microscopy. The density profiles of chimeric and L1/L2 VLP gradients were very similar as determined by the refractive index (data not shown). Consistent with the combined masses of L2 and E7, we detected a band of ≈100 kDa that was specifically recognized by both the L2 and E7 antisera in a dense fraction of the gradient (Fig. 3, rows B and C, fraction 10, data shown for BPV VLPs). An E7-reactive band was not detected in gradients of VLPs consisting of only L1 and L2, although the expected L2 immunoreactive protein with an apparent molecular mass of 70 kDa was detected in fraction 10 (data not shown). This same fraction (lane 10) contained the majority of the L1 protein, as determined by Coomassie-stained SDS/PAGE (Fig. 3, row A) and the majority of the well-formed VLPs as determined by TEM (data not shown). From these results, we conclude that the L2-E7 fusion proteins are incorporated into VLPs when coexpressed with L1 in insect cells. Rabbit antisera to the BL1/L2–16E7 chimeric VLPs recognized only one (peptide B) of six overlapping L2 peptides (A–F) that together spanned the entire L2 protein (data not shown). Because, in a previous study (7), this L2 peptide was found to have epitopes on the surface of infectious virions, these results support the conclusion that the L2 chimera is positioned normally in the chimeric VLPs.
Figure 3.

Cosedimentation of BL2–16E7 fusion protein with BL1 when coexpressed in Sf9 insect cells. Coexpressed BL1 and BL2–16E7 were purified on CsCl gradients and the proteins were separated on a 12–45% linear sucrose gradient. Gradient fractions (numbered at the top) were analyzed by Coomassie staining of SDS/PAGE for L1 (row A) and by Western blotting with GST-BPV1 L2 antiserum (row B), or trp-HPV16 E7 antiserum (row C).
Surface Properties of Chimeric VLPs.
Papillomavirus virions bind avidly to cell surfaces (15). Because this feature might potentiate the activity of VLPs as vehicles for generating a cell-mediated immune response (CMI), we determined whether this activity was retained by the chimeric VLPs. It has been shown previously that infectious virions, L1 VLPs and L1/L2 VLPs, can agglutinate mouse erythrocytes and that this hemagglutination (HA) correlates with cell surface binding to nucleated cells (27). In contrast, denatured VLPs or virions do not have HA activity. Both the BPV and HPV16 chimeric VLPs agglutinated mouse erythrocytes in a similar fashion to L1/L2 VLPs, requiring 30–60 ng of VLPs to induce HA in our standard assay (27). These results confirm the assembly state of the chimeric VLPs and indicate that they interact normally with cell surfaces.
Because we wished to produce a vaccine candidate with prophylactic as well as therapeutic potential against HPV16 infection, the ability of the chimeric particles to induce neutralizing antibodies was tested. Chimeric VLP antisera were prepared in rabbits and virion antibody titers were compared with those of antisera generated against the parental L1/L2 VLPs in two assays. In a HA inhibition assay (28), which measures the subset of neutralizing antibodies that inhibit virion cell surface binding, the sera elicited by both the BPV and HPV chimeras, and their corresponding parental VLPs, completely inhibited HA to a dilution of 1:1,600 (Fig. 4, data shown for HPV VLPs).
Figure 4.

Antibodies to chimeric VLPs inhibit VLP-mediated HA. A total of 60 ng of HPV16 L1/L2 VLPs were incubated for 1 h at ambient temperature with two-fold dilutions (1:400 to 1:3200, left to right) of prebleed serum or rabbit anti-VLP sera. Antisera were made either to native VLPs (N) or denatured VLPs (D). The samples were mixed with mouse erythrocytes at a final concentration of 0.5% (vol/vol) in 100 μl of PBS/0.1% BSA per well of a 96-well plate, incubated for 3 h at 4°C, and photographed.
Sera to the BL1/L2-16E7 chimera were also directly tested for their ability to neutralize BPV infection of mouse C127 cells, as measured by a reduction in the number of foci induced by a standard amount of BPV virions (5). The immune sera generated by inoculation with baculovirus-derived chimeric VLPs were able to reduce the infectivity of the BPV by 50% at a dilution of at least 1:30,000 (a titer of 30,000), which was equivalent to the neutralizing titer of the control serum raised against BPVL1/L2 VLPs (data not shown). The preimmune sera did not inhibit focal transformation at a dilution of 1:30, the lowest dilution tested.
The ability of anti-16L1/L2–16E7 serum to inhibit HPV16 infection was examined also by using in vitro-generated HPV16 pseudotype virus (29). As with BPV, the sera to the HPV16 chimera and wild-type VLP had similar neutralizing activities; both were able to reduce the infectivity of the pseudotype HPV16 virus by 50% at a dilution of at least 1:10,000 (a titer of 10,000) (Fig. 5). Taken together, these results indicate that the incorporation of the L2-E7 fusion protein into VLPs does not affect the presentation of conformationally dependent immunodominant epitopes on L1.
Figure 5.

Rabbit antiserum to 16L1/L2-16E7 VLPs neutralizes in vitro generated pseudotype HPV16 virions. HPV16L1/L2 recombinant SFV stock was used to infect BPHE-1 cells harboring the BPV1 genome (29). Cell lysates containing the resulting pseudotype HPV16 virions were incubated with preimmune at a dilution of 1:50 (A) or immune serum to HPV16 L1/L2 VLPs at dilutions of 1:10,000 (B) or 1:30,000 (C), or preimmune at a dilution of 1:50 (D) or immune serum to HPV16 L1/L2-16E7 VLPs at dilutions of 1:10,000 (E) or 1:30,000 (F), and placed onto monolayers of C127 cells. After 3 weeks, the cells were stained with 0.5% (wt/vol) methylene blue/0.25% (wt/vol) carbol fuschin in methanol, and the number of foci was scored (45).
The sera generated to intact and denatured chimeric VLPs were also screened for antibodies to HPV16 E7. No E7 antibodies were detected in the sera of rabbits inoculated with intact HPV16 or BPV chimeras as assayed by immunoblotting against a bacterially derived 6-His-E7 fusion protein. However, E7 antibodies were detected when the chimeras were denatured prior to inoculation (data not shown). These results suggest that E7 has an internal location in the chimeric particles and is not accessible for generating an antibody response unless the particles are disassembled.
Generation of an L2-E2 Chimera.
To examine the potential for incorporating larger proteins into chimeric VLPs, an L2-E2 chimera was generated by fusing the full-length CRPV E2 (391 aa) to the C-terminus of HPV16 L2 (16L2-CE2), and the chimera was coexpressed with HPV16 L1. Expression of the chimera was verified on immunoblots by the detection of a protein product with the expected apparent molecular weight of ≈120 kDa that reacted with HPV16 L2 antibodies (Fig. 1B). As with the E7 chimeras, the L2-E2 chimera coassembled with L1 into VLPs, as shown by TEM (Fig. 2C), cosedimented with L1 in a sucrose gradient (data not shown), and coimmunoprecipitated with L1 antisera (Fig. 1B). The HA activity of the L2-E2 chimera was also similar to the L1/L2 wild type, requiring 60 ng of protein in a standard assay.
Antitumor Responses Elicited by E7 Chimeric VLPs.
To examine whether the chimeric VLPs could generate protective CMI, C57BL/6 (B6) mice immunized with either wild-type or HPV16 E7 chimeric VLPs were tested for protection against subsequent tumor challenge by using a B6 mouse tumor cell line, TC-1, that was developed to examine E7 specific antitumor activity (16). TC-1 expresses E7, but not L1 or L2. Protection from challenge with TC-1 was demonstrated previously when HPV16 E7 was expressed via a recombinant vaccinia virus and targeted to lysosomes via a lysosomal-associated membrane protein (LAMP) sorting signal to increase CD4+ T helper responses (16). When injected without adjuvant, the 16L1/L2-16E7 chimeric VLPs were able to induce protection from tumor challenge (Table 1 and Fig. 6A). In contrast, aggressively growing tumors were observed in most animals vaccinated with the wild-type 16L1/L2 VLPs. Therefore, the results indicate that chimeric papillomavirus VLPs can induce antitumor immunity that is specific for the inserted polypeptide. Complete protection was obtained after a single vaccination with 10 μg of the E7 chimeric VLPs (Table 1).
Table 1.
HPV16 L1/L2-16E7 VLPs protect mice against challenge from TC-1 tumor cells
| Mouse strain | Immunogen | Exp. 1 | Exp. 2 | P value |
|---|---|---|---|---|
| B6 | PBS | 3/4 | 4/5 | Ref |
| B6 | L1/L2 | 4/5 | 5/5 | 0.5 |
| B6 | L1/L2-E7 | 0/5 | 1/5 | 0.005 |
| B6 | L1/L2-E7* | ND | 0/5 | 0.05 |
| β2m KO | L1/L2-E7 | 5/5 | 5/5 | 1.0 |
| Perforin KO | L1/L2-E7 | 3/5 | 5/5 | 1.0 |
| Class II KO | L1/L2-E7 | 1/5 | 1/5 | 0.02 |
| NK depleted | L1/L2-E7 | 1/5 | 0/5 | 0.005 |
The results for experiments 1 and 2 are reported as the number of tumor-positive animals per total number of animals challenged. The P values were determined for the comparison of tumor take in the VLP-immunized mice to that of PBS-vaccinated controls and was calculated using the sum of the results of the two experiments. B6, C57Bl/6; β2m KO, β2-microglobulin knockout; Class II, MHC class II knockout; Perforin KO, perforin knockout; NK depleted, C57Bl/6 depleted of natural killer cells; L1/L2, L1/L2 virus-like particles; L1/L2-E7, L1/L2-E7 chimeric virus-like particles.
Vaccinated only once with 10 μg VLPs in PBS; ND, not determined.
Figure 6.

Growth of TC-1 tumor cells in VLP-vaccinated mice. The mean tumor area in the days following tumor challenge is shown for experiment 1 of Table 1. (A) Results from C57BL/6 mice immunized with L1/L2 or L1/L2-E7 VLPs are compared with results from mice injected with PBS (naive). (B) Results after L1/L2-E7 vaccination of C57BL/6 derived β2-microglobulin knockout (B2M KO), perforin knockout, MHC class II knockout, or NK cell-depleted C57BL/6 mice are shown.
To examine the mechanism of protection from tumor challenge elicited by the E7 chimeric VLPs, we tested the ability of the chimeric VLPs to protect B6-derived mice with germ-line defects in β2-microglobulin, perforin, or MHC class II expression (23–25). Good protection was observed in the MHC class II-deficient mice, which lack mature CD4+ T cells (Table 1 and Fig. 6B). In contrast, no significant protection was observed in the β2-microglobulin knockouts (P = 0.0001 by using the vaccinated B6 mice as the reference), implying that protection requires the activity of MHC class I-restricted cytotoxic lymphocytes (CTLs). Protection is, at least in part, perforin-mediated because the perforin knockout mice were also not protected (P = 0.005 compared with the vaccinated B6 mice). NK cell-depleted B6 mice (NK depletion; data not shown) were protected from tumor challenge (Fig. 6B), supporting the idea that CD8+ CTLs were the prime mediators of protection. It is very unlikely that antibody responses play a critical role in protection from tumor challenge. The unprotected animals inoculated with parental VLPs and the protected animals inoculated with the chimeric VLPs generated similar titers of VLP antibodies (mean titer of 100,000 for both L1/L2 and L1/L2-E7 injected mice); none of the animals produced antibodies to E7.
DISCUSSION
This study used the L2 minor capsid protein for the incorporation of heterologous proteins into papillomavirus VLPs. Our generation of E7 and E2 chimeric papillomavirus VLPs demonstrates that entire proteins at least as large as the 391-aa CRPV E2 can be incorporated into papillomavirus VLPs as fusions with L2. Because E7 and E2 are not structurally related, these findings raise the possibility that papillomavirus VLPs could generally act as vaccine vehicles for delivery of protein antigens in their entirety, thus incorporating all of a protein’s potentially immunogenic epitopes.
Chimeric VLPs incorporating foreign antigens have been generated previously by using capsid proteins from other viruses, including parvovirus, HIV-1, and hepatitis virus (30–32), but they have not been shown to generate antitumor immunity. Furthermore, the foreign peptides have usually been inserted into the major capsid protein, whose ability to self-assemble limited the size of the insert to relatively small peptides. For instance, a maximum of 21 aa from HPV16 E7 was inserted into hepatitis B core antigen particles (31), and similar size constraints have been noted for HPV L1 (33). The insertion of a larger polypeptide, the 147-aa hen egg white lysozyme protein, into parvovirus capsids was presumably accomplished because it was fused to VP1, the parvovirus minor capsid protein (32).
Our results strongly suggest that incorporation of the extra polypeptides did not perturb the outer structure of the papillomavirus VLP. The morphology of the parental L1/L2 and chimeric VLPs were indistinguishable in electron micrographs, and the chimeric and parental particles possessed indistinguishable HA activities. Most importantly, the chimeric VLPs also had wild-type activity in inducing neutralizing antibodies. It was critical to demonstrate the retention of this activity because one of the primary goals of the study was to produce a candidate HPV vaccine that has the potential of generating effective immunoprophylactic as well as immunotherapeutic responses in human trials.
In outbred human populations, chimeric papillomavirus VLP-based vaccines could have a potential advantage over peptide-based strategies because protection would not be HLA allele restricted. Also, peptide vaccination can, in some instances, unexpectedly lead to enhanced tumor growth through the induction of specific T cell tolerance (34, 35). Chimeric VLPs also have a theoretical advantage over “naked” DNA or recombinant virus mediated strategies for inducing cytotoxic T cell responses in that there is no possibility for stable expression of the target protein. This characteristic is particularly advantageous in the case where oncoproteins, such as E7, are the immunogen because oncogenes are unacceptable vaccine candidates in many situations.
MHC class I-restricted responses by CTLs are the principal effectors of protective immunity to noncytopathic viruses and tumors (36, 37). It is highly likely that the antitumor response to the E7 chimeric VLPs measured here is mediated by CD8+ CTLs because protection was not seen in the β2-microglobulin or perforin knockout mice but was observed in the MHC class II-deficient mice or after NK cell depletion. It is conceivable that vaccination with VLPs could induce MHC class I-restricted CTLs by two mechanisms. Both L1 and L1/L2 VLPs mimic authentic virions in the avid binding to the papillomavirus cell surface receptor and subsequent internalization (14, 15). Therefore, they may also mimic authentic virus in the escape of endocytic vesicles into the cytoplasm where uncoating must occur to initiate the viral replication cycle. This result would permit presentation of the viral capsid proteins by the normal endogenous route. Alternatively, the VLPs may be presented via an exogenous pathway that preferentially processes particulate antigen through macrophages, or other phagocytic cells, such as subsets of dendritic cells (38, 39).
At the present time, it is unclear what physical aspects of VLPs lead to their effective induction of an antitumor response in the absence of adjuvant or MHC class II-restricted functions. It is possible that multiple features, including their ability to bind to cell surfaces, their particulate and repetitive structure (40, 41), and their presumed ability to escape endocytic vesicles into the cytoplasm, play a role in their ability to elicit CMI that appears to be T helper-independent. Our results with the chimeric VLPs are in contrast to those obtained after vaccination with an E7 expressing vaccinia virus. With the vaccinia vector, protection against TC-1 challenge was not observed unless E7 was targeted to lysosomes via a LAMP sorting signal, with the resulting enhancement of CD4+ T helper responses (16). We have found that class II-restricted functions are not required for efficacy of the VLP-based vaccine in this tumor model. Therefore, VLPs may be particularly advantageous for generating CTL responses in situations where chronic exposure to the target tumor antigen(s), in the absence of costimulation, has resulted in T helper tolerance or exhaustion (42).
Practically, the ability to induce CMI in the absence of adjuvant reduces the likelihood that vaccine administration will produce adverse inflammatory side effects. In addition, the ability to induce potent antigen-specific CMI in the absence of adjuvant suggests that the potential for inducing undesirable autoimmune reactions might be less by using chimeric VLPs relative to strategies employing potent nonspecific immune modulators. These are especially important considerations in the development of a combined prophylactic/therapeutic genital HPV vaccine, because the target population for the vaccine is predominantly healthy adolescents, relatively few of which are destined to develop HPV-induced pathologies.
Although protection from experimental tumor challenge in mice was elicited by a single 10 μg VLP inoculation, multiple boosts will probably be required to induce optimal immunity for eradication of established disease such as micrometastases following debulking of a local pelvic tumor by surgery or irradiation. An attractive feature of a papillomavirus VLP-based therapeutic vaccine strategy is the potential for multiple boosts without the possibility that antibody responses to previous inoculations will influence antigen presentation. Many papillomavirus types have been identified (more than 70 in humans) and their virions are remarkably type specific in their ability to generate neutralizing antibodies (43, 44). As a consequence, antibodies generated by one papillomavirus VLP type do not prevent normal cell surface interactions by other VLP types (28). Therefore, multiple papillomavirus VLP types incorporating the same chimeric protein could be used to boost the CMI to the inserted protein, whereas the immune system of the host remains naive to the vehicle in each successive round of inoculation. Our incorporation of HPV16 E7 into both HPV16 and BPV1 VLPs demonstrates the technical feasibility of this approach. Its feasibility is further supported by the fact that no antibody response to the E7 insert was detected after chimeric VLP vaccination, presumably because the inserted polypeptide projects into the interior of the empty capsid. Several types of CRPV E2 chimeric VLPs, including the HPV16L1/L2-CE2 chimera reported here, have been generated for use in therapeutic vaccine trials to examine regression of established CRPV papillomas after multiple rounds of inoculation with different chimeric VLP types (our unpublished results).
Several human clinical trials to examine the safety and immunogenicity of HPV L1 or L1/L2 VLP-based vaccines will almost certainly be conducted within the next few years (12). The results of this study suggest that human testing of E7 and/or E2 chimeric VLPs should also be considered. If the human safety profiles and serologic responses to the chimeric and parental particles are equivalent and evidence for CMI responses to the inserted protein are obtained, then the chimeric VLPs’ greater potential for inducing therapeutic responses against established HPV-induced lesions would make them an attractive candidate in future efficacy trials of genital HPV VLP-based vaccines.
Acknowledgments
We thank Chenglong Han for help with the statistical analysis, Patricia Day and Frank Booy for assistance with microscopy, T. C. Wu for providing the TC-1 cell line, and Dan Quinn for providing the β2-microglobulin and perforin knockout mice. This work was supported in part by National Institutes of Health Grants PO1CA74182 and RO1 CA74397 to W.M.K.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviations: BPV, bovine papillomavirus; HPV, human papillomavirus; CRPV, cottontail rabbit papillomavirus; VLP, virus-like particle; CMI, cell-mediated immune response; GST, glutathione S-transferase; MHC, major histocompatibility complex; TEM, transmission electron microscopy; HA, hemagglutination; CTL, cytotoxic lymphocyte.
References
- 1.World Health Organization. Human Papillomaviruses, IARC Monographs on the Evaluation of Carcinogenic Risks to Humans. Vol. 64. Lyons, France: World Health Organization; 1995. [PMC free article] [PubMed] [Google Scholar]
- 2.Bosch F X, Manos M M, Munoz N, Sherman M, Jansen A M, Peto J, Schiffman M H, Moreno V, Kurman R, Shah K V. J Natl Cancer Inst. 1995;87:796–802. doi: 10.1093/jnci/87.11.796. [DOI] [PubMed] [Google Scholar]
- 3.Pisani P, Parkin D M, Ferlay J. Int J Cancer. 1993;55:891–903. doi: 10.1002/ijc.2910550604. [DOI] [PubMed] [Google Scholar]
- 4.Schiller J T, Roden R B S. In: Papillomavirus-Like Particles: Basic and Applied Studies. Lacey C, editor. Leeds, U.K.: Leeds Medical Information; 1996. pp. 101–112. [Google Scholar]
- 5.Kirnbauer R, Booy F, Cheng N, Lowy D R, Schiller J T. Proc Natl Acad Sci USA. 1992;89:12180–12184. doi: 10.1073/pnas.89.24.12180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kirnbauer R, Taub J, Greenstone H, Roden R B S, Durst M, Gissmann L, Lowy D R, Schiller J T. J Virol. 1993;67:6929–6936. doi: 10.1128/jvi.67.12.6929-6936.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roden R B S, Weissinger E M, Henderson D W, Booy F, Kirnbauer R, Mushinski J F, Lowy D R, Schiller J T. J Virol. 1994;68:7570–7574. doi: 10.1128/jvi.68.11.7570-7574.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lowy D R, Kirnbauer R, Schiller J T. Proc Natl Acad Sci USA. 1994;91:2436–2440. doi: 10.1073/pnas.91.7.2436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Breitburd F, Kirnbauer R, Hubbert N L, Nonnenmacher B, Trin-Dinh-Desmarquet C, Orth G, Schiller J T, Lowy D R. J Virol. 1995;69:3959–3963. doi: 10.1128/jvi.69.6.3959-3963.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirnbauer R, Chandrachud L, O’Neil B, Wagner E, Grindlay G, Armstrong A, McGarvie G, Schiller J, Lowy D, Campo M. Virology. 1996;219:37–44. doi: 10.1006/viro.1996.0220. [DOI] [PubMed] [Google Scholar]
- 11.Suzich J A, Ghim S, Palmer-Hill F J, White W I, Tamura J K, Bell J, Newsome J A, Jenson A B, Schlegel R. Proc Natl Acad Sci USA. 1995;92:11553–11557. doi: 10.1073/pnas.92.25.11553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steller M A, Schiller J T. J Natl Cancer Inst Monogr. 1996;21:145–148. [PubMed] [Google Scholar]
- 13.Stoler M H, Rhodes M H, Witbeck A, Wolinsky S M, Chow L T, Broker T R. Hum Pathol. 1992;23:117–128. doi: 10.1016/0046-8177(92)90232-r. [DOI] [PubMed] [Google Scholar]
- 14.Muller M, Gissmann L, Cristiano R J, Sun X Y, Frazer I H, Jenson A B, Alonso A, Zentgraf H, Zhou J. J Virol. 1995;69:948–954. doi: 10.1128/jvi.69.2.948-954.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Roden R B S, Kirnbauer R, Jenson A B, Lowy D R, Schiller J T. J Virol. 1994;68:7260–7266. doi: 10.1128/jvi.68.11.7260-7266.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin K-Y, Guarnieri F G, Staveley-O’Carroll K F, Levitsky H I, August J T, Pardoll D M, Wu T-C. Cancer Res. 1996;56:21–26. [PubMed] [Google Scholar]
- 17.Feltkamp M C W, Smits H L, Vierboom M P M, Minnaar R P, deJongh B M, Drijfhout J W, Ter Schegget J, Melief C J M, Kast W M. Eur J Immunol. 1993;23:2242–2249. doi: 10.1002/eji.1830230929. [DOI] [PubMed] [Google Scholar]
- 18.Feltkamp M C W, Vreugdenhil G R, Vierboom M P M, Ras E, Van der Burg S H, Ter Schegget J, Melief C J M, Kast W M. Eur J Immunol. 1995;25:2638–2642. doi: 10.1002/eji.1830250935. [DOI] [PubMed] [Google Scholar]
- 19.Innis M A, Gelfand D H, Sninsky J J, White T J. PCR Protocols: A Guide to Methods and Applications. New York: Academic; 1990. [Google Scholar]
- 20.Barbosa M S, Vass W C, Lowy D R, Schiller J T. J Virol. 1991;65:292–298. doi: 10.1128/jvi.65.1.292-298.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Christensen N D, Dillner J, Eklund C, Carter J J, Wipf G C, Reed C A, Cladel N M, Galloway D A. Virology. 1996;223:174–184. doi: 10.1006/viro.1996.0466. [DOI] [PubMed] [Google Scholar]
- 22.Montross L, Watkins S, Moreland R B, Mamon H, Caspar D L, Garcea R L. J Virol. 1991;65:4991–4998. doi: 10.1128/jvi.65.9.4991-4998.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kagi D, Ledermann B, Burki K, Seiler P, Odermatt B, Podack E R, Zinkernagel R M. Nature (London) 1994;369:31–37. doi: 10.1038/369031a0. [DOI] [PubMed] [Google Scholar]
- 24.Grusby M J, Johnson R S, Papaioannou V E, Glimcher L H. Science. 1991;253:1417–1420. doi: 10.1126/science.1910207. [DOI] [PubMed] [Google Scholar]
- 25.Zijlstra M, Li E, Sajjadi F, Subramani S, Jaenish R. Nature (London) 1989;342:435–438. doi: 10.1038/342435a0. [DOI] [PubMed] [Google Scholar]
- 26.Karlhofer F M, Yokoyama W M. J Immunol. 1991;149:3662–3673. [PubMed] [Google Scholar]
- 27.Roden R B S, Hubbert N L, Kirnbauer R, Breitburd F, Lowy D R, Schiller J T. J Virol. 1995;69:5147–5151. doi: 10.1128/jvi.69.8.5147-5151.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Roden R B S, Hubbert N L, Kirnbauer R, Christensen N D, Lowy D R, Schiller J T. J Virol. 1996;70:3298–3301. doi: 10.1128/jvi.70.5.3298-3301.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roden R B S, Greenstone H L, Kirnbauer R, Booy F P, Jessie J, Lowy D R, Schiller J T. J Virol. 1996;70:5875–5883. doi: 10.1128/jvi.70.9.5875-5883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wagner R, Deml L, Schirmbeck R, Niedrig M, Reimann J, Wolf H. Virology. 1996;220:128–140. doi: 10.1006/viro.1996.0293. [DOI] [PubMed] [Google Scholar]
- 31.Tindle R W, Herd K, Londono P. Virology. 1994;200:547–557. doi: 10.1006/viro.1994.1217. [DOI] [PubMed] [Google Scholar]
- 32.Miyamura K, Kajigaya S, Monoeda M, Smith-Gill S J, Young N S. Proc Natl Acad Sci USA. 1994;91:8507–8511. doi: 10.1073/pnas.91.18.8507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Muller M, Zhou J, Reed T D, Rittmuller C, Burger A, Gabelsberger J, Braspenning J, Gissmann L. Virology. 1997;234:93–111. doi: 10.1006/viro.1997.8591. [DOI] [PubMed] [Google Scholar]
- 34.Toes R E M, Offringa R, Blom R J J, Melief C J M, Kast W M. Proc Natl Acad Sci USA. 1996;93:7855–7860. doi: 10.1073/pnas.93.15.7855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Toes R E M, Blom R J J, Offringa R, Kast W M, Melief C J M. J Immunol. 1996;156:3911–3918. [PubMed] [Google Scholar]
- 36.Zinkernagel R M, Bachmann M F, Kundig T M, Oehen S, Pirchet H, Hengartner H. Annu Rev Immunol. 1996;14:333–367. doi: 10.1146/annurev.immunol.14.1.333. [DOI] [PubMed] [Google Scholar]
- 37.Kast W M, Melief C J M. Immunol Lett. 1991;30:229–232. doi: 10.1016/0165-2478(91)90030-e. [DOI] [PubMed] [Google Scholar]
- 38.Bachmann M F, Lutz M B, Layton G T, Harris S J, Fehr T, Rescigno M, Ricciardi-Castagnoli P. Eur J Immunol. 1996;26:2595–2600. doi: 10.1002/eji.1830261109. [DOI] [PubMed] [Google Scholar]
- 39.Kovacsovics-Bankowski M, Clark K, Benacerraf B, Rock K L. Proc Natl Acad Sci USA. 1993;90:4942–4946. doi: 10.1073/pnas.90.11.4942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Baker T S, Newcomb W W, Olson N H, Cowsert L M, Olson C, Brown J C. Biophys J. 1991;60:1445–1456. doi: 10.1016/S0006-3495(91)82181-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bachmann M F, Zinkernagel R M. Immunol Today. 1996;17:553–558. doi: 10.1016/s0167-5699(96)10066-9. [DOI] [PubMed] [Google Scholar]
- 42.Matzinger P. Annu Rev Immonol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- 43.Roden R B S, Greenstone H L, Kirnbauer R, Booy F P, Jessie J, Lowy D R, Schiller J T. J Virol. 1996;70:5875–5883. doi: 10.1128/jvi.70.9.5875-5883.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Christensen N D, Kirnbauer R, Schiller J T, Ghim S J, Schlegel R, Jenson A B, Kreider J W. Virology. 1994;205:329–335. doi: 10.1006/viro.1994.1649. [DOI] [PubMed] [Google Scholar]
- 45.Dvoretzky I, Shober R, Chattopadhyay S K, Lowy D R. Virology. 1980;103:369–375. doi: 10.1016/0042-6822(80)90195-6. [DOI] [PubMed] [Google Scholar]