

Abstract
Human hereditary tumor syndromes serve as an ideal model for studying molecular pathways regulating tumorigenesis. Multiple endocrine neoplasia type 1 (MEN1), a human familial tumor syndrome, results from mutations in the Men1 gene. Men1 encodes a novel tumor suppressor, menin, of unknown biochemical function. Recently, significant progress has been made in identifying menin as a regulator of gene transcription, cell proliferation, apoptosis, and genome stability, leading to a new model of understanding menin's tumor-suppressing function. These findings suggest that menin's diverse functions depend on its association with chromatin and its control over gene transcription. This knowledge will likely be translated into new strategies to improve therapeutic interventions against MEN1 and other related cancers.
Keywords: menin, MEN1, cell proliferation, apoptosis, genome stability, transcription, epigenetics
Introduction
Multiple endocrine neoplasia type 1 (MEN1) is an inherited tumor syndrome characterized by development of tumors in multiple endocrine organs including the parathyroid glands, pancreatic islets, and the pituitary gland, and also in some non-endocrine organs (Marx et al., 1999b, Pannett and Thakker, 1999). MEN1 was first described as an autosomal dominant familial syndrome, and the gene mutated in MEN1 patients, MEN1, was identified in 1997 (Chandrasekharappa et al., 1997, Lemmens et al., 1997). Men1 encodes a novel protein, menin, of unknown biochemical function. Because menin does not display an obvious homology to any known protein motifs, it is challenging to elucidate how menin functions as a tumor suppressor.
To date, over 300 germline mutations in MEN1 patients have been identified (Leotlela et al., 2003, Marx et al., 1999b, Pannett and Thakker, 1999). Tumors from MEN1 patients with one mutated germline MEN1 allele often lose the normal MEN1 allele (loss of heterozygosity, LOH). Mice heterozygous for the disrupted Men1 allele also develop tumors in various endocrine glands with LOH of the Men1 allele in the tumors, closely resembling the human MEN1 syndrome (Crabtree et al., 2001, Bertolino et al., 2003). These observations indicate that menin is a bona fide tumor suppressor.
Emerging evidence suggests that menin plays a vital role in regulation of gene transcription, cell proliferation, apoptosis, and genome stability, which are among the hallmarks dysregulated in cancer cells. These observations provide novel insights into how menin suppresses tumorigenesis. In the present review, we will focus on the recent progress in understanding how menin regulates cellular homeostasis and functions as a tumor suppressor. In particular, how menin modulates gene transcription, cell proliferation, apoptosis and genome stability will be critically evaluated.
Regulation of gene transcription
Numerous studies demonstrate a crucial role for menin in regulating gene transcription. For instance, menin interacts with a number of transcriptional factors such as JunD, NF-κB, Smad3, and homeobox-containing DNA binding protein Pem (Poisson et al., 2003), and inhibits the activities of JunD and NF-κb (Agarwal et al., 1999, Heppner et al., 2001). Ectopic expression of menin inhibits promoter activity of the prolactin and insulin genes in pituitary tumor cells or insulinoma cells, respectively (Namihira et al., 2002, Sayo et al., 2002). Ectopic menin expression also inhibits insulin-induced endogenous c-Fos expression (Yumita et al., 2003). Several reports have recently shown that menin binds to the loci of several menin-dependent genes, including p18ink4c, p27kip1, Hoxa9, and Hoxc8, and regulates the transcription of those genes (Chen et al., 2006, Hughes et al., 2004, Karnik et al., 2005, Milne et al., 2005, Yokoyama et al., 2005). Genetic evidence also reinforces an essential role for menin in regulation of various endogenous genes. For example, ablation of Men1 reduces the expression of p27kip1, p18ink4c, caspase 8 and Hoxc8 in mouse embryonic fibroblasts (MEF)(Hughes et al., 2004, Milne et al., 2005, Schnepp et al., 2004b), but enhances the expression of insulin-like growth factor binding protein 2 (IGFBP-2) (La et al., 2004a), a gene involved in regulation of cell proliferation (Hoeflich et al., 2001). Complementing the menin-null cells with menin restores optimal expression of caspase 8 and represses the IGFBP-2 expression. Together, these studies strongly suggest an essential role for menin in regulating the transcription of endogenous genes.
Menin has been shown to associate with chromatin (Farley et al., 2006, Jin et al., 2003) and bind multiple endogenous genes including hTERT and Hoxc8 (Hughes et al., 2004, Lin and Elledge, 2003). Menin also interacts with nuclear proteins such as transcription factors, histone methyltransferases (HMT) (Hughes et al., 2004, Kim et al., 2003) and histone deacetylases (HDAC). Thus, menin may function as a scaffold protein to regulate transcription of its target genes by associating with one of several of these various interacting proteins. These multiple interactions of menin and the various nuclear proteins may facilitate regulation of gene transcription and cellular homeostasis (Fig. 1).
Figure 1.

A schematic model explaining how menin regulates gene transcription. (a) Menin and a hypothetically specific DNA binding protein (TF1), together with transcription-activating histone methyltransferases (HMTs), such as MLL or MLL2, target to the loci of p18ink4c, p27kip1 and Hoxc8 genes in chromatin. This HMT-containing complex methylates lysine 4 on histone H3, and changes chromatin structure and subsequently activates gene transcription. Activation of p18ink4c, p27kip1 and Hoxc8 genes leads to cell growth inhibition or cell differentiation. (b) Menin and a hypothetically specific DNA binding protein (TF2), together with a histone deacetylase (HDAC), may target the loci of menin target genes such as hTERT and IGFBP-2, to remove the acetyl group on histones and thus repress the target gene transcription. Inhibition of hTERT and IGFBP-2 may result in reduced cell proliferation and maintenance of genomic stability. Interaction of menin and HDACs in regulating endogenous genes remains to be determined.
Regulation of gene transcription by menin via associating with histone methyltransferases
Menin regulates gene transcription at least in part by modulating chromatin structure. Menin has been shown to associate with a protein complex containing Drosophila trithorax-like histone lysine methyltransferases, the mixed lineage leukemia (MLL) gene products, MLL and MLL2, both of which are SET domain-containing methyltransferases (Hughes et al., 2004, Yokoyama et al., 2004). This complex contains multiple proteins that are homologous to the members of the yeast SET1 complex (COMPASS) and three mammalian SET1-like complexes, including activating signal cointegrator 2 complex (ASCOM), the HCF-1 complex and the MLL complex (Hughes et al., 2004), which had previously been found to methylate histone H3 lysine 4 (H3K4) and activate gene transcription. The menin-interacting complex isolated from mouse embryonic fibroblasts (MEF) also methylates H3K4 in vitro (Hughes et al., 2004). These results support a model that menin recruits histone methyltransferases (HMT) and thus upregulates gene transcription (Fig.1A).
The menin complex has recently been shown to upregulate p18ink4c and p27kip1 transcription by upregulating H3K4 methylation at the p18ink4c and p27kip1 loci in both cultured cells and the murine pancreatic islets (Karnik et al., 2005, Milne et al., 2005). We and others have also showed that menin-HMT complex bind to the Hoxa9 locus in vivo and promotes H3K4 methylation at the Hoxa9 locus (Chen et al., 2006, Yokoyama et al., 2005). The trimethylated H3K4 recruits chd1, a methylated H3K4-specific binding protein of a chromatin remodeling complex, and activates gene transcription via chromatin remodeling (Chen et al., 2006, Pray-Grant et al., 2005).
The interaction between menin and HMT suggests that menin facilitates epigenetic regulation of gene transcription by histone modifications. It is likely that the cooperation between menin and MLL enhances the activity of menin in epigenetic control of gene expression (Fig. 1A). However, it is still unclear whether caspase 8, one of menin's target genes, is also regulated by H3K4 methylation. It is noteworthy that menin may regulate gene transcription in an MLL-independent manner (Scacheri et al., 2006), and how menin regulates gene transcription independent of MLL needs to be further investigated.
Regulation of gene transcription by menin via association with histone deacetylases
Acetylation of histones, which usually leads to activation of gene transcription, can be reversed by histone deacetylases that remove the acetyl group on histones, resulting in repression of the gene locus (Jenuwein and Allis, 2001). In contrast to the role of menin as an activator of caspase 8 and Hoxc8, menin inhibits the activity of transcription factor JunD (Agarwal et al., 1999). Interestingly, this inhibition can be relieved by a histone deacetylase (HDAC) inhibitor (Gobl et al., 1999, Kim et al., 2003). This suggests that menin may inhibit the transcription of certain genes via recruiting histone deactylases to the promoter regions. Consistent with this notion, menin has been shown to associate with several HDACs and represses the expression of a reporter gene driven by a JunD binding site (Kim et al., 2003).
Menin represses expression of endogenous genes such as hTERT and IGFBP-2 and associates with the JunD binding site in the hTERT promoter. Thus, it is likely that menin associates with JunD on the hTERT promoter, further recruits histone deacetylases to the promoter and downregulates the hTERT transcription (Fig. 1B) (Lin and Elledge, 2003, Kim et al., 2003). However, it is not yet clear whether HDACs inhibit menin's endogenous target genes in a menin-dependent manner. It is also possible that menin represses promoter regions through other histone modifications. For instance, methylation of lysine 9 and lysine 27 of histone H3 or acetylation of lysine 12 of histone H4 has been shown to change histone codes and repress gene transcription (Cao and Zhang, 2004, Jenuwein and Allis, 2001). Thus, detailed mechanisms underlying menin-mediated histone modifications and gene repression remain to be investigated.
Regulation of cell proliferation
Ectopic expression of menin inhibits proliferation of oncogenic Ras-transformed NIH3T3 cells, and suppresses tumorigenesis in nude mice transplanted with Ras-transformed cells (Kim et al., 1999). Overexpression of menin also represses proliferation of insulinoma cells and human endocrine tumor cells (Stalberg et al., 2004, Sayo et al., 2002). Moreover, immortalized Men1−/− MEFs display enhanced cell proliferation while complementing the Men1−/− cells with menin reduces cell proliferation, providing genetic evidence for menin's role in suppressing cell proliferation (La et al., 2004b, Schnepp et al., 2004a). In these studies, menin's effect on cell proliferation was measured by monitoring the accumulation of cell number after stable ectopic menin expression. It is still not clear whether transient or stable menin expression in these cells arrests or slows down cell cycle progression at a particular phase of the cell cycle.
Consistent with a role for menin in repressing cell proliferation, knock-down of menin expression increases cell proliferation in rat intestinal epithelial cells (Ratineau et al., 2004). Cell cycle profile analysis on these menin knockdown cells reveals that the number of cells in G1 phase reduces, while the number of cells in S-phase increases. In agreement with a role for menin in suppressing G1 to S phase transition, menin knockdown increases gene expression of cyclin D1, cyclin D3 and cyclin D-dependent kinase 4 (CDK4) (Ratineau et al., 2004), which form a functional protein kinase that promotes cell cycle transition from G1 to S phase (Sherr, 1996) (Fig. 2). Further evidence supporting menin as a regulator of cell proliferation is that menin is also essential for JunD-mediated inhibition of cell proliferation (Agarwal et al., 2003). However, how menin affects the kinetics of G1-S phase transition, and whether restoration of menin in menin knockdown cells can block or decrease the G1-S phase transition remains unclear. To definitively determine that menin is vital for regulating G1 to S-phase transition, ideally it should be shown that ablation of Men1 in cells accelerates G1 to S transition while complementing Men1−/− cells with wild type menin represses this G1 to S- transition.
Figure 2.
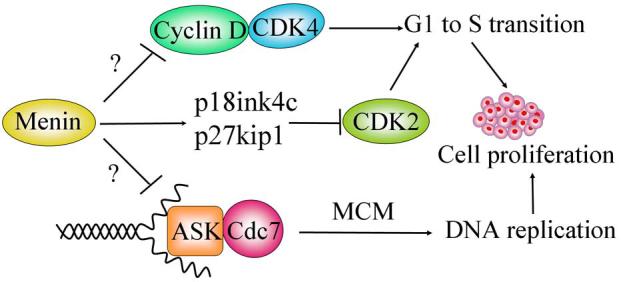
A schematic model showing how menin regulates cell proliferation via distinct mechanisms. Menin represses expression of Cyclin D1 and D3, which form an active kinase with cyclin-dependent kinase (CDK4), promoting cell cycle progression from G1 to S phase. Menin also upregulates the expression of p18ink4c and p27kip1, and represses cyclin-dependent kinase 2 (CDK2) activity and G0/1 to S transition. Cdc7/ activator of S-phase kinase (ASK) complex possesses protein kinase activity phosphorylating mini-chromosome maintenance (MCM) protein, and is essential for DNA replication and S phase progression. Menin interacts with ASK and inhibits ASK-induced cell proliferation. It is likely that, through either transcriptional or post-transcriptional regulation, menin represses CDK4/Cyclin D, CDK2/Cyclin A or E, and Cdc7/ASK, leading to repression of cell cycle transition from G1 to S phase or progression through the S phase.
Menin has been shown to functionally interact with activator of S-phase kinase (ASK), a mammalian homologue of yeast Dbf-4 that is an essential regulatory component of cdc7 protein kinase (Schnepp et al., 2004a). Cdc7/ASK is essential for DNA replication, most likely because it phosphorylates certain mini-chromosome maintenance (MCM) proteins (Masai et al., 2000). MEN1-derived mutations abolish not only menin's binding to ASK but also menin's repression on ASK- induced cell proliferation (Schnepp et al., 2004a). Thus, menin can repress cell proliferation by inhibiting ASK (Fig. 2). It is not yet exactly clear how menin regulates ASK. It is possible that menin affects the distribution of Cdc7/ASK to the origin of DNA replication. On the other hand, we and others have recently shown that menin plays a crucial role in upregulating expression of cyclin-dependent kinase inhibitors p18ink4c and p27kip1, and menin is recruited to the loci of these genes (Karnik et al., 2005, Milne et al., 2005, Schnepp et al., 2006). We further demonstrated that Men1 abrogation increased cyclin-dependent kinase 2 (CDK2) activity and accelerated S phase entry in MEFs (Schnepp et al., 2006). The crucial role of menin in repressing S-phase entry has also been demonstrated in pancreatic islets in vivo, and increased S phase entry was observed as early as 7 days post Men1 excision (Schnepp et al., 2006). Thus, menin has a crucial role in repressing G0/1 to S phase in pancreatic islet cells, perhaps through multiple mechanisms involving p18ink4c, p27kip1, CDK2 and ASK (Fig. 2).
Menin has also been shown to cooperate with transforming growth factor-β (TGF-β), a potent inhibitor of epithelial cells, and suppress cell proliferation of a rat pituitary cell line (Kaji et al., 2001). Menin interacts with Smad3 protein, which is activated by TGF-β and plays an important role in TGF-β-induced growth inhibition (Kaji et al., 2001). Consistent with these observations, reduced menin expression also leads to loss of TGF-β-mediated growth inhibition on primary parathyroid cells (Sowa et al., 2004). These results suggest that menin also inhibits cell proliferation by cooperating with other signaling pathways. However, it is unclear whether targeted disruption of Men1 attenuates or blocks TGF-β-induced anti-proliferation. Adding more complexity to the menin-mediated repression on cell proliferation, JunD, a menin-interacting partner, has been shown to inhibit cell growth in a menin-dependent manner in MEFs (Agarwal et al., 2003). It remains unclear whether this function depends on regulating p27kip1-CDK2 axis or not.
Thus, menin may regulate cell cycle progression in several distinct ways. Menin may regulate the p18ink4c, p27kip1/CDK2 pathway as well as the CDK4/cylin D pathway. In addition, it can also functionally repress ASK to regulate DNA replication as well as TGF-β-mediated suppression of cell proliferation (Fig. 2). However, whether all these pathways are crucial in regulating endocrine cells in vivo remains to be further investigated.
Regulation of apoptosis
Menin mediates apoptosis in MEFs (Schnepp et al., 2004b). Many tumor suppressors, including p53 and BRCA1, also regulate apoptosis. Infection of cells using menin-expressing adenoviruses, but not control viruses, triggers apoptosis (Schnepp et al., 2004b). Menin-induced apoptosis requires Bax and Bak, two proapoptotic proteins that form a gateway for multiple apoptotic pathways (Lindsten et al., 2000), suggesting that menin activates an apoptotic pathway that depends on Bax and Bak (Lindsten et al., 2000). Targeted deletion of Men1 causes increased resistance to TNF-α-induced apoptosis, further supporting a vital role for menin in regulating apoptosis, (Schnepp et al., 2004b).
Interestingly, menin induces expression of caspase 8, an essential component in death receptor-related apoptotic pathways (Varfolomeev et al., 1998). Ablation of Men1 in MEFs diminishes the caspase 8 expression, while complementing menin-null cells with menin enhances the caspase 8 enzymatic activities in response to TNF-α treatment (Schnepp et al., 2004b). As a result, the menin-expressing cells have greater sensitivity to TNF-α mediated apoptosis, as compared to the menin-null cells (Schnepp et al., 2004b). Consistent with these findings, ectopic expression of menin in an insulinoma cell line also increases the number of the cells stained by Annexin V, a hallmark for apoptosis (Sayo et al., 2002). These results suggest that menin suppresses tumorigenesis by promoting apoptosis, at least in part, by modulating caspase 8 expression.
Caspase 8 is an essential initiator caspase that is activated by multiple death-receptors (Chen and Goeddel, 2002). Upon death ligand-mediated ligation of the death receptors, caspase 8 forms oligomers and then autocleaves itself into active form (Chen and Goeddel, 2002). The activated caspase 8 can cleave various effector caspases and initiate apoptosis, while targeted disruption of caspase 8 in mouse leads to resistance to T cell apoptosis induced by death-receptors (Varfolomeev et al., 1998).
The promoter of human caspase 8 is activated by transcription factors Sp1, ETS-like proteins, and tumor suppressor p53 (Liedtke et al., 2003). The caspase 8 gene is frequently inactivated in neuroblastoma, in which caspase 8 is either deleted or silenced by DNA methylation (Teitz et al., 2000). Thus, these tumors are often resistant to death receptor-mediated apoptosis, and reintroduction of the caspase 8 cDNA into the tumor cells restores their sensitivity to death receptor-mediated apoptosis (Hopkins-Donaldson et al., 2000, Hopkins-Donaldson et al., 2003, Teitz et al., 2000). In addition, caspase 8 expression is also differentially inactivated by DNA methylation in its promoter region in many types of pediatric neuroblastomas, small cell lung cancer, their derived cell lines, and neuroendocrine lung cancers (Harada et al., 2002). Treating these cells with 5-aza-2 deoxycytidine, a nucleotide analog that inhibits DNA methylation, restores both t expression of caspase 8 in the cells and their sensitivity to caspase 8-dependent apoptosis (Fulda et al., 2001, Hopkins-Donaldson et al., 2003). Collectively, these studies suggest a vital role for caspase 8 in suppression of tumorigenesis.
Menin may potentiate apoptosis induced by various death receptor ligands, such as TNF-α, TRAIL, and Fas ligand, which promotes UV-induced skin cancer in mice (Hill et al., 1999). Menin may sensitize apoptosis by lowering the threshold for death receptor-mediated apoptosis and other apoptotic signals via maintaining optimal caspase 8 expression, although it is not yet clear whether menin prevents the caspase 8 promoter DNA from being methylated. Consistent with this notion, menin activates expression of the caspase 8 gene and enhances the cell sensitivity to TNF-α-induced apoptosis, but does not activate the basal level of apoptosis dramatically (Schnepp et al., 2004b).
Regulation of genome instability
Peripheral lymphocytes from MEN1 patients display increased chromosome breakage (Gustavson et al., 1983, Scappaticci et al., 1991). Moreover, these lymphocytes undergo extensive chromosomal breakage after treatment with diepoxybutane, an agent crosslinking double stranded DNA (Itakura et al., 2000, Tomassetti et al., 1995). A genome-wide screening of loss of heterozygosity of MEN1 on pancreatic tumors indicates that the tumor cells fail to maintain DNA integrity and chromosomal stability (Hessman et al., 2001). However, no obvious chromosomal instability was observed in the islet cells in which Men1 was lost (Scacheri et al., 2004). Thus, it is likely that Men1 probably plays an important role in maintenance of genomic stability when the cells are under stress (Itakura et al., 2000).
Replication protein A2 (RPA2), a subunit of a trimeric protein binding to single stranded DNA, has been shown to interact with menin (Sukhodolets et al., 2003). RPA is involved in DNA replication, DNA repair, DNA recombination and gene transcription. Because RPA is crucial in DNA repair and in ATR-mediated sensing of DNA damage (Zou and Elledge, 2003), the interaction between menin and RPA2 suggest a potential mechanism for menin in DNA repair. Menin also interacts with FancD2, a protein involved in a BRCA1-mediated DNA repair pathway (Garcia-Higuera et al., 2001), and the interaction between menin and FancD2 is enhanced by γ-irradiation (Jin et al., 2003). Furthermore, targeted disruption of Men1 increased sensitivity to DNA damage induced by an intra-stranded DNA crosslinking agent (Jin et al., 2003). Consistent with this observation, FancD2 also associates with chromatin (Wang et al., 2004). Recently, distribution of menin in cells has been shown to be regulated by UV irradiation (Farley et al., 2006). Thus, these reports suggest a crucial role for menin in repair of DNA damage in concert with RPA2 and FancD2. Menin also functionally interacts with ASK (Schnepp et al., 2004a), which plays a crucial role in response to DNA damage. Therefore, menin may participate in DNA repair in part by modulating the activity of Cdc7/ASK and/or transcription of other DNA repair-related genes.
Fruit flies with the disrupted Men1 homologous gene are viable, but are hypersensitive to ionizing irradiation and DNA-crosslinking agents (Bale, 2004). Consistent with this observation, menin has been found to regulate stress responses including heat shock (Papaconstantinou et al., 2005). These results further support that menin plays an important role in maintaining genome stability and cellular integrity. The precise role of menin in DNA repair and its mechanisms of action remain to be elucidated. Linking menin to RPA2, FancD2, hTERT, and cdc7/ASK points to an interesting direction for elucidating how menin participates in maintenance of genome stability.
Suppression of the MEN 1 development by menin
Menin represses cell proliferation, potentiates apoptosis (Sayo et al., 2002, Schnepp et al., 2004b), suppresses growth of transplanted tumors (Kim et al., 1999), and maintain the genome stability in culture cells (Itakura et al., 2000, Hessman et al., 2001, Jin et al., 2003, Scappaticci et al., 1992, Scappaticci et al., 1991). However, it is not yet clear whether these tumor-suppressing roles in culture cells are crucial for suppressing the development of MEN1. Hence, it is important to determine whether various MEN1 disease-related menin mutations compromise menin's various tumor-suppressing functions in both cell culture and mouse models.
Mutations in Men1, together with functional perturbations of other oncogenes and tumor suppressor genes, may orchestrate the development of the MEN1 tumors. Men1 mutation is a relatively early event in MEN1 tumorigenesis since MEN1 tumors arise in a later stage following hyperplasia in human MEN1 patients and MEN1 mice (Crabtree et al., 2001). p53 is mutated in atypical carcinoids and hormone-secreting tumors that also overexpress Bcl-2 (Schnirer et al., 2003). However, it is unclear whether p53 and Bcl-2 are mutated in the MEN1 tumors. If so, it is important to determine whether the mutations take place prior to LOH of Men1. Thus, temporary molecular changes in various common oncogenes and tumor suppressor genes, in reference to the LOH of Men1 during the MEN1 tumor development, should be further investigated. These studies should provide a temporary molecular order of mutations in various oncogenes and tumor suppressor genes, during the MEN1 tumor development.
Tumors in MEN1 patients preferentially develop in the endocrine organs (Marx et al., 1999a). Although the underlying mechanism is still poorly understood, several hints regarding their preference for endocrine organs exist. For example, like MEN1, patients with inherited multiple endocrine neoplasia type 2 (MEN2) also develop tumors in endocrine organs, primarily in the thyroid gland and the parathyroid gland (Eng, 1999); the parathyroid gland tumor is the most common tumor in MEN1 patients. In MEN2 patients, c-ret, a cell surface protein tyrosine kinase receptor for GDNF (glial cell-derived neurotrophic factor), is mutated, resulting in a constitutively active tyrosine kinase receptor (Eng, 1999), and triggers tumorigenesis in MEN2 patients. The constitutively active receptor induces cell proliferation and activation of the AKT pathway even in the absence of its ligand, GNDF. It is noteworthy that c-ret is highly expressed in neuroganglias and neuroendocrine tissues (Santoro et al., 1999). Thus, it is possible that the tumor-promoting effects of normal c-retin endocrine organs could be antagonized by menin's tumor-suppressing function. If this is the case, breeding mice expressing the oncogenic c-ret in endocrine cells (Cranston and Ponder, 2003) with mice with the Men1+/− genotype will accelerate the tumorigenesis in the endocrine organs.
Other evidence also exists that may explain why there is a high incidence of tumor development in the endocrine organs of MEN1 patients. Menin may cooperate with endocrine organ-specific factors and suppress cell proliferation, induce apoptosis, or regulate other genes functioning at crucial steps of tumorigenesis. Furthermore, it is also likely that menin transcriptionally regulates a subset of target genes in a tissue-specific manner to suppress tumorigenesis.
Perspectives
Numerous studies suggest a crucial role for menin in regulation of gene transcription, cell proliferation, apoptosis and genome stability. However, the precise underlying mechanisms for these functions remain to be elucidated. For instance, it is not clear whether transcriptional regulation is the most important means by which menin suppresses the MEN1 tumor development. How does menin precisely upregulate a group of genes, including p27kip1, p18ink4c, caspase 8 and Hoxc8, but downregulates hTERT, cyclin D1 and CDK4? Are these menin target genes altered in MEN1 tumors? How does menin precisely regulate histone modifications and change chromatin structure? Is it possible to explore specific effect of menin inhibition on enhancing pancreatic β cell proliferation to ameliorate diabetes? Answers to these questions should not only provide novel insights into treating MEN1 syndrome and other related tumors, but also uncover the novel molecular circuitry to modulate function of multiple endocrine cells including pancreatic β cells.
Figure 3.
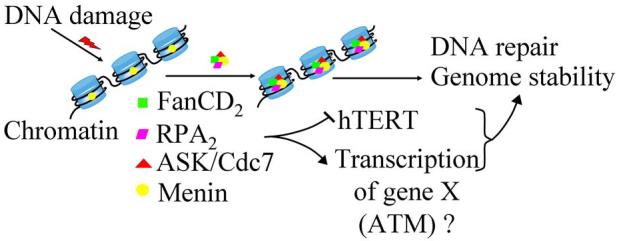
A model for regulation of genome stability by menin via association with various nuclear proteins. Menin associates with chromatin and, upon DNA damage induced by ionizing irradiation or DNA crosslinking, increases its affinity with FancD2, a protein involved in DNA repair and genome stability. In addition, menin also interacts with Cdc7/ASK (activator of S-phase kinase), which is also involved in DNA replication and repair, and with monomeric RPA2 (replication protein A2). DNA damage may increase menin's affinity with these interacting proteins and thus form a supper complex on the chromatin. Although menin alone interacts with each of the above proteins, it remains to be shown whether it forms a supper complex with the other four proteins simultaneously. Menin also represses hTERT (human telomere reverse transcriptase) and thus may protect genome stability indirectly. Because menin's prominent role in regulating gene transcription, it is also possible some of its target genes are involved in maintaining genome stability (Scacheri et al., 2006).
ACKNOWLEDGMENTS
This work is in part supported by an American Cancer Society Research Scholar award and NIH grants (R01 CA100912 and CA113962). We thank Robert Schnepp, Drs. Alan Diehl and Zhao-yuan Hou for stimulating discussions and critically reading of the manuscript. We also thank Ms. Lauren Meade for excellent assistance in preparing the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Agarwal SK, Guru SC, Heppner C, Erdos MR, Collins RM, Park SY, Saggar S, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. Menin interacts with the AP1 transcription factor JunD and represses JunD-activated transcription. Cell. 1999;96:143–152. doi: 10.1016/s0092-8674(00)80967-8. [DOI] [PubMed] [Google Scholar]
- Agarwal SK, Novotny EA, Crabtree JS, Weitzman JB, Yaniv M, Burns AL, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. Transcription factor JunD, deprived of menin, switches from growth suppressor to growth promoter. Proc Natl Acad Sci U S A. 2003;100:10770–10775. doi: 10.1073/pnas.1834524100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bale AE. Tumor models in Drosophila: application to MEN1. Journal of Internal Medicine. 2004;255:705. [Google Scholar]
- Bertolino P, Radovanovic I, Casse H, Aguzzi A, Wang ZQ, Zhang CX. Genetic ablation of the tumor suppressor menin causes lethality at mid-gestation with defects in multiple organs. Mech Dev. 2003;120:549–560. doi: 10.1016/s0925-4773(03)00039-x. [DOI] [PubMed] [Google Scholar]
- Cao R, Zhang Y. The functions of E(Z)/EZH2-mediated methylation of lysine 27 in histone H3. Curr Opin Genet Dev. 2004;14:155–164. doi: 10.1016/j.gde.2004.02.001. [DOI] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi SE, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science. 2002;296:1634–1635. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- Chen YX, Yan J, Keeshan K, Tubbs AT, Wang H, Silva A, Brown EJ, Hess JL, Pear WS, Hua X. The tumor suppressor menin regulates hematopoiesis and myeloid transformation by influencing Hox gene expression. Proc Natl Acad Sci U S A. 2006;103:1018–1023. doi: 10.1073/pnas.0510347103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crabtree JS, Scacheri PC, Ward JM, Garrett-Beal L, Emmert-Buck MR, Edgemon KA, Lorang D, Libutti SK, Chandrasekharappa SC, Marx SJ, Spiegel AM, Collins FS. A mouse model of multiple endocrine neoplasia, type 1, develops multiple endocrine tumors. Proc Natl Acad Sci U S A. 2001;98:1118–1123. doi: 10.1073/pnas.98.3.1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cranston AN, Ponder BA. Modulation of medullary thyroid carcinoma penetrance suggests the presence of modifier genes in a RET transgenic mouse model. Cancer Res. 2003;63:4777–4780. [PubMed] [Google Scholar]
- Eng C. RET proto-oncogene in the development of human cancer. J Clin Oncol. 1999;17:380–393. doi: 10.1200/JCO.1999.17.1.380. [DOI] [PubMed] [Google Scholar]
- Farley SM, Chen G, Guo S, Wang M, A J, Lee F, Lee F, Sawicki M. Menin localizes to chromatin through an ATR-CHK1 mediated pathway after UV-induced DNA damage. J Surg Res. 2006;133:29–37. doi: 10.1016/j.jss.2006.02.021. [DOI] [PubMed] [Google Scholar]
- Fulda S, Kufer MU, Meyer E, van Valen F, Dockhorn-Dworniczak B, Debatin KM. Sensitization for death receptor- or drug-induced apoptosis by re-expression of caspase-8 through demethylation or gene transfer. Oncogene. 2001;20:5865–5877. doi: 10.1038/sj.onc.1204750. [DOI] [PubMed] [Google Scholar]
- Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D'Andrea AD. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell. 2001;7:249–262. doi: 10.1016/s1097-2765(01)00173-3. [DOI] [PubMed] [Google Scholar]
- Gobl AE, Berg M, Lopez-Egido JR, Oberg K, Skogseid B, Westin G. Menin represses JunD-activated transcription by a histone deacetylase- dependent mechanism. Biochim Biophys Acta. 1999;1447:51–56. doi: 10.1016/s0167-4781(99)00132-3. [DOI] [PubMed] [Google Scholar]
- Gustavson KH, Jansson R, Oberg K. Chromosomal breakage in multiple endocrine adenomatosis (types I and II) Clin Genet. 1983;23:143–149. doi: 10.1111/j.1399-0004.1983.tb01863.x. [DOI] [PubMed] [Google Scholar]
- Harada K, Toyooka S, Shivapurkar N, Maitra A, Reddy JL, Matta H, Miyajima K, Timmons CF, Tomlinson GE, Mastrangelo D, Hay RJ, Chaudhary PM, Gazdar AF. Deregulation of caspase 8 and 10 expression in pediatric tumors and cell lines. Cancer Res. 2002;62:5897–5901. [PubMed] [Google Scholar]
- Heppner C, Bilimoria KY, Agarwal SK, Kester M, Whitty LJ, Guru SC, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ, Burns AL. The tumor suppressor protein menin interacts with NF-kappaB proteins and inhibits NF-kappaB-mediated transactivation. Oncogene. 2001;20:4917–4925. doi: 10.1038/sj.onc.1204529. [DOI] [PubMed] [Google Scholar]
- Hessman O, Skogseid B, Westin G, Akerstrom G. Multiple allelic deletions and intratumoral genetic heterogeneity in men1 pancreatic tumors. J Clin Endocrinol Metab. 2001;86:1355–1361. doi: 10.1210/jcem.86.3.7332. [DOI] [PubMed] [Google Scholar]
- Hill LL, Ouhtit A, Loughlin SM, Kripke ML, Ananthaswamy HN, Owen-Schaub LB. Fas ligand: a sensor for DNA damage critical in skin cancer etiology. Science. 1999;285:898–900. doi: 10.1126/science.285.5429.898. [DOI] [PubMed] [Google Scholar]
- Hoeflich A, Reisinger R, Lahm H, Kiess W, Blum WF, Kolb HJ, Weber MM, Wolf E. Insulin-like growth factor-binding protein 2 in tumorigenesis: protector or promoter? Cancer Res. 2001;61:8601–8610. [PubMed] [Google Scholar]
- Hopkins-Donaldson S, Bodmer JL, Bourloud KB, Brognara CB, Tschopp J, Gross N. Loss of caspase-8 expression in highly malignant human neuroblastoma cells correlates with resistance to tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. Cancer Res. 2000;60:4315–4319. [PubMed] [Google Scholar]
- Hopkins-Donaldson S, Ziegler A, Kurtz S, Bigosch C, Kandioler D, Ludwig C, Zangemeister-Wittke U, Stahel R. Silencing of death receptor and caspase-8 expression in small cell lung carcinoma cell lines and tumors by DNA methylation. Cell Death Differ. 2003;10:356–364. doi: 10.1038/sj.cdd.4401157. [DOI] [PubMed] [Google Scholar]
- Hughes CM, Rozenblatt-Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA, Kay GF, Hayward NK, Hess JL, Meyerson M. Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell. 2004;13:587–597. doi: 10.1016/s1097-2765(04)00081-4. [DOI] [PubMed] [Google Scholar]
- Itakura Y, Sakurai A, Katai M, Ikeo Y, Hashizume K. Enhanced sensitivity to alkylating agent in lymphocytes from patients with multiple endocrine neoplasia type 1. Biomed Pharmacother. 2000;54(Suppl 1):187s–190s. doi: 10.1016/s0753-3322(00)80041-4. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jin S, Mao H, Schnepp RW, Sykes SM, Silva AC, D'Andrea AD, Hua X. Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 2003;63:4204–4210. [PubMed] [Google Scholar]
- Kaji H, Canaff L, Lebrun JJ, Goltzman D, Hendy GN. Inactivation of menin, a Smad3-interacting protein, blocks transforming growth factor type beta signaling. Proc Natl Acad Sci U S A. 2001;98:3837–3842. doi: 10.1073/pnas.061358098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnik SK, Hughes CM, Gu X, Rozenblatt-Rosen O, McLean GW, Xiong Y, Meyerson M, Kim SK. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc Natl Acad Sci U S A. 2005;102:14659–14664. doi: 10.1073/pnas.0503484102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Lee JE, Cho EJ, Liu JO, Youn HD. Menin, a tumor suppressor, represses JunD-mediated transcriptional activity by association with an mSin3A-histone deacetylase complex. Cancer Res. 2003;63:6135–6139. [PubMed] [Google Scholar]
- Kim YS, Burns AL, Goldsmith PK, Heppner C, Park SY, Chandrasekharappa SC, Collins FS, Spiegel AM, Marx SJ. Stable overexpression of MEN1 suppresses tumorigenicity of RAS. Oncogene. 1999;18:5936–5942. doi: 10.1038/sj.onc.1203005. [DOI] [PubMed] [Google Scholar]
- La P, Schnepp RW, C DP, A CS, Hua X. Tumor suppressor menin regulates expression of insulin-like growth factor binding protein 2. Endocrinology. 2004a;145:3443–3450. doi: 10.1210/en.2004-0124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La P, Silva AC, Hou Z, Wang H, Schnepp RW, Yan N, Shi Y, Hua X. Direct binding of DNA by tumor suppressor menin. J Biol Chem. 2004b;279:49045–49054. doi: 10.1074/jbc.M409358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmens I, Van de Ven WJ, Kas K, Zhang CX, Giraud S, Wautot V, Buisson N, De Witte K, Salandre J, Lenoir G, Pugeat M, Calender A, Parente F, Quincey D, Gaudray P, De Wit MJ, Lips CJ, Hoppener JW, Khodaei S, Grant AL, Weber G, Kytola S, Teh BT, Farnebo F, Thakker RV, et al. Identification of the multiple endocrine neoplasia type 1 (MEN1) gene. The European Consortium on MEN1. Hum Mol Genet. 1997;6:1177–1183. doi: 10.1093/hmg/6.7.1177. [DOI] [PubMed] [Google Scholar]
- Leotlela PD, Jauch A, Holtgreve-Grez H, Thakker RV. Genetics of neuroendocrine and carcinoid tumours. Endocr Relat Cancer. 2003;10:437–450. doi: 10.1677/erc.0.0100437. [DOI] [PubMed] [Google Scholar]
- Liedtke C, Groger N, Manns MP, Trautwein C. The human caspase-8 promoter sustains basal activity through SP1 and ETS-like transcription factors and can be up-regulated by a p53-dependent mechanism. J Biol Chem. 2003;278:27593–27604. doi: 10.1074/jbc.M304077200. [DOI] [PubMed] [Google Scholar]
- Lin S, Elledge SJ. Multiple tumor suppressor pathways negatively regulate telomerase. Cell. 2003;113:881–889. doi: 10.1016/s0092-8674(03)00430-6. [DOI] [PubMed] [Google Scholar]
- Lindsten T, Ross AJ, King A, Zong WX, Rathmell JC, Shiels HA, Ulrich E, Waymire KG, Mahar P, Frauwirth K, Chen Y, Wei M, Eng VM, Adelman DM, Simon MC, Ma A, Golden JA, Evan G, Korsmeyer SJ, MacGregor GR, Thompson CB. The combined functions of proapoptotic Bcl-2 family members bak and bax are essential for normal development of multiple tissues. Mol Cell. 2000;6:1389–1399. doi: 10.1016/s1097-2765(00)00136-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marx SJ, Agarwal SK, Heppner C, Kim YS, Kester MB, Goldsmith PK, Skarulis MC, Spiegel AM, Burns AL, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Emmert-Buck MR, Guru SC, Manickam P, Crabtree JS, Collins FS, Chandrasekharappa SC. The gene for multiple endocrine neoplasia type 1: recent findings. Bone. 1999a;25:119–122. doi: 10.1016/s8756-3282(99)00112-x. [DOI] [PubMed] [Google Scholar]
- Marx SJ, Agarwal SK, Kester MB, Heppner C, Kim YS, Skarulis MC, James LA, Goldsmith PK, Saggar SK, Park SY, Spiegel AM, Burns AL, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Emmert-Buck MR, Guru SC, Manickam P, Crabtree J, Erdos MR, Collins FS, Chandrasekharappa SC. Multiple endocrine neoplasia type 1: clinical and genetic features of the hereditary endocrine neoplasias. Recent Prog Horm Res. 1999b;54:397–438. [PubMed] [Google Scholar]
- Masai H, Matsui E, You Z, Ishimi Y, Tamai K, Arai K. Human Cdc7-related kinase complex. In vitro phosphorylation of MCM by concerted actions of Cdks and Cdc7 and that of a criticial threonine residue of Cdc7 bY Cdks. J Biol Chem. 2000;275:29042–29052. doi: 10.1074/jbc.M002713200. [DOI] [PubMed] [Google Scholar]
- Milne TA, Hughes CM, Lloyd R, Yang Z, Rozenblatt-Rosen O, Dou Y, Schnepp RW, Krankel C, Livolsi VA, Gibbs D, Hua X, Roeder RG, Meyerson M, Hess JL. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc Natl Acad Sci U S A. 2005;102:749–754. doi: 10.1073/pnas.0408836102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namihira H, Sato M, Murao K, Cao WM, Matsubara S, Imachi H, Niimi M, Dobashi H, Wong NC, Ishida T. The multiple endocrine neoplasia type 1 gene product, menin, inhibits the human prolactin promoter activity. J Mol Endocrinol. 2002;29:297–304. doi: 10.1677/jme.0.0290297. [DOI] [PubMed] [Google Scholar]
- Pannett AA, Thakker RV. Multiple endocrine neoplasia type 1. Endocr Relat Cancer. 1999;6:449–473. doi: 10.1677/erc.0.0060449. [DOI] [PubMed] [Google Scholar]
- Papaconstantinou M, Wu Y, Pretorius HN, Singh N, Gianfelice G, Tanguay RM, Campos AR, Bedard PA. Menin is a regulator of the stress response in Drosophila melanogaster. Mol Cell Biol. 2005;25:9960–9972. doi: 10.1128/MCB.25.22.9960-9972.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poisson A, Zablewska B, Gaudray P. Menin interacting proteins as clues toward the understanding of multiple endocrine neoplasia type 1. Cancer Letters. 2003;189:1–10. doi: 10.1016/s0304-3835(02)00509-8. [DOI] [PubMed] [Google Scholar]
- Pray-Grant MG, Daniel JA, Schieltz D, Yates JR, 3rd, Grant PA. Chd1 chromodomain links histone H3 methylation with SAGA- and SLIK-dependent acetylation. Nature. 2005;433:434–438. doi: 10.1038/nature03242. [DOI] [PubMed] [Google Scholar]
- Ratineau C, Bernard C, Poncet G, Blanc M, Josso C, Fontaniere S, Calender A, Chayvialle JA, Zhang CX, Roche C. Reduction of menin expression enhances cell proliferation and is tumorigenic in intestinal epithelial cells. J Biol Chem. 2004 doi: 10.1074/jbc.M401835200. [DOI] [PubMed] [Google Scholar]
- Santoro M, Carlomagno F, Melillo RM, Billaud M, Vecchio G, Fusco A. Molecular mechanisms of RET activation in human neoplasia. J Endocrinol Invest. 1999;22:811–819. doi: 10.1007/BF03343650. [DOI] [PubMed] [Google Scholar]
- Sayo Y, Murao K, Imachi H, Cao WM, Sato M, Dobashi H, Wong NC, Ishida T. The multiple endocrine neoplasia type 1 gene product, menin, inhibits insulin production in rat insulinoma cells. Endocrinology. 2002;143:2437–2440. doi: 10.1210/endo.143.6.8950. [DOI] [PubMed] [Google Scholar]
- Scacheri PC, Davis S, Odom DT, Crawford GE, Perkins S, Halawi MJ, Agarwal SK, Marx SJ, Spiegel AM, Meltzer PS, Collins FS. Genome-wide analysis of menin binding provides insights into MEN1 tumorigenesis. PLoS Genet. 2006;2:e51. doi: 10.1371/journal.pgen.0020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scacheri PC, Kennedy AL, Chin K, Miller MT, Hodgson JG, Gray JW, Marx SJ, Spiegel AM, Collins FS. Pancreatic insulinomas in multiple endocrine neoplasia, type I knockout mice can develop in the absence of chromosome instability or microsatellite instability. Cancer Res. 2004;64:7039–7044. doi: 10.1158/0008-5472.CAN-04-1648. [DOI] [PubMed] [Google Scholar]
- Scappaticci S, Brandi ML, Capra E, Cortinovis M, Maraschio P, Fraccaro M. Cytogenetics of multiple endocrine neoplasia syndrome. II. Chromosome abnormalities in an insulinoma and a glucagonoma from two subjects with MEN1. Cancer Genet Cytogenet. 1992;63:17–21. doi: 10.1016/0165-4608(92)90057-f. [DOI] [PubMed] [Google Scholar]
- Scappaticci S, Maraschio P, del Ciotto N, Fossati GS, Zonta A, Fraccaro M. Chromosome abnormalities in lymphocytes and fibroblasts of subjects with multiple endocrine neoplasia type 1. Cancer Genet Cytogenet. 1991;52:85–92. doi: 10.1016/0165-4608(91)90057-2. [DOI] [PubMed] [Google Scholar]
- Schnepp RW, Chen YX, Wang H, Cash T, Silva A, Diehl JA, Brown E, Hua X. Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 2006;66:5707–5715. doi: 10.1158/0008-5472.CAN-05-4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnepp RW, Hou Z, Wang H, Petersen C, Silva A, Masai H, Hua X. Functional interaction between tumor suppressor menin and activator of S-phase kinase. Cancer Res. 2004a;64:6791–6796. doi: 10.1158/0008-5472.CAN-04-0724. [DOI] [PubMed] [Google Scholar]
- Schnepp RW, Mao H, Sykes SM, Zong WX, Silva A, La P, Hua X. Menin induces apoptosis in murine embryonic fibroblasts. J Biol Chem. 2004b;279:10685–10691. doi: 10.1074/jbc.M308073200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnirer II, Yao JC, Ajani JA. Carcinoid--a comprehensive review. Acta Oncol. 2003;42:672–692. doi: 10.1080/02841860310010547. [DOI] [PubMed] [Google Scholar]
- Sherr CJ. Cancer cell cycles. Science. 1996;274:1672–1677. doi: 10.1126/science.274.5293.1672. [DOI] [PubMed] [Google Scholar]
- Sowa H, Kaji H, Kitazawa R, Kitazawa S, Tsukamoto T, Yano S, Tsukada T, Canaff L, Hendy GN, Sugimoto T, Chihara K. Menin inactivation leads to loss of transforming growth factor beta inhibition of parathyroid cell proliferation and parathyroid hormone secretion. Cancer Res. 2004;64:2222–2228. doi: 10.1158/0008-5472.can-03-3334. [DOI] [PubMed] [Google Scholar]
- Stalberg P, Grimfjard P, Santesson M, Zhou Y, Lindberg D, Gobl A, Oberg K, Westin G, Rastad J, Wang S, Skogseid B. Transfection of the multiple endocrine neoplasia type 1 gene to a human endocrine pancreatic tumor cell line inhibits cell growth and affects expression of JunD, delta-like protein 1/preadipocyte factor-1, proliferating cell nuclear antigen, and QM/Jif-1. J Clin Endocrinol Metab. 2004;89:2326–2337. doi: 10.1210/jc.2003-031228. [DOI] [PubMed] [Google Scholar]
- Sukhodolets K, Hickman A, Agarwal SK, Sukhodolets M, Obungu V, Novotny E, Crabtree J, Chandrasekharappa SC, Collins FS, Spiegel AM, Burns AL, Marx SJ. The 32-kilodalton subunit of replication protein A interacts with menin, the product of the MEN1 tumor suppressor gene. Molecular and Cellular Biology. 2003;23:493–509. doi: 10.1128/MCB.23.2.493-509.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000;6:529–535. doi: 10.1038/75007. [DOI] [PubMed] [Google Scholar]
- Tomassetti P, Cometa G, Del Vecchio E, Baserga M, Faccioli P, Bosoni D, Paolucci G, Barbara L. Chromosomal instability in multiple endocrine neoplasia type 1. Cytogenetic evaluation with DEB test. Cancer Genet Cytogenet. 1995;79:123–126. doi: 10.1016/0165-4608(95)98126-j. [DOI] [PubMed] [Google Scholar]
- Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D. Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally. Immunity. 1998;9:267–276. doi: 10.1016/s1074-7613(00)80609-3. [DOI] [PubMed] [Google Scholar]
- Wang X, Andreassen PR, D'Andrea AD. Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin. Mol Cell Biol. 2004;24:5850–5862. doi: 10.1128/MCB.24.13.5850-5862.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama A, Somervaille TC, Smith KS, Rozenblatt-Rosen O, Meyerson M, Cleary ML. The Menin Tumor Suppressor Protein Is an Essential Oncogenic Cofactor for MLL-Associated Leukemogenesis. Cell. 2005;123:207–218. doi: 10.1016/j.cell.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W, Cleary ML. Leukemia proto-oncoprotein MLL forms a SET1-like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol. 2004;24:5639–5649. doi: 10.1128/MCB.24.13.5639-5649.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumita W, Ikeo Y, Yamauchi K, Sakurai A, Hashizume K. Suppression of insulin-induced AP-1 transactivation by menin accompanies inhibition of c-Fos induction. Int J Cancer. 2003;103:738–744. doi: 10.1002/ijc.10885. [DOI] [PubMed] [Google Scholar]
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science. 2003;300:1542–1548. doi: 10.1126/science.1083430. [DOI] [PubMed] [Google Scholar]