

Abstract
Model-based, three-dimensional (3D) image reconstruction procedures require a starting model to initiate data analysis. We have designed an ab initio method, which we call the random model (RM) method, that automatically generates models to initiate structural analysis of icosahedral viruses imaged by cryo-electron microscopy. The robustness of the RM procedure was demonstrated on experimental sets of images for five representative viruses. The RM method also provides a straightforward way to generate unbiased starting models to derive independent 3D reconstructions and obtain a more reliable assessment of resolution. The fundamental scheme embodied in the RM method should be relatively easy to integrate into other icosahedral software packages.
Keywords: 3D image reconstruction, Icosahedral virus, Origin and orientation determination, Electron cryo-microscopy, Resolution determination, Model-based refinement, automated image processing
1. Introduction
Advances in cryo-electron microscopy (cryoEM) and three-dimensional (3D) image reconstruction techniques as well as increased computer speed and storage during the last decade have led to the determination of numerous macromolecular structures at subnanometer resolutions (Orlova, et al., 2004; Jiang, et al., 2005). A 3D density map of a single-particle type specimen at subnanometer resolution requires averaging hundreds or thousands of noisy images of individual particles that are all assumed to have identical structures. Typical cryoEM samples contain particles whose orientations are random, a prerequisite for high resolution work. The primary challenge in 3D reconstruction studies, to determine the correct orientation and a common reference point (origin) for each particle, is a direct consequence of the low signal-to-noise ratio in each image.
The first methods devised to solve this problem for analysis of icosahedral particles involved the use of common lines and cross-common lines (Crowther, 1971; Fuller, et al., 1996). These Fourier-based methods rely on identifying symmetry-related lines in the Fourier transform of each particle image, and the locations of these lines define the particle view direction. Common-line type analyses only use a portion of the image transform and significant expertise is required to successfully interpret the results (Fuller, et al., 1996; Baker, et al., 1996). Recognizing such limitations, a number of robust, model-based methods have been developed to analyze the images of symmetric as well as asymmetric particles (Frank, et al., 1996). All these methods make use of a larger portion of the data in real or Fourier space. Various model-based methods have been used to solve numerous different structures to 10Å or better, including two-dimensional crystals (Gonen, et al., 2005), helical particles (Miyazawa, et al., 2003; Samatey, et al., 2004), single particles with high (icosahedral) symmetry (Zhou, et al., 2001; Zhang, et al., 2005a), single particles with lower symmetry (Cheng, et al., 2004; Fotin, et al., 2004; Ludtke, et al., 2004) and asymmetric particles (Mueller, et al., 2000; Valle, et al., 2003). Model-based methods have become the de facto standard procedure in most labs for refining orientations and origins of very large numbers of particles.
Any model-based method requires a starting model to initiate data analysis. The model is used to generate a database of evenly distributed projections to which each individual particle image is compared. The particle is assigned the view orientation of the projection it best matches. There are two basic strategies to obtain a starting model. Ideally, an ab initio model is generated directly from the images, but more commonly, the model is derived indirectly from related or simulated data. An indirect model is typically derived from any of several sources: an existing reconstruction of a vitrified (e.g. Olson, et al., 1992) or negatively stained (e.g. Cheng, et al., 2006) specimen related to the current sample, a map of a related X-ray structure (e.g. Cheng, et al., 1994) or one calculated from pseudo atomic coordinates (e.g. Wikoff, et al., 1994), or a simple, geometric structure constructed in silico (e.g. Cheng, et al., 1992). The potential for model-induced bias in the reconstruction process always exists with indirect models since they are not derived from the images being analyzed.
Whatever the nature of the starting model, it should not bias the final result (Grigorieff, 2000; Stewart, et al., 2004; Yang, et al., 2003; Shaikh, et al., 2003). An advantage of ab initio starting models is that they are less likely to introduce such bias. These direct methods include common lines (Fuller, et al., 1996; Thuman-Commike, et al., 1997), angular reconstitution (van Heel, 1987), one particle reconstruction (Caston, et al., 1999; Cantele, et al., 2003), random conical tilt (Radermacher, et al., 1987), orthogonal tilt (Leschziner, et al., 2006), maximum likelihood (Provencher, et al., 1988; Yin, et al., 2001), and reconstruction of particles viewed along symmetry axes (Ludtke, et al., 1999). The main disadvantage of most ab intio methods is that they can be difficult to implement. They may, for example, require significant user expertise, intensive computation, additional data manipulation, or large data sets, and additionally may not be amenable to automation.
The challenges identified above motivated us to develop the random model (RM) method to study particles with icosahedral symmetry. This method fulfills most, if not all, of the requirements for an ideal procedure to generate an ab initio starting model: quick and reliable, requiring minimal user input and intervention, and readily automated. The RM method can be used to obtain two unique starting models that enable independent 3D reconstructions to be computed from a data set of icosahedral particle images. It is widely accepted that the analysis of independent data sets ensures more accurate resolution assessment (Grigorieff, 2000). However, any split-data processing scheme, especially with very large data sets, necessarily at least double the computational and human efforts and hence becomes an impediment to many researchers. To address these issues, the RM method, coupled with the split-data scheme, has been incorporated into an automated processing package driven by the program AUTO3DEM (Yan, et al., 2006). This program was designed to minimize the need for extensive user input or intervention and significantly relieves the user of the increased workload. The robustness of the RM method is demonstrated on five experimental sets of virus images. In addition, a data set of previously unprocessed baboon reovirus (BRV) images served to test out our automated, independent processing strategy.
2. Material and Methods
2.1 Images of virus samples
All image data used in this study were recorded on Kodak SO-163 films by standard low-dose, cryoEM methods (Baker, et al., 1999) (Table 1). With the exception of the sea bass nodavirus (SBNV) data, all images were recorded in FEI/Philips CM200 FEG transmission electron microscopes at nominal magnifications of ×38,000 or ×50,000. The SBNV images were recorded in a Philips CM120 microscope equipped with a LaB6 filament. All micrographs were digitized at 7μm intervals on a Zeiss SCAI microdensitometer and the scanned images were bin-averaged or interpolated to yield effective pixel sizes as listed in Table 1. For the analyses of Dengue virus (DENV), Sindbis virus (SINV), and Paramecium chlorella virus, type 1 (PBCV-1), the particle images were from previously published data (Zhang, et al., 2003a; Zhang, et al., 2002; Yan, et al., 2000). Samples of SBNV and baboon reovirus (BRV) were isolated as described (Thiery, et al., 2004; Duncan, et al., 1995).
Table 1.
Comparison of random model computations for five test sets of virus image data.
| Virus | Family | D* (nm) | Radii^ (nm) | M+ | Defocus (μm) | P3D/PB† | Pixel Size (Å) | Map size‡ | Map Resolution (Å) | Time # (mins) | Cycles § | Search models & |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SBNV | Nodaviridae | 37 | 9–19 | 1 | 5.6 | 111/161 | 2.8 | 1653 | 41 | 3 | 4 | 4 |
| DENV | Flaviviridae | 46 | 16–24 | 10 | 3.4–4.3 | 90/144 | 2.8 | 2213 | 39 | 4 | 10 | 2 |
| SINV | Alphaviridae | 75 | 16–35 | 1 | 3.6 | 93/127 | 1.8 | 4413 | 30 | 15 | 5 | 6 |
| BRV | Reoviridae | 90 | 27–45 | 1 | 3.4 | 50/76 | 3.7 | 3113 | 35 | 10 | 5 | 2 |
| PBCV-1 | Phycodnaviridae | 190 | 74–96 | 6 | 1.4–1.9 | 138/221 | 4.0 | 5113 | 38 | 40 | 4 | 6 |
D = maximum diameter
Radii = the range of radii (r1 and r2) selected for origin and orientation determinations
M = number of micrographs
P3D = number of particles used to compute 3D reconstruction
PB = total number of particles boxed from micrographs
Map size = 3D map dimension in voxels
Time = elapsed time for each iteration
Cycles = number of iterations needed to reach the resolution listed in this table
Search models = the number of suitable models obtained from total of ten 10 different random models, each processed for ten iterations.
2.2 Image processing
2.2.1 Generating a file of random orientations
The program randGen was used to assign a random orientation (θ, φ, ω) within the icosahedral asymmetric unit to each particle in a data set. At execution time, the numerical values that define the process id and program start time were used to provide a seed for the random number generator to ensure that a given set of image data would never receive the same set of random orientations regardless of how many times the same data were processed. A unique (θ,φ) value was assigned to each particle image. Each (θ,φ) was selected from a grid of 2860 possible combinations within the asymmetric unit, with the θ values sampled at 0.5° intervals and the φ values sampled at 0.5°/sin(θ) intervals. An integer value of ω, randomly selected from the 360 potential values ranging between 0° and 359°, was assigned to each particle image. RandGen outputs an ASCII file that contains five floating point values for each particle, representing the (θ, φ, ω, x, y) parameters needed to compute a 3D reconstruction from the set of images. The origin (x, y) of each particle was set equal to the geometrical center of the box used to extract the particles from the digitized micrograph. Hence, all particles are initially assigned the same (x, y) coordinates.
2.2.2 Generation of an initial random model (MODELrand)
The basic steps involved in producing a 3D density map (MODELsrch(n)) that can be used as a search model for an entire data set of images are summarized as a flow chart in Fig. 1. In the first step, an initial 3D model (MODELrand) is computed using the program P3DR (Marinescu, et al., 2003) from a subset of the images (usually 100–200 particles) that have been assigned the (θ, φ, ω, x, y) values as described above (§2.2.1). As expected, the greater the number of images included in the computed map, the more closely MODELrand approximates a spherically symmetric object.
Fig. 1.
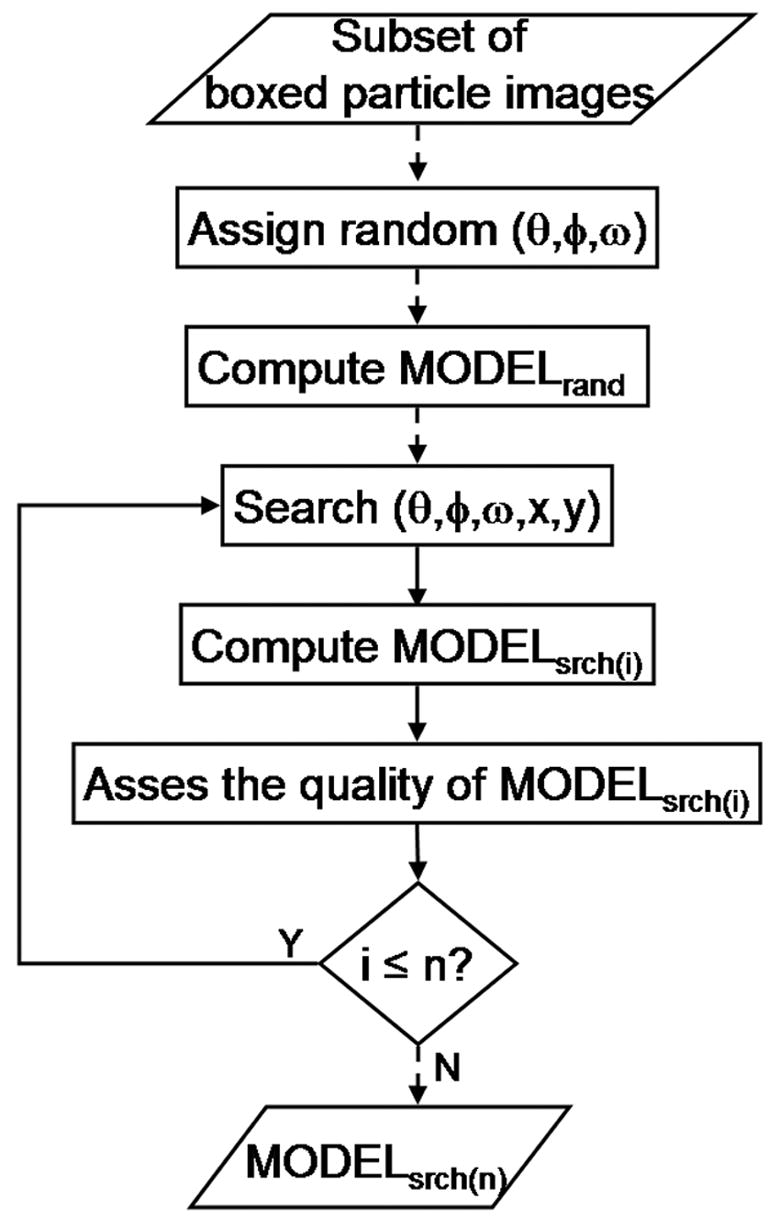
Random model method. The flowchart illustrates the primary computational operations involved in generating a suitable search model (MODELsrch(n)) from an initial random model (MODELrand). MODELrand, the initial 3D model, is computed from a subset of particle images that are assigned random orientations and an origin at the center of the square box used to window the images from a digitized micrograph. A new set of origin and orientation parameters for each particle are determined with program PFTsearch by comparing each image against a database of projections computed at regular angular intervals from MODELrand (Baker, et al., 1996). Based on this updated set of parameters, a new model, MODELsrch(1), is computed and the above process is repeated with a database of projections computed from MODELsrch(1). Each new search model is tested for its suitability in starting the process of determining parameters for a complete data set of particle images. The procedure depicted here is accomplished with the program AUTO3DEM, which, by default, executes ten iterations of the process to yield MODELsrch(10). Dashed lines indicate one time operation and solid ones indicate iterative operations. All models were computed with icosahedral symmetry imposed using program P3DR.
Tests were conducted both with and without corrections being made to image data to compensate in part for the effects of the microscope contrast transfer function (CTF). The defocus level and astigmatism in each micrograph was estimated using the interactive graphics program, RobEM (http://cryoem.ucsd.edu/programDocs/runRobem.txt). CTF corrections (Bowman, et al., 2002) were implemented in the 3D reconstruction program P3DR during the generation of initial random models. Tests performed with uncorrected data generally led to a similar rate of success if the particle images were all recorded at the same or very similar defocus levels (data not shown). However, as CTF correction has become a standard part of many image processing procedures, it is used by default in the RM method. All of the results reported here have been generated with the use of corrected images. As expected, CTF correction becomes mandatory when the images are derived from multiple micrographs recorded at different defocus levels.
2.2.3 Generation of search model (MODELsrch(n))
Program PFTsearch (Baker, et al., 1996) uses MODELrand to produce a database of uniformly spaced projections in the asymmetric unit. Raw particle images from the data subset were CTF corrected (§2.2.2) and compared to the model projections to generate an estimate of the true (θ, φ, ω, x, y) parameters for each particle. Generally, about 80% of the particle images were used to compute a new 3D map (MODELsrch(1)) after the first iteration of the search procedure (Fig. 1). These particle images were selected on the basis of their correlation to MODELrand, with a default threshold set equal to a value one standard deviation below the mean correlation coefficient computed for all images. MODELsrch(1) was then used to initiate the next (i.e. second) cycle of origin and orientation determination for each particle in this subset. This process was repeated and we estimated the quality of the model generated after each cycle by conventional FSC (Fourier shell correlation) criteria (Harauz, et al., 1986). The quality of MODELsrch(i) was estimated by comparing FSC values for spatial frequencies from ~1/60 to ~1/30 Å−1 between two 3D maps (‘odd’ and ‘even’), each combining half the input data. The number of iterations (n) used in the search procedure is a user defined variable. Ten iterations generally lead to a model that does not change appreciably with further iterations. Hence, this number is the default used for automation of the method as described below (§2.2.4).
2.2.4 Automated processing and selection of best MODELsrch
The procedure used to generate search models is tedious and repetitious, and furthermore, not all search models are suitable for processing of complete image data sets (see Results). In practice, several random models must be generated from the same subset of images to produce at least one reliable search model, and this necessitates the use of an automated processing scheme. For this purpose, the program AUTO3DEM was adapted to generate any number of random models from which the ‘best’ (defined below) search model is then selected for processing the complete image data set. AUTO3DEM iteratively executes the programs used to globally search particle parameters. At each iteration, it also optimizes a number of program variables that previously had to be manually adjusted by the user (see §2.2.5). For the examples described in Results, the default conditions for AUTO3DEM were used. For each subset of virus images, the process (Fig. 1) was repeated to generate ten different random models, each of which was subjected to ten search iterations to produce a new MODELsrch(10). In our experience, the mean FSC value for spatial frequency data ranging from ~1/60 to ~1/30 Å−1 provides a reliable metric to distinguish among all the search models. On this basis, AUTO3DEM was used to automatically select the best MODELsrch(10) for subsequent model-based determination and refinement of parameters for all images in the complete data set (Ji, et al., 2006; Baker, et al., 1996).
The quality of each of the ten MODELsrch(10) maps was also inspected visually with the program RobEM and compared with published or known structures determined at higher resolution and derived from more extensive data sets. In several instances, a number of the MODELsrch(10) maps would have made suitable search models and led to correct, refined 3D maps. The number of suitable (‘successful’) models generated in each of our five examples that started with ten different random models range from two to six (Table 1).
2.2.5 Real and reciprocal space filtering
Success of the PFTsearch program in producing a reliable search model is dictated in large part by the values assigned to a number of user-specified input variables. The most influential of these variables include specification of inner and outer particle radii (r1,r2) and selection of spatial frequencies (R1,R2) at which Fourier filtering is performed.
The ability of PFTsearch to determine correct (θ, φ, ω, x, y) parameters for each particle image is enhanced when the analysis is restricted to an annular window. The values chosen for variables r1 and r2 are specimen dependent, but they are generally selected to include the outer capsid, where the viral components are most likely to be organized with icosahedral symmetry. Image data masked outside the particle periphery (>r2) clearly removes any non-particle ‘noise’; data masked inside (<r1) mainly removes contributions to the image dominated by the asymmetric viral genome. The inner and outer radii used for each of the five examples presented in this study are listed in Table 1 and identified in the figures (see Results).
Low (<R2) and high (>R1) pass Fourier filtering was also used to mask out an annular window in the Fourier transform of each particle image. This restricted data analysis to those spatial frequencies dominated by signal from the icosahedrally-ordered viral components. As a first approximation, R1 was set equal to the reciprocal of one-fifth the diameter (D) of the particle (i.e. R1 = 5/D Å−1). The default value for D is set equal to the width of the square region boxed from the digitized micrograph, which is generally just larger than the particle diameter. A high-pass filter with this property assures that most of the spherically-symmetric, high amplitude part of the Fourier transform is excluded from analysis. In this study, the value set for R1 for each set of virus images was not changed during analysis. In contrast, the value of R2 chosen to create the low-pass filter (designed to remove high frequency noise from the analysis), was gradually increased as the resolution of successive search models improved. The resolution limit, and hence value for R2, was adjusted upward depending on the results of the FSC test (see §2.2.3) obtained after each cycle of processing. In AUTO3DEM, R2 was automatically set to the highest spatial frequency at which the FSC value never drops below 0.3.
2.2.6 Unbiased resolution assessment
AUTO3DEM was used to process a test data set of 702 BRV particle images to produce two independent 3D reconstructions. An unbiased measure of the data resolution was then assessed by AUTO3DEM. The details of this assessment with the BRV data set are presented in Results (see §3.5).
2.2.7 Hardware
Programs were executed on a Linux PC cluster consisting of ten 2.4-GHz Pentium IV processors. AUTO3DEM can also run on a single CPU Linux workstation.
3. Results
3.1 Test data
The robustness of our RM method was tested using cryoEM image data sets from five different icosahedral viruses. These include SBNV (Thiery, et al., 2004), PBCV-1 (Yan, et al., 2000), DENV (Zhang, et al., 2003a), SINV (Zhang, et al., 2002), and BRV (Duncan, et al., 1995) (Table 1). These viruses span a large size range and their outer capsids exhibit a variety of surface features. For example, the diameters of the virions range from a low of 30nm for SBNV to a maximum of 190nm for PBCV-1, and the pixel dimensions of the reconstructed 3D maps of these viruses vary between 1653 (SBNV) and 5113 (PBCV-1). Regarding surface features, SINV displays prominent trimeric ‘spikes’ but DENV has a relatively smooth and featureless exterior. In addition, the test data include micrographs recorded over a significant range of defocus values (1.4–5.6 μm; Table 1).
3.2 Need to generate multiple random models
As illustrated in five separate examples (Figs. 2–6), when the RM procedure (Fig. 1) leads to a suitable search model (i.e. one that can be used to determine reliable origin and orientation values for all images in a particular data set), often only four or five iterations within the procedure are needed. However, in less favorable cases, such as with DENV, ten or more iterations are needed (Table 1). In any event, ten iterations did not always lead to a suitable search model each time the procedure was run. Thus, with every test set of test images, the procedure was repeated ten times to generate ten separate random models and the success rate at obtaining suitable search models was assessed (Table 1). For example, six out of ten such trials led to suitable search models for the SINV and PBCV-1 data, but only two of ten trials worked for DENV and BRV (Table 1). It is important to note that, even for those real image data that proved most problematic, suitable search models did emerge. The use of AUTO3DEM, which generates ten random models by default, certainly makes it easier to run more, or as many tests as necessary, to yield at least one reliable search model.
Fig. 2.
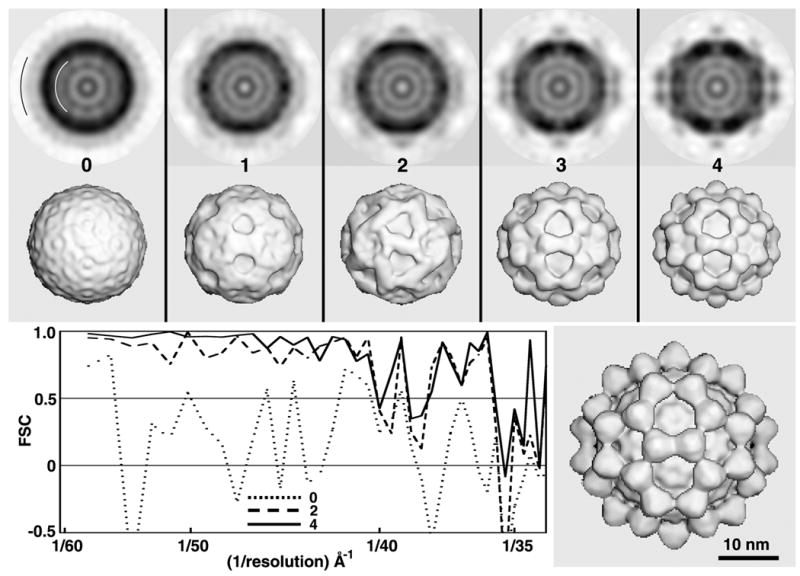
Test of RM method with SBNV images. Central sections (top row; darker shades correspond to higher particle densities) and shaded-surface representations (2nd row) computed from 3D maps corresponding to MODELrand (cycle 0) and MODELsrch(1-4) (cycles 1–4). All 3D maps shown in the top two rows were corrected for effects of the microscope CTF as described in Methods and were computed to a resolution limit of 35 Å. The black and white arcs in the first central section (top left) identify the upper and lower limits of particle radii included in the calculations. FSC plots (lower left) provide a quantitative measure of the quality of each search map. Lower right: CTF-corrected, 3D cryo-EM reconstruction of SBNV, computed to 25Å resolution from 1500 particle images extracted from 29 micrographs.
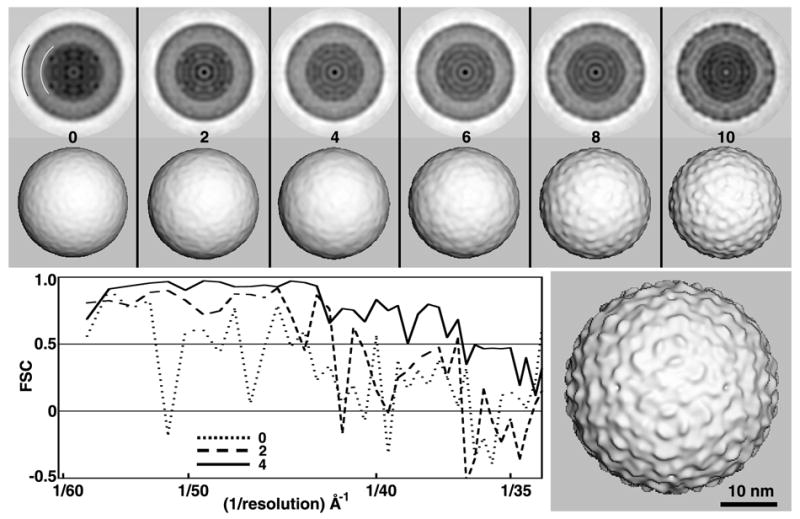
Fig. 6.
Test of RM method with BRV images. All 3D maps shown in the top two rows were CTF-corrected and computed to a resolution limit of 30 Å. The lower right panel shows a CTF-corrected 3D reconstruction of BRV, calculated from 500 particles extracted from 27 micrographs with the effective resolution determined to be 16Å based on the split data set processing scheme illustrated in Fig. 7 (see Results §3.6).
3.3 Selection of best search model
FSC criteria provided the metrics to distinguish suitable and unsuitable search models for each experiment. In our experience, the mean FSC value for data between spatial frequencies ranging from ~1/60 to ~1/30 Å−1 provided a reliable measure to determine if a particular search model was suitable for origin and orientation refinement of the entire image data set. AUTO3DEM selects as best the MODELsrch(n) that has the highest mean FSC value. In each of the five examples reported, AUTO3DEM selected a search model that would lead to an accurate and reliable 3D reconstruction using larger numbers of images.
3.4 Examples
The most successful results for each of the five test cases are documented in Figs. 2–6. The top row of each figure shows a series of central sections and the second row shows a corresponding series of shaded-surface representations, both obtained from the 3D reconstructions generated at the end of one cycle of the RM procedure (Fig. 1). The initial random model is generated in cycle zero when a 3D map is computed from a subset of virus images to which random orientations have been assigned. The radii chosen to mask the images of each virus are highlighted by white (r1) and black (r2) arcs in the top left panel of each figure. Ten full cycles were employed each time a search model was produced, but only a few, representative results are shown for each virus. Likewise, the lower left panel of each figure portrays FSC plots for the random model (cycle 0) and for two of the subsequent search models. Finally, the lower right panel in each figure is an enlarged view of either a published (Zhang, et al., 2003a; Zhang, et al., 2002; Yan, et al., 2000) or unpublished (SBNV and BRV) 3D map reconstructed from a more extensive data set at 25Å or higher resolution. These maps provide a qualitative check of the results obtained using the RM method and demonstrate that the number of iterations needed to derive a reliable search model varies for different data sets.
3.4.1 Sea bass nodavirus (SBNV)
Fish nodaviruses are small (~30nm diameter), non-enveloped viruses (Family Nodaviridae; Genus Betanodavirus) that contain a bipartite genome comprised of single-stranded, positive-sense RNA molecules (Thiery, et al., 2004). The structure of recombinant, virus-like particles of malabaricus grouper nervoius necrosis virus was recently determined by cryoEM and image reconstruction and showed that it has a T=3 capsid with 60 prominent protrusions that encircle the three- and five-fold axes of the icosahedral particles. Structural results on a different fish nodavirus (SBNV) illustrated here are based on a 161-particle data set from one cryoEM micrograph. These data were used to derive a search model, reliable to ~40Å resolution, after just four cycles of computation and about 12 minutes of elapsed time (Fig. 2 and Table 1). After cycle one, MODELsrch(1) already exhibited distinct features. After cycle three, the features resolved into 60 recognizable domains, which clearly resemble those present in the correct structure. Interestingly, the FSC plot after cycle two already indicated that MODELsrch(2) would lead to a suitable search model.
3.4.2 Dengue virus (DENV)
DENV (Family Flaviviridae) has a relatively smooth outer capsid that surrounds a lipid bilayer and a poorly-defined nucleocapsid core (Zhang, et al., 2003a). Samples of many enveloped viruses like DENV are often too low in concentration to give a sufficient number of particle images in a single micrograph to carry out a rigorous test of the RM method. Consequently, we tested the procedure with 144 particle images extracted from 10 micrographs (Table 1). Even so, the DENV images proved to be the most challenging ones we analyzed in this study. We believe this is primarily a consequence of the rather featureless nature of the DENV capsid, and the fact that images with well-defined features are more amenable to analysis by the PFTsearch program (Baker, et al., 1996). For the example illustrated in Figure 3, about 40 minutes of elapsed time were required to generate a suitable search model after 10 cycles. The smooth DENV capsid morphology also made it difficult to visually assess whether the 3D map generated at any cycle would make a suitable search model.
Fig. 3.
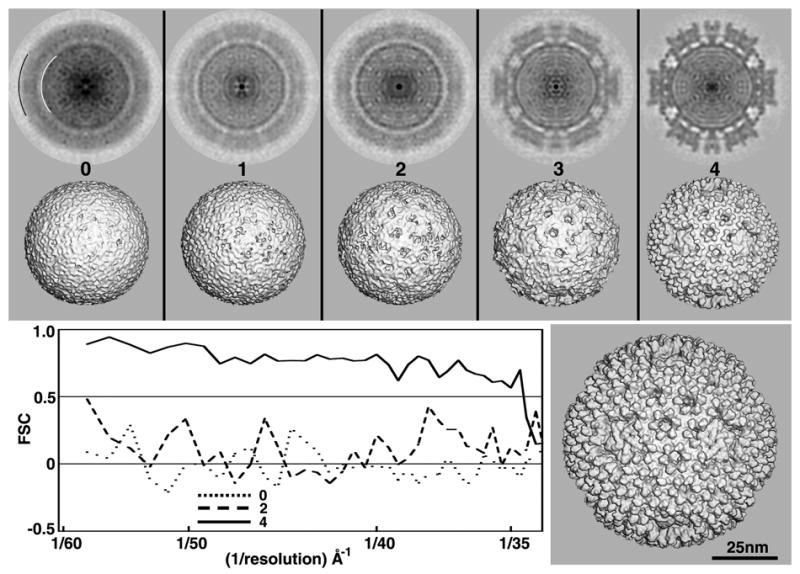
Test of RM method with DENV images. Except for obvious differences, descriptions for all panels in this as well as Figs. 4–6 parallel those given in the caption to Fig. 2. All 3D maps shown in the top two rows were CTF-corrected and computed to a resolution limit of 25 Å. The lower right panel shows a CTF-corrected 3D reconstruction of DENV, computed to 25Å resolution from 1691 particle images extracted from 78 micrographs (Zhang, et al., 2003a).
Interestingly, in contrast to the analysis of SBNV data (Fig. 2), the FSC criteria generated in the analysis of DENV appear to be misleading. Indeed, the FSC plot indicates that MODELsrch(4) is reliable to about 45Å resolution, though the map is essentially a featureless sphere resembling the initial random model from which it arose. Thus, in this particular example, the FSC criteria overestimated the quality of the map produced early in the search procedure. This may arise because at low resolution the two maps used to compute the FSC plot both contain similar, smooth protein shells that correlate well within the annulus chosen for analysis (arcs in Fig. 3).
Of the ten random models generated from the subset of DENV images, two led to suitable search models (Table 1). In each of these instances, a full ten cycles were needed. The resultant search models exhibited genuine substructural features both within and at the surface of the outer capsid as seen in published 3D reconstructions (Zhang, et al., 2003a). Despite the low (20%) success rate in our experiments with DENV images, it is nonetheless noteworthy that the RM method did work with images of relatively featureless particles including the 31 nm diameter Adeno associated virus (Yan, et al., 2006). Analysis of similar specimens may therefore necessitate use of ten or more random models to assure that at least one suitable search model is obtained.
3.4.3 Sindbis virus (SINV)
SINV (Family Alphaviridae) contains 240 copies each of three structural proteins (glycoproteins E1 and E2 and nucleocapsid protein C), a lipid bilayer membrane, and a single-stranded RNA genome (Zhang, et al., 2002). The outer capsid of SINV exhibits prominent and characteristic surface features (‘spikes’), which arise from eighty E1-E2 heterotrimers, organized with T=4 quasi-symmetry(Zhang, et al., 2002). In this study, 127 SINV images from a single micrograph were analyzed. Five cycles of computation and about 75 minutes of elapsed time were required to generate a reliable search model in the experiment illustrated in Figure 4. After two cycles, the search model (MODELsrch(2)) already exhibited outer capsid features characteristic of alphaviruses (Zhang, et al., 2002), but the FSC plot showed little or no evidence that a suitable search model would be produced (data not shown). However, after five cycles, visual and FSC comparisons both clearly indicated that a reliable search model had emerged (Fig. 4). In contrast to the relatively featureless DENV structure, SINV contains many distinct features over a wide range of radii, but these internal features only begin to appear at cycle four. Indeed, after cycle four the FSC plot significantly improved relative to that of cycle three (data not shown). For the subset of SINV images used in this study, six out of ten initial random models led to suitable search models (Table 1).
Fig. 4.
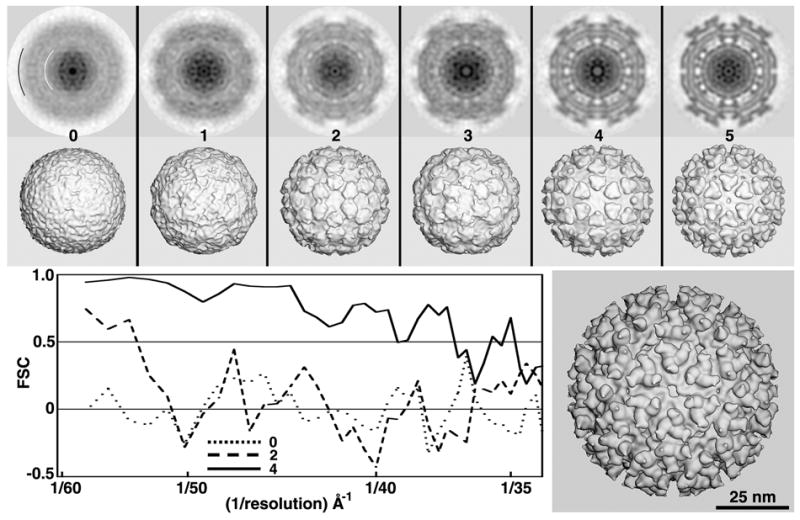
Test of RM method with SINV images. All 3D maps shown in the top two rows were CTF-corrected and computed to a resolution limit of 35 Å. The lower right panel shows a CTF-corrected 3D reconstruction of SINV computed from a large data set (~15,000 particles from 76 micrographs) with the map only rendered at 25Å resolution.
3.4.4 Paramecium bursaria chlorella virus, type 1 (PBCV-1)
PBCV-1 (Family Phycodnaviridae) is the largest and most complex virus among those tested, with a maximum diameter of 1900 Å and a map dimension of 511 pixels (Table 1). The outer capsid of PBCV-1, which contains 5,040 protein subunits arranged with T=169 quasi-symmetry, has a distinct icosahedral shape and encapsulates a lipid bilayer membrane, a ~330 kbp double-stranded DNA genome and about 50 other structural proteins (Yan, et al., 2000). Typical micrographs of vitrified PBCV-1 samples contain too few (~40) virion images to compute reliable 3D reconstructions even at low (40–50 Å) resolution. Hence, for this study we used a set of 221 particle images, obtained from six micrographs recorded at defocus settings ranging between 1.4 and 1.9 μm underfocus (Table 1). Four cycles and about 3 hours of computation led to a search model deemed reliable to about 38Å resolution (Fig. 5). Like SINV, six of ten random models generated from the PBCV-1 images led to suitable search models (Table 1). With PBCV-1, except during the first two cycles, visual and FSC plot analyses correlated well. At very low resolution (< 1/52 Å−1), MODELrand yielded higher FSC values than those, for example, from MODELsrch(2). This apparent anomaly merely reflects the dominant spherical nature of MODELrand as well as the odd and even maps generated to compute the FSC curve. Hence, the structure of the random model is predominantly artificial, since the PBCV-1 shell has a distinct icosahedral morphology (Fig. 4, top row, cycle four).
Fig. 5.
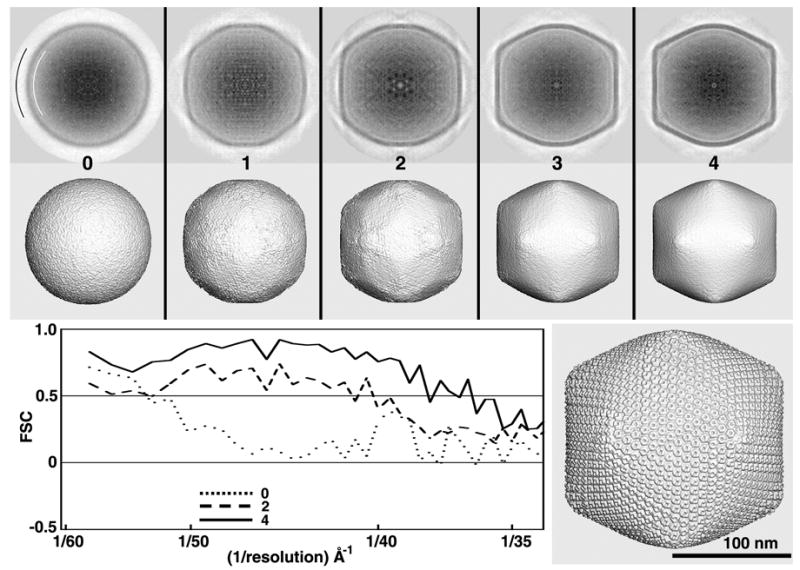
Test of RM method with PBCV-1 images. All 3D maps shown in the top two rows were CTF-corrected and computed to a resolution limit of 30 Å. The lower right panel shows a CTF-corrected 3D reconstruction of PBCV-1, computed to 25Å resolution from about 1,000 particle images extracted from 45 micrographs (Yan, et al., 2000).
3.4.5 Baboon Reovirus (BRV)
BRV (Family Reoviridae; Genus Orthoreovirus) is a non-enveloped virus with a multi-shelled protein capsid and a segmented, double-stranded RNA genome (Duncan, et al., 1995). At the time of this study the structure of BRV was unknown, but its relation to other orthoreoviruses indicated that it might share similar distinct features. Consequently, new cryoEM images of BRV provided a particularly good test of the RM procedure. A data set of 76 boxed particle images was obtained from a single micrograph. About 50 minutes of computation and five cycles were required to obtain each of two suitable search models (Table 1), both of which were reliable to ~35Å resolution. One of these is shown in Figure 6. Surprisingly, although the BRV structure has prominent surface features, only two of the ten RM procedures generated suitable search models. We attribute this to the limited number of particles included in this initial experiment with BRV images, because further tests with more particle images led to higher success rates. For example, when 350 particle images from 10 micrographs recorded at 3.4 to 4.9 μm underfocus, six out of ten random models led to suitable search models (data not shown).
3.5 Unbiased resolution assessment
A goal for many investigators has been to produce an automated image processing procedure that enables scientists of diverse background and level of expertise to produce reliable 3D reconstructed maps at the highest possible resolutions from images of vitrified specimens. In addition, it is important to have a reliable measure of map resolution (Grigorieff, 2000). The resolution achieved in 3D cryoEM density maps has traditionally been evaluated by computing the FSC between two separate reconstructions (Harauz, et al., 1986; van Heel, et al., 2005). Frequently, the whole data set is refined against the same model and is split into two subsets only to calculate resolution (Baker, et al., 1999), or, if the data set was split initially, the subsets are analyzed with the same starting model (Dryden, et al., 1998). Therefore, in either instance, the FSC method will generally overestimate the similarity of two reconstructions (Grigorieff, 2000; Stewart, et al., 2004; Shaikh, et al., 2003). To obtain a more accurate resolution assessment, the whole data should be divided initially and each subset start with an independent, ab initio model. The new BRV images provided an excellent data set to assess our implementation of an unbiased 3D image reconstruction scheme for icosahedral particles (Fig. 7).
Fig. 7.
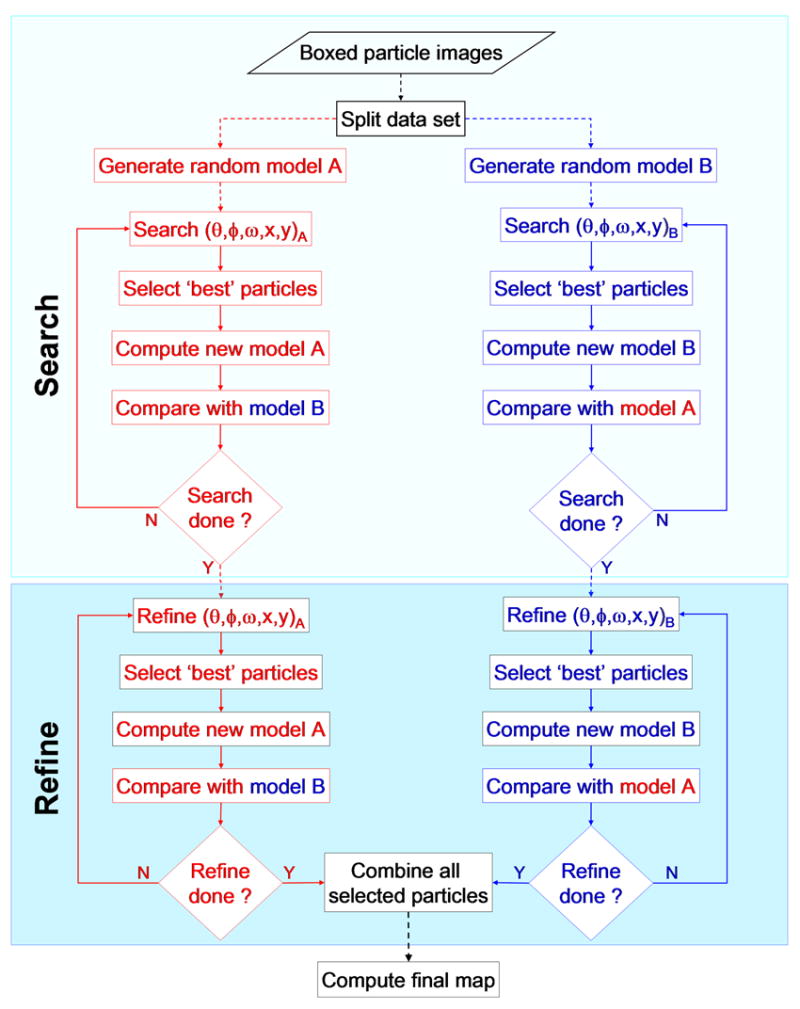
Split data processing scheme used to generate a reliable estimate of 3D map resolution. An entire set of boxed particle images is evenly divided into two subsets (A and B) and two independent processing pathways are followed to compute separate 3D reconstructions from images selected from each subset. Each subset is used to generate its own initial (random) model, and computations on the separate image subsets are run in parallel and include iterations of global search (top boxed region) and local refinement (bottom boxed region). The two data sets are treated independently until the last step when a final map is produced either by averaging the two independent maps or by computing a 3D map directly from images selected from the two subsets. Prior to the last step, images from a given subset are only compared to projections obtained from the corresponding models generated from the images in that subset. Progress is monitored during any iteration of SEARCH or REFINE by comparing corresponding models in opposite branches. Comparisons may be performed by qualitative (e.g. by direct visualization of density or difference density maps) and quantitative (e.g. by computing FSC plots) means. Dashed lines indicate one time operation and solid ones indicate iterative operations.
The BRV data set, consisting of 702 particle images (27 micrographs; defocus range = 2.5–6.5 μm), was evenly divided into two subsets, A and B. Rather than further subdividing these data sets, a separate MODELrand was calculated with all 351 particles in each subset, and each model was used to initiate an independent 3D structure determination as illustrated in Fig. 7. For both of these subsets, because the very first random model led to search models that were clearly suitable after the fifth cycle, we did not need to generate nine additional models as was done for all the previous test data sets (see §3.4). However, the A random model and images yielded a suitable model one or two cycles before the B model and images. It is uncertain whether this different converging was a consequence of the random model or the images. The two MODELsrch(5) maps were then used to compute independent 3D image reconstructions.
Independent processing of the two data sets was carried out by AUTO3DEM for additional cycles in SEARCH mode (Fig. 7, top) to further improve the two reconstructions. In SEARCH mode, the program PFTsearch compares each particle image with a database of projected views of the current search model and the best match defines the approximate view orientation for that particle. An unbiased assessment of the similarity of the two maps obtained at the end of each cycle was determined by FSC analysis (Fig. 8). After five cycles, the two search models showed no further improvement, and AUTO3DEM switched to REFINE mode.
Fig. 8.
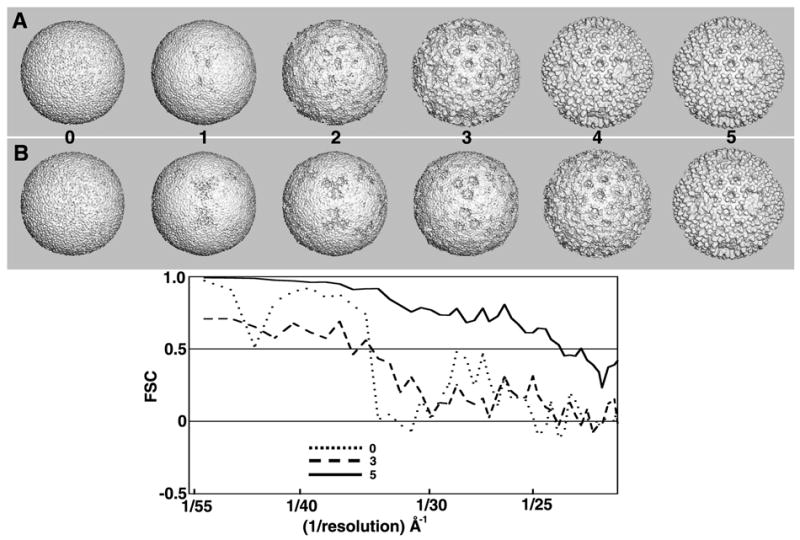
Split data processing of BRV images. The rows of images at the top show shaded-surface representations of the 3D maps (MODELrand and MODELsrch(1-5)) generated from two independent subsets (A and B) of BRV images when processed using the two-branch SEARCH mode scheme depicted in Fig. 7. The processing of subset A images converges towards a suitable search model faster than the subset B images, but after five cycles both processing branches lead to models of similar quality (~24 Å resolution). By chance, the absolute handedness of both maps yielded T=13d quasi-symmetric structures (i.e. the dextro enantiomers). To be consistent with all other orthoreoviruses studied to date (Zhang, et al., 2005b), we reversed the hands of the two BRV maps to make each a T=13l structure (the laevo enantiomer) consistent with that shown in Fig. 6.
In REFINE mode the program OOR (Zhang, et al., 2003b) was used to refine the orientations on a smaller angular grid that spans a limited region of the asymmetric unit and was centered at the orientation determined in the previous cycle of analysis (Fig. 7, bottom). Refinement of these data yielded a final 3D reconstruction at about 16Å resolution (FSC = 0.5 threshold) when the best 500 BRV images were combined (Fig. 6, lower right panel). The same set of BRV images, when processed according to traditional procedures (Baker, et al., 1999), also yielded a reconstruction at about 16Å resolution (data not shown). A detailed analysis of the BRV structure will be published separately.
4. Discussion
A primary objective in any image reconstruction project is to generate a reliable 3D density map of the sample under investigation and to do so rapidly and with minimal user intervention. The RM method was designed to help initiate the process of calculating a low resolution map from a small subset of images of icosahedral particles, and additionally may be an effective tool for analyzing images of non-icosahedral particles.
The low resolution map obtained from the RM method serves as an unbiased, reliable starting model for determining initial origin and orientation parameters for a complete data set of images. The effectiveness of this straightforward method has been demonstrated in analyses of five different data sets of icosahedral virus images. To assure that optimal results are obtained, several issues must be addressed. These include knowing how to assess if a particular model is suitable for data refinement, knowing how to judge the likelihood that the method will work with a particular data set, dividing the data in half and processing each independently to provide an accurate measure of resolution achieved, checking for consistency of handedness in independent reconstructions, and making use of efficient, automated processing procedures that enhance data processing for researchers regardless of their level of expertise.
4.1 Assessing the suitability of search models
As our tests have demonstrated, not all search models generated by our RM procedure prove suitable for refinement of particle origins and orientations (Table 1). Thus, it is important not simply to identify the best among a set of generated search models, but also to assure that the chosen model is adequate to initiate a successful refinement of the full data set of particle images. Experience indicates, as expected, that a model exhibiting genuine structural features is one most likely to yield a successful refinement. As shown in the five examples, the suitability of a particular search model is routinely evaluated both qualitatively by visual assessment and quantitatively by FSC analysis. However, subjective and objective criteria such as these sometimes yield a contradictory evaluation. We therefore recommend using an objective, quantitative approach. Even experienced researchers, who routinely may be able to correctly determine the suitability of a given search model based solely on a visual assessment may be misled when studying a new specimen of unknown structure.
We have used standard FSC criteria (Harauz, et al., 1986) to assess quantitatively the quality of models generated by the RM procedure. Generally, when a set of n (usually ten) search models emerges from the RM computations, the model with the highest mean FSC value is judged the best one. However, if the history of each search model is monitored as it emerges during iterations of the RM procedure, the FSC metric can be unreliable, particularly during the first few cycles of computation. Indeed, the FSC curves obtained in the initial cycles of the RM procedure are extremely noisy and it is nearly impossible to accurately judge at this stage whether a particular random model will lead to a suitable search model. However, if after the third or fourth cycle the FSC curve exhibits a more predictable, monotonic behavior (i.e. with high values at low spatial frequencies followed by progressively decreasing values at higher frequencies), the resulting search model is more likely to be suitable for subsequent data refinement (Figs. 2–6, 8). Conversely, when the FSC curves continue to show erratic fluctuations at all spatial frequencies even after several cycles, the RM procedure is less likely to yield a suitable search model.
The erratic character of the FSC plots generated during initial cycles of the RM procedure are consistent with the expectation that the odd and even 3D maps being compared will necessarily be dominated by high levels of noise. These maps are noisy because they are derived from a limited set of particle images, the majority of whose origins and orientations are certainly incorrect. In fact, during the initial cycles of computations with the DENV, SINV, and PBCV-1 test data, the FSC metric was either unreliable or inconsistent with judgments based on visual assessments made by experienced researchers. For the DENV and PBCV-1 data, the initial reconstructed maps only exhibited dominant, spherically symmetric features, and this led to potentially misleading but high FSC values at low spatial frequencies. An opposite result was obtained in the analysis of the SINV data. Here, despite visual cues that the procedure was already converging by the second cycle, the FSC values remained quite low and noisy. The SINV structure has distinct features that span a wide range of radii and the FSC curves simply provide a global measure of 3D map consistency averaged over all selected radii. An experienced observer may, however, focus attention only on specific features such as the prominent spikes on the outer capsid surface, and these might be well represented even in the earliest reconstructed maps. For the SINV example, the qualitative and quantitative measures of map quality concur after cycle four, at which time internal features are better resolved.
Based on the above observations with our test data, any measure that monitors search model suitability is likely to be specimen dependent. In particular, the FSC metric is most reliable if used after at least four cycles of the RM procedure have been completed. Prior to this, any qualitative and quantitative measure of success is likely to yield inconsistent or misleading results.
The RM protocol, as implemented in the AUTO3DEM environment, was designed to allow experienced users to exit the process after any cycle, if for example the specimen is a familiar one and it is obvious that a suitable search model has already emerged. In fact, AUTO3DEM provides this flexibility to any user since it continuously evaluates and ranks all of the n potential search models and identifies the best one. Again, we would caution against using subjective, visual comparisons to monitor the process and instead recommend that the process be allowed to proceed to completion so final evaluation can be based on more rigorous, quantitative criteria.
In situations where the specimen is new and of unknown structure, additional criteria may be used to judge the suitability of generated search models. For example, existing knowledge about similar specimens such as biochemical data on subunit composition and stoichiometry would likely provide useful clues to evaluate models. Also, if an incorrect starting model is used to analyze an entire data set of images, refinement will often stall at relatively low resolution (usually no better than 35Å) despite exhaustive computation. Such behavior signals a need to try one or more alternative starting models. Lastly, it is useful to recognize that the standard deviation of density values in maps of reliable models generally far exceeds that in maps of erroneous models (Cantele, et al., 2003). Hence, standard deviation can be an additional metric to help assess the suitability of different search models.
4.2 Reliability of the random model method
Like most algorithms of its kind, the RM procedure is unlikely to succeed with every experimental data set. Though we have tested the procedure using a relatively diverse range of icosahedral virus images, some data will prove to be more challenging than others. Based on our experience, the robustness of the RM method is influenced by several factors, the most significant of which include the nature of the virus structure, the defocus levels of the particle images, and the number of particles included in the data subset. In addition, we presume that a skewed or non-uniform distribution of particle orientations represented in the data subset would negatively influence the success of the procedure, though we have yet to test this hypothesis.
The morphology of the specimen under investigation not surprisingly has a dominant influence on the success rate of the RM procedure. In the examples tested, viruses with prominent features, such as SBNV and SINV, showed higher success rates compared to less distinct viruses like DENV. The number of suitable search models that emerge from multiple runs of the RM procedure provides one metric for estimating the difficulty in generating a reliable starting model from a given set of images.
The defocus levels of the particle images also directly influence the success rate of the RM procedure. Indeed, particles selected from micrographs imaged such that the first maximum of the CTF occurred within the range of spatial frequencies used for analysis (~1/30–1/60Å) were more likely to generate positive results. The defocus levels of the experimental images used in this study ranged between 3.4 and 5.6 μm underfocus (yielding CTF maxima at 1/40 to 1/50Å for 200 keV electrons), except for PBCV-1, which was recorded closer to focus (1.4 to 1.9 μm) (Table 1). The PFTsearch program is most effective in determining origin and orientation parameters of particle images that contain strong icosahedral signal. That signal generally arises from large morphological features on viral surfaces (e.g. capsomers) and the contrast of these features is enhanced by defocusing to a larger extent. For PBCV-1, the sharply defined icosahedron morphology of the 190nm diameter capsid remains dominant in images recorded over a wide range of defocus settings, and includes those at < 1.5 μm underfocus. In contrast, a preliminary examination of 100 SINV particle images extracted from a micrograph recorded 1.4 μm underfocus did not lead to any suitable search models even after ten trials of the RM procedure (data not shown). Based on the above observations and additional experience, it is generally best to work initially with images recorded at high levels of defocus (first CTF maxima at ~1/40Å or smaller). These images most likely lead to a suitable search model that can subsequently be used to analyze all images including those recorded much closer to focus.
The success of the RM procedure is also influenced by the number of particle images included in the data subset. The number need not be large. A starting model computed at moderate resolution (30–40Å) from a small number of images usually contains sufficient detail to enable PFTsearch to correctly define an initial set of origins and orientations. Ideally, one would like to use as few particle images as possible, even one, to generate a suitable search model as rapidly as possible. In practice however, a minimum number of particles is required for each particular data subset and the number is influenced by the nature of the virus and the imaging conditions. If too few particle images are included, the reconstructed map may suffer from limited resolution and the densities will exhibit high levels of noise due to inadequate averaging and possible artifacts arising from under-sampling of the 3D Fourier transform. In this situation, the FSC metric will be unreliable and can easily lead to important variables being set incorrectly in the program PFTsearch.
The number of particle images included in the RM calculations are also bounded by an upper limit. Just as a MODELrand generated from a single particle image might fail because it deviates significantly from the true structure, a MODELrand computed from an infinite number of images might also fail because it would be perfectly spherically symmetric. All projected views of such a symmetric model are, in principle, identical, and hence incapable of discriminating different orientations for the experimental images. For each particular set of images there will be an optimum number of particles that will maximize the success of the RM procedure. With the BRV data, separate analyses including 76, 200, 350, 500 or 702 particle images and ten trials in each case led to success rates of 20%, 40%, 60%, 30% and 20%, respectively. Experience shows that data subsets containing 100–200 particle images at high defocus lead to at least one suitable search model.
Though it has yet to be tested, the RM procedure we describe here would likely fail if the subset of images is derived from particles that preferentially orient in the vitreous sample. Under such circumstances it would be necessary to collect images from tilted specimens (Dryden, et al., 1993) to provide a more random sampling of views within the asymmetric unit. This strategy should then enhance success with the RM procedure.
The five experimental sets of virus images used in this study all led to suitable search models. There is of course no guarantee that the method will work for every data set examined. Success is dictated by the nature of the data set itself, including sample consistency, micrograph quality, and method of data preprocessing. In challenging situations, where initially no suitable search model emerges, success might be achieved by working with particle images selected from higher defocus micrographs, by increasing or decreasing the number of images in the subset, or by generating more MODELrand maps.
4.3 Independent reconstruction protocol improves accuracy of resolution assessment
The resolution achieved in a 3D reconstruction study provides an important quantitative measure of the quality of the density map and reliability of features representative of the specimen structure. However, it is widely recognized that the FSC criterion used for estimating resolution in commonly practiced model-based procedures is imperfect (Grigorieff, 2000). This is in part because, during processing, images are not subdivided into subsets and processed strictly independently with different starting models, but instead all images are refined against a single model. Therefore, when resolution is assessed by splitting a set of images refined in this way followed by computing and comparing odd and even reconstructed maps, any bias introduced by use of a single model will overestimate the agreement between maps often cited as being ‘independent’.
The independent processing protocol we have described eliminates model bias by segregating the analysis into two separate pathways, each of which uses a different subset of images to generate a new starting model via the RM method. In this way, FSC comparisons of truly independent reconstructions provide a more accurate estimate of resolution, which is expected to be, at best, equal to that given by the traditional method. Indeed, in our analysis of 702 BRV images, the final reconstructions obtained using the two different methods were both estimated to be limited at 16Å resolution. At low resolutions such as this, the traditional and independent protocols are expected to lead to comparable results, especially when two starting models are very similar. However, at significantly higher resolutions (< 10 Å) where signal-to-noise in the image data rapidly drops, a larger discrepancy may be expected to occur in the estimates owing to a higher likelihood of noise bias being present in the single model processing scheme (Grigorieff, 2000). Despite these differences, in both processing schemes the signal-to-noise of the final map ought to exceed by √2 that in each of the reconstructions computed from half the images. Hence, in both instances the cited resolution probably underestimates the resolution achieved in the final reconstruction.
4.4 Resolution assessment requires consistent handedness in reconstructions
An accurate and reliable assessment of map resolution is only possible if the reconstructions being compared represent structures of the same, though not necessarily correct, enantiomer. For example, in our analysis of the BRV data, the two independent sets of images by chance both led to reconstructions that exhibited incorrect handedness (dextro) for the quasi T=13 lattice (Fig. 8, top panels). To be consistent with the known laevo hand of the mammalian reovirus capsid (Zhang, et al., 2003b), we reversed the handedness of the final BRV reconstruction (Fig. 6, lower right panel). In our split data processing scheme, there is an even chance that the two independent reconstructions will differ in their handedness. Hence, for resolution assessment, it is necessary to compute two FSC plots, with one map compared to each of the two possible enantiomers of the second map. The comparison that yields better FSC statistics signifies the correct relative hand of the second map, which is then used as the model for the next cycle of computation with its respective set of images. In any reconstruction study where the absolute hand of the structure under investigation is unknown, the true hand must be determined separately, for example by means of tilting (e.g. (Rosenthal, et al., 1984; Belnap, et al., 1997) or metal-shadowing (Stark, et al., 1984) experiments, or by fitting models of atomic structures to reconstructions representing both enantiomers (Zhang, et al., 2005a).
4.5 Enhanced automated processing
We continuously strive to streamline and make objective the process of generating 3D reconstructions at moderate to high resolution from a set of raw, boxed particle images. An ultimate goal is to maximize time focused on interpreting and analyzing structural results while minimizing time spent preprocessing data and setting up and running programs. As part of our efforts, we have developed the AUTO3DEM program and recently modified it to automatically calculate independent reconstructions and thereby obtain more reliable measures of map resolution. AUTO3DEM is designed to provide users of varying levels of expertise a flexible environment for efficiently producing reliable 3D density maps. Any of the many different input parameters can be set manually by the user or automatically by the program. Users also have the option of being able to modify the parameters during data processing.
AUTO3DEM automatically controls data flow between several sub-programs in the reconstruction procedure, and together these require the setting of ~80 different input variables. Thirty additional variables are intrinsically needed to control AUTO3DEM operation. At the end of each cycle of AUTO3DEM, about ten of these variables are reset to optimal values. The success of the RM method is sensitive to the setting of these parameters. Though AUTO3DEM allows parameter values to be manually reset at any point during the procedure, inexperienced users are discouraged from exercising this option as they are more likely than not to set them inappropriately. Hence, such users are encouraged to let AUTO3DEM automatically update and optimize parameter values. As one example, AUTO3DEM can automatically set the real and Fourier space limits used in the PFTsearch routine (§2.2.5). These limits may be relatively easy to define for a familiar structure but they will be less obvious for a new structure and particularly for less experienced users.
AUTO3DEM is currently being developed to completely free users, if they choose to, from having to define input parameters to the sub-programs. Ideally then, the user is simply left with the task of preprocessing the digitized images, which includes boxing individual particles, determining defocus values, and placing necessary files into a project directory. Preliminary tests on a variety of experimental data sets have provided proof of principle that AUTO3DEM can generate suitable search models with no user intervention.
Acknowledgments
We thank Drs. T. Richard and A. Schneemann for providing the SBNV clone, for particle purification and for sharing unpublished SBNV data; Dr. W. Zhang for providing DENV and SINV image data; Drs. M. Nibert and R. Duncan, and N. Olson and J. Kaufman for sharing unpublished BRV data; and R. Ashmore and Dr. C. Xiao for help with programming. This work was supported in part by research grants from the National Institutes of Health, USA, to T.S.B (GM R37-033050 and AI-055672) and to M. Yeager (GM-066087). We appreciate the reviewers’ insightful comments and suggestions, which have greatly enhanced the clarity and accuracy of this paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baker TS, Cheng RH. A model-based approach for determining orientations of biological macromolecules imaged by cryoelectron microscopy. J Struct Biol. 1996;116:120–130. doi: 10.1006/jsbi.1996.0020. [DOI] [PubMed] [Google Scholar]
- Baker TS, Olson NH, Fuller SD. Adding the third dimension to virus life cycles: three-dimensional reconstruction of icosahedral viruses from cryo-electron micrographs. Microbiol Mol Biol Rev. 1999;63:862–922. doi: 10.1128/mmbr.63.4.862-922.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belnap DM, Olson NH, Baker TS. A method for establishing the handedness of biological macromolecules. J Struct Biol. 1997;120:44–51. doi: 10.1006/jsbi.1997.3896. [DOI] [PubMed] [Google Scholar]
- Bowman VD, Chase ES, Franz AW, Chipman PR, Zhang X, Perry KL, Baker TS, Smith TJ. An antibody to the putative aphid recognition site on cucumber mosaic virus recognizes pentons but not hexons. J Virol. 2002;76:12250–12258. doi: 10.1128/JVI.76.23.12250-12258.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantele F, Lanzavecchia S, Bellon PL. The variance of icosahedral virus models is a key indicator in the structure determination: a model-free reconstruction of viruses, suitable for refractory particles. J Struct Biol. 2003;141:84–92. doi: 10.1016/s1047-8477(02)00577-4. [DOI] [PubMed] [Google Scholar]
- Caston JR, Belnap DM, Steven AC, Trus BL. A strategy for determining the orientations of refractory particles for reconstruction from cryo-electron micrographs with particular reference to round, smooth-surfaced, icosahedral viruses. J Struct Biol. 1999;125:209–215. doi: 10.1006/jsbi.1999.4085. [DOI] [PubMed] [Google Scholar]
- Cheng RH, Olson NH, Baker TS. Cauliflower mosaic virus: a 420 subunit (T = 7), multilayer structure. Virology. 1992;186:655–668. doi: 10.1016/0042-6822(92)90032-k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng RH, Reddy VS, Olson NH, Fisher AJ, Baker TS, Johnson JE. Functional implications of quasi-equivalence in a T = 3 icosahedral animal virus established by cryo-electron microscopy and X-ray crystallography. Structure. 1994;2:271–282. doi: 10.1016/s0969-2126(00)00029-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, Wolf E, Larvie M, Zak O, Aisen P, Grigorieff N, Harrison SC, Walz T. Single particle reconstructions of the transferrin-transferrin receptor complex obtained with different specimen preparation techniques. J Mol Biol. 2006;355:1048–1065. doi: 10.1016/j.jmb.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- Crowther RA. Procedures for three-dimensional reconstruction of spherical viruses by Fourier synthesis from electron micrographs. Philos Trans R Soc Lond B Biol Sci. 1971;261:221–230. doi: 10.1098/rstb.1971.0054. [DOI] [PubMed] [Google Scholar]
- Dryden KA, Farsetta DL, Wang G, Keegan JM, Fields BN, Baker TS, Nibert ML. Internal/structures containing transcriptase-related proteins in top component particles of mammalian orthoreovirus. Virology. 1998;245:33–46. doi: 10.1006/viro.1998.9146. [DOI] [PubMed] [Google Scholar]
- Dryden KA, Wang G, Yeager M, Nibert ML, Coombs KM, Furlong DB, Fields BN, Baker TS. Early steps in reovirus infection are associated with dramatic changes in supramolecular structure and protein conformation: analysis of virions and subviral particles by cryoelectron microscopy and image reconstruction. J Cell Biol. 1993;122:1023–1041. doi: 10.1083/jcb.122.5.1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan R, Murphy FA, Mirkovic RR. Characterization of a novel syncytium-inducing baboon reovirus. Virology. 1995;212:752–756. doi: 10.1006/viro.1995.1536. [DOI] [PubMed] [Google Scholar]
- Fotin A, Cheng Y, Sliz P, Grigorieff N, Harrison SC, Kirchhausen T, Walz T. Molecular model for a complete clathrin lattice from electron cryomicroscopy. Nature. 2004;432:573–579. doi: 10.1038/nature03079. [DOI] [PubMed] [Google Scholar]
- Frank J, Radermacher M, Penczek P, Zhu J, Li Y, Ladjadj M, Leith A. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Fuller SD, Butcher SJ, Cheng RH, Baker TS. Three-dimensional reconstruction of icosahedral particles--the uncommon line. J Struct Biol. 1996;116:48–55. doi: 10.1006/jsbi.1996.0009. [DOI] [PubMed] [Google Scholar]
- Gonen T, Cheng Y, Sliz P, Hiroaki Y, Fujiyoshi Y, Harrison SC, Walz T. Lipid-protein interactions in double-layered two-dimensional AQP0 crystals. Nature. 2005;438:633–638. doi: 10.1038/nature04321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorieff N. Resolution measurement in structures derived from single particles. Acta Crystallogr D Biol Crystallogr. 2000;56:1270–1277. doi: 10.1107/s0907444900009549. [DOI] [PubMed] [Google Scholar]
- Harauz G, van Heel M. Exact filters for general geometry three dimensional reconstruction. Optik. 1986;73:146–156. [Google Scholar]
- Ji Y, Marinescu DC, Zhang W, Zhang X, Yan X, Baker TS. A model-based parallel origin and orientation refinement algorithm for cryoTEM and its application to the study of virus structures. J Struct Biol. 2006;154:1–19. doi: 10.1016/j.jsb.2005.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Ludtke SJ. Electron cryomicroscopy of single particles at subnanometer resolution. Curr Opin Struct Biol. 2005;15:571–577. doi: 10.1016/j.sbi.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Leschziner AE, Nogales E. The orthogonal tilt reconstruction method: an approach to generating single-class volumes with no missing cone for ab initio reconstruction of asymmetric particles. J Struct Biol. 2006;153:284–299. doi: 10.1016/j.jsb.2005.10.012. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Chen DH, Song JL, Chuang DT, Chiu W. Seeing GroEL at 6 A resolution by single particle electron cryomicroscopy. Structure (Cambridge) 2004;12:1129–1136. doi: 10.1016/j.str.2004.05.006. [DOI] [PubMed] [Google Scholar]
- Marinescu DC, Ji Y. A computational framework for the 3D structure determination of viruses with unknown symmetry. Journal of Parallel and Distributed Computing. 2003;63:738–758. [Google Scholar]
- Miyazawa A, Fujiyoshi Y, Unwin N. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 2003;423:949–955. doi: 10.1038/nature01748. [DOI] [PubMed] [Google Scholar]
- Mueller F, Sommer I, Baranov P, Matadeen R, Stoldt M, Wohnert J, Gorlach M, van Heel M, Brimacombe R. The 3D arrangement of the 23 S and 5 S rRNA in the Escherichia coli 50 S ribosomal subunit based on a cryo-electron microscopic reconstruction at 7.5 Å resolution. J Mol Biol. 2000;298:35–59. doi: 10.1006/jmbi.2000.3635. [DOI] [PubMed] [Google Scholar]
- Olson NH, Baker TS, Willingmann P, Incardona NL. The three-dimensional structure of frozen-hydrated bacteriophage phi X174. J Struct Biol. 1992;108:168–175. doi: 10.1016/1047-8477(92)90016-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova EV, Saibil HR. Structure determination of macromolecular assemblies by single-particle analysis of cryo-electron micrographs. Curr Opin Struct Biol. 2004;14:584–590. doi: 10.1016/j.sbi.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Provencher SW, Vogel RH. Three-dimensional reconstruction from electron micrographs of disordered specimens. I Method Ultramicroscopy. 1988;25:209–221. doi: 10.1016/0304-3991(88)90016-2. [DOI] [PubMed] [Google Scholar]
- Radermacher M, Wagenknecht T, Verschoor A, Frank J. Three-dimensional reconstruction from a single-exposure, random conical tilt series applied to the 50S ribosomal subunit of Escherichia coli. J Microsc. 1987;146 ( Pt 2):113–136. doi: 10.1111/j.1365-2818.1987.tb01333.x. [DOI] [PubMed] [Google Scholar]
- Rosenthal DI, Christensen S, Emerson RH. "Handedness" of spiral fractures of the tibia. Skeletal Radiol. 1984;11:128–132. doi: 10.1007/BF00348801. [DOI] [PubMed] [Google Scholar]
- Samatey FA, Matsunami H, Imada K, Nagashima S, Shaikh TR, Thomas DR, Chen JZ, Derosier DJ, Kitao A, Namba K. Structure of the bacterial flagellar hook and implication for the molecular universal joint mechanism. Nature. 2004;431:1062–1068. doi: 10.1038/nature02997. [DOI] [PubMed] [Google Scholar]
- Shaikh TR, Hegerl R, Frank J. An approach to examining model dependence in EM reconstructions using cross-validation. J Struct Biol. 2003;142:301–310. doi: 10.1016/s1047-8477(03)00029-7. [DOI] [PubMed] [Google Scholar]
- Stark W, Kuhlbrandt W, Wildhaber I, Wehrli E, Muhlethaler K. The structure of the photoreceptor unit of Rhodopseudomonas viridis. Embo J. 1984;3:777–783. doi: 10.1002/j.1460-2075.1984.tb01884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart A, Grigorieff N. Noise bias in the refinement of structures derived from single particles. Ultramicroscopy. 2004;102:67–84. doi: 10.1016/j.ultramic.2004.08.008. [DOI] [PubMed] [Google Scholar]
- Thiery R, Cozien J, de Boisseson C, Kerbart-Boscher S, Nevarez L. Genomic classification of new betanodavirus isolates by phylogenetic analysis of the coat protein gene suggests a low host-fish species specificity. J Gen Virol. 2004;85:3079–3087. doi: 10.1099/vir.0.80264-0. [DOI] [PubMed] [Google Scholar]
- Thuman-Commike PA, Chiu W. Improved common line-based icosahedral particle image orientation estimation algorithms. Ultramicroscopy. 1997;68:231–255. doi: 10.1016/s0304-3991(97)00033-8. [DOI] [PubMed] [Google Scholar]
- Valle M, Zavialov A, Li W, Stagg SM, Sengupta J, Nielsen RC, Nissen P, Harvey SC, Ehrenberg M, Frank J. Incorporation of aminoacyl-tRNA into the ribosome as seen by cryo-electron microscopy. Nat Struct Biol. 2003;10:899–906. doi: 10.1038/nsb1003. [DOI] [PubMed] [Google Scholar]
- van Heel M. Angular reconstitution: a posteriori assignment of projection directions for 3D reconstruction. Ultramicroscopy. 1987;21:111–123. doi: 10.1016/0304-3991(87)90078-7. [DOI] [PubMed] [Google Scholar]
- van Heel M, Schatz M. Fourier shell correlation threshold criteria. J Struct Biol. 2005;151:250–262. doi: 10.1016/j.jsb.2005.05.009. [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Wang G, Parrish CR, Cheng RH, Strassheim ML, Baker TS, Rossmann MG. The structure of a neutralized virus: canine parvovirus complexed with neutralizing antibody fragment. Structure. 1994;2:595–607. doi: 10.1016/s0969-2126(00)00062-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Olson NH, Van Etten JL, Bergoin M, Rossmann MG, Baker TS. Structure and assembly of large lipid-containing dsDNA viruses. Nat Struct Biol. 2000;7:101–103. doi: 10.1038/72360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X, Sinkovits RS, Baker TS. AUTO3DEM - an automated and high throughput program for 3D image reconstruction of icosahedral particles. J Struct Biol. 2006 doi: 10.1016/j.jsb.2006.08.007. (this issue), submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Yu X, Galkin VE, Egelman EH. Issues of resolution and polymorphism in single-particle reconstruction. J Struct Biol. 2003;144:162–171. doi: 10.1016/j.jsb.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Yin Z, Zheng Y, Doerschuk PC. An ab initio algorithm for low-resolution 3-D reconstructions from cryoelectron microscopy images. J Struct Biol. 2001;133:132–142. doi: 10.1006/jsbi.2001.4356. [DOI] [PubMed] [Google Scholar]
- Zhang W, Chipman PR, Corver J, Johnson PR, Zhang Y, Mukhopadhyay S, Baker TS, Strauss JH, Rossmann MG, Kuhn RJ. Visualization of membrane protein domains by cryo-electron microscopy of dengue virus. Nat Struct Biol. 2003a;10:907–912. doi: 10.1038/nsb990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Mukhopadhyay S, Pletnev SV, Baker TS, Kuhn RJ, Rossmann MG. Placement of the structural proteins in Sindbis virus. J Virol. 2002;76:11645–11658. doi: 10.1128/JVI.76.22.11645-11658.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Ji Y, Zhang L, Harrison SC, Marinescu DC, Nibert ML, Baker TS. Features of reovirus outer capsid protein μ1 revealed by electron cryomicroscopy and image reconstruction of the virion at 7.0 Å resolution. Structure. 2005a;13:1545–1557. doi: 10.1016/j.str.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Tang J, Walker SB, O'Hara D, Nibert ML, Duncan R, Baker TS. Structure of avian orthoreovirus virion by electron cryomicroscopy and image reconstruction. Virology. 2005b;343:25–35. doi: 10.1016/j.virol.2005.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Walker SB, Chipman PR, Nibert ML, Baker TS. Reovirus polymerase λ3 localized by cryo-electron microscopy of virions at a resolution of 7.6 Å. Nat Struct Biol. 2003b;10:1011–1018. doi: 10.1038/nsb1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou ZH, Baker ML, Jiang W, Dougherty M, Jakana J, Dong G, Lu G, Chiu W. Electron cryomicroscopy and bioinformatics suggest protein fold models for rice dwarf virus. Nat Struct Biol. 2001;8:868–873. doi: 10.1038/nsb1001-868. [DOI] [PubMed] [Google Scholar]