

Abstract
Sulfotransferases (SULTs) catalyzed sulfation is important in the regulation of biological activities of hormones and neurotransmitters, the metabolism of drugs, and the detoxification of xenobiotic toxicants. Sulfation also leads to the bioactivation of procarcinogens. Human dehydroepiandrosterone sulfotransferase (hSULT2A1) is a major SULT catalyzing the sulfation of hydroxysteroids and xenobiotic alcohols. Our previous studies had shown that the anti-folate drug methotrexate (MTX) can up-regulate several major isoforms of human SULTs. To determine the mechanisms controlling the regulation of hSULT2A1, the 5′-flanking region of hSULT2A1 was constructed into the pGL3-Basic luciferase reporter vector. The transcriptional regulation mechanism of hSULT2A1 promoter was studied using Caco-2 cell line based on the reporter gene assay. Nuclear receptor co-transfection results indicated that human constitutive androstane receptor (hCAR) and human retinoid X receptor α (hRXRα) were involved in the transcriptional regulation of hSULT2A1. RNA interference experiments further proved the role of hCAR in hSULT2A1 regulation. Progressive promoter deletion, DNA sequence alignment, and site directed promoter mutation results suggested that an imperfect inverted repeat DNA motif, IR2 (-186AGCTCAGATGACCC-173), within the hSULT2A1 promoter region mediated the hSULT2A1 induction by MTX. Furthermore, electrophoretic mobility shift assay and super shift assay were employed to characterize the interactions of hCAR and hRXRα with the IR2 element. In summary, we identified an IR2 DNA cis-element located at -186/-173 of hSULT2A1 promoter region; the IR2 element mediates the MTX induction of hSULT2A1 through interacting with hCAR and hRXRα.
Keywords: Sulfotransferase, SULT2A1, constitutive androstane receptor, CAR, methotrexate, induction
Introduction
Cytosolic sulfotransferases (SULTs) are one of the major families of phase II drug metabolizing enzymes. SULTs catalyze the sulfation (sulfuryl transfer) of hydroxyl-containing molecules (Gamage et al., 2006, Pacifici, 2004, Coughtrie, 2002, Duffel et al., 2001, Glatt, 2000). The co-substrate (sulfuryl group donor) for all SULTs is adenosine 3′-phosphate 5′-phosphosulfate (PAPS) (Klaassen and Boles, 1997). Sulfation is widely observed in various biological processes. Bio-signaling molecules including hormones, neurotransmitters, bile acids, and peptides can be sulfated to alter their biological activities. Some SULT isoforms have a broad range of substrate specificities and catalyze the sulfation of many xenobiotics (Duffel et al., 2001). Sulfation of drugs and xenobiotics is mainly associated with detoxification - biotransformation of a relatively hydrophobic xenobiotic into a more water-soluble sulfuric ester that is readily excreted. However, there are numerous important exceptions wherein the formation of chemically reactive sulfuric esters is an essential step in the metabolic pathways leading to toxic or carcinogenic responses (Falany and Wilborn, 1994, Hengstler et al., 1998).
Studies on SULT2A1 regulation have been primarily focused on rats and mice. The primary bile acid, chenodeoxycholic acid (CDCA), is a potent inducer of rSULT2A1, and its inducing effect is mediated through farnesoid X receptor (FXR) (Song et al., 2001). The ligand-activated FXR forms a heterodimer with the retinoid X receptor (RXR) and regulates rSULT2A1 by binding to the IR0 element located in the 5′ flanking region of rSULT2A1. Pregnane X receptor (PXR) was reported to mediate the induction of mSULT2A1 (Sonoda et al., 2002). The results indicated that co-transfection of PXR, RXRα, and the IR0 element in the promoter region is all necessary for the gene stimulation. It was proposed that PXR serves as a master regulator of phase I and II responses to facilitate rapid and efficient detoxification and elimination of xenobiotics. A study on the suppression of mSULT2A1 during the acute phase response also suggested that PXR and FXR are responsible for the induction of mSULT2A1(Kim et al., 2004). It was reported that SULT2A1 is a target for transcriptional activation by vitamin D3 mediated through vitamin D receptor (VDR) in rats and mice (Echchgadda et al., 2004b). FXR and PXR inhibited the vitamin D3 induction of SULT2A1. Another report also suggested a repressive role of PXR and FXR on basal mSULT2A1 expression (Kitada et al., 2003). Using knockout mice, it was reported that mSULT2A1 can be induced by TCPOBOP and phenobarbital (PB) through constitutive androstane receptor (CAR) (Assem et al., 2004). This suggests that SULT2A1 participates in an integrated pathway mediating the elimination of sulfated steroid and bile acid metabolites from the liver. Both steroidogenic factor 1 (SF1) and estrogen-related receptor alpha (ERRα) were reported to regulate hSULT2A1 activity through the same DNA cis-element located in the proximal promoter region of hSULT2A1 (Saner et al., 2005, Seely et al., 2005). Peroxisome proliferator-activated receptor alpha (PPARα) was also found to involve in the transcriptional regulation of hSULT2A1 through the DNA response element located in - 5949 to -5929 upstream of the promoter region (Fang et al., 2005). The vitamin D receptor (VDR) was reported to target hSULT2A1 promoter through interaction with CAAT/Enhancer Binding Protein-alpha (C/EBPα) (Song et al., 2006).
Methotrexate (MTX) is a widely used drug against cancer and other diseases (Walling, 2006, Green et al., 2006). The efficiency of this drug is through tightly binding to dihydrofolate reductase (DHFR), a key enzyme for DNA and several amino acids synthesis. As a result, MTX decreases the rapid proliferation of cancer cells. MTX enters cells primarily by a carrier-mediated active transport system. The metabolites of MTX include 7-hydroxymethotrexate (7-OH-MTX), 2,4-diamino-N10-methylpteroic acid (DAMPA), and polyglutamate derivatives [MTX-(Glu)n, n=2–7] (Kuo et al., 2003, Durand et al., 1983). In general, MTX induction on drug metabolizing enzymes is not well studied. In male rat livers, MTX lacks a significant effect on the expression of CYP3A2 and CYP2C22 at both protein and mRNA level (Cheung et al., 1996). MTX did not significantly induce CYP3A4 in primary human hepatocytes (Abbas-Terki et al., 2002).
We previously reported that MTX induces SULT differently in rat liver/intestine (Maiti and Chen, 2003) and human cell lines (Chen et al., 2005). In this report, we investigated the mechanism of hSULT2A1 induction by MTX. Our results suggest that MTX induction of hSULT2A1 is mediated through hCAR. We have identified an imperfect invert repeat IR2 sequence responsible for the transactivation of hSULT2A1 through the interaction with nuclear receptor hCAR and hRXRα.
Materials and Methods
Materials
DNA restriction enzymes, Wizard® SV Genomic DNA Purification System and Access RT-PCR System were from Promega (Madison, WI). One Shot® Top 10 competent cells and Lipofectamine™ 2000 were from Invitrogen (Carlsbad, CA). 9-cis-Retinoic acid was from Sigma Chemical Co. (St. Louis, MO). Methotrexate was from ICN pharmaceuticals (Aurora, OH). The plasmid extraction kit and total RNA extraction kit were from QIAGEN (Valencia, CA). The DNA gel purification kit was from Q-Biogene (Carlsbad, CA). Protein assay reagent was from Bio-Rad (Hercules, CA). The DIG Gel Shift Kit, 2nd Generation (Cat. No. 03353591910) was from Roche Applied Science. Human retinoid X receptor (hRXRα) plasmid was from Dr. Ronald M. Evans’s laboratory (Howard Hughes Medical Institute, La Jolla, CA). hCAR plasmid was from Dr. Steven A Kliewer’s laboratory (University of Texas Southwestern Medical Center, Dallas, TX).
hSULT2A1 Promoter Reporter Construction and Mutagenesis in Caco-2 Cells
Luciferase reporter constructs were used in the transfection studies. Primer design for the hSULT2A1 promoter sequence was based on that previously described by Duanmu, et. al (Duanmu et al., 2002, Otterness et al., 1995a). Briefly, a fragment encoding the 5′-flanking region (−1463 to +48) of hSULT2A1 was generated by PCR using specific primers and genomic DNA extracted from Hep G2 cells. The fragment was inserted into the luciferase reporter vector, pGL3-Basic (Promega, Madison, WI) at the MluI and XhoI sites to drive the promoterless firefly luciferase gene.
Reporter plasmids containing nested deletions of the hULT2A1 5′-flanking region were all generated by PCR reactions. Specifically, constructs –713, -414, -354, -235, -188, -130 and –65 were generated by using the −1463 to +48 fragment of the hSULT2A1 gene as template. A series of 5′ primers were designed to incorporate a SacI site for sub-cloning (5′-TTACATACACGTCAGCCATCAA - 3′ for construct -713; 5′ – TGTGGTCTTTTGGATTTGGAG - 3′ for construct -414; 5′-GCACGATTGCAGGATTATTT - 3′ for construct -354; 5′-TTGTCCTCGTGTTTGTTATTCG - 3′ for construct -235; 5′-CAAGCTCAGATGACCCCTAAA - 3′ for construct -188; 5′-CAATCTTTTGAGTATGG GTCACA - 3′ for construct –130; and 5′-GTGACATGCTGGGACAAGG - 3′ for construct -65). The 3′ primers were designed with a SmaI site that was identical for all of the constructs (5′-GCGTGGTGTGAGGGTTTC - 3′). These amplified fragments were initially ligated into the pUC19 vector and then cloned into the SacI and SmaI sites of the pGL3-Basic vector.
A site-directed mutagenesis construct (construct IR2-Mut) was prepared using overlap PCR. In initial step of overlap PCR, the left arm of the PCR product was generated from the wild type template, using the same sense primer as deleted construct -414 and the antisense primer (5′-GCAAGCTCAGAACTCCCCTAAAATGG-3′) containing desired base changes corresponding to the hCAR binding site of the hSULT2A1 promoter; similarly, the right arm of the PCR product was generated using the sense primer (5′-CCATTTTAGGGGAGTTCTGAGCTT GC-3′) containing the mutant oligo sequence and the antisense primer was the same as deleted construct -414. Amplified DNAs were gel-purified, and construct -414 sense and antisense primers were used to splice the left arm and right arm DNA products by overlap PCR. The PCR product was initially ligated to pUC19 vector and then subcloned to the upstream of the luciferase gene in pGL3-Basic vector at SacI and SmaI sites. DNA sequencing at the Oklahoma State University core facility verified all constructs.
Transfections and Reporter Gene Assays in Caco-2 Cells
Human colon adenocarcinoma, Caco-2 cells (ATCC, Manassas, VA) were grown in Dulbecco’s Modified Eagle’s Medium supplemented with 20% fetal bovine serum (FBS). Caco-2 cells at 1 × 105/well were seeded onto a 48-well plate and transfected after 16 h with 50 ng of reporter plasmid, 25 ng of nuclear receptor expression vector and 10 ng of the pRL-TK plasmid (Promega, Madison, WI) with 5% charcoal stripped FBS. The transfection agents contained 49 μl of Opti-MEM and 1 μl of Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA). The pRL-TK plasmid, which expresses Renilla luciferase, was used as an internal standard for transfection efficiency. The pUC19 vector DNA was used as an empty vector to keep the total transfected DNA at a fixed value of 210 ng. hCAR and hRXRα nuclear receptor agonists was added 6 hours after transfection with a final concentration of 0.1 μM MTX, 0.1 μM CITCO, 1 μM retinoic acid or 0.1% (V/V) ethanol. Culture medium supplemented with drug was replaced 12 h post-transfection to remove the Lipofectamine™ 2000. Cells were collected 48 h after transfection and firefly and Renilla luciferase activities were measured using the Dual-Luciferase® Reporter Assay System (Promega, Madison, WI). Each experiment was repeated three times with each performed in duplicate. Results are given as means ± S.E.
hCAR RNA Interference in Caco-2 Cells
Caco-2 cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 20% fetal bovine serum (FBS) to about 40% confluence. Just before transfection, the cell culture medium was replaced with 5% charcoal stripped FBS which contain reduced level of hormone that might cause induction effect of target gene. The siRNA targeting hCAR (siRNA ID: 5535) and the siRNA negative control (Cat # AM4611) was chemically synthesized by Ambion (Austin, US). The transfection agents containing 300 μl of Opti-MEM and 3 μl of Lipofectamine™ 2000 were added to Caco-2 cells according to the manufacturer’s instructions in 6-well plates containing 125 nM siRNA per well. Negative control siRNA was used in control experiments to exclude the possibility of cytotoxicity caused by siRNA transfection. Forty eight hours after transfection, cells were harvested for RNA analysis. For dual luciferase assay, plasmid DNA was first transfected to Caco-2 cells and siRNA was transfected 6 hours later with refreshed 5% charcoal stripped FBS medium. Cells were collected 48 h after transfection and firefly and Renilla luciferase activities were measured using the Dual-Luciferase® Reporter Assay System.
Quantitative Real-Time PCR
Total RNA was prepared from hCAR vector or hCAR specific siRNA treated Caco-2 cells. Superscript II (Invitrogen) reverse transcriptase with 50 to 100 ng of total RNA was used to synthesize cDNA, and 1 μl of reverse-transcribed product served as the template in polymerase chain reactions. Real-time PCR was performed using QuantiTect SYBR Green PCR kit (Qiagen, Valencia, CA) following the manufacturer’s instruction. Primers were designed with Primer Express as follow: ACTBF321: 5′-AGAAAATCTGGCACCACACC -3′, ACTBR462: 5′-GGGGTGTTGAAGGTCTCAAA -3′, GI, L5016088; hSULT2A1F163: 5′-TGAGTTCGTGATAAGGGATGAA -3′, hSULT2A1R294: 5′-CAGATGGGCACAGATTGGAT -3′, GI, 29540544; hCARF959: 5′-CTTCTCTCCTGACCGACCTG -3′, hCARR1089: 5′-TCGCATACAGAAACCGATCC -3′, GI: 32189358. Real-time PCR was performed on ABI PRISM 7500 (Applied Biosystems, Foster City, CA). Initially, regular PCR products were purified with GENECLEAN Turbo (Qbiogene, Carlsbad, CA) for constructing standard curves (10–108 copies). A standard curve was plotted with the threshold cycle (CT) vs. the logarithmic value of the gene copy number. The gene copy number of unknown samples was generated directly from the standard curve by the software Sequence Detector 1.7. At least two repeats were run for each sample; each experiment was repeated 3 times. All gene copy numbers were normalized to human beta actin mRNA.
Electrophoretic Mobility Shift Assay (EMSA) and Super Shift Assay
EMSA was performed using digoxigenin-11-ddUTP labeled oligonucleotides which contain the IR2 element DF-191: 5′-GGAACGCAAGCTCAGATGACCCCTAAAATGG -3′ or DF-191m: 5′ –GGAACGCAAGCTCAGAACTCCCCTAAAATGG -3′ (mutated nucleotides underlined). A standard gel shift binding reaction (20 μl) contained 20 mM Hepes, pH 7.6, 1 mM EDTA, 10 mM (NH4)2 S04, 1 mM DTT, 0.2 % (w/v) Tween 20, 30 mM KCl, 1 μg poly [d(I-C)], 1 μg poly L-lysine, and 5 μg Caco-2 nuclear extract. Reactions were incubated at room temperature for 20 min after the addition of double-stranded oligonucleotide probe (0.4 ng). Competitions were performed with 125-fold molar excess of unlabeled oligonucleotides. In the super shift assay, the nuclear extract was pre-incubated with the antibody at room temperature for 20 min before the addition of DNA probes. Antibodies to MB67 (C-20): sc-8541 and hRXRα (D-20): sc-553 X were from Santa Cruz Biotechnology (Santa Cruz, CA). The protein-DNA complexes were resolved on a pre-electrophoresed 5% native polyacrylamide gel in 0.5× TBE (45 mM Tris borate, 1 mM EDTA) at room temperature and then blotted to positively charged nylon membrane. The DIG labeled oligonucleotides are visualized by an enzymatic immunoassay using anti-Digoxigenin-AP Fab-fragments and the chemiluminescence’s substrate CSPD. The generated chemiluminescence’s signals are recorded on X-ray film.
Statistical Analysis
Luciferase activities and cDNA copy number of hCAR and hRXR were expressed as median ± SE (standard error). The data distribution was checked with Q-Q plot, MTX and nuclear receptor effect on hSULT2A1 in reporter gene assay, siRNA effect on reporter gene assay and real-time PCR assay were analyzed for significance by one-way ANOVA, Dunnett’s test. In all cases, */#, P< 0.05 was considered significant; **/##, P< 0.01 was considered very significant.
Results
hCAR Transactivation of hSULT2A1 Promoter in Caco-2 Cells
To explore the molecular mechanism of hSULT2A1 regulation, the 1.5-Kb fragment of hSULT2A1 (−1463 to +48) was generated by PCR with primer pairs using genomic DNA extracted from Hep G2 cells. Our sequencing result of the PCR product was the same as the published sequence (L36191 and U13056) (Otterness et al., 1995a, Otterness et al., 1995b). The generated promoter sequence was inserted into pGL3-Basic vector (Promega) at the MluI and XhoI sites. The constructed reporter vector was used for transfection into Caco-2 cells.
CITCO (6-(4-chlorophenyl)imidazo [2,1-b][1,3] thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime) is a well-known agonist of hCAR and was reported to induce human CYP2B6 in human primary cells (Maglich et al., 2003). Results shown in Figure 1 demonstrate that CITCO can also induce hSULT2A1 in Caco-2 cells. The induction activity of CITCO to hSULT2A1 is similar to that of MTX. Transfection of hCAR or hRXRα alone caused 2 fold activation of hSULT2A1 promoter. For both CITCO and MTX, when the drug treated cells were co-transfected with hCAR, the luciferase activity increased 4 fold. Co-transfection of hCAR, hRXRα together with 9-cis-retinoic acid (9-cis-RA), caused the activation of hSULT2A1 promoter more than 8 fold for both MTX and CITCO. This suggests that the hCAR transactivation may occur via the interaction with hRXRα. This agrees with P-450s and other drug metabolizing enzymes induction mechanisms: most nuclear receptors, which induce drug-metabolizing enzymes, form heterodimers with hRXRα (Hughes et al., 2006, Xu et al., 2005, Tan et al., 2005, Noreault et al., 2005). This suggests that MTX can function as an agonist of hCAR and induce the expression of hSULT2A1. All these results suggest a functional DNA cis-element is present in the cloned promoter region of hSULT2A1, and it can be activated by hCAR in both ligand-dependent and ligand-independent manner.
Figure 1. Effect of hCAR and hRXRα on CITCO and MTX induction of hSULT2A1 promoter activity in Caco-2 cells.
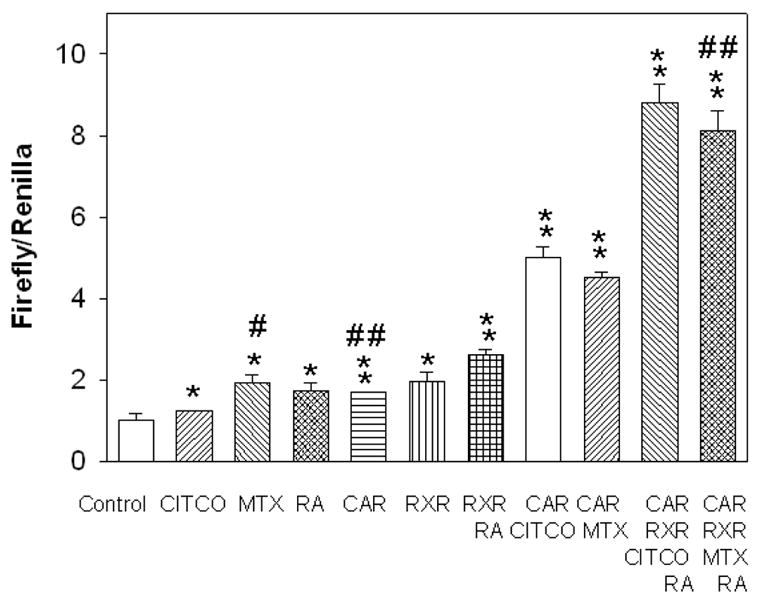
Luciferase constructs containing 5′-flanking sequences of hSULT2A1 were co-transfected with expression vector of hCAR, hRXRα and treated with MTX, CITCO, retinoic acid or alcohol (control) as described in the methods section. pRL-TK was used as internal control for transfection assay. Fold inductions were calculated relative to the promoter activity in vehicle-treated cells. Concentrations used: hCAR (25 ng), hRXRα (25 ng), MTX (100 nM), CITCO (100 nM) and 9-cis-RA (1 μM). Dual-luciferase activities were measured according to the manufacturer’s recommendations. The histograms with standard deviations are average values from three independent transfections; each independent transfection was performed in duplicate. One way ANOVA/Dunnett’s test was used in the data analysis, * was used when all treatment was compared with control and # was used when treatments were compared with MTX + CAR group. */#, p < 0.05; and **/##, p < 0.01.
To further investigate the role of hCAR in hSULT2A1 regulation, the RNA interference experiments were carried out. Negative control siRNA (Ambion Inc., ID: 4611) and hCAR specific siRNA (provided by Ambion Inc., ID: 5535) was used in both reporter gene assay and endogenous study. In the reporter gene assay experiments, the hSULT2A1 promoter regulated luciferase reporter vector was transfected into Caco-2 cells. The luciferase expression of control cells reflects the basal promoter activity of hSULT2A1. Upon the transfection of hCAR specific siRNA, the luciferase expression was reduced to approximately 50% of the control value (Figure 2A). This strongly suggests that hCAR was involved in the transcriptional regulation of hSULT2A1. As previously discussed, the co-transfection of hCAR can induce hSULT2A1 promoter activity 2 fold; this induction can also be partly knocked down by the hCAR specific siRNA (Figure 2A).
Figure 2. Effect of RNA interference of hCAR on hSULT2A1 transcriptional activity.
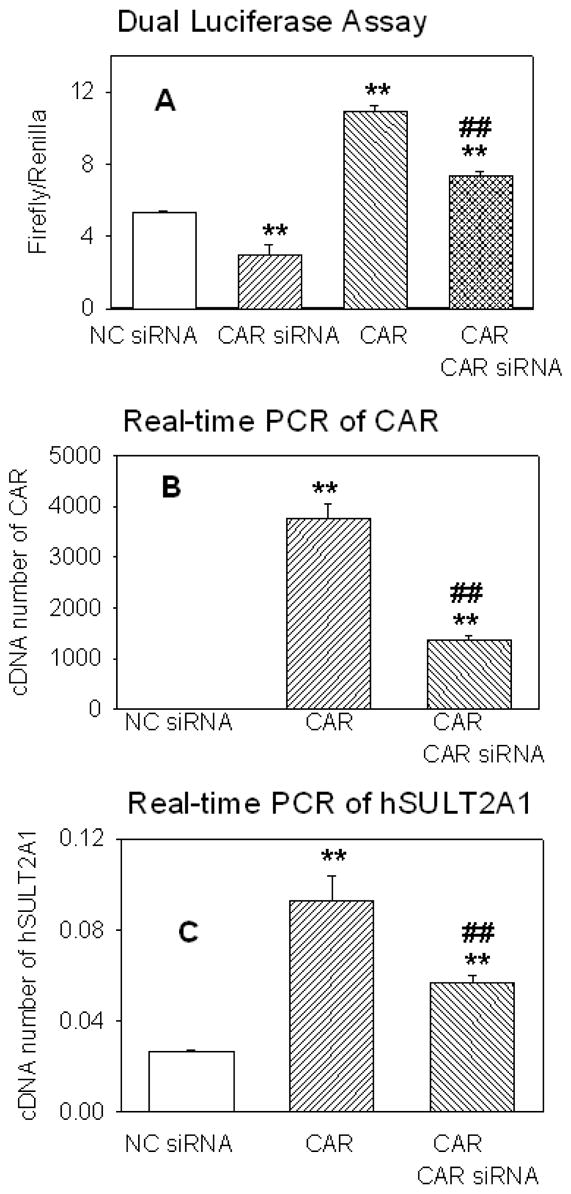
Figure 2A, the luciferase reporter vector regulated by hSULT2A1 promoter was transfected into Caco-2 cells and dual luciferase activity was measured according to manufacture’s recommendation. Negative control siRNA (NC siRNA) or hCAR specific siRNA (CAR siRNA) was transfected into Caco-2 cells with (column 4) or without (column 2) the co-transfection of hCAR expression vector. The control cells for reporter gene assay received only the reporter vectors. For figure 2B and figure 2C, Caco-2 cells were transfected with hCAR expression vector, and then negative control siRNA or hCAR specific siRNA were transfected after 6 hours. Cells were harvested after 48 hours and analyzed for real-time PCR with gene specific primers for human β-actin, hCAR and hSULT2A1 as described under Methods section. Relative concentrations were calculated using standard curves method. Each treatment group was analyzed in triplicate and the data shown were average of three independent experiments. One way ANOVA/Dunnett’s test was used in the data analysis, * was used when all treatment was compared with control and # was used when CAR+CAR siRNA group was compared with CAR group. */#, p < 0.05; and **/##, p < 0.01.
Real-time RT-PCR method was used to check the mRNA copy number of hCAR and hSULT2A1 in Caco-2 cells. The results shown in figure 2B and figure 2C suggest that the expression of hSULT2A1 is related to the expression of hCAR. The control cells in figure 2B contain very low copy numbers of hCAR, when the hCAR expression vector was transfected, the mRNA copy number of hCAR dramatically increased. When the hCAR transfected cells were treated with hCAR specific siRNA, the mRNA copy number of hCAR decreased about 70%. This indicates the luciferase expression in figure 2A is closely related to the mRNA copy numbers of hCAR. When the mRNA amount of hCAR was increased, the promoter activity of hSULT2A1 was also increased. When the mRNA amount of hCAR was decreased, the promoter activity of hSULT2A1 was also decreased. Results shown in Figure 2C illustrate that the mRNA copy number of hSULT2A1 was closely related to the expression of hCAR. When the mRNA amount of hCAR increased through the transfection of hCAR, the mRNA copy number of hSULT2A1 was also increased. When the hCAR mRNA level was knocking down by the siRNA, the hSULT21 expression level also decreased. These results strongly support that hCAR is responsible for hSULT2A1 gene regulation.
DNA Response Element Responsible for hCAR and hRXRα Mediated Induction of hSULT2A1
To identify the DNA response element, functional promoter assay with progressively 5′ deleted hSULT2A1 promoter fragments was performed. Deletion analysis results are shown in Figure 3. The results suggest that the deletion between -1463 and -713 did not significantly change the promoter activity. Deletion from -731 to -414 decreased the relative promoter activity, but the promoter activity was still elevated. Deletion between -414 and -188 (C-F) did not significantly change the promoter induction ability by hCAR and MTX. When the promoter sequence was deleted to 130 upstream of the transcription start site, the reporter vector guided by hSULT2A1 promoter suddenly lost its induction activity mediated by hCAR and MTX. This suggests that the key element responsible for the promoter activity is located between -188 and -130. Although, the DNA cis-acting regulatory elements located between -713 and -414 may also be necessary for the full responsiveness of hCAR mediated MTX induction of hSULT2A1.
Figure 3. Deletion analysis of hSULT2A1 promoter.
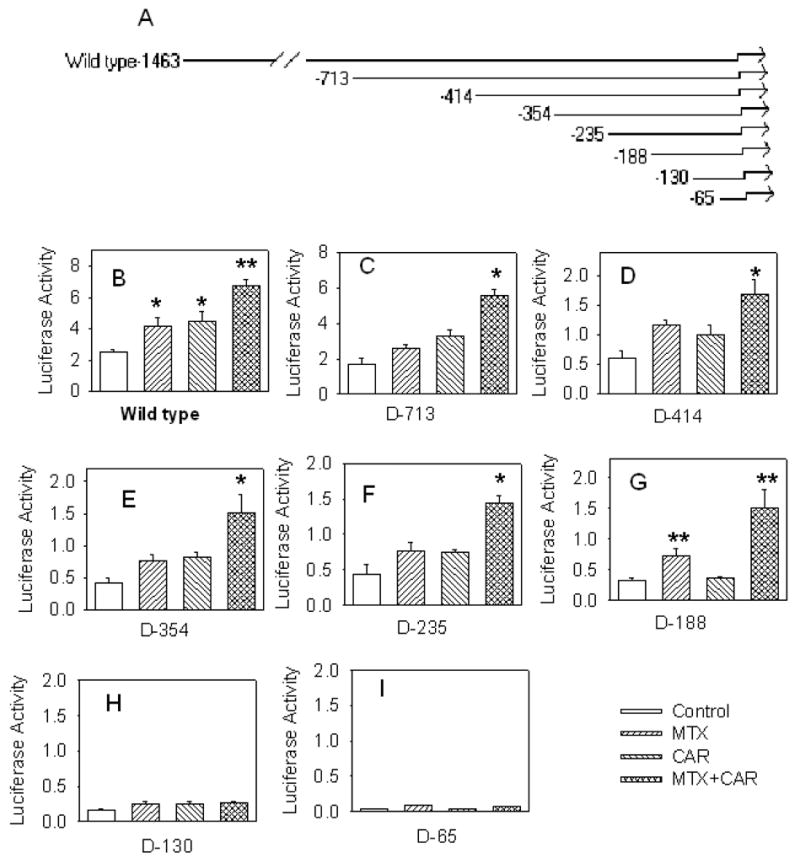
Figure 3A, schematic diagram of deleted hSULT2A1 promoter constructs. Figure 3B–3I, transactivation of deleted promoter constructs by hCAR and MTX. The luciferase reporter vector was regulated by hSULT2A1 5′-flanking region from the number indicated in the figure to +48. 25 ng hCAR was used for transfection. 100 nM MTX was used for treatment. Promoter activity is expressed as normalized luciferase activity. The histograms with standard deviations are average values from three independent transfections; each independent transfection was performed in duplicate. ANOVA/Dunnett’s test was used in the data analysis, *, p < 0.05; and **, p < 0.01; compared with control.
Careful analysis of the DNA sequence (Figure 4) between -188 and -130 revealed a hexameric core sequence AGCTCA between -186 and -181. Two base pairs downstream from this half-site is an imperfect inverted repeat sequence TGACCC (-178 to –173). This forms an imperfect inverted repeat motif IR2 (-186AGCTCAGATGACCC-173). This IR2 sequence may be the binding site for the hCAR and hRXRα. This IR2 element in hSULT2A1 does not exist in rat and mouse SULT2A1, but sequence alignment results (Figure 4) show that the IR2 in hSULT2A1 is located in a close position as the IR0 in rat and mouse SULT2A1.
Figure 4. Sequence alignments of rat, mouse, and human SULT2A1 promoter regions.

MultAlin program was used for the alignment of rat (GenBank accession no. M29301), mouse (Echchgadda et al., 2004a), and human (GenBank accession no. U54701) SULT2A1 promoter regions.
Mutation results shown in Figure 5 demonstrate that this IR2 is the element responsible for induction of hSULT2A1 via hCAR. Compared with the wild type promoter (Figure 3 D), the mutated 5′-flanking region of hSULT2A1 (−414 to +48) lost about 50% of it basal luciferase activity. When treated with MTX or hCAR, the mutated promoter can’t be activated by hCAR or MTX. Interestingly, the transfected hCAR can even cause the inhibition of the basal luciferase activity. This suggests that there may be multiple pathways involved in hSUST2A1 induction.
Figure 5. Point mutation analysis of the IR2 DNA cis-element.
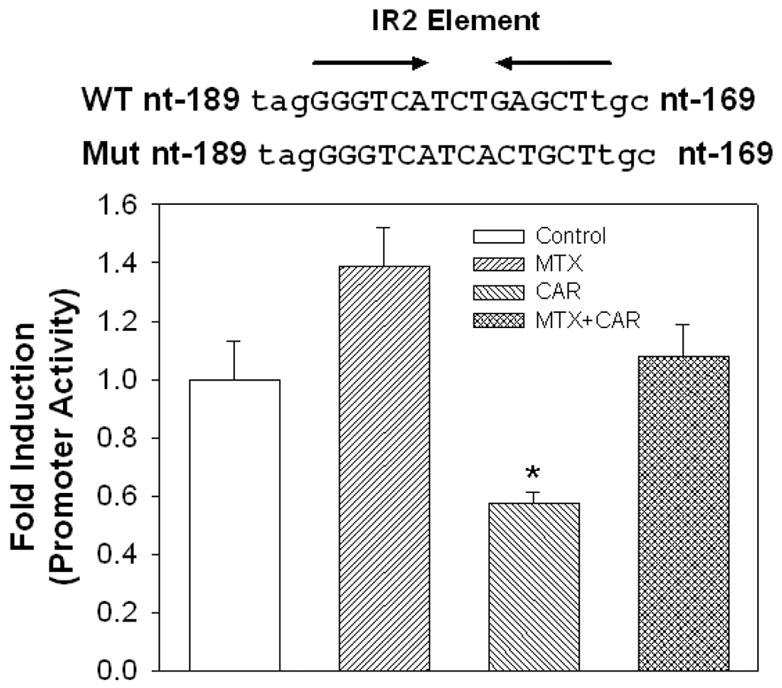
Mutated 5′-flanking region of hSULT2A1 (−414 to +48) as shown in the figure was inserted into pGL3-Basic vector. Luciferase expression regulated by this mutated promoter was determined in Caco-2 cells. The histograms are average values from three independent experiments (each performed in duplicate). ANOVA/Dunnett’s test was used in the data analysis, *, p < 0.05 compared with control.
Electrophoretic mobility shift assay (EMSA) and super shift assay were used to prove the interaction of hCAR and hRXRα with the identified IR2 element in the promoter region of hSULT2A1. The 31 bp oligonucleotide (5′-GGA ACG CAA GCT CAG ATG ACC CCT AAA ATG G - 3′) including the IR2 sequence in the middle of the sequence and its mutant (5′-GGA ACG CAA GCT CAG AAC TCC CCT AAA ATG G - 3′) were ordered from IDT. Both the IR2 probe and IR2 mutant probe were labeled with digoxigenin (DIG). The DIG labeled probes were visualized by an enzymatic immunoassay using anti-digoxigenin-AP Fab-fragments and the chemiluminescence’s substrate CSPD. Experimental results show that nuclear extract from Caco-2 cells caused the IR2 probe shift (Figure 6, lane 2). Addition of 125 fold excess cold IR2 probe inhibited this shift (lane 3) and mutated cold IR2 probe (125 fold) did not inhibit this shift (lane 4). Also, mutated IR2 labeled probe was not shifted by the nuclear extract (lane 5). These results strongly suggest that certain nuclear receptors can bind to the IR2 elements. Super shift assay (lane 6 and 7) results show that hCAR and hRXRα antibodies caused the nuclear extract shift to disappear (or tremendously decrease). The super shift band was not found. There are two possible causes for the disappearance of the shift band. The antibody may cause the corresponding receptor-oligonucleotide complex to precipitate; therefore not appear in the gel or not run into the gel. The second possibility is that the interaction between antibody and nuclear receptor prevents the receptor to bind to the IR2 element. Either hCAR antibody (lane 6) or hRXRα antibody (lane 7) alone can almost completely make the shift band disappear. This suggests that the hCAR and hRXRα is required for the interaction with the IR2 element.
Figure 6. EMSA of Caco-2 nuclear extract and super shift assay of hCAR and hRXRα with the IR2-containing oligonucleotide (31 bp).
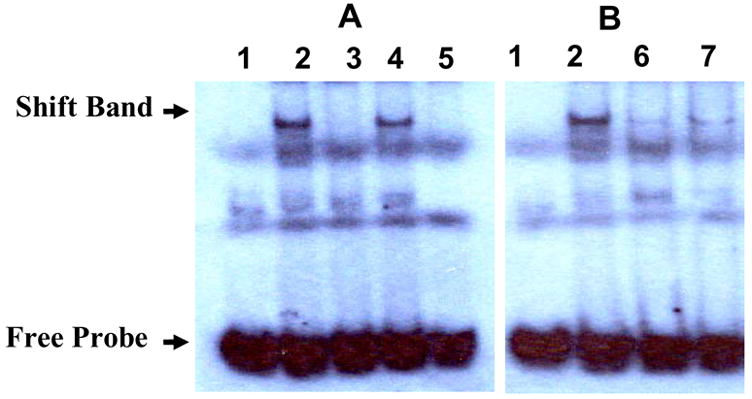
Figure 6A, gel shift assay of Caco-2 nuclear extract with the IR2-containing oligonucleotide. The labeled IR2 wild type oligonucleotide (lane 1 – 4) or labeled mutated IR2 oligonucleotide (lane 5) was incubated with Caco-2 nuclear extract (lane 2–5, lane 1 for control without nuclear extract) for 20 minutes at room temperature to form possible DNA protein complexes. 125-fold excess of wild type cold competitors (lane 3) and mutated cold competitors (lane 4) was added to test the specificity of the IR2. Figure 6B, super shift assay of hCAR and hRXR. Antibody specific to hCAR (lane 6) and hRXRα (lane 7) was added to the nuclear extract and incubated for 20 minutes at room temperature, then followed with the shift reaction.
Discussion
Phase I and phase II drug metabolizing enzymes, such as cytochrome P-450 and UDP-glucuronosyltransfersase (UGT), are well known to be regulated by endogenous hormones as well as by therapeutic drugs and other xenobiotics. SULTs, one of the major families of phase II drug metabolizing enzymes, are also well known to be regulated by hormones. However, xenobiotic induction of SULTs is not well studied (Runge-Morris, 1997, Gaworecki et al., 2004). Nuclear receptors, which mediate the induction of some phase I and phase II enzymes, have recently been shown to mediate the induction of SULTs. Reports on the nuclear receptor mediated induction mechanisms of SULTs focused primarily on SULT2A1 from different species. An IR0 (-189GGGTCATGAACT-178) in rats and mice was proposed to be the DNA binding site for different nuclear receptor activators of SULT2A1, such as, FXR/RXRα (Aste et al., 2001), PXR/RXRα (Duanmu et al., 2002), VDR/RXRα (Echchgadda et al., 2004a), and CAR/RXRα (Saini et al., 2004). FXR and PXR were also reported to repress basal mSULT2A1 expression using FXR-null or PXR-null mice (Kitada et al., 2003) and to inhibit vitamin D3 induction of SULT2A1 via VDR (Echchgadda et al., 2004a).
In the present study, we have shown that the promoter of hSULT2A1 was induced by the highly selective hCAR agonist CITCO in Caco-2 cells. Most importantly, we found that the widely used anti-folate drug MTX induced hSULT2A1 similarly as CITCO. Our results indicate that hCAR transactivates the induction of hSULT2A1. hRXRα enhances hCAR transactivation of hSULT2A1. This agrees with earlier reports on nuclear receptor transactivation of various drug-metabolizing enzymes. Almost all known nuclear receptors form heterodimers with RXR to transactivate drug metabolizing enzyme genes (Hughes et al., 2006, Xu et al., 2005, Tan et al., 2005, Noreault et al., 2005). MTX induced hSULT2A1 similar to CITCO, which suggests that MTX can function as an agonist of hCAR. The RNA interference experiments further support that the expression of hSULT2A1 was closely related to the expression level of hCAR. When hCAR expression vector was transfected into Caco-2 cells, both hCAR and hSULT2A1 levels were significantly increased, which suggests that hCAR can mediate the induction of hSULT2A1. When hCAR was knocked down by siRNA, the mRNA level of hSULT2A1 was also decreased. RNA interference experimental results agree with hCAR vector transfection results.
Our deletion analysis suggests that the binding site for hCAR/hRXRα is between -188 to -130 in the 5′-flanking region of hSULT2A1. Sequence alignment results (Figure 4) indicate that the IR0 sequence, which is responsible for transactivation of rat and mouse SULT2A1, does not exist in the hSULT2A1 5′-flanking region. Analysis of -188 to -130 DNA sequence of hSULT2A1 5′-flanking region revealed an IR2 sequence. Our mutation results (Figure 5) further show the importance of this IR2 sequence in the transactivation of hSULT2A1 via the hCAR and hRXRα during MTX induction. The identified IR2 in this report agrees with a previous prediction of several putative (A/G)G(G/T)TCA nuclear receptor half-site motifs in the 5′-flanking region of hSULT2A1 (Duanmu et al., 2002). Our results also agree with a recent report on VDR mediated bile acid induction of hSULT2A1 through interaction with CAAT/Enhancer Binding Protein-alpha (C/EBPα) (Song et al., 2005).
It is well known that nuclear receptors can bind to a broad range of ligands and regulate extensive arrays of genes that are involved in drug clearance and disposition. These nuclear receptors share partner, ligands, DNA response elements and target genes. Our results show that hCAR can induce the transcriptional activity of hSULT2A1. When the IR2 sequence was mutated, the co-transfection of hCAR repressed the mutated promoter activity, which suggests that there may be other nuclear receptors and DNA binding elements that also involved in transactivation of hSULT2A1 by MTX. The mutation affects the binding of hCAR and hRXRα to the mutated IR2 sequence and abolishes the induction of hCAR. The co-transfection of extra exogenous hCAR may compete for limited endogenous hRXR with other nuclear receptors and cause a final inhibition effect of the promoter activity.
In conclusion, our results demonstrate the role of hCAR in the induction of hSULT2A1 by MTX. We identified an hCAR-binding element in the promoter region of hSULT2A1, IR2. This IR2 in hSULT2A1 is different from the reported IR0 in rat and mouse SULT2A1 genes. The relative position of IR2 in the hSULT2A1 gene and IR0 in rat and mouse SULT2A1 genes is very close (Figure 4). These results could explain the differences and similarities between hSULT2A1 induction and rat and mouse SULT2A1 induction. Further characterization of the role of MTX involved in the hCAR signaling pathway in hSULT2A1 gene expression may be of considerable therapeutic significance. We are currently investigating the roles of other nuclear receptors in the regulation of hSULT2A1 expression.
Acknowledgments
The authors appreciate the generous gift of human RXRα plasmid from Ronald M. Evans, Howard Hughes Medical Institute, Gene Expression Laboratory, La Jolla, California, and human CAR plasmids from Dr. Steven A. Kliewer, Department of Molecular Biology, University of Texas Southwestern Medical Center, Dallas, Texas.
Abbreviations
- SULT
sulfotransferase
- hSULT
humansulfotransferase
- mSULT
mouse sulfotransferase
- rSULT
rat sulfotransferase
- SULT2A1
dehydroepiandrosterone sulfotransferase
- SULT1A1
simple phenol sulfotransferase
- DHEA
dehydroepiandrosterone
- MTX
methotrexate
- CAR
constitutive androstane receptor
- RXR
retinoid X receptor
- PXR
pregnane X receptor
- FXR
farnesoid X receptor
- VDR
vitamin D receptor
- IR
inverted repeat hormone response element
- EMSA
electrophoretic mobility shift assay
- DIG
digoxigenin
Footnotes
This work was supported in part by National Institutes of Health (NIH) R01 Grant GM59873 (G.C.) and the Oklahoma Center for the Advancement of Science and Technology (OCAST) grant HR05-015 (G.C.)
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- ABBAS-TERKI T, BLANCO-BOSE W, DEGLON N, PRALONG W, AEBISCHER P. Lentiviral-mediated RNA interference. Hum Gene Ther. 2002;13:2197–201. doi: 10.1089/104303402320987888. [DOI] [PubMed] [Google Scholar]
- ASSEM M, SCHUETZ EG, LEGGAS M, SUN D, YASUDA K, REID G, ZELCER N, ADACHI M, STROM S, EVANS RM, MOORE DD, BORST P, SCHUETZ JD. Interactions between hepatic Mrp4 and Sult2a as revealed by the constitutive androstane receptor and Mrp4 knockout mice. J Biol Chem. 2004;279:22250–7. doi: 10.1074/jbc.M314111200. Epub 2004 Mar 5. [DOI] [PubMed] [Google Scholar]
- ASTE N, COZZI B, STANKOV B, PANZICA G. Sexual differences and effect of photoperiod on melatonin receptor in avian brain. Microsc Res Tech. 2001;55:37–47. doi: 10.1002/jemt.1154. [DOI] [PubMed] [Google Scholar]
- CHEN X, BAKER SM, CHEN G. Methotrexate induction of human sulfotransferases in Hep G2 and Caco-2 cells. J Appl Toxicol. 2005;28:28. doi: 10.1002/jat.1071. [DOI] [PubMed] [Google Scholar]
- CHEUNG RL, LEE C, JONES EJ, RIDDICK DS. Lack of effect of methotrexate on the expression of constitutive hepatic cytochromes P450 in the male rat. Xenobiotica. 1996;26:503–14. doi: 10.3109/00498259609046728. [DOI] [PubMed] [Google Scholar]
- COUGHTRIE MW. Sulfation through the looking glass--recent advances in sulfotransferase research for the curious. Pharmacogenomics J. 2002;2:297–308. doi: 10.1038/sj.tpj.6500117. [DOI] [PubMed] [Google Scholar]
- DUANMU Z, LOCKE D, SMIGELSKI J, WU W, DAHN MS, FALANY CN, KOCAREK TA, RUNGE-MORRIS M. Effects of Dexamethasone on Aryl (SULT1A1)- and Hydroxysteroid (SULT2A1)-Sulfotransferase Gene Expression in Primary Cultured Human Hepatocytes. Drug Metab Dispos. 2002;30:997–1004. doi: 10.1124/dmd.30.9.997. [DOI] [PubMed] [Google Scholar]
- DUFFEL MW, MARSHAL AD, MCPHIE P, SHARMA V, JAKOBY WB. Enzymatic aspects of the phenol (aryl) sulfotransferases. Drug Metab Rev. 2001;33:369–95. doi: 10.1081/dmr-120001394. [DOI] [PubMed] [Google Scholar]
- DURAND R, FABRE G, CANO JP, CATALIN J, AHMED OA, JUST S. High-performance liquid chromatographic analysis of MTX, 7-OH-MTX and MTX derivatives: application to intracellular metabolism in tumor cells (HT 29) J Appl Toxicol. 1983;3:189–95. doi: 10.1002/jat.2550030406. [DOI] [PubMed] [Google Scholar]
- ECHCHGADDA I, SONG CS, OH TS, CHO SH, RIVERA OJ, CHATTERJEE B. Gene regulation for the senescence marker protein DHEA-sulfotransferase by the xenobiotic-activated nuclear pregnane X receptor (PXR) Mech Ageing Dev. 2004a;125:733–45. doi: 10.1016/j.mad.2004.08.008. [DOI] [PubMed] [Google Scholar]
- ECHCHGADDA I, SONG CS, ROY AK, CHATTERJEE B. Dehydroepiandrosterone sulfotransferase is a target for transcriptional induction by the vitamin D receptor. Mol Pharmacol. 2004b;65:720–9. doi: 10.1124/mol.65.3.720. [DOI] [PubMed] [Google Scholar]
- FALANY CN, WILBORN TW. Biochemistry of cytosolic sulfotransferases involved in bioactivation. Advances in Pharmacology (New York) 1994;27:301–29. doi: 10.1016/s1054-3589(08)61037-6. [DOI] [PubMed] [Google Scholar]
- FANG HL, STROM SC, CAI H, FALANY CN, KOCAREK TA, RUNGE-MORRIS M. Regulation of human hepatic hydroxysteroid sulfotransferase gene expression by the peroxisome proliferator-activated receptor alpha transcription factor. Mol Pharmacol. 2005;67:1257–67. doi: 10.1124/mol.104.005389. [DOI] [PubMed] [Google Scholar]
- GAMAGE N, BARNETT A, HEMPEL N, DUGGLEBY RG, WINDMILL KF, MARTIN JL, MCMANUS ME. Human sulfotransferases and their role in chemical metabolism. Toxicol Sci. 2006;90:5–22. doi: 10.1093/toxsci/kfj061. [DOI] [PubMed] [Google Scholar]
- GAWORECKI KM, RICE CD, VAN DEN HURK P. Induction of phenol-type sulfotransferase and glucuronosyltransferase in channel catfish and mummichog. Mar Environ Res. 2004;58:525–8. doi: 10.1016/j.marenvres.2004.03.041. [DOI] [PubMed] [Google Scholar]
- GLATT H. Sulfotransferases in the bioactivation of xenobiotics. Chem Biol Interact. 2000;129:141–70. doi: 10.1016/s0009-2797(00)00202-7. [DOI] [PubMed] [Google Scholar]
- GREEN MR, CHOWDHARY S, LOMBARDI KM, CHALMERS LM, CHAMBERLAIN M. Clinical utility and pharmacology of high-dose methotrexate in the treatment of primary CNS lymphoma. Expert Rev Neurother. 2006;6:635–52. doi: 10.1586/14737175.6.5.635. [DOI] [PubMed] [Google Scholar]
- HENGSTLER JG, ARAND M, HERRERO ME, OESCH F. Polymorphisms of N-acetyltransferases, glutathione S-transferases, microsomal epoxide hydrolase and sulfotransferases: influence on cancer susceptibility. Recent Results in Cancer Research. 1998;154:47–85. doi: 10.1007/978-3-642-46870-4_4. [DOI] [PubMed] [Google Scholar]
- HUGHES PJ, ZHAO Y, CHANDRARATNA RA, BROWN G. Retinoid-mediated stimulation of steroid sulfatase activity in myeloid leukemic cell lines requires RARalpha and RXR and involves the phosphoinositide 3-kinase and ERK-MAP kinase pathways. J Cell Biochem. 2006;97:327–50. doi: 10.1002/jcb.20579. [DOI] [PubMed] [Google Scholar]
- KIM MS, SHIGENAGA J, MOSER A, GRUNFELD C, FEINGOLD KR. Suppression of DHEA sulfotransferase (Sult2A1) during the acute-phase response. Am J Physiol Endocrinol Metab. 2004;287:E731–8. doi: 10.1152/ajpendo.00130.2004. [DOI] [PubMed] [Google Scholar]
- KITADA H, MIYATA M, NAKAMURA T, TOZAWA A, HONMA W, SHIMADA M, NAGATA K, SINAL CJ, GUO GL, GONZALEZ FJ, YAMAZOE Y. Protective role of hydroxysteroid sulfotransferase in lithocholic acid-induced liver toxicity. J Biol Chem. 2003;278:17838–44. doi: 10.1074/jbc.M210634200. [DOI] [PubMed] [Google Scholar]
- KLAASSEN CD, BOLES JW. Sulfation and sulfotransferases 5: the importance of 3′-phosphoadenosine 5′-phosphosulfate (PAPS) in the regulation of sulfation. FASEB Journal. 1997;11:404–18. doi: 10.1096/fasebj.11.6.9194521. [DOI] [PubMed] [Google Scholar]
- KUO CY, WU HL, KOU HS, CHIOU SS, WU DC, WU SM. Simultaneous determination of methotrexate and its eight metabolites in human whole blood by capillary zone electrophoresis. J Chromatogr A. 2003;1014:93–101. doi: 10.1016/s0021-9673(03)00776-3. [DOI] [PubMed] [Google Scholar]
- MAGLICH JM, PARKS DJ, MOORE LB, COLLINS JL, GOODWIN B, BILLIN AN, STOLTZ CA, KLIEWER SA, LAMBERT MH, WILLSON TM, MOORE JT. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J Biol Chem. 2003;278:17277–83. doi: 10.1074/jbc.M300138200. [DOI] [PubMed] [Google Scholar]
- MAITI S, CHEN G. Methotrexate is a novel inducer of rat liver and intestinal sulfotransferases. Arch Biochem Biophys. 2003;418:161–8. doi: 10.1016/j.abb.2003.08.019. [DOI] [PubMed] [Google Scholar]
- NOREAULT TL, KOSTRUBSKY VE, WOOD SG, NICHOLS RC, STROM SC, TRASK HW, WRIGHTON SA, EVANS RM, JACOBS JM, SINCLAIR PR, SINCLAIR JF. Arsenite decreases CYP3A4 and RXRalpha in primary human hepatocytes. Drug Metab Dispos. 2005;33:993–1003. doi: 10.1124/dmd.105.003954. [DOI] [PubMed] [Google Scholar]
- OTTERNESS DM, HER C, AKSOY S, KIMURA S, WIEBEN ED, WEINSHILBOUM RM. Human dehydroepiandrosterone sulfotransferase gene: molecular cloning and structural characterization. DNA Cell Biol. 1995a;14:331–41. doi: 10.1089/dna.1995.14.331. [DOI] [PubMed] [Google Scholar]
- OTTERNESS DM, HER C, AKSOY S, KIMURA S, WIEBEN ED, WEINSHILBOUM RM. Human dehydroepiandrosterone sulfotransferase gene: molecular cloning and structural characterization. DNA & Cell Biology. 1995b;14:331–41. doi: 10.1089/dna.1995.14.331. [DOI] [PubMed] [Google Scholar]
- PACIFICI GM. Inhibition of human liver and duodenum sulfotransferases by drugs and dietary chemicals: a review of the literature. Int J Clin Pharmacol Ther. 2004;42:488–95. doi: 10.5414/cpp42488. [DOI] [PubMed] [Google Scholar]
- RUNGE-MORRIS MA. Regulation of expression of the rodent cytosolic sulfotransferases. Faseb J. 1997;11:109–17. doi: 10.1096/fasebj.11.2.9039952. [DOI] [PubMed] [Google Scholar]
- SAINI SP, SONODA J, XU L, TOMA D, UPPAL H, MU Y, REN S, MOORE DD, EVANS RM, XIE W. A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol Pharmacol. 2004;65:292–300. doi: 10.1124/mol.65.2.292. [DOI] [PubMed] [Google Scholar]
- SANER KJ, SUZUKI T, SASANO H, PIZZEY J, HO C, STRAUSS JF, 3RD, CARR BR, RAINEY WE. Steroid sulfotransferase 2A1 gene transcription is regulated by steroidogenic factor 1 and GATA-6 in the human adrenal. Mol Endocrinol. 2005;19:184–97. doi: 10.1210/me.2003-0332. [DOI] [PubMed] [Google Scholar]
- SEELY J, AMIGH KS, SUZUKI T, MAYHEW B, SASANO H, GIGUERE V, LAGANIERE J, CARR BR, RAINEY WE. Transcriptional regulation of dehydroepiandrosterone sulfotransferase (SULT2A1) by estrogen-related receptor alpha. Endocrinology. 2005;146:3605–13. doi: 10.1210/en.2004-1619. [DOI] [PubMed] [Google Scholar]
- SONG CS, ECHCHGADDA I, BAEK BS, AHN SC, OH T, ROY AK, CHATTERJEE B. Dehydroepiandrosterone sulfotransferase gene induction by bile acid activated farnesoid X receptor. J Biol Chem. 2001;276:42549–56. doi: 10.1074/jbc.M107557200. [DOI] [PubMed] [Google Scholar]
- SONG CS, ECHCHGADDA I, SEO YK, OH T, KIM S, KIM SA, CHO S, SHI L, CHATTERJEE B. An Essential Role of the CAAT/Enhancer Binding Protein-{alpha} in the Vitamin D Induced Expression of the Human Steroid/Bile Acid-sulfotransferase (SULT2A1) Mol Endocrinol. 2005 doi: 10.1210/me.2005-0428. [DOI] [PubMed] [Google Scholar]
- SONG CS, ECHCHGADDA I, SEO YK, OH T, KIM S, KIM SA, CHO S, SHI L, CHATTERJEE B. An essential role of the CAAT/enhancer binding protein-alpha in the vitamin D-induced expression of the human steroid/bile acid-sulfotransferase (SULT2A1) Mol Endocrinol. 2006;20:795–808. doi: 10.1210/me.2005-0428. [DOI] [PubMed] [Google Scholar]
- SONODA J, XIE W, ROSENFELD JM, BARWICK JL, GUZELIAN PS, EVANS RM. Regulation of a xenobiotic sulfonation cascade by nuclear pregnane X receptor (PXR) Proc Natl Acad Sci U S A. 2002;99:13801–6. doi: 10.1073/pnas.212494599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAN NS, MICHALIK L, DESVERGNE B, WAHLI W. Multiple expression control mechanisms of peroxisome proliferator-activated receptors and their target genes. J Steroid Biochem Mol Biol. 2005;93:99–105. doi: 10.1016/j.jsbmb.2004.12.025. [DOI] [PubMed] [Google Scholar]
- WALLING J. From methotrexate to pemetrexed and beyond. A review of the pharmacodynamic and clinical properties of antifolates. Invest New Drugs. 2006;24:37–77. doi: 10.1007/s10637-005-4541-1. [DOI] [PubMed] [Google Scholar]
- XU C, LI CY, KONG AN. Induction of phase I, II and III drug metabolism/transport by xenobiotics. Arch Pharm Res. 2005;28:249–68. doi: 10.1007/BF02977789. [DOI] [PubMed] [Google Scholar]