


Abstract
R1Bm is a long interspersed element (LINE) inserted into a specific sequence within 28S rDNA of the silkworm genome. Of two open reading frames (ORFs) of R1Bm, ORF2 encodes a reverse transcriptase (RT) and an endonuclease (EN) domain which digests specifically both top and bottom strand of the target sequence in 28S rDNA. To elucidate the sequence specificity of EN domain of R1Bm (R1Bm EN), we examined the cleavage tendency for the target sequences, and found that 5′-A(G/C)(A/T)!(A/G)T-3′ is the consensus sequence (! = cleavage site). We also determined the crystal structure of R1Bm EN at 2.0 Å resolution. Its structure was basically similar to AP endonuclease family, but had a special β-hairpin at the edge of the DNA binding surface, which is a common feature among EN of LINEs. Point-mutations on the DNA binding surface of R1Bm EN significantly decreased the cleavage activities, but did not affect the sequence recognition in most residues. However, two mutants Y98A and N180A had altered cleavage patterns, suggesting an important role of these residues (Y98 and N180) for the sequence recognition of R1Bm EN. In addition, Y98A mutant showed another cleavage pattern, that implies de novo design of novel sequence-specific EN.
INTRODUCTION
Non-long terminal repeat (non-LTR) retrotransposons, also called long interspersed nuclear elements (LINEs), are the most abundant family among mobile elements. LINEs have been identified in all major groups of eukaryotes, with the exception of the bdelloid rotifers (1). In human, up to 21% of the genome is comprised of LINEs (2), which are implicated to be involved in the gene evolution and genome reconstruction (3–5).
LINEs are divided into two subtypes. The first has only one open reading frame (ORF) that encodes a type-2 restriction-enzyme-like endonuclease (EN), whereas the other has two ORFs including an apurine/apyrimidine endonuclease (APE)-like EN at the N-terminus of the second ORF (ORF2) (6). ORF2 of the latter members of the LINE family, which include L1 (7,8), TRAS1 (9), SART1 (10), Tx1L (11) and R1 (12), typically encodes an EN domain at the N-terminus, a reverse transcriptase (RT) domain in the middle region and a zinc-finger-like domain at the C-terminus. The first ORF (ORF1) has no sequence similarity to other known sequences, although some ORF1 proteins have been reported with RNA-binding (13–16) activities.
The retrotransposition process of LINEs is known as target-primed reverse transcription (TPRT). In the TPRT model, the EN domain first recognizes and cleaves the target DNA and the RT domain immediately proceeds with reverse transcription using its own mRNA as the template (17). Although most LINEs insert randomly throughout the genome, some elements show high-target sequence specificity. For example, TRAS1 (9,18) and SART1 (10) insert into the telomeric repeats of Bombyx mori, Tx1L (11) inserts into the Tx1D sequence of Xenopus laevis and R1 inserts into 28S rDNA sequences of many insect species (12,19). Of these target-specific LINEs, TRAS1, SART1, and R1 belong to the R1 clade. Most retrotransposons in this group are target specific (20). Previous studies have demonstrated that the EN itself has sequence specificity (11,18,19,21). Furthermore, a study of EN swapping between TRAS1 and SART1 showed that the target specificity of EN determines its insertion site (22). Structural studies of TRAS1 (21) and human L1 (23) have revealed that the LINE EN has a folding pattern similar to that of apurinic/apyrimidinic (AP) EN, but has an extra β-hairpin at the DNA-binding surface. A mutagenesis study of the TRAS1 EN indicated that the β-hairpin region is necessary for sequence recognition (21). In spite of these reports, molecular basis for the sequence specificity of EN encoded in LINEs are still unclear.
R1Bm, the R1 retrotransposon species of B. mori, inserts into a 14-bp region of 28S rDNA. Biochemical analysis has shown that R1Bm EN cleaves the sequence ACA!GTG (! = cleavage site) on the bottom strand followed by the sequence ACT!ATC on the top strand (19), although the detailed mechanism remains unknown. In this study, to elucidate how R1Bm EN achieves such complicated sequence recognition, we performed a substituted oligonucleotide cleavage study for the target DNA sequence and determined the crystal structure of R1Bm EN at 2.0 Å resolution. Furthermore, mutagenesis studies of the DNA-binding surface of R1Bm EN based on the crystal structure revealed some amino acids involved in the sequence specificity.
MATERIALS AND METHODS
Plasmid construction
The R1Bm EN domain was amplified by PCR with Pfu Turbo DNA polymerase (Stratagene) using primers R1EN-NdeI-s (5′-AAAAACATATGGATATTAGGCCCCGACTTCG-3′) and R1EN + 19aa-XhoI-a (5′-AA AAACTCGAGTTACGGCTCGCCCGGCCTCAAATCGC-3′) with R1WT-pAcGHLTB as a template (24). The first primer contains an NdeI site. The second primer includes a stop codon followed by an XhoI site. The PCR product was digested with NdeI and XhoI, and subcloned between NdeI and XhoI sites of the pET16b expression vector (Novagen). The resulting plasmid, named pR1EN, includes the first 717 bp of ORF2, corresponding to the first 239 amino acids, and the sequence encoding 10-His tag at the N terminus of R1Bm EN. All point mutations were generated by a QuickChange site-directed mutagenesis Kit (Stratagene) according to manufacturer's instructions. The sequences of the primers used for the introduction of these mutations are available on request. The mutation of each plasmid was confirmed by DNA sequencing.
Expression and purification of R1Bm-EN
The pR1EN and its mutant derivatives were transformed into E. coli BL21(DE3)/pLysS strain. The transformants were cultured at 37°C in 50 ml of Luria broth until the optical density at 600 nm reached ∼0.8. Isopropyl- β-d-thiogalactopyranoside (IPTG) was then added to a final concentration of 1 mM, followed by further incubation at 25°C overnight. Cells were pelleted by centrifugation and frozen in liquid nitrogen. Purification of R1EN was conducted following the protocol from Qiagen (catalog number 30210). Cell pellets were thawed at 4°C for 10 min, suspended in 0.6 ml of sonication buffer [50 mM sodium phosphate (pH 6.0), 0.5 M NaCl, 100 mM imidazole, 20 mM 2-mercaptoethanol, 2% triton X-100] and sonicated for 1 min on ice. The cell extracts were clarified by centrifugation at 20 000 × g for 10 min and filtration through a 0.45 μm membrane (Millipore). Because the total volume of cell extracts increased when the mass of cell pellets are taken into account, the imidazole concentration was readjusted to 100 mM, the supernatant was mixed with 30 μl of pre-equilibrated nickel NTA agarose (Qiagen) at 4°C for 2 h. The resin was washed three times with 1 ml of sonication buffer, three times with 1 ml of washing buffer [50 mM sodium phosphate (pH 6.0), 1 M NaCl, 100 mM imidazole] and once with 1 ml of washing buffer containing 0.35 NaCl. Finally, the protein was eluted with 0.25 ml of elution buffer [50 mM sodium phosphate (pH 6.0), 0.35 M NaCl, 0.3 M imidazole]. The eluted protein was ultrafiltered and concentrated in storage buffer [50 mM sodium phosphate (pH 7.0), 0.35 M NaCl, 10% glycerol, 10 mM 2-mercaptoethanol] with Microcon YM-10 (Millipore). The purified protein, the concentration of which was determined by SDS–PAGE, was diluted with storage buffer at 0.5 mg/ml and stored at −80°C. The concentration of R1Bm EN was determined by comparing the intensity of the band with an analytical curve obtained from Coomassie blue stained SDS–PAGE gel with that of known amounts of bovine serum albumin.
Crystallographic study
For crystallization, BL21(DE3)/pLysS cells carrying pR1EN were cultured in LB medium at 37°C. When OD600 had reached to 0.6, IPTG was added to the medium at final concentration as 0.75 mM, and cells were further incubated for 6 h at 26°C. Cells were collected by centrifugation and stored at −80°C.
Stored cells were thawed and sonicated in lysis buffer [40 mM Tris–Cl (pH 7.5), 0.5 M NaCl, 50 mM imidazole, 0.1% triton X-100 and 0.1 mM PMSF], centrifuged and the supernatants were subjected to Nickel-trapped HiTrap Chelating column (GE healthcare) and eluted by elution buffer [40 mM Tris, 0.5 M NaCl, 0.35 M imidazole, (pH7.5)]. The proteins were precipitated by adding ammonium sulfate and collected by centrifugation, and the pellet was dissolved with 10 ml of digestion buffer [50 mM Tris–Cl (pH 7.5), 0.3 M NaCl, 1 mM DTT, 2 mM CaCl2], followed by Factor Xa (New England Biolab) digestion for overnight at 10°C.
The sample solution was loaded on HiTrap SP column (GE healthcare) followed by Superdex 200 (GE healthcare), resulting the highly purified R1Bm EN. The protein was concentrated in sample buffer (40 mM HEPES-Na pH 7.2, 200 mM NaCl, 1 mM DTT, 1 mM EDTA) with 10 kDa Microcon (Millipore) and protein solution was prepared to 6 mg/ml. In that case, the concentration of R1Bm EN was determined by absorption of 280 nm spectra with extinction coefficients of 27 310 M−1 cm−1.
Crystals of the R1Bm EN were obtained by hanging drop vapor diffusion method at 283 K. Two microliters of protein solution were mixed with 1.5 μl of reservoir solution containing 2.4 M sodium acetate (pH 6.9), 10 mM ammonium sulfate, and 1–2% Jeffamine M-600 reagent. The hexagonal rod-shaped crystals, whose size of 0.2 × 0.2 × 0.5 mm3, were gown within 2–3 weeks.
Prior to the data collection, crystals were transferred into cryoprotectant solutions containing 9% (v/v) ethylene glycol and 9% (v/v) PEG-200 in reservoir solution and flash frozen in cryo nitrogen stream. Dataset was collected at the Photon Factory (Tsukuba, Japan) on beamline BL-6A. Data processing and reduction were carried out with the programs MOSFLM (25) and SCALA (26). Phases were calculated by molecular replacement method with the MolRep (27) program using TRAS1-EN structure (PDB entry: 1WDU) as a search model. Further model was built with program O (28) and structure refinement calculations including simulated annealing and B-factor refinement were done with CNS (29). Summary of the crystallographic statistics are shown in Table 1.
Table 1.
Summary of crystallographic statisticsa
| Data collection | |
|---|---|
| X-ray source | Photon Factory BL-6A |
| Wavelength (Å) | 1.000 |
| Space group | P321 |
| Unit cell parameters | a = b = 141.3 Å, c = 37.5 Å, α = β = 90°, γ = 120° |
| Resolution range (Å) | 40–2.0 (2.1–2.0) |
| Observed reflections | 622 185 (81 080) |
| Redundancy | 21.3 (19.3) |
| Completeness (%) | 100 (100) |
| 〈 I / σ I 〉 | 6.8 (2.1) |
| R-symb | 0.088 (0.35) |
| Structure refinement | |
| Resolution range (Å) | 40–2.0 (2.1–2.0) |
| No. of reflections (work/free) | 27 763 (2713)/1432 (161) |
| R-factor c (work/free) | 0.195 (0.212)/0.211 (0.250) |
| No. of atoms (protein/solvent) | 1672/128 |
| 〈 B 〉 (protein/solvent) (Å2) | 23.68/29.75 |
| RMSD Bonds (Å) | 0.0052 |
| RMSD Angles (°) | 1.304 |
| Ramachandran plot analysisd (%) | |
| Most favored | 91.4 |
| Additional allowed | 7.6 |
| Generously allowed | 1.1 |
| Disallowed | 0 |
| PDB entry | 2EI9 |
aValues in parentheses correspond to the outer shell.
bR-sym = Σ | Ii- 〈 Ii 〉 |/ Σ Ii, where Ii is the observed intensity and 〈 Ii 〉 is the average intensity obtained from multiple observations of symmetry-related reflections.
cR-work = Σhkl||Fobs| − |Fcalc||/Σhkl|Fobs|. Five percent of the reflections were excluded for R-free calculation.
dAnalyzed with PROCHECK (41).
Oligonucleotide cleavage assay
The 32P-labeled substrates containing the R1Bm target site were prepared exactly as previously described (18). The 40-bp top or bottom strand oligonucleotides were radio-labeled, annealed with the complementary non-labeled oligonucleotides and gel-purified. The cold substrates were also prepared by annealing of non-labeled top and bottom strand oligonucleotides. The mixture of labeled and non-labeled substrates was used for the cleavage reaction. The reaction mixture containing 50 mM PIPES-NaOH at pH 6.0, 17.5 mM NaCl, 1 mM MgCl2, 200 ng of purified proteins and 1 or 3 pmol of substrate DNA (the molar ratio of protein:DNA is 15:2 or 15:6) in a total volume of 10 μl was incubated at 25°C for 60 min. The reaction was stopped by the addition of 10 μl of denaturing solution (95% formamide, 50 mM EDTA, 0.01% bromophenol blue). The reaction product was denatured for 3 min at 95°C, immediately chilled on ice, and separated on a 30% polyacrylamide denaturing gel. The cleavage efficiency was quantified with BAS 5000 imaging analyzer system (Fujifilm). In the experiment of Figure 6E, oligonucleotide cleavage assays were performed with the reaction mixture containing 10 mM MgCl2 and 0.1 pmol of substrate DNA, and incubation time was increased from 1 to 5 h.
Figure 6.

Mutagenesis study of R1Bm EN. (A) Positions of residues mutated in this study. The positions of residues substituted with alanine are indicated as line models (magenta). His-209 is also depicted to indicate the catalytic site. (B) Bottom strand cleavage by R1Bm EN mutants. Three picomoles of the substrate was cleaved with wild-type R1Bm EN and the mutants. The solid arrowheads indicate the target site cleavage of the bottom strand. (C) Top strand cleavage by R1Bm EN mutants. One picomole of the substrate was used. The open arrowheads indicate the target site cleavage of the top strand. (D) Nicking activities of R1Bm EN and the mutants. The cleavage products nicked at the target site were quantified and the percentage of the cleavage product relative to that of wild-type R1Bm EN was shown in each strand cleavage. The mutants representing <20% of wild-type activity in both strand cleavage are underlined in magenta and the mutants showing the great reduction of the nicking activity only in bottom strand cleavage are underlined in blue. The results of three independent experiments were averaged and error bars show S.E. (E) Cleavage of the bottom strand by R1Bm EN mutants under the vigorous condition. The cleaved bands corresponding to Figure 1B are indicated. Asterisk indicates the abnormal band observed in Y98A mutant.
Radio-labeled oligonucleotides with the same sequence as the expected cleavage products were used for the size markers: 5′-ACGAGATTCCCACTGTCCCTATCTACT-3′ and 5′-GGTTTCGCTAGATAGTAGATAGGGACA-3′ as for top and bottom strand cleavage, respectively. To determine cleavage sites precisely, sequencing markers were used in some experiments. Sequencing markers were generated by primer extension with TaKaRa Taq Cycle Sequencing Kit (Takara) using the manufacturer's suggested protocol except for the PCR condition. The reaction mixture was denatured at 94°C for 20 s, followed by 25 cycles of 94°C for 20 s, 30°C for 20 s and 72°C for 1 min, and then another 15 cycles of 94°C for 20 s and 72°C 20 s. To generate the sequencing template, 247 bp of 28S rDNA sequence containing the R1Bm target site was amplified by PCR with Pfu Turbo DNA polymerase (Stratagene) with the primers 28SrDNA-XhoI-s (5′-AAAAACTCGAGGCGCGGGTAAACGGCGGG-3′) and 28SrDNA-BamHI-a (5′-AAAAAGGATCCCGCGAAACGATCTCCC-3′) using pBmR161 (30) as template. The PCR product was purified with GenElute PCR Clean-up Kit (Sigma) prior to use. The sequencing primers used for the bottom and the top strands are 5′-GGTTTCGCTAGAT-3′ and 5′-ACGAGATTCCCAC-3′, respectively.
RESULTS
R1Bm EN cleaves both strands of the 28S rDNA sequence at the target site
We expressed R1Bm EN in Escherichia coli with an N-terminal His10 tag and purified it by nickel chelation chromatography. It has been reported that the EN domain of R1Bm cleaves the target sequence (19). The bottom and top strands of the target sequence are shown in Figure 1A, and the cleavage sites are indicated as X and Y. To confirm the sequence specificity of R1Bm EN, the activity of R1Bm EN was tested using a double-stranded oligonucleotide substrate that contained the target sequence. This substrate was radiolabeled on either the top or bottom strand and digested with R1Bm EN in the reaction mixture for different reaction times. The reaction products were separated on a polyacrylamide denaturing sequencing gel with sequencing markers. As shown in Figure 1B, the cleavage of the target sequence was detected on both strands of the substrate (band-X and Y). This demonstrates that the R1Bm EN purified in this study precisely generates a nick on the target sequence. As well as X and Y, a few other bands (bands 1–7) were observed from both strands. This was because cleavage by R1Bm EN is not absolutely sequence specific in vitro, as shown previously (19). By comparing the gel migration length with the sequencing ladders, these cleavage sites were confirmed as shown in Figure 1A.
Figure 1.

Target sequence cleavage of R1EN. (A) Substrate DNA used for oligonucleotide cleavage study. The substrate consists of the 40-bp long 28S rDNA sequence containing the target site of R1Bm. The sequence of target site duplication (TSD) is boxed. The X and Y indicate the original target sites on the bottom and top strands, respectively. One to seven represent cleaved sites other than the target sites (A and B). (B) Sequence-specific cleavage of both strands. Three picomoles of the substrates was incubated with 200 ng of H209A and R1EN at 25°C for 0, 30, 60 and 120 min and separated on a 30% polyacrylamide denaturing gel with sequencing markers. Cleavage sites are summarized in (A).
A mutated protein, H209A, was also purified and used as a negative control because His-209 is essential for the catalysis of TRAS1 EN and L1 EN (18, 8). H209A exhibited no detectable cleavage activity on the top or bottom strand, indicating that there was no contaminating nuclease activity from E. coli in the purified protein (Figure 1B). The time course analysis showed the cleavage of bottom strand is more efficient than the top strand cleavage (Figure S1).
We next determined the sequences required for the sequence-specific nicks on the bottom or top strand. A series of mutated substrates was prepared. Each substrate contained a 2-bp substitution, TT or AA, on the bottom or top strand, respectively. The mutated site was gradually moved in 2-bp steps from 5′ to 3′ around the cleavage site. The nicking activity on the mutated substrate was tested and compared with that on the non-mutated substrate. During bottom-strand cleavage, no significant change in nicking activity was detected on six substrates (lanes B1 to B3 and B9 to B11, Figure 2A). However, mutations within 5 bp of the cleavage site, either upstream or downstream, resulted in a severe reduction in nicking activity (lanes B4 to B8, Figure 2A), suggesting that this 10-bp region is important for the sequence-specific cleavage on the bottom strand. On the substrates of lanes B5 and B8, we observed strong signals at positions 4-bp shorter (B5#) and 2-bp longer (B8*) than the original cleavage site, respectively. This indicates that these mutations result in novel cleavage sites, other than the original site targeted by R1Bm EN.
Figure 2.

Identification of sequences involved in the cleavage of the target sites. Nicking activities of the target sites on mutated substrates were examined (A, bottom strand cleavage; B, top strand cleavage). Mutated substrates contained TT or AA substitution (boxed) and a mutated position of the substrates was sequentially changed. Each substrate was numbered and CTRL represents a non-mutated control substrate. Nicked strands of mutated substrates are shown at the lower left of each panel and mutated bases were boxed. The solid and open arrowheads indicate the cleavage sites on the top and bottom strands, respectively. Three picomoles (for bottom strand cleavage) or 1 pmol (for top strand cleavage) of the mutated substrate was incubated with R1EN and electrophoresed. Gel bands around the cleavage products were shown at the top of each panel. The size marker has the same sequence as the expected cleaved product. The cleavage products nicked at the target site were quantified and the percentage of the cleavage product relative to that of a control substrate was shown at the lower bottom of each panel. The results of three independent experiments were averaged and error bars show S.E.
In top-strand cleavage, the nicking activity on four mutated substrates was reduced to about 6–38% of that on the original substrate (lanes T3, T4, T6 and T8, Figure 2B). A modest decrease in nicking activity was observed on the substrate mutated at the center of the cleavage site (lane T5, Figure 2B). These results suggest that 5-bp upstream and 3-bp downstream of the cleavage site are involved in sequence-specific cleavage of the top strand. Curiously, the substrates of lanes T7 and T9 increased the cleavage activity, whereas T8 reduced it to 32% again. We also found on the bottom strand that the activity on substrate B8 was lower than that on substrate B7 (Figure 2A). Both B8 and T8 substrates have a series of G:C pairs substituted with A:T pairs. Hence, reduction of the nicking activities on B8 and T8 may have resulted from instability of double-strand DNA rather than sequence specificity.
A previous study has shown that bottom-strand cleavage occurs faster than top-strand cleavage in the target-sequence cleavage by R1Bm EN (19). Considering this result, it is possible that the cleavage of the top strand does not occur by sequence recognition. For example, R1Bm EN may have binding affinity for the nicked DNA structure. To address this issue, we studied the top-strand cleavage of the B6 sequence. This sequence is mutated in the original bottom strand cleavage site X, and thus its cleavage on site X has been severely reduced (Figure 2A, lane B6), with no large difference in cleavage of other sites (bands 1–7) (Figures 2A and S1). Unexpectedly, when we tested for the top strand cleavage, the B6 substrate was cleaved about twice as efficiently as the control (Figure 2B, lane B6). This was probably because the incompetence of the bottom-strand target site enhanced the top-strand cleavage relative to that of the original sequence. Nevertheless, this result indicates that the top strand cleavage site is also recognized sequence specifically by R1Bm EN.
DETERMINATION OF THE CLEAVAGE SEQUENCE OF R1Bm EN
Figure 1B shows that the 28S rDNA sequence was cleaved by R1Bm EN not only at the target site, but also at other sites. Because the signal patterns for the non-specific products were not random, we focused on them and tried to determine the sequence tendency recognized and digested by R1Bm EN. The cleavage activity was estimated from the density of each band, and they were aligned by cleavage site to allow comparison (Figure 3). In all those in the high-cleavage group (>50% that of the wild-type sequence), which included X, Y, 7, 4, 5 (Figure 1), and B8* (Figure 2), adenine is located at the −3 position from the cleavage site. In five of the six highly cleaved sequences, thymine is located at the +2 position. Furthermore, guanine or adenine is located at the +1 position and thymine or adenine at the −1 position in all sequences, and five of the six sequences have cytosine or guanine at the −2 position. Taken together, these results suggest that the consensus sequence for the high-cleavage group is 5′-A(G/C)(A/T)!(G/A)T-3′ (! = cleavage site).
Figure 3.
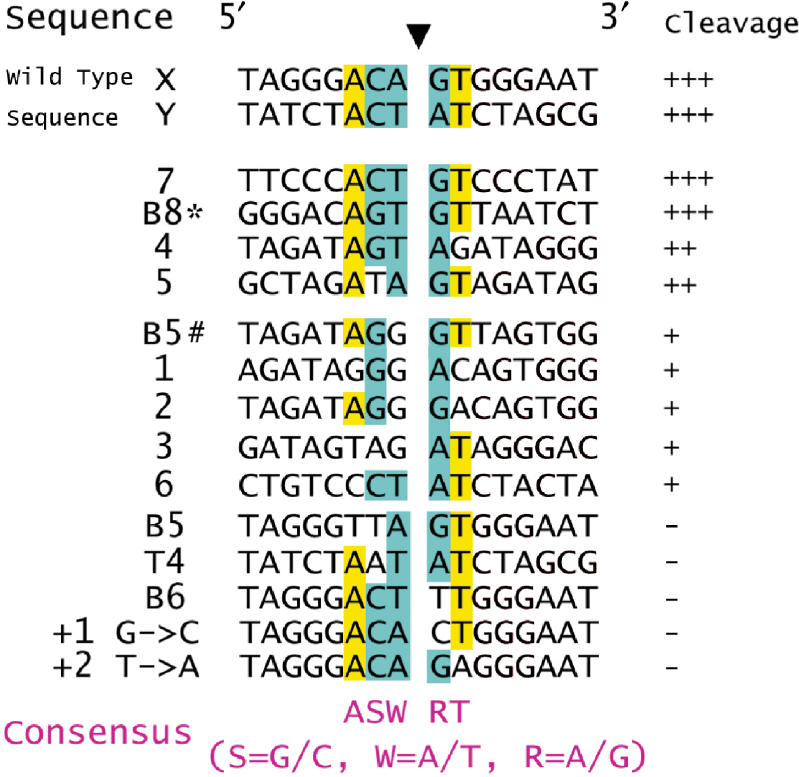
Sequence comparison of the cleavage sites of R1Bm EN. The cleaved sites shown in Figures 1 and 2 were classified into four classes (+++ >70%; ++ >50%; + >10%; − <10% of wild-type cleavage, respectively) according to the density of each band, and aligned by the cleavage site (filled arrowhead). The conserved bases among the substrates are highlighted with yellow (one base species) and cyan (two bases species).
The weakly cleaved sequences (<50% that of the wild-type sequence), which included 1, 2, 3, 6 and B5#, also all have guanine or adenine at the +1 position, but the sequence identities to 5′-A(G/C)(A/T)!(G/A)T-3′ are low. However, several sequences with single or double substitutions, such as B5, B6 and T4, were almost uncleaved (<10% that of the wild-type sequence). As the different contributions of each nucleotide to R1Bm EN cleavage were considered, we tested two other oligonucleotides substituted at +1 with cytosine and +2 with adenine. These sequences were almost uncleaved (Figures 3 and S1). Substitution at the +1 position to thymine or cytosine led to a severe reduction in cleavage. This suggests that a purine base at the +1 position is essential for R1Bm EN recognition and cleavage.
Structural analysis of R1Bm EN
To clarify its sequence specificity at the atomic level, R1Bm EN (residues 1–220) were expressed, purified and crystallized (‘Materials and Methods’). The crystal structure of R1Bm EN, the final model for which contained 215 amino acids (residues 5–219), was resolved by molecular replacement at 2.0 Å resolution, with values for R-factor and R-free of 19.5% and 21.1%, respectively (Figures 4A and B). The asymmetric component of the crystal is a monomer. Furthermore, in gel filtration chromatography during the purification, R1Bm EN eluted with a fraction corresponding to 22 kDa, therefore, R1Bm EN does not multimerize under this condition (Figure S3).
Figure 4.

Crystal structure of R1Bm EN. (A) Cartoon representation of R1Bm EN (5–219). The characteristic β-hairpin region (β8–β9) and α3 are indicated. (B) The sigma-weighted 2Fo-Fc electron-density map showing around the active site of R1Bm EN. The map was contoured at 1.5σ. (C) The sequence-alignment of R1Bm EN, TRAS1-EN, L1-EN (human) and human APE1. The secondary structure of R1Bm EN is indicated above the amino acid sequence. Residues conserved among all or most APE family are indicated as red or black bold cases, respectively. Residues substituted to alanine in mutagenesis study are highlighted by green. All figures were prepared by PyMOL (DeLano Scientific, http://pymol.sourceforge.net/).
The overall structure of R1Bm EN is a four-layered α/β sandwiched fold (Figure 4A), and is almost identical to that of other known EN domains of the APE-type LINEs, TRAS1 EN and L1 EN. The root-mean-square differences of the Cα atoms of the core β-sheets (71 residues) between R1Bm EN versus TRAS1 EN and L1 EN are 0.68 Å and 2.21 Å, respectively. Furthermore, three α-helices located outside the β-sheets also fit well between R1Bm EN and TRAS1 EN (Figure 5A), indicating that the R1Bm EN structure resembles the TRAS1 EN structure more than the L1 EN structure. This observation is consistent with previous phylogenetic analyses of the LINE family, which demonstrated that TRAS1 and R1Bm are in the same clade, whereas L1 is not (20).
Figure 5.

Structural comparison of R1Bm EN. (A) Stereoview representation of superposed backbone model of R1Bm EN (green), TRAS1-EN (PDB entry:1WDU, magenta) and L1-EN (PDB entry:1VYB, cyan). β-hairpin region (β8–β9) and α3 are indicated. (B) Close up view of superposed active site of R1Bm EN (green), TRAS1-EN (magenta), L1-EN (cyan) and human APE1 (PDB entry: 1DEW, pale orange). Residue names and numbers of R1Bm EN are shown.
R1Bm EN has a characteristic β-hairpin region (residues 176–183), stretched out to the edge of the DNA-binding surface (Figure 4A). This β-hairpin structure is not observed in human AP endonuclease I (hAPE1) or other AP ENs. However, it also occurs in TRAS1 EN (Figure 5A; 21) and L1 EN (Figure 5A; 23), so it is probably a common feature among the LINE ENs. The R1Bm EN also has a short α-helix (α3; residues 138–144) stretched out to the DNA-binding surface. The amino acid sequence of α3 is Arg–His–Tyr–Val–Gly–Arg, and it thus has basic properties. Interestingly, this α-helix does not occur in the TRAS1 EN or L1 EN structures (Figure 5A), whereas a similar helix is found in hAPE1 (residues 221–228) (31). In hAPE1, the amino acid sequence of this region is Arg–Asn–Pro–Lys–Gly–Asn–Lys–Lys, which is also thus a basic region. In the crystal structure of the hAPE1:DNA complex, this α-helix interacts with the DNA backbone phosphate (32).
Looking at the R1Bm EN structure in detail, all the catalytic residues, including Asn-13, Glu-40, Tyr-95, Asp-126, Asn-128, Thr-175, Phe-176, Asp-186, Ser-207, Asp-208 and His-209, are conserved relative to those of the highly ordered arrangement among the AP EN family (Figure 5B). This indicates that R1Bm EN should cleave dsDNA in a similar manner to the AP ENs. From this perspective, R1Bm EN and other sequence-specific LINE ENs should have extra regions that function in sequence recognition, because the AP ENs do not recognize DNA sequences, but only the AP site (32), for which the β-hairpin region is one candidate (21).
Mutagenesis study of R1Bm EN
We performed mutagenesis studies to investigate which residues of R1Bm EN are important for its sequence specificity. The crystal structure of R1Bm EN gave us information about the residues of the protein that potentially interact with DNA. Thus, we chose 18 polar residues located at the DNA-binding surface that have not been reported to be involved in EN activity (Figure 6A). Each residue was substituted with alanine and the mutated proteins were expressed and purified. We tested the nicking activity of these mutants on both the bottom and top strands using a double-stranded oligonucleotide substrate (Figures 6B and C). Although the nicking activities of several mutants were reduced, the nicking pattern of no mutant changed relative to that of the wild-type R1Bm EN in the cleavage of either strand. Nicking activities on the target sequences of both strands were measured, and are expressed as a percentage of the wild-type activities in Figure 6D. Among the mutants, S43A, S99A and E147A did not exhibit a significant reduction in nicking activity, compared with that of the wild-type R1Bm EN. However, Y42A, H78A, Q97A, Y98A, R139A, H140A, R144A, T178A and N180A showed <20% of the wild-type activity for the cleavage of both strands, suggesting that these residues are significant for both strand cleavages. K56A and H130A also moderately decreased the nicking activity. Many mutants exhibited no great difference in nicking activity between the bottom- and top-strand cleavages. However, in E18A, D19A, H54A and S206A, the relative activity of bottom-strand cleavage was less than half that of top-strand cleavage, whereas the activity of top-strand cleavage was >80% that of the wild-type R1Bm EN. This suggests that these four residues are important only for bottom-strand cleavage. Looking at the R1Bm EN structure, His-78, Gln-97 and His-130 interact with other residues directly or via water-mediated hydrogen bonding (Figures 6A and S4). Therefore, their substitution with alanine made the domain structurally unstable, leading to the reduction of cleavage activity.
In several mutants (Y42A, H78A, Q97A, Y98A, R139A, H140A, R144A, T178A and N180A) the amounts of cleavage products were <1% of the total substrates. We could not detect the nicking pattern of each R1Bm EN mutant in Figures 6B and C. Therefore, we re-examined the bottom-strand cleavage by R1Bm EN under high-magnesium conditions, so that the R1Bm EN mutants more efficiently cleaved the target sequence (Figure 6E; ‘Materials and Methods’). The cleavage activities of all mutants whose substituted positions were located at α3 (R139A, H140A and R144A) were significantly reduced (<10%). This result suggests that the α3 region is involved in phosphate-backbone interactions rather than in base recognition, as is observed for the hAPE:DNA complex (32).
When the nicking patterns were compared, that of Y98A was particularly different from the other patterns. Y98A gave a strong signal that was identical to that of band 4 and above the original target site (shown by a white asterisk). At the same time, signals for bands X, 3 and 5 were markedly reduced. We analyzed the Y98A cleavage product mixture with sequencing-gel electrophoresis, which revealed that this oligonucleotide is 2-bp longer than band X (data not shown). Therefore, the sequence cleaved by Y98A is suggested to be 5′-ACAGT!GGGAA-3′ (! = cleavage site). Compared with 5′-A(G/C) (A/T)!(G/A)T-3′, the thymine at the +2 position is changed to guanine. Furthermore, a slightly increased signal for band 2 and a reduced signal for band 5 were observed for N180A. This residue is located in the β-hairpin region (Figure 4C and 6A), as which the same structure was shown to be important for specific cleavage in TRAS1 EN (21).
DISCUSSION
R1Bm EN has been considered to have high sequence specificity, because the R1Bm element is found at precisely the same target sequence of 28S rDNA in many insect species (33). Although TRAS1 EN (18) and Tx1L EN (11) have been reported to recognize a region of about 10 bp around the cleavage site in bottom-strand cleavage, the recognition sequence for top-strand cleavage has not been identified. In this study, we found that R1Bm EN recognizes 8 to10-bp regions around the cleavage sites (Figure 2). We also identified the target sequence of R1Bm EN as 5′-A(G/C)(A/T)!(A/G)T-3′. This result immediately prompts the question: how can R1Bm insert precisely at a 28S rDNA site? Referring to previous results, we predict that the target-sequence cleavage process of R1Bm is as follows. ORF2p binds to ORF1p and its own mRNA to form the ribonuclear protein complex (13,34,35), which is recruited to the 28S rDNA region of the genome. We assume that ORF1p guides this recruiting process, as in some telomere specific non-LTR retrotransposons such as SART1, HeT-A and TART, ORF1p localize in a dotted pattern (presumably telomeres) and which suggested that the ORF1p has a role in non-LTR retrotransposon ribonucleoprotein complex localization. The region required for this localization pattern is essential for retrotransposition in SART1 (36,37).
We demonstrated that the modified oligonucleotide substrate that is not nicked at the bottom-strand target site is cleaved at the top strand (B6 in Figure 2B). This result indicates that the top-strand cleavage does not depend on bottom-strand cleavage. Top-strand cleavage does not occur by the recognition of a nicked DNA structure by R1Bm EN, but R1Bm EN separately recognizes both target sequences. From this perspective, the difference of nicking efficiency of R1Bm EN between bottom and top strand (Figure S1) may be achieved merely by the level of sequence specificity. The fact that four residues (Glu-18, Asp-19, His-54 and Ser-206) are specifically involved in top-strand cleavage also supports this hypothesis. Interestingly, all these residues are located on the opposite to α3, and would interact with the region upstream from the target DNA.
Feng et al. (19) reported that R1Bm EN forms multimer, on the other hand, our structural analysis and gel filtration chromatography during purification indicated that tag-removed R1Bm EN is a monomer (Figure S3). In previous study, gel-filtration chromatography and equilibrium sedimentation experiments were done with extra C-terminal hexa-His tag and 33 amino acids (19). This difference may rely on the C-terminal non-native peptide, as there are some cases of protein oligomerization caused by histidine tags (38,39).
The crystal structure of R1Bm EN indicates that the characteristic β-hairpin and positively charged α-helix (α3) occur on the DNA-binding surface. The mutagenesis experiment showed that the α3 region is probably not involved in DNA base recognition, but in backbone interactions, as observed for the hAPE1:DNA structure. We found that mutations at Tyr-98 reduced the target-site cleavage instead of increasing the site 4 and 5′-ACAGT!GGGAA-3′ cleavage. Compared with the consensus sequence 5′-A(G/C)(A/T)!(A/G)T-3′, thymine at the +2 position is changed to guanine. Furthermore, the amounts of bands X, 3 and 5, which have thymine at the +2 position, were reduced and, at the same time, band 4, which has guanine at the +2 position, was increased in the Y98A mutant (Figure 6F). Putting these data together, Tyr-98 may interact with the thymine moiety at the +2 position (or adenine on the opposite strand). Although the cleavage rate was low, it is noteworthy that Y98A efficiently cleaves a novel site, which implies that the LINE EN could be used for the de novo design of target-site-specific ENs. It would be interesting to examine the effect of mutation at Y98 for the insertion site using in vivo retrotransposition assay system (24).
When the sequence of R1Bm is compared with those of its other subfamily members, the position of Tyr-98, which seems to recognize the thymine at +2 position, is phenylalanine whereas this position contains proline in TRAS1 and TRAS3, and asparagines in L1 and APE1 [Figure S6 and (40)]. Both tyrosine and phenylalanine have aromatic rings, so that a ring structure at this position might be critical for the recognition of the target sequence in the R1 family. Compared with the TRAS1 EN structure, the position of Tyr-98 is very close to Asp-130 of TRAS1 EN, which is one of the key residues for sequence specificity (21).
The β-hairpin region (β8–β9) is conserved in all the structurally determined LINE ENs. Mutation in the β-hairpin region of R1Bm EN decreased its cleavage activity (T178A and N180A in Figure 6D), and affected sequence-specific cleavage under high-magnesium conditions (N180A in Figure 6E). The band patterns of most mutants, except Y98A and N180A, did not differ from that of the wild-type under high-magnesium conditions (Figure 6E). This indicates that recognition of the 28S rDNA sequence is achieved by the cooperation of many residues and/or main-chain atoms, which explains the limited effects of single mutations. In the case of TRAS1 EN, the β-hairpin deletion (Δ-loop) caused greater alteration of the cleavage pattern than single mutations of amino acids in the β-hairpin region (Anzai and Fujiwara, unpublished data). Hence, the backbone atoms also contribute to sequence recognition.
To investigate the interactions between each residue and DNA, R1Bm EN was superimposed onto the hAPE1:DNA complex structure (PDB entry: 1dew; Figure S7). In this model, Tyr-98 is located near the −1 to +1 region of DNA, so it is reasonable to predict that Tyr-98 interacts with the +2 position thymine base. The β-hairpin region could attach to the −1 to +3 region on the minor groove side of the DNA in the predicted model. This model suggests that the DNA-binding surface of R1Bm EN extends from the −6 to the +4 position on the DNA which is in good correspondence with the substitution experiment (Figure 2). However, residues that seem to be involved in sequence recognition occur mainly at the −1 to +3 positions of the target sequence. This is not fully consistent with the finding that R1Bm EN recognizes 5′-A(G/C)(A/T)!(A/G)T-3′, because this sequence ranges from −3 to +2. The substitution of a series of G:C pairs with A:T pairs led to a reduction in nicking activity. This suggests that the stability of dsDNA, that is, the DNA structure, may be important for R1Bm EN cleavage, together with the sequence itself. The target-sequence recognition of R1Bm seems to be more complex than we had considered, and it has been difficult to clarify with the limited biochemical experiments used so far. Moreover, if we consider the LINE EN as a gene engineering tool, the Y98A mutant has a novel sequence-specific cleavage activity. Therefore, experiments that address structural studies of the R1Bm:DNA complex should provide further details about the mechanisms underlying its recognition and nicking of the target DNA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
Authors are grateful to Drs S. Wakatsuki, N. Igarashi and N. Matsugaki for diffraction data collection at PF BL-6A. This work was partly supported by grants from the Japan New Energy and Industrial Technology Development Organization (NEDO) (to N.M.), and supported in part by grants from the Ministry of Education, Culture, Sports, Science and Technology of Japan (MEXT) (to N.M and H.F.), and the Program for Promotion of Basic Research Activities for Innovative Biosciences (PROBRAIN) (to H.F.). Funding to pay the Open Access publication charges for this article was provided by MEXT. H.A. and M.O. are recipients of Research fellowships of the Japan Society for the Promotion of Science (JSPS) for Young Scientists. The coordinate of R1Bm EN has been deposited to Protein Data Bank with the entry ID 2EI9.
Conflict of interest statement. None declared.
REFERENCES
- 1.Arkhipova I, Meselson M. Transposable elements in sexual and ancient asexual taxa. Proc. Natl Acad Sci. USA. 2000;97:14473–14477. doi: 10.1073/pnas.97.26.14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, et al. Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- 3.Moran JV, DeBerardinis RJ, Kazazian HH., Jr Exon shuffling by L1 retrotransposition. Science. 1999;283:1530–1534. doi: 10.1126/science.283.5407.1530. [DOI] [PubMed] [Google Scholar]
- 4.Gilbert N, Lutz-Prigge S, Moran JV. Genomic deletions created upon LINE-1 retrotransposition. Cell. 2002;110:315–325. doi: 10.1016/s0092-8674(02)00828-0. [DOI] [PubMed] [Google Scholar]
- 5.Symer DE, Connelly C, Szak ST, Caputo EM, Cost GJ, Parmigiani G, Boeke JD. Human L1 retrotransposition is associated with genetic instability in vivo. Cell. 2002;110:327–338. doi: 10.1016/s0092-8674(02)00839-5. [DOI] [PubMed] [Google Scholar]
- 6.Kojima KK, Fujiwara H. An extraordinary retrotransposon family encoding dual endonucleases. Genome Res. 2005;15:1106–1117. doi: 10.1101/gr.3271405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fanning T, Singer M. The LINE-1 DNA-sequences in 4 mammalian orders predict proteins that conserve homologies to retrovirus proteins. Nucleic Acids Res. 1987;15:2251–2260. doi: 10.1093/nar/15.5.2251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feng Q, Moran JV, Kazazian HH, Jr, Boeke JD. Human L1 retrotransposon encodes a conserved endonuclease required for retrotransposition. Cell. 1996;87:905–916. doi: 10.1016/s0092-8674(00)81997-2. [DOI] [PubMed] [Google Scholar]
- 9.Okazaki S, Ishikawa H, Fujiwara H. Structural analysis of TRAS1, a novel family of telomeric repeat-associated retrotransposons in the silkworm, Bombyx mori. Mol. Cell. Biol. 1995;15:4545–4552. doi: 10.1128/mcb.15.8.4545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takahashi H, Okazaki S, Fujiwara H. A new family of sitespecific retrotransposons, SART1, is inserted into telomeric repeats of the silkworm, Bombyx mori. Nucleic Acids Res. 1997;25:1578–1584. doi: 10.1093/nar/25.8.1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Christensen S, Pont-Kingdon G, Carroll D. Target specificity of the endonuclease from the Xenopus laevis non-long terminal repeat retrotransposon, Tx1L. Mol. Cell. Biol. 2000;20:1219–1226. doi: 10.1128/mcb.20.4.1219-1226.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Xiong Y, Eickbush TH. The site-specific ribosomal DNA insertion element R1Bm belongs to a class of non-long-terminal-repeat retrotransposons. Mol. Cell. Biol. 1988;8:114–123. doi: 10.1128/mcb.8.1.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hohjoh H, Singer MF. Cytoplasmic ribonucleoprotein complexes containing human LINE-1 protein and RNA. EMBO J. 1996;15:630–639. [PMC free article] [PubMed] [Google Scholar]
- 14.Hohjoh H, Singer MF. Sequence specific single-strand RNA-binding protein encoded by the human LINE-1 retrotransposon. EMBO J. 1997;16:6034–6043. doi: 10.1093/emboj/16.19.6034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kolosha VO, Martin SL. In vitro properties of the first ORF protein from mouse LINE-1 support its role in ribonucleoprotein particle formation during retrotransposition. Proc. Natl Acad. Sci. USA. 1997;94:10155–10160. doi: 10.1073/pnas.94.19.10155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kolosha VO, Martin SL. High affinity, non-sequence-specific RNA binding by the open reading frame 1 (ORF1) protein from long interspersed nuclear element 1 (LINE-1) J. Biol. Chem. 2003;278:8112–8117. doi: 10.1074/jbc.M210487200. [DOI] [PubMed] [Google Scholar]
- 17.Luan DD, Korman MH, Jakubczak JL, Eickbush TH. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: a mechanism for non-LTR retrotransposition. Cell. 1993;72:595–605. doi: 10.1016/0092-8674(93)90078-5. [DOI] [PubMed] [Google Scholar]
- 18.Anzai T, Takahashi H, Fujiwara H. Sequence-specific recognition and cleavage of telomeric repeat (TTAGG)(n) by endonuclease of non-long terminal repeat retrotransposon TRAS1. Mol. Cell. Biol. 2001;21:100–108. doi: 10.1128/MCB.21.1.100-108.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feng Q, Schumann G, Boeke JD. Retrotransposon R1Bm endonuclease cleaves the target sequence. Proc. Natl Acad. Sci. USA. 1998;95:2083–2088. doi: 10.1073/pnas.95.5.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kojima KK, Fujiwara H. Evolution of target specificity in R1 clade non-LTR retrotransposons. Mol. Biol. Evol. 2003;20:51–61. doi: 10.1093/molbev/msg031. [DOI] [PubMed] [Google Scholar]
- 21.Maita N, Anzai T, Aoyagi H, Mizuno H, Fujiwara H. Crystal structure of the endonuclease domain encoded by the telomere-specific long interspersed nuclear element, TRAS1. J. Biol. Chem. 2004;279:41067–41076. doi: 10.1074/jbc.M406556200. [DOI] [PubMed] [Google Scholar]
- 22.Takahashi H, Fujiwara H. Transplantation of target site specificity by swapping the endonuclease domains of two LINEs. EMBO J. 2002;21:408–417. doi: 10.1093/emboj/21.3.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weichenrieder O, Repanas K, Perrakis A. Crystal structure of the targeting endonuclease of the human LINE-1 retrotransposon. Structure (Camb.) 2004;12:975–986. doi: 10.1016/j.str.2004.04.011. [DOI] [PubMed] [Google Scholar]
- 24.Anzai T, Osanai M, Hamada M, Fujiwara H. Functional roles of 3′-terminal structures of template RNA during in vivo retrotransposition of non-LTR retrotransposon, R1Bm. Nucleic Acids Res. 2005;33:1993–2002. doi: 10.1093/nar/gki347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Leslie AG. Integration of macromolecular diffraction data. Acta Crystallogr. Section D. 1999;55:1696–1702. doi: 10.1107/s090744499900846x. [DOI] [PubMed] [Google Scholar]
- 26.Collaborative Computational Project Number 4. Acta Crystallogr. Section D. 1994;50:760–763. [Google Scholar]
- 27.Vagin A, Teplyakov A. MOLREP: an automated program for molecular replacement. J. Appl. Cryst. 1997;30:1022–1025. [Google Scholar]
- 28.Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr. Section A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 29.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. Section D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 30.Fujiwara H, Ogura T, Takada N, Miyajima N, Ishikawa H, Maekawa H. Introns and their flanking sequences of Bombyx mori rDNA. Nucleic Acids Res. 1984;12:6861–6869. doi: 10.1093/nar/12.17.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gorman MA, Morera S, Rothwell DG, de La Fortelle E, Mol CD, Tainer JA, Hickson ID, Freemont PS. The crystal structure of the human DNA repair endonuclease HAP1 suggests the recognition of extra-helical deoxyribose at DNA abasic sites. EMBO J. 1997;16:6548–6558. doi: 10.1093/emboj/16.21.6548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mol CD, Izumi T, Mitra S, Tainer JA. DNA-bound structures and mutants reveal abasic DNA binding by APE1 and DNA repair coordination. Nature. 2000;403:451–456. doi: 10.1038/35000249. [DOI] [PubMed] [Google Scholar]
- 33.Jakubczak JL, Burke WD, Eickbush TH. Retrotransposable elements R1 and R2 interrupt the rRNA genes of most insects. Proc. Natl Acad. Sci. USA. 1991;88:3295–3299. doi: 10.1073/pnas.88.8.3295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Martin SL. Ribonucleoprotein particles with LINE-1 RNA in mouse embryonal carcinoma cells. Mol. Cell. Biol. 1991;11:4804–4807. doi: 10.1128/mcb.11.9.4804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Matsumoto T, Hamada M, Osanai M, Fujiwara H. Essential domains for ribonucleoprotein complex formation required for retrotransposition of telomere-specific non-long terminal repeat retrotransposon SART1. Mol. Cell. Biol. 2006;26:5168–5179. doi: 10.1128/MCB.00096-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsumoto T, Takahashi H, Fujiwara H. Targeted nuclear import of open reading frame 1 protein is required for in vivo retrotransposition of a telomere-specific non-long terminal repeat retrotransposon, SART1. Mol. Cell. Biol. 2004;24:105–122. doi: 10.1128/MCB.24.1.105-122.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rashkova S, Athanasiadis A, Pardue ML. Intracellular targeting of Gag proteins of the Drosophila telomeric retrotransposons. J. Virol. 2003;77:6376–6384. doi: 10.1128/JVI.77.11.6376-6384.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wu J, Filutowicz M. Hexahistidine (His6)-tag dependent protein dimerization: a cautionary tale. Acta Biochim Pol. 1999;46:591–599. [PubMed] [Google Scholar]
- 39.Zhukovsky EA, Lee JO, Villegas M, Chan C, Chu S, Mroske C. TNF ligands: is TALL-1 a trimer or a virus-like cluster? Nature. 2004;427:413–414. doi: 10.1038/427413a. [DOI] [PubMed] [Google Scholar]
- 40.Kubo Y, Okazaki S, Anzai T, Fujiwara H. Structural and phylogenetic analysis of TRAS, telomeric repeat-specific non-LTR retrotransposon families in Lepidopteran insects. Mol. Biol. Evol. 2001;18:848–857. doi: 10.1093/oxfordjournals.molbev.a003866. [DOI] [PubMed] [Google Scholar]
- 41.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993;26:283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}