

Abstract
The brain is unique among organs in many respects, including its mechanisms of lipid synthesis and energy production. The nervous system-specific metabolite N-acetylaspartate (NAA), which is synthesized from aspartate and acetyl-coenzyme A in neurons, appears to be a key link in these distinct biochemical features of CNS metabolism. During early postnatal CNS development, the expression of lipogenic enzymes in oligodendrocytes, including the NAA-degrading enzyme aspartoacylase (ASPA), is increased along with increased NAA production in neurons. NAA is transported from neurons to the cytoplasm of oligodendrocytes, where ASPA cleaves the acetate moiety for use in fatty acid and steroid synthesis. The fatty acids and steroids produced then go on to be used as building blocks for myelin lipid synthesis. Mutations in the gene for ASPA result in the fatal leukodystrophy Canavan disease, for which there is currently no effective treatment. Once postnatal myelination is completed, NAA may continue to be involved in myelin lipid turnover in adults, but it also appears to adopt other roles, including a bioenergetic role in neuronal mitochondria. NAA and ATP metabolism appear to be linked indirectly, whereby acetylation of aspartate may facilitate its removal from neuronal mitochondria, thus favoring conversion of glutamate to alpha ketoglutarate which can enter the tricarboxylic acid cycle for energy production. In its role as a mechanism for enhancing mitochondrial energy production from glutamate, NAA is in a key position to act as a magnetic resonance spectroscopy marker for neuronal health, viability and number. Evidence suggests that NAA is a direct precursor for the enzymatic synthesis of the neuron specific dipeptide N-acetylaspartylglutamate, the most concentrated neuropeptide in the human brain. Other proposed roles for NAA include neuronal osmoregulation and axon-glial signaling. We propose that NAA may also be involved in brain nitrogen balance. Further research will be required to more fully understand the biochemical functions served by NAA in CNS development and activity, and additional functions are likely to be discovered.
Keywords: NAA, Canavan disease, myelination, dysmyelination, lipid synthesis, aspartoacylase, energy metabolism, N-acetyl-L-aspartic acid, acetate, acetyl coenzyme A
1. Introduction
N-Acetylaspartate (NAA, Figure 1) is an enigmatic molecule present at exceptionally high concentrations in the brain (Tallan et al., 1956; Tallan, 1957). The levels found in various areas of the brain can reach 10mM or greater (Bluml, 1999; Miyake et al., 1981; Pan and Takahashi, 2005), making it one of the most concentrated molecules in the CNS. After five decades of research by neuroscientists and clinicians into the roles played by NAA, its metabolic and neurochemical functions remain controversial, and its connections to several disease states are uncertain. For three decades after its discovery, NAA research was limited to a few laboratories scattered around the world, and progress was slow. There were two seminal findings that brought NAA to the attention of the general neuroscience and neurology communities, dramatically accelerating the pace of research into the neurochemistry and neurobiology of this unique molecule. First was the prominence of the NAA proton signal in magnetic resonance spectroscopy (MRS), making NAA one of the most reliable markers for brain MRS studies (Barany et al., 1987; Fan et al., 1986; Luyten and den Hollander, 1986). The second was the connection to the rare but fatal hereditary genetic disorder known as Canavan disease (Bartalini et al., 1992; Divry and Mathieu, 1989; Hagenfeldt et al., 1987; Matalon et al., 1988).
Figure 1.
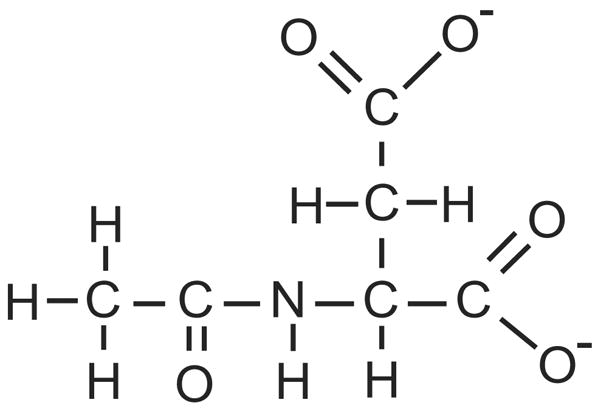
Chemical structure of NAA (mw ~173 Da in the ionic form). The acetate moiety (CH3CO) is on the left, and is attached through the amine nitrogen of aspartate. The 3 roughly equivalent methyl hydrogen atoms on the acetate group resonate with a frequency shift of 2.02 parts per million relative to an MRS standard.
In the case of Canavan disease, it was found that a mutation in the gene for the enzyme aspartoacylase (ASPA; EC 3.5.1.15) resulted in an inability to catabolize NAA, leading to a progressive, fatal leukodystrophy in affected infants. In the case of the prominent NAA signal in MRS, it was found that the levels of NAA in various parts of the brain correlate with neuronal health or integrity. Decreased levels of NAA as detected by MRS have been interpreted to indicate neuronal/axonal loss, or compromised neuronal metabolism. In contrast, high levels of NAA were found in the brains of many Canavan disease patients, who lacked the degradative enzyme ASPA, suggesting that excess NAA levels may have detrimental effects in the CNS. These findings provided further impetus for investigators to pursue intensified research into NAA biochemistry and the correlations between altered NAA levels and various neuropathological conditions. Subsequent studies led to important discoveries as diverse as links between NAA catabolism and myelin lipid metabolism, and reversible decreases in brain NAA levels associated with pathologies ranging from hypoxia to multiple sclerosis.
The most intense research focus on NAA in recent years has involved MRS-based and magnetic resonance imaging (MRI)-based analyses of changing NAA levels under numerous neuropathological and psychopathological conditions. Unfortunately, spectroscopic studies have dramatically outnumbered studies into the basic biochemistry of NAA in the brain, and this disparity has complicated the interpretation of MRS results in various disease states due to the lack of basic knowledge on NAA function and metabolism. Nonetheless, much important information on NAA has been garnered by means of spectroscopic studies.
1.1. Magnetic resonance spectroscopy and imaging of NAA
NAA is unique not only by virtue of its exceptionally high concentration in the brain, but also due to the powerful signal that it gives off in water-suppressed proton MRS spectrograms (Luyten and den Hollander, 1986). The acetate moiety of NAA is coupled through the amine nitrogen of aspartate (Figure 1), and the 3 equivalent hydrogen atoms of the acetate group resonate in NMR with a single, sharp peak having a chemical shift of 2.02 ppm relative to the standard tetramethylsilane. While the peak at 2.02 ppm is prominently attributable to NAA, this signal includes smaller contributions from other acetylated compounds, such as from the neuron-specific dipeptide, N-acetylaspartylglutamate (NAAG) (Caramanos et al., 2005), N-acetylneuraminic acid (Varho et al., 1999), and underlying coupled resonances of glutamate and glutamine. NAAG, the most concentrated peptide in the brain, may contribute 15% to 25% (Pouwels and Frahm, 1997; Pouwels and Frahm, 1998) to the acetate signal that is usually ascribed to NAA (Barker et al., 2006), so the reductions in the NAA peak associated with various brain disorders either involve coincident reductions in NAAG, or they may underestimate the drop in NAA in situations where NAAG levels remain constant.
Despite technical differences in the methods applied to acquire localized proton spectra from the human brain on different MR scanners and at different magnetic field strengths, the resulting spectra demonstrate substantial visual similarities from one MR scanner to another and between different brain regions. MRS reliably quantifies NAA directly from the number of moles per unit brain volume and can therefore provide valuable insights into its variations regionally, between white and gray matter, and in a wide variety of clinical brain disorders. However, magnetic resonance techniques have substantial limitations, including the fact that a relatively large volume of tissue must be sampled to obtain a reliable signal to noise ratio. This lack of spatial resolution often means that the signals acquired tend to average metabolite concentrations over gray matter, white matter and CSF (Barker et al., 2006). Further, MRS can only detect compounds present at high concentrations, and therefore most metabolites can not be analyzed by these techniques. Despite these limitations, MRS and MRI provide a great deal of information about several important metabolites of interest to clinicians and neuroscientists.
In MRS spectra of normal human brain the major peaks observed from left to right on the spectrogram include myo-inositol, choline (including glycerophosphocholine, phosphocholine and free choline), total creatine (creatine and phosphocreatine), and NAA (including NAAG) (Figure 2). NAA represents the largest peak in spectra of healthy brain tissue. In pathological conditions ranging from hypoxia to brain injury, additional peaks are seen representing lipids and lactate. In practice, while the cerebral concentrations of total creatine (creatine + phosphocreatine) remain relatively constant, changes in NAA, either as an absolute concentration or as a ratio between NAA versus total creatine (NAA/Cr), have proved diagnostically important. Similarly, increased signals for lactate and lipid have substantial diagnostic value.
Figure 2.
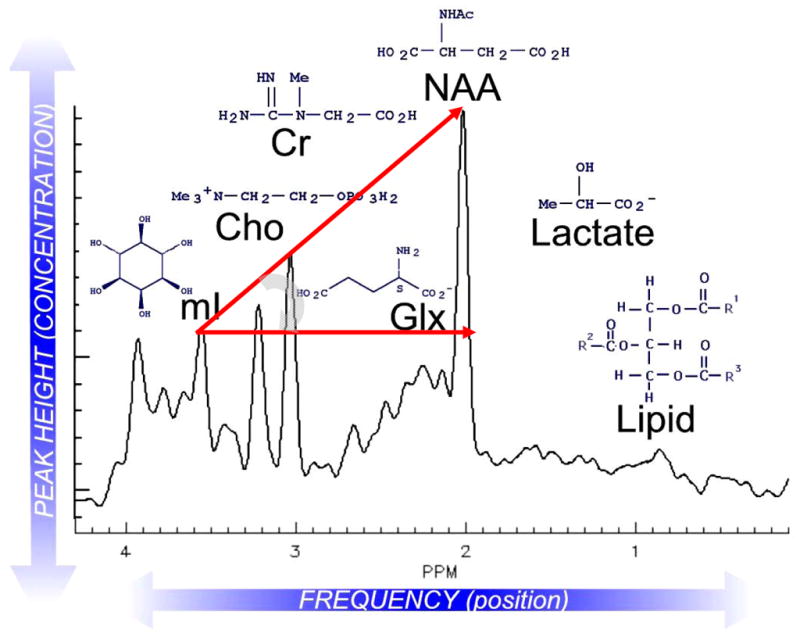
Representative proton MRS spectrum of normal human brain with major peaks of interest depicted. Lactate and lipid signals are absent from this spectrum of a healthy individual. Hunter’s angle (HA; curved gray arrow) refers to the approximate 45-degree angle formed by the peaks myo-inositol (mI), creatine (Cr), choline (Cho), and NAA, when they are present in normal proportions (NAA/Cr ~1.5, Cho/Cr ~0.75; mI/Cr ~0.5) using short-echo-stimulated echo acquisition mode (STEAM) spectroscopy. Changes in HA can be applied to such common MRS diagnoses as tumor (HA < − 50°), stroke, Alzheimer disease (HA ~ 15°), neonatal hypoxia (HA ~ − 45°) or AIDS-related progressive multifocal leukomalacia (HA ~ 0°). Glx = glutamine and glutamate. Reprinted with permission, NeuroRx (Lin et al., 2005).
Comparisons of the various MRS and MRI techniques currently available to neuroscientists and clinicians have been published (Bammer et al., 2005; Barker et al., 2006; Di Costanzo et al., 2003). NAA levels measured by MRS have been shown to be changed in a number of neurological disorders and conditions. Most of these studies have detected decreases in NAA concentrations in the affected brain areas, with the notable exception of Canavan disease which involves accumulation of NAA throughout the brain (Wittsack et al., 1996). In earlier studies, the decreases in NAA associated with various neuropathologies were interpreted to represent irreversible loss of neurons. However, accumulating evidence indicates that these decreases in regional NAA levels can also represent reversible neuronal or mitochondrial dysfunction (Bates et al., 1996; Clark, 1998; De Stefano et al., 1995; Demougeot et al., 2004; Gasparovic et al., 2001; Kalra et al., 1998; Narayanan et al., 2001). This issue will arise later in the discussion in a number of contexts.
Reports on conditions other than Canavan disease that result in raised NAA levels are relatively few. An early investigation reported that anesthetics and GABA administration caused NAA levels to increase in rodent brains (Buniatian et al., 1965), but to our knowledge, these findings have not been confirmed elsewhere. Two recent reports have shown that NAA levels in the brain are increased under certain circumstances other than Canavan disease. First, striatal NAA levels as measured by MRS were reported to be elevated between 7% and 12% in children with sickle cell disease (Steen and Ogg, 2005). Second, administration of the anti-psychotic drug haloperidol has been reported to elevate NAA levels in the striatum of rats by 23% as measured by high performance liquid chromatography (HPLC) (Harte et al., 2005). No changes were observed in other brain areas. Reports from another group did not support these results, and instead these researchers detected no changes in NAA levels in frozen rat brain micro-punches after haloperidol treatment using HPLC (Bustillo et al., 2004) or high field MRS (Bustillo et al., 2005). Further studies are required to determine the effects of haloperidol on brain NAA levels.
The diseases and disorders in which brain NAA levels are decreased include brain ischemia, brain injury, brain cancer, multiple sclerosis and Alzheimer disease, among others (Danielsen and Ross, 1999). Several chemical treatments have been reported to reduce brain NAA levels, including a report that 4 day treatment with ethanol or alcohol dehydrogenase inhibitors reduced whole brain NAA levels by between 5% and 20% (Baslow et al., 2000). In a subsequent study, 5 day administration of lithium chloride was reported to reduce whole brain NAA levels by approximately 13% in the rat model of Canavan disease (Baslow et al., 2002). Selected MRS studies detailing changes in brain NAA levels associated with various conditions are discussed below.
1.1.1. Ischemia and stroke
The current state of knowledge on the use of MRS to analyze changing NAA levels in experimental ischemia in animals has been reviewed (Demougeot et al., 2004; Hoehn et al., 2001). The use of MRS in medical diagnosis of human neurological disease has also been described (Danielsen and Ross, 1999; Gillard et al., 2005; Lin et al., 2005). In human and animal studies, proton MRS and MRI have consistently demonstrated large increases in lactate levels, and significant decreases in NAA levels in ischemic brain tissue several hours after onset (Bruhn et al., 1989b; Di Costanzo et al., 2003; Fenstermacher and Narayana, 1990; Franke et al., 2000; Graham et al., 1994). However, there is a paucity of published information concerning the early response of neuronal NAA metabolism in human stroke patients. Localized decreases in NAA are seen within a few hours of ischemic onset in patients with clinical evidence of stroke, and the levels as measured by MRS are typically low or absent in chronic infarcts (Gideon et al., 1994; Gillard et al., 2005). Several key issues concerning spectroscopic examination of NAA remain unresolved. These include the basis of the relatively slow observed decreases in NAA signal after the onset of stroke, the clinical and functional correlates of NAA loss, and the issue of whether NAA levels can recover over time under certain circumstances. To the extent that satisfactory answers to these issues can be obtained from animal research, MRS of NAA can profoundly alter the way we diagnose, monitor and treat human stroke. First we will examine MRS studies of stroke and hypoxia in humans, and then we will explore findings from animal studies of ischemia to focus on possible mechanisms underlying the neurochemical changes observed by spectroscopy.
Parsons and coworkers have shown that the ratios of both NAA to creatine (NAA/Cr) and lactate to creatine (Lac/Cr) are diagnostic of the long term outcome in stroke patients (Parsons et al., 2000). High Lac/Cr ratios (indicating anaerobic glycolysis) and low NAA/Cr ratios are indicative of poor outcome after stroke. However, animal and human studies have shown that the loss of NAA is relatively slow such that NAA levels may be only partially reduced by the time lactate levels begin to return to normal (Graham et al., 1993; Petroff et al., 1988). The Lac/Cr ratio during the subacute phase of stroke in humans ha been particularly useful in determining long term outcome, and is more predictive than diffusion weighted imaging of infarct size alone (Parsons et al., 2000; Walker et al., 2004). Kreis and colleagues provide evidence from studies after near-drowning that NAA levels in the human brain respond to hypoxia in a similar way to that described for animals (Kreis et al., 1996). In children examined repeatedly with quantitative MRS, T½ for NAA was estimated at about 48 hours after near-drowning. Mean NAA concentrations were reduced by 25% after 1–2 days, and by 35% to 50% after 3–4 days. Five or more days after the hypoxic insult, NAA levels reached a minimum of about 25% of control. The degree and the rate of decline of NAA occurred considerably slower in a predominantly white matter region of the brain than that described above for a predominantly gray matter region. The reason for the slower reductions in the concentration of NAA in white matter remains unknown.
With regard to the parallels between changes in brain NAA levels as measured by MRS and the clinical outcome in stroke patients, much anecdotal evidence and a few controlled studies have equated water suppressed proton MRS measurements of NAA with neurological outcome. Neurological status on admission and neurological outcome a few weeks or months later do indeed correlate with NAA concentration and the ratio of NAA to creatine/phosphocreatine (NAA/Cr) in gray matter after near-drowning. The use of NAA as a surrogate for neuronal cell number and viability, and thus for diagnosis and prognosis in these clinical settings has been supported by clinical research (Danielsen and Ross, 1999). Finally, the question of whether NAA concentrations ever recover is crucial from the clinical management perspective, since the interpretation of NAA loss as representing neuronal cell death rests strongly on this premise. Several reports have documented NAA concentration recovery in gray matter after stroke, suggesting that some degree of reversible neuronal injury accompanies ischemia (Walker et al., 2004). Reports of recovery of NAA in white matter regions are more numerous, and in MELAS, a metabolically induced regional stroke characterized by accumulation of lactate and loss of NAA, recovery of NAA is regarded as the norm (Lin et al., 2003b). In a recent study of focal ischemia (1 hour middle cerebral artery occlusion) in baboons, Coon et al. showed that lactate was significantly elevated 3 days and 10 days post injury, and NAA was reduced significantly at both time points (Coon et al., 2006). In less severe injuries encompassing less than 30% of the affected hemisphere, NAA levels partially normalized by day 10. High lactate levels correlated negatively with functional outcome, whereas high NAA correlated positively with functional outcome. In a large study of 71 stroke patients, a substantial recovery of NAA levels was seen in patients with small strokes 100 days after the ischemic event (Walker et al., 2004).
A recent investigation on patients with ischemic injuries in the region of the middle cerebral artery examined risk factors associated with stroke, such as age, hypertension and diabetes to determine if metabolite ratios were affected in contralateral structures (Walker et al., 2006). These investigators found that of all the assessed risk factors, only hypertension correlated significantly with reduced NAA/Choline ratios in the basal ganglia and periventricular white matter in normal appearing regions contralateral to the injury. They concluded that hypertension affects metabolite ratios in normal brain tissue making MRS measurements after stroke problematic in hypertensive patients when using contralateral tissue as a control. These findings highlight the sensitivity of brain NAA to perturbations, and the ability of MRS to detect these changes, but also emphasize the many caveats associated with the use of these techniques to monitor ischemia and other brain injuries.
Animal studies provide a much more detailed picture of the progression of the pathophysiology associated with stroke and ischemia, but there are reasons for considering animal models as imperfect surrogates for studying stroke and hypoxia in humans. For example, animal models of stroke are done under highly controlled conditions, and the injuries are relatively uniform in extent and severity. Further, experimental ischemia in animals is induced under anesthesia, rather than in unanaesthetized animals, which may affect the outcomes significantly. In contrast, human stroke is highly heterogeneous in nature and extent, and occurs under extremely varied conditions. Nonetheless, experimental models of ischemia have been very useful in following the evolution of ischemic injury using MRS, and in confirming the results by independent methods.
Barker and Gillard have recently summarized evidence from numerous animal studies showing that the decreases in NAA occur more slowly than increases in lactate after focal ischemia, and that larger drops in NAA are seen in the core of the infarct than in peripheral areas (Barker and Gillard, 2005). There is an approximate 10% decline in NAA over the first 6–8 hours after occlusive stroke, with a T ½ of about 15 hours for the remaining 50%. The lowest in vivo levels of NAA are observed only after 50–70 hours in animal studies (Higuchi et al., 1996; Sager et al., 1995). Animal studies into the early response to experimental ischemia have shown that increases in lactate can be observed by MRS within 40 minutes of onset, and small decreases in NAA/Cr were observed at one hour (Yi et al., 2002). Studies in animals have demonstrated that the concentration of NAA, as measured by HPLC, may not always be a good surrogate for neuronal viability after experimental stroke (Demougeot et al., 2003). In these studies, NAA was dramatically reduced in the core infarct at 3 days (approximately 90%), but was partly recovered after 8 days (approximately 60% of control), and returned to near normal by 30 days post-ischemia (approximately 90% of control). Similar findings have been reported in rats subjected to a 15 minute middle cerebral artery occlusion, where NAA/Cr was decreased significantly at 1 day post-injury, but recovered to near normal levels two weeks after injury (Wang et al., 2006). Histology showed that the partial recovery of NAA concentrations correlated with strong microglial activation and proliferation in the core of the infarct, suggesting that activated microglia may be capable of synthesizing NAA (Demougeot et al., 2003). Large declines in NAA levels associated with brain ischemia parallel the onset of histopathological events including dramatically reduced neuronal numbers, reduced cell size, nuclear pyknosis and infiltration first of polymorphonuclear cells, followed by infiltration of mononuclear cells, which are most evident between 24 and 48 hours in the case of photochemically induced focal lesions in rats (Lee et al., 1996).
Animal studies of ischemia provide the opportunity to compare results from MRS with results from other more direct techniques for measuring NAA levels. Sager and colleagues have used multiple techniques to study NAA levels and the levels of other metabolites in experimental ischemic injury in rodents. HPLC analyses of focal and global ischemia in rats indicated a slightly more rapid decline in NAA levels in focal ischemia, reaching approximately 50% of controls in 8 hours (Sager et al., 1995). The most conspicuous difference between focal and global ischemia was that aspartate levels decreased in focal ischemia, but were increased significantly over the course of 24 hours after global ischemia, possibly indicating that global ischemia brings NAA and its degratory enzyme ASPA, normally segregated in distinct compartments, into contact. Using MRS, HPLC and microdialysis, Sager and colleagues showed that MRS and HPLC gave comparable results with respect to reductions in NAA levels, but that MRS tended to overestimate NAA concentrations, and underestimate NAA reductions during ischemia (Sager et al., 1999a). Clearance of NAA from the extracellular space, as measured by microdialysis, was observed after focal ischemia, but not after global ischemia. NAA export mechanisms may explain the differences in NAA loss between focal and global ischemia, where surrounding intact tissue in focal ischemia is able to remove NAA to the circulation and maintain a concentration gradient, whereas globally damaged brain may suffer from compromised glial clearance mechanisms.
In a subsequent study, Sager and colleagues used standard histopathological techniques in conjunction with HPLC analysis of NAA levels and NAA immunohistochemistry to examine ischemic injury (middle cerebral artery occlusion) in mice (Sager et al., 2000). NAA declined to 50% and 20% of control levels in infarcted tissue after 6 hours and 24 hours, respectively, and no further decrease was observed during the following 6 days. Six hours after ischemic injury, the number of normal-appearing neurons in infarct-damaged cortical tissue decreased to 70% of control, and the majority of neurons were eosinophilic, indicating damage. After 24 hours almost no normal-appearing neurons were seen. The number of eosinophilic neurons decreased steadily to virtually zero after 7 days. The number of immunopositive cells staining for standard neuronal markers within the infarct was progressively reduced, and after 3 to 7 days the staining was confined to discrete granulomatous material in the center of the infarct, which was infiltrated with macrophages. The central granulomatous material also stained positively for NAA. NAA levels as measured by HPLC 7 days after ischemia were still detectable, remaining between 10% and 20% of controls. Astrocyte markers progressively increased at the circumference of infarcted areas, and these areas also showed increased immunoreactivity against NAA. The authors concluded that NAA becomes trapped in dying neurons and in cell debris, thus restricting its use as a marker of neuronal density. There are reasons for questioning the “NAA trapping hypothesis” as an adequate explanation for the remaining NAA levels at 7 days post ischemic injury. First, it is possible that low levels of NAA production could be associated with macrophages or reactive microglia in the infarct and peri-infarct areas under pathological conditions. Further, other acetylated compounds may contribute 10% or more to the so-called “NAA signal” in MRS, and some of these compounds may be more resistant to decline after injury than NAA (Pouwels and Frahm, 1997; Sager et al., 2001). Finally, we have observed that the method used to perform NAA immunohistochemistry (Moffett et al., 1993; Moffett and Namboodiri, 1995) causes strong, non-specific antibody binding to damaged brain tissue (unpublished observations), as was observed by Sager and colleagues in areas bordering the ischemic injury (Sager et al., 2000).
In order to determine whether NAA levels more closely parallel neuronal loss in transient global ischemia, where overt tissue necrosis is avoided, Sager and coworkers studied the loss of CA1 hippocampal neurons in the 2 vessel occlusion model of global ischemia in gerbils (Sager et al., 2001). NAA levels as measured by single-voxel proton MRS and HPLC correlated with neuronal loss as determined by histological examination, with a reported 25% reduction in NAA and complete loss of CA1 pyramidal neurons 4 days after ischemic injury. The death of CA1 neurons and the loss of NAA were comparably slow in the global ischemia model as compared with the focal model. NAA could be released from dying neurons and taken up and excreted to the circulation by remaining, ischemic-resistant glia. In global ischemia all brain tissue is damaged to some extent, rather than a specific locus, thus increasing the efflux of NAA from neurons throughout the brain, while simultaneously impairing glial clearance.
In summary, despite complicating issues, reduced NAA levels as detected by MRS can be an extremely valuable marker of brain injury after stroke or hypoxia. In vivo MRS studies support the hypothesis of NAA as a surrogate for neuronal loss and dysfunction, and the clinically associated neurological deficits observed in patients after local or global hypoxia-ischemic incidents. Outcome predictions based upon residual NAA, and increased lactate levels in localized proton spectra have proved rather accurate in such varied ischemia settings as stroke (Barker and Gillard, 2005; Parsons et al., 2000; Wild et al., 2000), neonatal hypoxia (Barkovich et al., 1999; Cappellini et al., 2002), and near-drowning in children (Kreis et al., 1996).
1.1.2. Alzheimer disease
A reasonable working hypothesis, subsequently confirmed in myriad studies, suggests that regional reductions in NAA concentration and NAA-creatine ratio are to be found in individual patients and in patient groups who test positive for clinical dementia. Patients with Alzheimer disease consistently show a 15–20% reduction in NAA/Cr (or NAA concentration) in posterior cingulate gyrus gray matter (Kantarci and Jack, Jr., 2003; Moats et al., 1994; Shonk et al., 1995; Waldman et al., 2002). The gray matter in the region of the posterior cingulate gyrus has been found to be the most reliable for the diagnosis of this and other dementias, rather than hippocampus, the region selected for PET diagnosis, and postmortem histological examination of Alzheimer disease. In this regard, NAA levels may reveal something about Alzheimer disease and the metabolic basis of memory and attention. The finding that the posterior cingulate gyrus loses NAA early in the disease supports the notion that this brain region takes part in attention and memory (Raichle et al., 2001). It has proved technically more difficult to define significant loss of NAA from the hippocampus by MRS, the region most implicated in memory, and involved earliest in plaque deposition as demonstrated by histopathological studies of Alzheimer disease brain. This does not argue against existing theory; rather it indicates the technological limitations of in vivo MRS measurements in the temporal lobe. In the hippocampus, a relatively small structure in the magnetically inhomogeneous environs of the petrous temporal bone, localized short echo time MRS has measurement errors of 20% or more, sufficient to mask any biological differences in NAA associated with Alzheimer disease. On the other hand, use of the more robust MRS technique, long echo time chemical shift imaging confirms a loss of NAA from the hippocampus in Alzheimer disease which is directly proportional to the volume loss of that structure (Schuff et al., 2002). Schuff and colleagues have reported that NAA reductions and volume loss made independent contributions to the correct discrimination of Alzheimer disease patients from controls, with a better than 80% correct diagnosis using NAA declines alone, which improved to 90% when NAA loss and volume reductions were combined (Schuff et al., 2006).
In the familial form of Alzheimer disease NAA/Cr is markedly reduced from the posterior cingulate gyrus (Lin et al., 2003a), and pre-symptomatic carriers have significantly reduced NAA/Cr and NAA-myo-inositol (NAA/MI) metabolite ratios (Godbolt et al., 2006). A precursor of Alzheimer disease is the recently defined syndrome of mild cognitive impairment. Evolution to true Alzheimer disease occurs in a sizeable proportion of these patients and is also accompanied by a reduction in brain NAA/Cr (Kantarci et al., 2000; Kantarci et al., 2002; Kantarci et al., 2003). Perhaps most instructive in this exploration of a role for NAA in a disease for which histopathological confirmation is notoriously lacking, is the recent MRS data from the well defined knockout models of Alzheimer disease in mice (Jack, Jr. et al., 2004; Jack, Jr. et al., 2005). In mice that coexpress mutated forms of human presinilin-1 and amyloid –β precursor protein, accelerated amyloid deposition occurs, and the ratios of NAA/Cr, NAA/MI and glutamate/Cr are decreased with increasing age.
One plausible hypothesis places NAA at the end of the chain of events wherein dementia results from a critical reduction in the number of functioning neuronal units. That could mean that NAA is simply a direct means of measuring neuronal number and hence quantifying the degree of neurodegeneration. An alternative hypothesis associates NAA with neuronal energetics, suggesting that reduced NAA could simultaneously reflect both reduced neuronal number, and reduced neuronal energetics in the remaining neurons. [13C] MRS has been used to demonstrate that glutamate turnover falls in Alzheimer disease in proportion to the loss of NAA concentration (Lin et al., 2003a) and that both glutamate and NAA levels are reduced in the frontal lobes of Alzheimer mice (Dedeoglu et al., 2004). Human NAA-turnover rates in the brain as measured by MRS have recently been published (Harris et al., 2006) offering a glimpse of a role for NAA metabolism, rather than steady-state concentrations of NAA in the pathobiology of Alzheimer disease. The rate of NAA-synthesis, normally about 1% of the Krebs cycle, is decreased by approximately 60% in Canavan patients (9.2 + 3.9 nmol/min/g in controls vs. 3.6 + 0.1 nmol/min/g in Canavan disease) (Moreno et al., 2001). In contrast to Canavan disease where NAA synthesis rates are reduced, NAA synthesis rates appear to be modestly increased in Alzheimer disease brain (Harris et al., 2006). If confirmed, this would suggest an adaptive process which might represent a compensatory upregulation of NAA synthesis in remaining functional neurons in Alzheimer disease and perhaps other dementias.
1.1.3. Epilepsy
It has been known since the 1800’s that hippocampal damage is apparent in brain autopsies of epilepsy patients, a condition known as hippocampal sclerosis. In particular, neuronal loss is especially apparent in regions CA1, CA3 and the hilus of the dentate gyrus. Descriptive studies with [1H] MRS in patients with epilepsy of all types include the observation of focal or global reductions in NAA signal and NAA/Cr, regardless of etiology (Briellmann et al., 2005). However, seizures also occur in many patients with normal NAA concentrations and NAA/Cr, so that most investigations have explored focal epilepsies, including post-inflammatory (e.g., Rasmussen’s encephalitis), post tumoral and mesial temporal sclerosis induced seizures (temporal lobe epilepsy or TLE). In all of these, NAA concentrations are reduced. Perhaps the most challenging observations come from clinical studies of TLE. While NAA/Cr is systematically reduced in the affected hemisphere, the contralateral hemisphere is frequently affected as well (Vermathen et al., 2002). This certainly complicates the use of MRS-NAA data as a clinical lateralizing tool for epilepsy surgeons. But more interesting is the fact that after extirpation of the damaged tissue from the temporal lobe, NAA recovers both in the tissue behind the resection, and in the contralateral hemisphere (Serles et al., 2001). Serles and colleagues suggest that these postoperative increases in NAA levels may result from recovery of neuronal metabolism, and possibly increased dendritic sprouting, synaptogenesis, and neurogenesis. Such observations, supported by histological and anatomic MRI evidence, provide convincing evidence of NAA-recovery with time, and confirm that NAA is responsive to transient neuronal dysfunction (Briellmann et al., 2005). This aspect of NAA metabolism will be discussed further in section 6 below.
1.1.4. Mass lesions including brain cancer
There are 5 basic changes in the MRS signal associated with brain tumors; NAA is decreased, lactate is increased, lipid is increased, creatine plus phosphocreatine are decreased, and choline is increased (Danielsen and Ross, 1999). NAA is reduced or even absent from most brain tumor spectra, as well as from most space-occupying brain lesions, whether benign or malignant (Arnold et al., 1990b; Barker et al., 2006; Bruhn et al., 1989a; Danielsen and Ross, 1999; Fulham et al., 1992). The explanation for the lack of NAA signal lies in the cell lines of origin for almost all primary and secondary brain tumors, which do not express the biosynthetic enzyme for NAA, Asp-NAT (see section 2.1. below). Interestingly, central neurocytoma (a rare neuronal tumor accounting for less than 0.5% of CNS tumors) have been reported by some investigators to exhibit detectable NAA levels, but reduced NAA/Cr (Chuang et al., 2005; Jayasundar et al., 2003). Hypothalamic hamartomas are benign congenital malformations that are disorganized in cytological architecture, and can contain predominantly glial or neuronal populations. Amstutz and coworkers have recently shown that hypothalamic hamartomas exhibit reduced NAA/Cr and increased mI/Cr ratios as compared with normal gray matter (Amstutz et al., 2006). Further, using histological analyses of these tumors, they show that the NAA signal is greater in hamartomas with predominantly neuronal content, and lower in hamartomas with predominantly glial content. This finding confirms the utility of proton MRS to differentiate tumors with different cellular populations. One of the most promising uses for MRS in neuro-oncology may be to follow the course of recovery after radiation therapy for glioma, permitting the differentiation of active glioma from radiation effects in the surrounding tissue (Fan, 2006).
1.1.5. Multiple sclerosis
Multiple sclerosis is an autoimmune inflammatory demyelinating disease of the CNS which links axonal damage to reduced NAA levels in gray and white matter (Criste and Trapp, 2006). Clinical studies (Bruhn et al., 1992; Davie et al., 1994; Larsson et al., 1991; Leary et al., 1999) as well as a recent metanalysis of the use of MRS in multiple sclerosis (Caramanos et al., 2005) universally show decreased NAA levels associated with the progression of the disease. Reduced amounts of NAA and NAA/Cr in MR-visible lesions and in normal appearing white matter are readily documented (De Stefano et al., 2001; Fu et al., 1998; Larsson et al., 1991; Tedeschi et al., 2002). In this central inflammatory disease, additional MRS changes in cerebral choline, myoinositol, lactate and lipid have provided insights and diagnostic value. With the investigations of Trapp and colleagues demonstrating transection of axons throughout the brain of MS patients, these MRS observations are explained as axonal injury (Criste and Trapp, 2006; Trapp et al., 1998). NAA also falls in gray matter, perhaps accounting for cognitive defects often recorded in multiple sclerosis patients (Staffen et al., 2005). Assays of whole-brain NAA using MRI have shown that cognitive impairment in multiple sclerosis correlates with reductions in NAA content (Mathiesen et al., 2006b). While atrophy and loss of NAA are both features of multiple sclerosis, the degree of loss of whole-brain NAA exceeds the development of atrophy by several fold, encouraging the conclusion that neuronal dysfunction may precede tissue loss in multiple sclerosis (Mathiesen et al., 2006a). Studies employing NAA measurements appear to provide a better correlation with disability in MS patients than do MRI measures of lesion load (Wolinsky and Narayana, 2002). Other neurodegenerative diseases such as amyotrophic lateral sclerosis also show reductions in NAA in affected brain regions (Kalra and Arnold, 2004; Kalra and Arnold, 2006; Rooney et al., 1998), demonstrating the usefulness of MRS for clinical observations on the progression of neurodegenerative disorders of all types.
Axonal injury begins early in multiple sclerosis (De Stefano et al., 2002), and cumulative axon loss results in progressive disability. However, the disease often goes through phases of remission and relapse, and white matter plaques visible in magnetic resonance images can wane with remission of symptoms. MRS studies show that NAA levels can be associated with neuronal dysfunction, as well as neuronal death, because levels have been shown to recover when MRI visible plaques resolve (Arnold et al., 1990a). Partial recovery of NAA levels has also been reported after treatment of patients with interferon beta-1b (Narayanan et al., 2001) glatiramer acetate (Khan et al., 2005) or fluoxetine (Mostert et al., 2006) suggesting that NAA levels reflect not only neuronal and axonal integrity, but also may reflect improvements in neuronal energetics and possibly remyelination.
1.1.6. Neuro-AIDS and other infections
Human immunodeficiency virus (HIV) infection in and of itself does not result in reduced NAA levels in the brain, but many HIV-associated encephalopathies do result in altered brain metabolite levels, including reduced NAA (Paley et al., 1996). In addition to AIDS-dementia complex, a condition which significantly reduces regional concentrations of NAA (Meyerhoff et al., 1993; Paley et al., 1996; Vion-Dury et al., 1994), a number of associated conditions stemming from immune-incompetence also result in local or global reductions in NAA and NAA/Cr (Schuff et al., 2006). These include lymphoma, in which the brain spectrum closely resembles that from other brain neoplasms, and toxoplasmosis, an opportunistic intracellular parasitic infection which reduces all cerebral metabolite concentrations including NAA (Chinn et al., 1995). JC-virus induced progressive multifocal leukoencephalopathy is associated with moderate reductions in NAA, and increased choline and myo-inositol (Chang et al., 1997). Progressive multifocal leukoencephalopathy is a fatal demyelinating disease of the central nervous system that predominantly affects immunocompromised individuals (Hou and Major, 2000). This CNS viral disease may now be arrested or even reversed by introduction of effective treatments termed HAART (highly active anti-retroviral therapy), in which cases, the decline in cerebral NAA may also be arrested.
Viral encephalopathies including herpes encephalitis (Danielsen and Ross, 1999; Menon et al., 1990), and prion diseases such as Creutzfeldt-Jakob disease (Oppenheim et al., 2004), result in reductions in NAA and NAA/Cr over quite large volumes of the affected brain, and may provide clues as to the nature of the disease process. In hamsters inoculated with Creutzfeldt-Jakob disease, large reductions in NAA were observed in cortex only in later stages of the disease process (135 days post-inoculation) (Behar et al., 1998) suggesting that the reductions were associated with tissue loss. Bacterial and fungal abscesses all result in local reductions in NAA concentration and NAA/Cr ratio by the means of tissue destruction (Gujar et al., 2005; Harada et al., 1994; Kadota et al., 2001; Nakaiso et al., 2002).
1.1.7. Traumatic brain injury
NAA levels as measured by MRS have proven to be a sensitive measure of neuronal compromise after traumatic brain injury (TBI) (Danielsen and Ross, 1999). Appearances on CT and MRI scans after TBI, while of great assistance in management of individual clinical problems, do not always correlate well with neurological deficit during the acute phase after brain injury, and give even less guidance on long term outcome for patients. In contrast, the use of proton MRS to determine NAA content locally, and distant to the site of injury, has proved of considerable value in the clinical setting (Brooks et al., 2001) based upon the simplifying hypothesis that in white matter, loss of NAA defines diffuse axonal injury, and in gray matter, loss of neurons. However, the heterogeneous nature of the neuropathological response to trauma has stimulated interest in other interpretations. In infants subject to “shaken baby syndrome” loss of NAA appears to follow a cascade of neurochemical events in which activated phospholipases are hypothesized to release lipids and macromolecules visible in the proton brain spectrum (Haseler et al., 1997). Childhood traumatic brain injury may result in a reversible loss of NAA which recovers approximately with the time-scale of recovery from the syndrome of inappropriate anti-diuretic hormone (SIADH) (Ross et al., 1998). Endocrine response to trauma, in the form of SIADH, is seen in 40 to 50% of children with head injury, and often results in reduced blood and brain osmolytes, or hyponatremia. Effects of brain hyponatremia are seen in MRS in the form of decreased myo-inositol, choline, creatine, and NAA (Danielsen and Ross, 1999). Despite potential complicating factors such as hyponatremia, MRS measurements of NAA levels in the brain after TBI provide significant prognostic value relative to long-term outcome, wherein higher post-injury NAA levels correlate with significantly better neurological recoveries.
MRS observations in humans after TBI or hypoxia suggest a time course of hours or days for substantial loss of NAA from affected brain tissue. However, animal studies of moderate to severe TBI have shown more rapid declines in NAA levels which were paralleled by decreases in ATP levels. Using HPLC analyses of brain extracts, Signoretti and colleagues reported significant and concomitant drops in NAA and ATP within 10 minutes of injury, and partial recovery of both compounds by 5 days in less severe injuries (Signoretti et al., 2001). In more severe TBI injuries, and those exacerbated by hypoxia-hypotension, recovery of NAA and ATP levels was not observed. In accord with human MRS studies, the lowest NAA levels attained after severe TBI were detected at the longest time period examined (5 days). These data provide strong support for the idea that NAA levels are linked to ATP levels, and that both can recover after injuries that do not involve substantial, permanent brain tissue destruction. In summary, MRS measurements of NAA provide an invaluable tool for assessing the degree and potential recoverability of brain damage following head injury.
1.1.8. Schizophrenia
MRS is capable of detecting both endogenous compounds in the brain as well as consumed or administered drugs ranging from anti-cancer agents (Port and Wolf, 2003), to alcohol ingested in excess (Danielsen and Ross, 1999) to anti-epileptic medications (e.g., valproic acid) administered in therapeutic doses (Bluml et al., 2002). Chronic alcoholism is accompanied by loss of NAA (Jagannathan et al., 1996; Meyerhoff et al., 2004) which in some studies is reported to recover after prolonged abstinence (Parks et al., 2002).
In the great majority of neuropsychiatric disorders listed in DSM-IV 1R, the accepted compendium of clinical psychiatric diseases, there has been little incontrovertible evidence of abnormalities in the concentration of NAA in the brain. The neuropsychiatric condition most studied by MRS is schizophrenia, wherein the majority of published papers report small, regional reductions in NAA or NAA/Cr (Bertolino et al., 1996; Callicott et al., 2000a; Deicken et al., 2001). Often the targeted regions differ from one study to the next so that we obtain no clear picture of the full extent of the loss or its regional distribution (Deicken et al., 2000b), and some studies have found no significant alterations in NAA or NAA/Cr in schizophrenic patients. A number of groups have reviewed the use of MRS to analyze alterations in NAA and other metabolites in schizophrenia (Abbott and Bustillo, 2006; Bertolino and Weinberger, 1999; Keshavan et al., 2000; Marenco et al., 2004; Rowland et al., 2001; Stanley et al., 2000). A recent meta-analysis of MRS studies on NAA levels in schizophrenia indicated that most studies did not include enough subjects for substantial statistical power, but that the majority of studies indicated gray matter reductions of approximately 5% to 10% in the frontal lobes (Steen et al., 2005). A number of studies have found that NAA levels and NAA/Cr ratios in the medial temporal lobe and the prefrontal cortex are reduced in schizophrenia (Abbott and Bustillo, 2006), and that these reductions parallel alterations in cerebral blood flow measured with PET and functional MRI (Callicott et al., 2000b; Marenco et al., 2006a). In a recent MRS study comparing NAA concentrations in 14 schizophrenics with 13 control subjects it was found that NAA was decreased significantly in the frontal lobe of affected patients (average, 7.94 mmol/L, compared with healthy subjects average of 8.45 mmol/L, P < 0.05) (Tanaka et al., 2006). The reduced NAA levels correlated with the severity of negative symptoms and poor performance in the Wisconsin Card Sorting Test. Other brain regions where NAA reductions have been reported in schizophrenia include the thalamus (Deicken et al., 2000a; Ende et al., 2003; Jakary et al., 2005), anterior cingulate cortex (Deicken et al., 1997) and the cerebellum (Ende et al., 2005).
Because schizophrenia may be associated with volume reductions in certain brain regions (Selemon et al., 2002; Selemon and Goldman-Rakic, 1999; van Haren et al., 2003), such as the superior temporal gyrus, the medial temporal lobe, and prefrontal cortex (Shenton et al., 2001), MRS data are usually normalized for tissue volume (Ohrmann et al., 2006). Nonetheless, it is difficult to completely separate the issues of tissue volume and metabolite concentrations as measured by MRI and MRS, and some differences observed between schizophrenic patients and controls may have a volume component. Early signs of schizophrenia have been associated with reductions in NAA or NAA/Cr. For example, NAA/Cr was reduced in the left frontal lobe of patients who were considered at risk for developing schizophrenia, as well as in schizophrenics (Jessen et al., 2006). Wood et al. found that the only MRS measure which correlated with poor outcome 18 months after a first psychotic incident in schizophrenia patients was a low NAA/Cr ratio detected in the prefrontal cortex (Wood et al., 2006). Further, reductions in NAA levels in the dorsolateral prefrontal cortex of schizophrenic patients have been correlated with poorer performance in the Auditory Verbal Learning Task (AVLT) indicating connections between cognitive performance and NAA levels (Ohrmann et al., 2006).
Intriguing functional correlates of the observed NAA reductions in schizophrenia have emerged over the last several years. Ohrmann and coworkers used MRS to show that glutamate and glutamine levels in the frontal lobes of schizophrenia patients were reduced in conjunction with the loss of NAA, suggesting a connection between NAA levels and glutamatergic neurotransmission (Ohrmann et al., 2005). A number of studies have found that the expression or activity of proteins and genes associated with glutamatergic neurotransmission are altered in schizophrenia patients (Moghaddam, 2003; Tsai et al., 1995; Tsai et al., 1998). Metabotropic glutamate receptors are involved in regulating neurotransmitter release, including the release of glutamate (Cartmell and Schoepp, 2000), and disorders of the NMDA type of glutamate receptor have been strongly implicated in the etiology of schizophrenia (Moghaddam and Jackson, 2003). In individuals at risk for schizophrenia Egan and colleagues found variations in a particular allele of GRM3, the gene encoding the type 2/3 metabotropic glutamate receptor (mGluR2/3) (Egan et al., 2004). These GRM3 alleles were associated with reduced levels of NAA in prefrontal cortex, and reduced expression of a glial glutamate transporter. MRS studies which examined individuals with the schizophrenia associated GRM3 allele confirmed reductions in NAA levels in the right prefrontal cortex (Marenco et al., 2006b). NAA reductions in prefrontal cortex have been associated with a dysregulation of dopamine release in the striatum of schizophrenia patients (Bertolino et al., 2000). It is interesting to note that NAAG, which is synthesized from NAA, acts to regulate glutamate and dopamine release, most likely via activation of presynaptic mGluR2/3 receptors.
Additional work with larger groups will be required to confirm and expand these results and determine the full degree and localization of perturbations in NAA metabolism in schizophrenia patients. Further work will also be required to ascertain if NAA metabolic disturbances are etiologically involved in the development of schizophrenia, or are a secondary consequence of other factors. Nonetheless, as techniques improve, MRS will certainly become an invaluable tool in the diagnosis and treatment of schizophrenia (Abbott and Bustillo, 2006).
It is important to mention before concluding this section that NAA levels in the brain as measured by MRS, particularly levels in white matter, have been positively correlated with general measures of intellectual functioning (Yeo et al., 2006). In fact, the regions of white matter that were found to correlate with general measures of intelligence were reported to be different in the brains of men and women (Jung et al., 2005). These findings highlight the sensitivity and utility of MRS techniques for studying normal and pathological brain function non-invasively, but also raise additional questions concerning the role of NAA in brain activity and cognition.
1.2. Overview of Proposed functions of NAA in the nervous system
NAA has presented neuroscientists with a particularly perplexing subject of study in part because application of NAA to various cell types often does not elicit a detectable response. Based on a relatively small number of published papers, four primary hypotheses have been proposed for the role of NAA in the nervous system. These include: 1) NAA acts as an organic osmolyte that counters the “anion deficit” in neurons, or a co-transport substrate for a proposed “molecular water pump” that removes metabolic water from neurons, 2) NAA is an immediate precursor for the enzyme-mediated biosynthesis of the important neuronal dipeptide N-acetylaspartylglutamate (NAAG), 3) NAA provides a critical source of acetate for myelin lipid synthesis in oligodendrocytes, and 4) NAA is involved in facilitating energy metabolism in neuronal mitochondria.
In addition, a limited number of reports suggest other possible roles for NAA. In a single study, NAA was suggested to form a complex with transfer RNA, which then might be involved in the initiation of protein synthesis (Clarke et al., 1975). NAA has also been reported to increase cAMP and cGMP levels in minced cortical preparations (Burgal et al., 1982). In another more recent study, NAA was proposed to be an endogenous ligand for G protein-coupled metabotropic glutamate receptors (Yan et al., 2003). In this study, NAA acted in a dose-dependent manner to induce neuronal depolarization in dissociated hippocampal neurons, but this finding has not yet been confirmed in other laboratories. Finally, NAA has been reported to be present in and released by peritoneal mast cells, implicating it in possible immune functions (Burlina et al., 1997). It is interesting to note that NAAG has potent anti-allergic actions (Bonnet et al., 1985; Chevance and Etievant, 1986; Jambou and Lapalus, 1990; Lapalus et al., 1986; Miadonna et al., 1998).
It is likely that the list of biological functions served by NAA will grow with additional research. For further background information, two previous reviews on the state of knowledge about NAA are available (Baslow, 2003b; Tsai and Coyle, 1995), and a recent international symposium on NAA has been published (Moffett et al., 2006).
2. NAA Metabolism
2.1. NAA Synthesis
Studies on the biosynthesis of NAA had a controversial beginning. In 1959, Goldstein reported that in the brain NAA is synthesized through the acetylation of aspartate by a soluble enzyme, L-aspartate N-acetyltransferase (Asp-NAT; EC 2.3.1.17) (Goldstein, 1959). This result was subsequently disputed on the grounds of incomplete product identification and the fact that a membrane bound enzyme was found which could use acetyl CoA and aspartate to form NAA (Goldstein, 1969; Knizley, Jr., 1967). It was then shown that the enzyme that synthesizes NAA is highly specific to aspartate as the amino acid substrate, and that it is detectable only in the nervous system (Benuck and D’Adamo, Jr., 1968). A subsequent report suggested that NAA might be formed by acetylation of tRNA bound aspartate rather than free aspartate (Clarke et al., 1975), but this finding was never corroborated. A decade later, Truckenmiller et al. reexamined the biosynthesis of NAA in the nervous system and presented evidence that Asp-NAT is a membrane bound enzyme which could be solubilized by treatment with Triton X-100, and they confirmed the earlier findings that the enzyme is highly specific to aspartate, and that it is detectable only in the nervous system (Truckenmiller et al., 1985).
Patel and Clark used another approach to study NAA biosynthesis (Patel and Clark, 1979). They used isolated rat brain mitochondrial preparations to show that NAA is synthesized and exported from mitochondria. Production of aspartate and acetyl CoA were required for the synthesis of NAA in these preparations. Aspartate and NAA efflux from mitochondria were inversely related, such that as NAA efflux increased, aspartate efflux was reduced. Glutamate and malate provided sources for aspartate, and pyruvate or 3-hydroxybutyrate served as the sources for acetyl CoA. As expected, release of aspartate was decreased as the production of NAA increased under these conditions. Also, the efflux of NAA, which represents a composite rate of synthesis and efflux, was much faster in state 3 than in state 4, indicating an apparent dependence of NAA synthesis on mitochondrial energy state. In a subsequent study, these authors proposed that NAA is transported out of mitochondria by a dicarboxylic acid transporter and that this might be an additional mechanism to that of citrate for carbon transport from mitochondria to cytosol (Patel and Clark, 1981).
Asp-NAT has been partially purified from rat brain using combined chromatographic techniques including size-exclusion chromatography, which showed that the active enzyme is a high molecular weight protein (~670 kD) with multiple subunits (Madhavarao et al., 2003). Approximately 95% of Asp-NAT activity was lost when the enzyme preparation was treated with 10 mM CHAPS and subjected to size exclusion chromatography. In contrast, Asp-NAT was stable toward ionic perturbations as high as 2 M NaCl. The enzyme complex was confirmed to be very specific to aspartate, with 3% or less activity toward glutamate, asparagine and aspartylglutamate. These investigators reported that activity was predominantly localized in the crude mitochondrial pellet, with only about 10% of the total activity being detectable in the remaining supernatant. After Percoll gradient centrifugation of the crude mitochondrial fraction, Asp-NAT activity was found to be distributed in the pure mitochondrial, synaptosomal and myelin rich fractions.
A subsequent study by Lu et al. showed that rat brain Asp-NAT is bimodally distributed in two subcellular fractions, with approximately 3-fold higher total activity (5-fold higher specific activity) in the microsomal fraction than in highly purified mitochondria (Lu et al., 2004). Enzyme activity was not further purified from microsomes to establish which elements present in that fraction contained Asp-NAT, but endoplasmic reticulum is a likely candidate for the localization of enzyme activity. One major difference between this study and the prior study by Madhavarao and coworkers is in the distribution of total activity. The study by Madhavarao et al., showed about 90% of the total activity in the crude mitochondrial fraction and about 10% or less in the microsomal fraction. The study by Lu et al. found approximately 8 % Asp-NAT activity in the purified mitochondrial fraction and about 25% in the microsomal fraction. It is possible that methodological differences in the subcellular isolation techniques led to the reported discrepancies in subcellular localization. In view of the potential implications of microsomal (non-mitochondrial) Asp-NAT to the functional roles of NAA, this issue requires further investigation.
Magnetic resonance spectroscopy has provided opportunities to study NAA biosynthesis in broken cell preparations as well as in tissue extracts following a bolus injection of [1-13C] glucose into the brain. In extract studies, the water soluble metabolites are extracted from tissues, and subsequent MRS measurements of the extracts allow not only identification of various compounds within the tissue samples, but also identification of the intramolecular positions of the labeled atomic nuclei on the basis of their chemical shift. Tyson and Sutherland have used this approach to study the labeling of NAA and NAAG after infusion of [1-13C] glucose in rats, followed by dissection and homogenization of samples from cerebellum, cerebral cortex and hippocampus (Tyson and Sutherland, 1998). Their studies showed that label was incorporated into NAA from labeled acetate (acetyl CoA) and from labeled aspartate, while NAAG was labeled from labeled glutamate. In general, incorporation of label into NAA was slow, whereas incorporation into NAAG through glutamate was fast, suggesting slow turnover of NAA, and rapid turnover of NAAG. One finding that was particularly difficult to explain either by slow turnover rates or isotopic dilution effect was the fact that they did not see any incorporation of label from either acetate or aspartate into NAAG over a 200 minute infusion period.
Two in vivo [1-13C] glucose studies, one in human and the other in rat, also have provided interesting results with regard to NAA synthesis. The human study was done in Canavan disease patients and corresponding controls. The results showed that NAA synthesis is decreased by about 60% in the Canavan disease patients (Moreno et al., 2001). NAA concentrations were increased by about 50% while aspartate levels were decreased to a similar extent in these patients. Therefore, decreased synthesis of NAA in the Canavan patients could have resulted from limited substrate availability of aspartate or from product inhibition by NAA, or both. The rat study examined the relative incorporation label from [1-13C] glucose into the aspartyl and acetyl groups of NAA (Choi and Gruetter, 2004). Label incorporation was detected in the acetyl group of NAA about 1.5 hours earlier than in the aspartyl group of NAA reflecting the delayed labeling of aspartate compared to acetate (acetyl coenzyme A) from [1-13C] glucose. These investigators concluded that NAA was present in a single compartment, that it exhibits relatively low turnover rates (2–3 days for complete turnover) and that is not likely to be involved as a major source of energy when the brain is in a resting state.
2.1.1. NAA localization
In the first report on the distribution of NAA in the brain by Harris Tallan in 1957, it was found that NAA was present in the brains of all vertebrates studied, but the levels in amphibians were exceptionally low. Further, it was found that the levels of NAA in spinal roots, medulla, pons and cerebral white matter from bovine brain were substantially lower than the concentrations found in cerebral gray matter. This early study has been cited as the first which demonstrated a neuronal localization of NAA, but Tallan never proposed a strictly neuronal localization in this report. Rather, he wrote “There is a 2-fold difference between gray and white matter. Acetylaspartic acid is probably localized in the cells of nervous tissue, inasmuch as analysis of a sample of human cerebrospinal fluid showed no detectable amounts of this substance” (pp 42–43). In 1972, Nadler and Cooper showed that NAA content in glial tumors was much lower than the levels found in normal brain tissue, and proposed that NAA was predominantly localized in neurons, as opposed to glia (Nadler and Cooper, 1972b).
It was not until the 1990s that specific antibodies to protein-coupled NAA, including monoclonal antibodies (Simmons et al., 1991) and polyclonal antibodies (Moffett et al., 1991; Moffett et al., 1993; Moffett and Namboodiri, 1995; Moffett and Namboodiri, 2006), permitted the cellular localization of NAA. These studies demonstrated that NAA was present predominantly in neurons and their processes, and that the levels varied substantially between different neuronal populations. Immunohistochemical studies on the distribution of NAA in the rat brain showed notably high levels of NAA in cortical pyramidal neurons (see Figure 3), many granule cell populations throughout the brain, mitral cells of the olfactory bulb (Simmons et al., 1991), neurons of the olfactory tubercle (Moffett and Namboodiri, 1995), the dorsal column nuclei and many neurons throughout the brainstem and spinal cord (Moffett et al., 1993). Axons in many fiber tracts were moderately stained for NAA, including the corpus callosum, corticospinal tracts, optic nerves and tracts, lateral olfactory tracts and stria terminalis (Moffett and Namboodiri, 1995). HPLC studies have confirmed the presence of NAA in optic nerves (Bjartmar et al., 2002). High levels of NAA have also been observed in cerebellar cortex, including the Purkinje cell layer, the granule cell layer and the molecular layer (Simmons et al., 1991).
Figure 3.
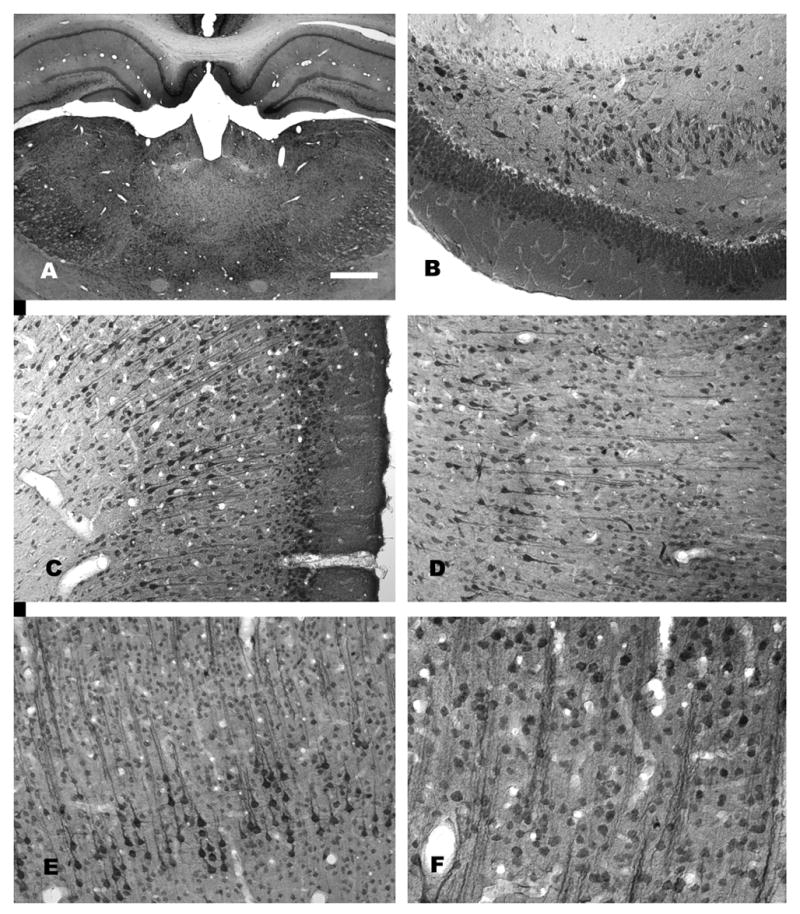
NAA-immunoreactivity in the rat forebrain. A low magnification photomicrograph of NAA staining in the thalamus and hippocampus is shown in (A). Staining is stronger in most gray matter areas as compared with white matter. Immunoreactivity in the hippocampus is strongest in pyramidal cells, polymorph cells and granule cells (B). Strong NAA-IR is also observed in cortical areas including retrosplenial granular cortex, where both pyramidal and granule cells are strongly immunoreactive (C). In neocortex, staining is particularly strong in layer 5 pyramidal cells, such as those in temporal cortex (D) and motor cortex (E). The columnar organization in these cortical areas can be discerned in NAA-stained sections wherein vertical columns of clustered apical dendrites stained for NAA can be seen (D–F). For methods, see (Moffett and Namboodiri, 1995). Bar = 600 μm A, 100 μm B-E, 50 μm F.
Based on immunohistochemical findings NAA is present in most neuronal cell populations, but the intracellular concentration appears to vary greatly between neuronal groups. For example, neocortical layer V pyramidal cells typically stain much more intensely for NAA than do neurons of layer VI, or neurons in the caudate nucleus (Moffett and Namboodiri, 1995). Immunohistochemical studies confirmed early reports that NAA concentrations were substantially higher in cerebral gray matter than in white matter. Further, they confirmed that NAA did not exhibit a significant concentration gradient along the rostro-caudal axis of the CNS. No immunocytochemical electron microscopic studies have been done on the subcellular localization of NAA, but light microscopic studies suggest that NAA is found diffusely throughout neuronal cytoplasm, and is also exceptionally concentrated in small punctate structures within neuronal cell bodies and basal dendrites, that may represent clustered neuronal mitochondria (Moffett and Namboodiri, 1995; Moffett and Namboodiri, 2006).
The neuronal localization of NAA has been confirmed using HPLC of extracts from various cell types in vitro (Urenjak et al., 1992). This investigation also found high NAA levels in immature oligodendrocytes and oligodendrocyte-type 2 astrocyte progenitor cells. Similar in vitro findings were reported by comparing results from HPLC and magnetic resonance spectroscopy of oligodendrocyte-type 2 astrocyte progenitor cells and differentiated oligodendrocytes in cell culture (Bhakoo and Pearce, 2000). They found the highest levels of NAA in mature oligodendrocytes differentiated with ciliary neurotrophic factor and in oligodendrocyte-type 2 astrocyte progenitor cells. Optic nerve transection studies in rats have shown that NAA levels in the nerves are completely eliminated 24 days post-transection (Bjartmar et al., 2002). In the same study, retinal ablation on postnatal day 4 suggested that approximately 20% of the NAA in the optic nerve at day 14, and 5% of the NAA at day 20 was derived from proliferating oligodendrocyte progenitor cells. In contrast, all NAA in adult optic nerves appeared to be synthesized exclusively by neurons.
Immunohistochemical studies have indicated very low levels of NAA immunoreactivity in oligodendrocytes in adult rats as compared with most neurons (Moffett and Namboodiri, 2006). Developmental studies using anti-NAA antibodies remain to be done, and no immunohistochemical studies have been published on the localization of the NAA synthetic enzyme, Asp-NAT, so it is not known what cell types in brain express it. However, studies on Asp-NAT enzyme activity in homogenates of 13 different brain regions showed an increasing activity gradient from the rostral to the caudal CNS (Truckenmiller et al., 1985). The highest activity levels were observed in the medulla, pons, cervical spinal cord and midbrain. Lowest activity levels for Asp-NAT were observed in the retina and amygdala, whereas the pituitary had undetectable levels. It is interesting to note that the expression of the neuronal dipeptide NAAG also exhibits an increasing concentration gradient from the rostral to the caudal CNS. NAA has not been reported to exhibit such an increasing concentration gradient (Moffett and Namboodiri, 2006), and in fact may have an opposite distribution pattern, with higher levels in forebrain, and lower levels in the hindbrain and spinal cord of adult animals (Miyake et al., 1981; Tallan, 1957).
2.2. NAA Catabolism
The enzyme that catalyzes the deacetylation of NAA is known as aspartoacylase (ASPA; EC 3.5.1.15), and is also known by several other names including acylase II and N-acetyl-L-aspartate amidohydrolase. The enzyme was first identified in supernatant fractions from hog kidney homogenates, and was shown to be very specific for NAA, with little or no activity toward other acetylated amino acids (Birnbaum et al., 1952). Another acylase in kidney, acylase I, was found to be responsible for the deacetylation of other acetylated amino acids. The “acylase II” enzyme was partially purified and characterized several years later (Birnbaum, 1955). It would be nearly two decades before the enzyme was studied in detail (D’Adamo, Jr. et al., 1973), and several more years before the partially purified enzyme was well characterized (D’Adamo, Jr. et al., 1977). ASPA was purified from bovine brain to apparent homogeneity by Kaul and colleagues, who also showed that it was expressed at higher levels in white matter than in gray matter (Kaul et al., 1991). They predicted that the active enzyme existed as a 58 kD monomer.
The cloning of the human ASPA gene followed, and the cDNA predicted an enzyme with 313 amino acids, which corresponds to a molecular weight of approximately 36 kD (Kaul et al., 1993). These investigators also found a common (85%) missense mutation of the ASPA gene in Canavan disease patients with an A to C substitution at nucleotide 854. This mutation was found to reduce ASPA activity to 2.5% of normal levels, while two other less common mutations (C to A mutations at nucleotides 693 and 914) led to a complete loss of ASPA activity (Kaul et al., 1994b). Subsequently, a number of additional mutations in the gene for ASPA were identified (Kaul et al., 1996; Zeng et al., 2002). The mouse ASPA gene was cloned and expressed in bacteria, with a predicted molecular weight of approximately 60 kD on SDS gels, which was thought to be an overestimation of the actual size of the enzyme, possibly due to phosphorylation or other influences on gel mobility (Namboodiri et al., 2000). The deduced amino acid sequence of murine ASPA is 86% identical to that of human ASPA, and the proposed catalytic domains are 100% identical (Namboodiri et al., 2000). Moore and coworkers expressed murine and human ASPA in bacteria and purified it to homogeneity by anion exchange chromatography (Moore et al., 2003). The purified enzyme was found to prefer N-trifluoroacetyl-aspartate over NAA by a factor of 25 fold, but was very highly specific for aspartate as the amino acid. Using ESI mass spectroscopy they determined the molecular weight of the bacterially expressed ASPA to be 35,171 ± 1 Daltons, which was thought to differ from the predicted mass of 35,304 Daltons due to removal of the N-terminal methionine in the bacterial expression system.
2.2.1. Cellular ASPA localization
Early work on the distribution of ASPA was done using assays for enzyme activity, and indicated that it was present in several tissues, especially CNS, liver and kidney, and that its expression profile in the CNS followed the time course of postnatal myelination (D’Adamo, Jr. et al., 1973). Much later, in vitro studies in cell culture showed that ASPA enzymatic activity was present in oligodendrocytes, but not neurons or astrocytes (Baslow et al., 1999). Bhakoo and colleagues also found ASPA activity to be expressed in white matter, and the increase in activity during brain maturation correlated with the time course of myelination in the rat brain (Bhakoo et al., 2001). These investigators tested several purified brain cell types in culture and found no ASPA activity in neurons, and the highest activity in mature oligodendrocytes (1.749 ± 0.0261 nmol/min/mg protein) and oligodendrocyte-type 2 (O2A) astrocyte progenitor cells (2.832 ± 0.785 nmol/min/mg protein). Another research group reported contradictory findings after measuring ASPA activity using a sensitive radiometric assay. They showed that primary cultures of oligodendrocytes exhibited 50 to 100 fold higher activity as compared to O2A progenitor cells or oligodendrocytes derived from the O2A progenitor line (Madhavarao et al., 2002). The reason for the discrepancy with ASPA activity assays in O2A cells is unclear, and requires further investigation.
In situ hybridization studies further confirmed that ASPA was present in oligodendrocytes, and that its expression pattern paralleled that of myelination (Kirmani et al., 2002; Kirmani et al., 2003). Immunohistochemical studies using polyclonal antibodies to ASPA confirmed that it was expressed in oligodendrocytes (Klugmann et al., 2003). Further, using dual-labeling immunohistochemistry it has been shown that ASPA is definitely expressed strongly in oligodendrocytes in the CNS, but that it is not expressed in astrocytes (Madhavarao et al., 2004). Interestingly, ASPA was also found to be expressed at low levels in microglia throughout the CNS, as well as in some large neurons and ascending and descending fiber pathways in the brainstem and spinal cord. Using the same polyclonal antibodies, it was also found that ASPA immunoreactivity in oligodendrocytes in the CNS was not restricted to their cytoplasm, but was also present in many oligodendrocyte nuclei (Hershfield et al., 2006; Madhavarao et al., 2004). ASPA immunoreactivity in the adult rat forebrain is shown in Figure 4A. The great majority of oligodendrocytes throughout fiber pathways such as the corpus callosum strongly express ASPA. ASPA expression in rat kidney is shown in Figure 4C, with the strongest expression being present in kidney proximal tubule cells.
Figure 4.
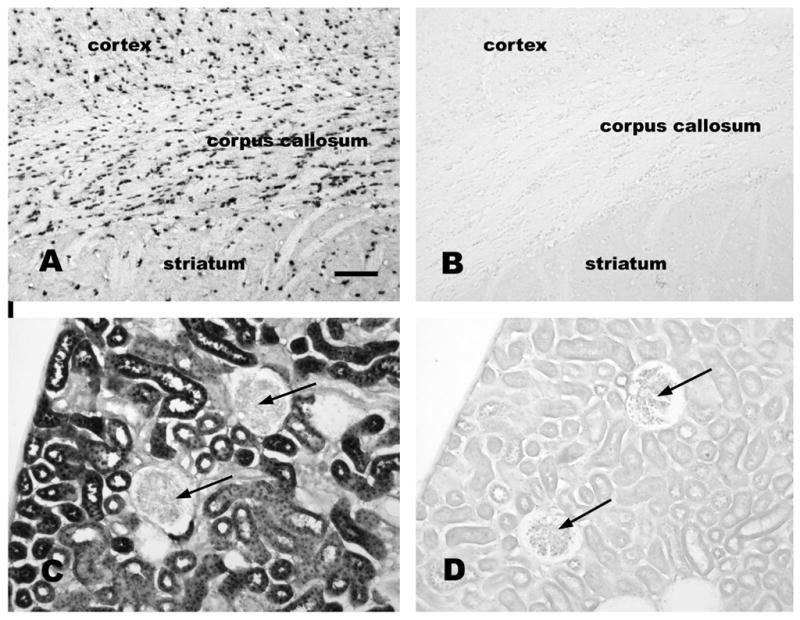
ASPA immunoreactivity in normal and Tremor mutant rat brain and kidney. A section of rat forebrain stained with anti-ASPA antibodies (1:5,000 dilution) is shown in A, whereas a similar section from a Tremor rat ASPA-null mutant is shown stained with the same antibody in B (1:1,000 dilution). In control rat kidney, ASPA expression was strong in proximal tubule cells (C; 1:5,000 dilution), but was not observed in glomeruli (arrows C and D). ASPA immunoreactivity was absent throughout the Tremor rat kidney (D; 1:2,000antibody dilution), Bar = 120 μm.
Immunohistochemical studies often suffer from the possibility that the antibodies might cross-react with similar epitopes on unrelated proteins. In order to address this possibility with our polyclonal ASPA antibodies, we tested antibody binding to brain and kidney tissues from the Tremor rat, an ASPA-null mutant (Kondo et al., 1991) (see section 3 below). This mutant lacks the entire ASPA gene sequence, so no ASPA protein is produced. Application of the polyclonal ASPA antibodies to tissues from the Tremor rat demonstrated a complete lack of staining (Figure 4B and 4D). This indicates that the immunoreactivity observed in the brains and kidneys of wild type rats was due to the presence of ASPA gene product or products, and was not due to cross-reactivity with other proteins.
2.3. NAA and osmoregulation
Tallan noted in his discussion from an early report on NAA: “Acetylaspartic acid serves to make up part of the anion deficit known to exist in nervous tissue” (page 44) (Tallan, 1957). He showed in birds and mammals that NAA accounted for more than 10% of the total anion content of brain tissue. In the 1960s, several research groups proposed that NAA might function in the brain to counter the so-called “anion deficit” in neurons, based on the high concentrations present (Margolis et al., 1960; McIntosh and Cooper, 1965). In the 1990’s, Taylor and colleagues proposed that NAA acted as a neuronal osmolyte involved in either neuronal volume regulation, or acid-base homeostasis.(Taylor et al., 1994) Using microdialysis, they demonstrated that NAA concentrations in the extracellular fluid increased in response to hypoosmotic stress (Taylor et al., 1995). Despite this, the changes in extracellular NAA concentrations were relatively modest (from ~14 μM to ~32 μM) as compared with the substantially larger increases in extracellular taurine concentrations (from ~ 12 μM to ~ 80 μM) under the same hypoosmotic conditions. Similar extracellular NAA concentration changes were observed using microdialysis under induced acidosis (Gotoh et al., 1997). Subsequently, Davies et al. showed that under hypoosmotic conditions extracellular taurine levels increased almost 20 fold, whereas extracellular NAA increased by only a few percent (Davies et al., 1998). As such, NAA may be a minor contributor to neuronal volume regulation, whereas ions including Na+, K+ and Cl−, and metabolites including taurine, glutamate, glutamine, choline and myo-inositol provide the predominant regulation of water homeostasis in the brain (Estevez et al., 1999; Heilig et al., 1989). It has been noted that NAA is only a minor contributor to the pool of osmotically active compounds in the brain which responds to hyperosmolarity or hypoosmolarity (Gullans and Verbalis, 1993; Verbalis, 2006).
2.3.1. In vitro MRS studies of NAA responses to osmotic stress
MRS has been reported to be an accurate method for measuring brain metabolite concentrations in vitro (Burri et al., 1990). These methods often employ extraction of solutes from tissues with perchloric acid, and subsequent analysis of the recovered metabolites by MRS. One study using MRS to detect metabolite concentrations in perchloric acid extracts from brain slices have shown a 60% decrease in NAA concentrations after hypo-osmotic shock (Bothwell et al., 2001). However, there are several considerations that call these findings into question. First, these investigators observed much larger changes in NAA in response to hypo-osmotic stress than were seen with taurine. Microdialysis studies have shown that taurine concentrations respond more to osmotic changes than have been observed with NAA (Taylor et al., 1995). Second, such large decreases in NAA levels after hypo-osmotic stress have not been observed using other methods (Verbalis, 2006). Finally, these investigators reported that the total dissolved species in the brain slices decreased from 500.6 ± 102.2 to 217.0 ± 60.0 (nmol/mg protein), or more than half of the solutes measured, a value that may not be physiologically relevant.
2.3.2. NAA molecular water pump hypothesis
More recently, Baslow and colleagues have proposed a modified version of the neuronal osmolyte hypothesis in which NAA acts as a cotransport substrate for a putative molecular water pump that removes excess metabolic water from neurons (Baslow, 2002; Baslow, 2003a; Baslow and Guilfoyle, 2002). The aspects of NAA that suggest that it could be involved in such a function include; 1) very high concentration, 2) strong intraneuronal to extracellular gradient, and 3) the fact that osmotic stress has been shown to increase the extracellular concentration of NAA. Despite these arguments, to date no proteins have been characterized that act to cotransport NAA and water out of neurons. However, it has been shown that the sodium-dependent transporter NaDC3 moves extracellular NAA into glial cells (Ganapathy and Fujita, 2006; George et al., 2004). In order for the molecular water pump hypothesis to be verified, a protein or protein complex that cotransports NAA and water from neurons needs to be identified and characterized. This issue will be discussed in greater detail in section 3.2.
2.3.3. NAA in axon-glial signaling
NAA has been purported to have an axon-glial signaling function by some investigators. Based on the localization of the synthetic and catabolic enzymes for NAA and NAAG, Baslow has proposed that these compounds and their intermediates, including acetate, aspartate and glutamate, are cycled between neurons and glia as a mechanism of intercellular signaling (Baslow, 2000b; Baslow and Guilfoyle, 2006). This model emphasizes the facts that both NAA and NAAG are synthesized primarily in neurons, but their corresponding catabolic enzymes are located in oligodendrocytes and astrocytes respectively. According to this hypothesis, NAA released from neurons could signal oligodendrocytes, and NAAG released from neurons could signal astrocytes. Then NAA and NAAG would be broken down by the target cells and the catabolites purportedly cycled back to neurons. This hypothesis assumes that the breakdown products of NAA and NAAG are passed from glia back to neurons, and then reutilized for re-synthesis of both compounds. It is well established that glutamine and glutamate are recycled between glia and neurons (see section 7. below), but there is strong evidence that the acetate and aspartate derived by catalysis of NAA in the brain are not used for the re-synthesis of NAA (Miller et al., 1996). However, it is not necessary for the breakdown products of NAA and NAAG to be re-cycled between glia and neurons in order for these compounds to act in a signaling capacity between neurons and glia. Indeed, other investigators have provided evidence for axon-glial signaling functions for NAA and NAAG (Ledeen et al., 2006; Lieberman et al., 2006).
2.4. NAA transporters and clearance
Little is known about the membrane transporters for NAA in different cell types in the brain, and in tissues such as kidney. Neurons are the primary producers of NAA in the CNS, and there are several possible routes for NAA efflux from these cells. The first is transport of NAA from neurons to oligodendrocytes, where ASPA removes the acetate moiety to be used for lipid synthesis (Chakraborty et al., 2001). To our knowledge, there are no reports that directly demonstrate NAA uptake by oligodendrocytes, although injection of radiolabeled NAA into the posterior chamber of the eye clearly shows that NAA is transported down axons and is passed from neurons to oligodendrocytes (Chakraborty et al., 2001), suggesting specific transport mechanisms between these cell types. In theory, to accomplish this task an export transporter would be required in the plasma membrane of neurons, and an uptake transporter like NaDC3 would be similarly required in oligodendrocytes. In addition to this route of NAA clearance, extracellular NAA can arise through efflux from neurons along the steep intraneuronal to extracellular concentration gradient, or through the enzymatic release of NAA from NAAG by the action of glutamate carboxypeptidase II on the surface of astrocytes (Berger et al., 1999; Blakely et al., 1988; Cassidy and Neale, 1993b; Slusher et al., 1992). Extracellular NAA has been shown to be preferentially taken up by astrocytes as opposed to neurons (Sager et al., 1999b) via the dicarboxylate transporter NaDC3, (Fujita et al., 2005; Ganapathy and Fujita, 2006; George et al., 2004; Huang et al., 2000), and NAA appears in urine at low micromolar concentrations, indicating that there is continuous efflux from the brain (Kelley and Stamas, 1992; Kvittingen et al., 1986; Miyake et al., 1982). Normal serum NAA levels are very low, and this may be due to glomerular filtration in the kidneys (Hagenfeldt et al., 1987). With regard to NAA clearance, it has been estimated that the human brain contains approximately 7 to 8 millimoles of NAA, and that normally about 1% is excreted in the urine per day (Miyake et al., 1982). In Canavan disease the amount of NAA excreted in the urine per day is increased to between 3 and 4 millimoles, or about half of the total NAA content of normal brain (Kvittingen et al., 1986). Increased excretion of NAAG has also been observed in Canavan patients (Burlina et al., 1994).
There has been some debate as to whether or not astrocytes express ASPA in vivo, and therefore whether or not they can deacetylate and metabolize NAA. Some investigators have found ASPA activity in oligodendrocyte-type 2 astrocyte progenitor cells (O2A) in culture (Bhakoo and Pearce, 2000). However, other results have failed to show significant ASPA activity in O2A cells in culture (Madhavarao et al., 2002), and immunohistochemical studies have shown a complete lack of ASPA expression in astrocytes in the brain (Madhavarao et al., 2004). Considering the facts that astrocytes form a significant portion of the blood-brain barrier, and that NAA is normally found in the urine (Hagenfeldt et al., 1987; Kvittingen et al., 1986), and that astrocytes express a sodium-dependent transporter for the uptake of extracellular NAA (Fujita et al., 2005), it seems likely that these glial cells take up extracellular NAA and excrete it to the circulation. In this regard it is interesting to note that we have observed NAA-immunoreactivity in a sub-population of brain endothelia (Figure 5). Only a very small proportion of brain endothelial cells displayed NAA-immunoreactivity, and the immunoreactivity was more intense in the nuclei of the cells than in the cytoplasm. Because small molecules can traverse nuclear pore complexes, it is difficult to explain the strong nuclear localization of NAA in select brain endothelial cells. The possible presence of NAA in some brain endothelial cells requires further investigation.
Figure 5.
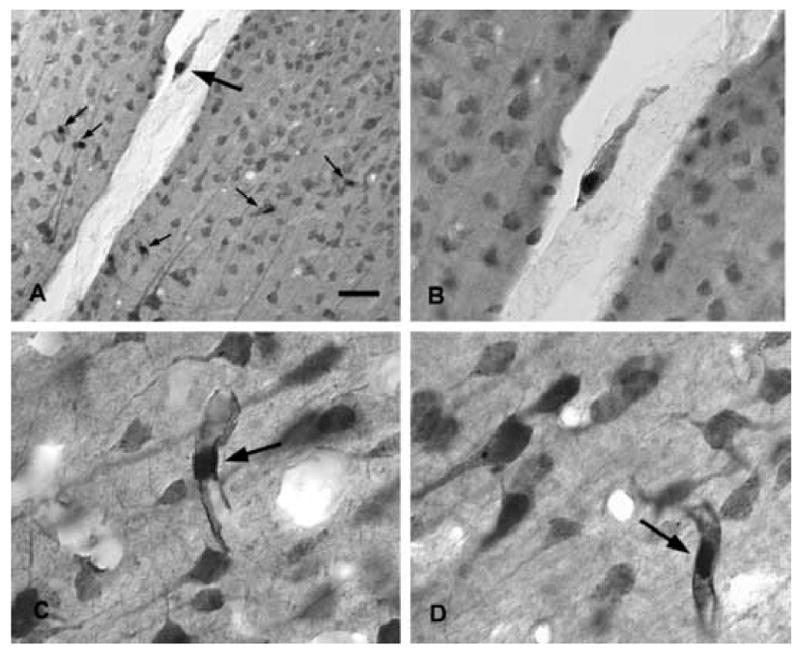
NAA immunohistochemistry in rat neocortex. NAA is expressed primarily in neurons throughout the brain, but a relatively small number of endothelial cells show strong staining (arrows in A). Stained endothelial cells were observed throughout cortex, including parietal cortex (A and B) and temporal cortex (C and D). Most endothelial cells were unstained for NAA (white holes in tissue, and unstained endothelia in the large vein in A and B). A single endothelial cell in a vein wall is strongly stained for NAA in (B), whereas the other endothelia are unstained. Staining was stronger in endothelial cell nuclei, as opposed to their cytoplasm (arrows C and D). Bar = 60 μm A, 30 μm B, 15 μm C and D.
Together, these findings suggest that in addition to the neuron to oligodendrocyte transfer of NAA, that there are other routes of NAA clearance from the brain. One probable route proceeds from neurons to the extracellular space, followed by uptake into astrocytes and release to the circulation, and finally reuptake in the kidney or excretion via urine. This possible route of NAA clearance from the brain will be discussed further in section 7.1.1. Also, because NAA appears at low micromolar levels in cerebrospinal fluid (Faull et al., 1999), this could represent another route of NAA clearance from brain. Finally, it is possible that NAA taken up from the extracellular space by astrocytes could be transferred to oligodendrocytes via specific gap junctions (connexons) composed of connexins Cx32 and Cx47 (Menichella et al., 2003; Odermatt et al., 2003).
3. NAA and Canavan Disease
A rare leukodystrophy, or spongy degenerative disease involving brain white matter was described in an infant by Myrtelle May Moore Canavan in 1931, which she tentatively identified as Schilder’s disease, or diffuse cerebral sclerosis of Schilder (Canavan, 1931). This fatal infantile condition was later determined to be a unique autosomal inheritable white matter degenerative disease by van Bogaert and Bertrand in 1949 (van Bogaert and Bertrand, 1949). Despite the fact that these clinicians were the first to correctly identify Canavan disease as a distinct disorder from Schilder’s sclerosis, the disease is nonetheless most often referred to as Canavan disease. The genetic disorder has been designated by other names in the literature, including “Canavan-van Bogaert-Bertrand disease” and “spongy degeneration of the brain, van Bogaert-Bertrand type”. A number of early reviews on Canavan disease are available (Adachi et al., 1973; Hogan and Richardson, Jr., 1965; Pratt, 1972). Adachi et al. recognized three forms of Canavan disease based on time of onset; congenital, infantile and juvenile (Adachi et al., 1973), however the vast majority of confirmed cases fit into the infantile category (Matalon et al., 1995).
Infants with Canavan disease appear normal at birth, but usually show signs of delayed development and decreased muscle tone (hypotonia), including head lag between 2 and 6 months of age. By one year, macrocephaly is often evident, and motor development is severely impaired. Infants later develop optic atrophy, and hypotonicity converts to limb stiffness and spasticity. The affected children become increasingly debilitated with age, often including seizures and an inability to move voluntarily or swallow. Death typically occurs before adolescence, but some Canavan patients with milder forms survive into their 20’s or beyond.
It was not until the 1980’s that the connection between elevated NAA levels and a progressive demyelinating cerebral atrophy was discovered. Kvittingen and coworkers first described a condition of “N-acetylaspartic aciduria” in a child with progressive cerebral atrophy, wherein the excretion of NAA was dramatically elevated in the urine (Kvittingen et al., 1986). They observed demyelination in cerebral CT scans from the child, and as the child aged they noted cerebral atrophy and enlarged ventricles. Based on the dramatic increase in urinary NAA excretion, these authors surmised that this infantile leukodystrophy might be due to either increased synthesis or reduced degradation of NAA. Shortly thereafter, N-acetylaspartic aciduria was identified as being due to a deficiency of ASPA activity by Hagenfeldt and colleagues, who found reduced ASPA activity in skin fibroblasts from a child with severe leukodystrophy. They proposed that the observed dysmyelination in the CNS was due to a failure of NAA to serve as an acetate carrier of acetyl groups from mitochondria to cytoplasm for lipogenesis (Hagenfeldt et al., 1987). Matalon and colleagues were the first to connect N-acetylaspartic aciduria and ASPA deficiency specifically to Canavan disease by showing high NAA levels in urine, and a lack of ASPA activity in skin fibroblasts in 3 children with Canavan disease (Matalon et al., 1988). They proposed three possible mechanisms whereby a lack of ASPA activity could lead to the spongy degeneration and dysmyelination observed in Canavan disease patients. First, because NAA had been shown to be involved in the production of cerebronic acid (Shigematsu et al., 1983), Matalon et al. proposed that a defect in myelination could be responsible for the progression of the leukodystrophy. Second, they proposed that because aspartate can act as a neurotransmitter (Bradford and Thomas, 1969), the lack of deacetylase activity might lead to a disruption of aspartate neurotransmission, resulting in neurological disturbances. Finally, they suggested that high levels of brain NAA might directly lead to myelin damage and spongy degeneration. Determining which of these mechanisms were involved in Canavan disease pathogenesis would require the cloning of the ASPA gene, and further basic research.
The human ASPA gene was cloned by Kaul and colleagues in 1993, as discussed above (section 2.2.) (Kaul et al., 1993). Once Canavan disease was determined to be a specific autosomal recessive genetic disorder, it was assigned a Mendelian Inheritance in Man (MIM) number (Hamosh et al., 2005) with the designation of #271900. Early genetic studies found that two mutations were prevalent among Ashkenazi Jewish patients with Canavan disease (Kaul et al., 1994a). A missense mutation at codon 285 resulted in a substitution of glutamate to alanine, and was found to account for 83.6 % mutations identified in 104 alleles from 52 unrelated Ashkenazi Jewish patients. A nonsense mutation was found in 13.4% of the alleles from Jewish patients at codon 231, which converts tyrosine to a stop codon. The incidence of mutations in the ASPA gene are far less common in non-Jewish patients, and the mutations are distinct, and more diverse (Kaul et al., 1996; Sistermans et al., 2000). The most prevalent non-Jewish mutation was identified in codon 305, a missense mutation substituting alanine for glutamic acid. This mutation was observed in 35.7% of the 70 alleles from 35 unrelated non-Jewish patients (Kaul et al., 1994). Fifteen other mutations were detected in 24 other Canavan disease patients. Recently additional mutations have been reported, in some cases with the children dying immediately after birth (Zeng et al., 2002). The diverse number of mutations associated with Canavan disease limits the usefulness of DNA analyses for prenatal diagnosis in non-Jewish parents. However, preimplantation genetic diagnosis has recently been successfully employed to screen in vitro embryos from Ashkenazi Jewish parents prior to implantation (Yaron et al., 2005).
Two animal models of Canavan disease exist (see Section 3.3. below); the so-called Tremor rat (Serikawa et al., 1987) and the ASPA knockout mouse (Matalon et al., 2000). The Tremor rat model is a stable line developed from a naturally occurring mutant with a genomic deletion on chromosome 11 spanning 4 genes, including the aspartoacylase gene, olfactory receptor gene, vanilloid receptor subtype I gene, and the calcium/calmodulin-dependent protein kinase IV gene (Kitada et al., 2000). The Tremor rat shows no ASPA activity in brain, and greatly reduced activity in kidney, and also exhibits increased brain NAA levels (Kitada et al., 2000). Tremor rats exhibit muscular tremors starting at about 2 weeks of age, which give way to absence-like seizure activity in later development. Pathology in the CNS involves white matter spongiform degeneration and hypomyelination (Kondo et al., 1991). Similarly, homozygous ASPA −/− knockout mice exhibit subcortical and white matter vacuolation. In addition, they fail to thrive, exhibit macrocephaly, display tremors, and some develop seizures by 6 months of age (Matalon et al., 2000). Homozygous ASPA −/− mice lack detectable ASPA activity, and show impaired motor activity as well as a nine fold increase in NAA levels in urine as compared with control mice. Because ASPA is strongly expressed in kidney proximal tubule cells it might be expected that ASPA deficiency would result in kidney pathologies, but this does not appear to be the case, as reports of Canavan disease patients or ASPA-deficient animals do not make note of observable kidney damage or disease.
Despite the great deal of work done over the last 5 decades to understand this fatal inheritable CNS leukodystrophy, there is still controversy over the specific etiology resulting in progressive white matter disease and subcortical vacuolation. Experimental and clinical attempts to halt or reverse the pathogenesis of Canavan disease will be discussed below (see sections 3.3. and 3.4.).
3.1. NAA neurotoxicity
An unresolved issue concerning NAA involves its potential toxicity to neurons or oligodendrocytes when the concentration is substantially elevated in the brain, as in the case in Canavan disease (Kitada et al., 2000; Leone et al., 1999). Despite the established connection between mutations in the gene for ASPA in Canavan disease, and the lost capacity to deacetylate NAA, the specific connection between ASPA deficiency and the failure of proper CNS development remains controversial (Matalon et al., 1995; Matalon and Michals-Matalon, 1998). It has been proposed that lack of deacetylase activity against NAA leads to toxic increases in the concentration of NAA in the brain (Akimitsu et al., 2000; Kitada et al., 2000; Leone et al., 1999; Leone et al., 2000). The level of extracellular NAA may be a critical factor in determining if it has toxic effects. Pliss and colleagues reported that injection of 0.25 micromoles of NAA into the lateral cerebral ventricles of rats did not induce any detectable neuronal death in the hippocampus, (Pliss et al., 2003) whereas Akimitsu et al. reported that injection of 8 micromoles of NAA into the lateral ventricle of rats induced strong seizures (Akimitsu et al., 2000). However, these same investigators observed no effects after injecting 2 micromoles of NAA into the lateral ventricles of normal rats (Kitada et al., 2000). Seizures are one of the symptoms of late stage Canavan disease, but it has not been conclusively shown that elevated NAA levels are responsible for the seizure activity. Akimitsu and colleagues used exceptionally high concentrations of NAA (~0.5M to 1M) to elicit seizure activity, so there is some question about physiological relevance to Canavan disease, where the concentration may be elevated no more than 2 fold (Kitada et al., 2000). This translates to approximately 15 to 25mM, which is far below the 500mM to 1M concentrations injected by Akimitsu and colleagues to induce seizures in rats.
Using organotypic hippocampal slice preparations, Tranberg and colleagues showed that the addition of 10 mM NAA to the culture medium had no effect on increasing cell death after 24 hours, indicating that NAA is not toxic to neurons or glia at extracellular concentrations far higher than normal (Tranberg et al., 2004). The potential toxic or excitotoxic actions of NAA have not been fully explored, but the evidence suggests that high levels of NAA in the brains of Canavan disease patients may be involved in some aspects of the pathogenesis, possibly by inducing seizure activity. However, it is also possible that the observed hypomyelination could also lead to seizure activity by way of disrupting normal neurotransmission.
3.2. NAA-related osmotic/hydrostatic pressure in Canavan Disease
A well-cited hypothesis concerning the etiology of Canavan disease is that the lack of deacetylase activity against NAA leads to osmotic dysregulation in the brain, which then results in a buildup of NAA-associated water in the extracellular fluid, resulting in the dysmyelination and subcortical vacuolation observed in Canavan patients (Baslow, 2000a; Baslow, 2002; Baslow, 2003a; Baslow, 2003b; Baslow and Guilfoyle, 2006). Currently to our knowledge, there is no direct evidence that has shown that excess NAA in the brain causes dysmyelination through osmotic damage, but nonetheless, this hypothesis is often invoked in discussions of the pathogenesis of Canavan disease. The osmotic damage hypothesis of Canavan disease may have had its origins in work on osmoregulation in ocular tissues of fish (Baslow and Yamada, 1997). N-acetylhistidine is present at high concentrations in the lens of poikilotherms including fish, and Baslow proposed that it could be involved in tissue fluid balance (Baslow, 1997). It was also proposed that what appeared to be an inverse phylogenetic relationship between the concentrations of NAA and N-acetylhistidine in brain, eye and other tissues might indicate that they served similar membrane transport or fluid balance functions in different physiological contexts. Baslow proposed that the synthetic and hydrolytic compartments for NAA and N-acetylhistidine were distinct and involved a cellular or fluid boundary, and therefore the two compounds served similar transport or fluid balance functions in different organisms.
Based on subsequent studies with 14C-isotopes and excised lens from goldfish, Baslow proposed that N-acetylhistidine acted as a “molecular water pump” in which the solute flowed down its concentration gradient through calcium channels, and removed 33 millimoles of water per millimole of N-acetylhistidine against a water gradient (Baslow, 1998). Further, he proposed that the N-acetylhistidine molecular water pump was an archetype for similar molecular water pump systems in derived vertebrates, including NAA synthesis, transport and breakdown in the brains of birds and mammals. This molecular water pump hypothesis was elaborated in later publications, wherein Baslow proposed that NAA is a significant neuronal osmolyte and that the inability to deacetylate NAA in oligodendrocytes leads to increased osmotic pressure in the intracellular space between axons and their myelin ensheathment, leading to the intramyelinic splitting and intralamellar edema observed in Canavan disease (Baslow, 1999; Baslow, 2000a). Baslow and Guilfoyle used magnetic resonance spectroscopy of samples of NAA in water to estimate the degree of hydration of NAA in order to determine the degree to which NAA could transport water across neuronal membranes (Baslow and Guilfoyle, 2002). They calculated a hydration factor of 32:1 for NAA, and suggested that NAA was a good candidate for a neuronal osmolyte which acted in concert with a transport protein as a molecular water pump.
Baslow outlined the criteria for meeting the status of a so-called molecular water pump. “In theory many solutes could be used for this purpose as long as certain conditions were met. These conditions include the requirement for a suitable cotransporter protein with associated gate and trigger mechanism for regulation of solute transport, a method for cycling the solute by means of which its intercompartmental gradient can be continuously reestablished, and a coupled energy source” (Baslow, 1999) (page 89). Currently, NAA does not meet all of these criteria for molecular water pump status, including the fact that no cotransport protein has been identified, and there is only scant evidence on possible gating mechanisms that specifically regulate NAA release from neurons. Results with organotypic hippocampal slices have been instrumental in demonstrating that NAA is not released under hyper-osmotic conditions, but that it is released in a calcium-dependent manner upon stimulation of neuronal NMDA receptors (Tranberg et al., 2004). NAA release in this system was not stimulated by potassium depolarization, but was prolonged for 20 minutes after a 5 minute application of 60 μM NMDA. The authors noted that the efflux was specific to selected organic ions, including NAA, taurine, glutathione and phosphoethanolamine, but they did not propose a physiological role for the regulated release. Receptor-meditated, calcium-dependent release of NAA from neurons has not been reported by other research groups, but if confirmed, these findings suggest that glutamatergic neuronal activity is linked to NAA efflux from neurons, and that this may be a regulated physiological process (see section 7.1.1. below).
Many different mechanisms are involved in brain water regulation, and cotransport associated with the active or passive movements of various solutes into or out of neurons is one key factor. As such, the solutes that are most actively redistributed in response to osmotic stress must be the same hydrated solutes involved in moving water into and out of neurons. As mentioned above (section 2.3.) the major solutes that are redistributed in response to osmotic stress in the brain include glutamine, glutamate, taurine, choline and myo-inositol (Verbalis, 2006). For this reason, it seems likely that NAA is only a minor contributor to the osmotically active pool of solutes present in neurons that can respond to changes in osmolarity. Finally, until an osmotically-sensitive NAA export protein is found in neurons, the NAA molecular water pump hypothesis remains speculative.
3.3. Gene Transfer Therapy for Canavan Disease
Based on the assumption that the primary etiology of Canavan disease involves toxic NAA concentration increases in the brain (Leone et al., 1999), possibly causing osmotic dysregulation and intramyelinic water accumulation, adenoviral transfer of the ASPA gene to the brains of humans has been performed in an to attempt to reverse brain edema and vacuolation (Janson et al., 2002; Leone et al., 2000). No follow up studies showing significant myelination or motor improvements in these children have been published to date, so it is difficult to assess to what extent any pathologies were ameliorated. However, a number of studies have been done using the ASPA-null mutant Tremor rat (see section 3. above), and the ASPA −/− knockout mouse model of Canavan disease, and these studies have yielded somewhat conflicting results with regard to the efficacy of this approach.
The Tremor rat has been proposed to be an animal model for petit mal seizures due to the fact that the rats exhibit so-called absence-like seizures, which are characterized by sudden immobility, with the coincident appearance of 5–7 Hz spike–wave-like complexes in cortical and hippocampal electroencephalograms (Seki et al., 2002; Serikawa et al., 1987). The Tremor rat ASPA gene transfer studies that have been done in the last several years have made use of adenoviral gene vectors to deliver the ASPA gene to the brain. The investigations by Seki and colleagues involved delivery of replication-deficient recombinant adenoviral gene vectors containing the ASPA gene into the lateral cerebral ventricles, which was reported to primarily induce expression in ependymal cells of the ventricle lining (Seki et al., 2002; Seki et al., 2004). These two studies were done on different mutant rat strains, both of which lacked the gene for ASPA, and which display absence-like seizures. In both studies, a very transient reduction in seizure activity was noted at 1 week after gene transfer, but this effect was not maintained by 10 days after transfer, and no improvement in animal survival rates was observed. The authors noted that the viral vector they employed induced immune system responses which may have interfered with the efficacy of the treatment.
The most recent adenoviral gene transfer studies employed improved gene transfer technology using the recombinant adeno-associated virus serotype 2 vector (AAV-2) (Klugmann et al., 2005; McPhee et al., 2005). In one of these gene transfer studies in Tremor rats, NAA levels were reduced, and seizure activity was diminished, but brain vacuolation and dysmyelination were unaffected, suggesting that these pathological features of the disease are not mediated by excessive NAA concentrations (Klugmann et al., 2005). More importantly, no motor improvements were observed in the Tremor rats after gene transfer. In the other study, reduced NAA levels and somewhat improved motor functions were reported (McPhee et al., 2005). However, motor improvements were modest, and statistical significance in motor performance improvement was determined between Tremor rats which had received adenovirally transferred green fluorescent protein gene, versus Tremor rats which had received adenovirally transferred ASPA gene. Statistical significance between naïve (no gene transfer) Tremor rats and those that received adenovirally transferred ASPA was not reported. Because Tremor rats with green fluorescent protein gene transfer performed more poorly on the motor tests than the naïve Tremor rats, the significance of the reported improvements in motor function may be questionable.
An ASPA −/− knockout mouse Canavan disease model has been developed (Matalon et al., 2000), but somewhat less work has been done with this model to date. A single reported trial of recombinant AAV-ASPA treatment has been attempted with this model (Matalon et al., 2003). In that study, recombinant AAV-ASPA gene transfer provided excellent ASPA expression and activity in and around vector injection sites, and reduced NAA levels as detected by magnetic resonance spectroscopy. Further, histopathological examinations revealed locally reduced brain vacuolation, particularly in thalamus, in animals that received AAV-ASPA. In spite of the histopathological improvements, no improvements in motor performance, brain myelination or development were reported after ASPA gene transfer in ASPA −/− mice.
A potential problem with gene transfer therapy for ASPA deficiency is that current technology is limited by the available vectors for introducing genes into different brain cell populations. ASPA is present in the brain primarily in oligodendrocytes, but NAA is produced primarily in neurons, so the anabolic and catabolic compartments are distinct. Adenoviral gene transfer (AAV-2) involves the use of a neurotrophic viral vector to deliver a specific expression cassette to target cells, and it was shown that the majority of cells which expressed delivered genes were neuronal (McPhee et al., 2005). As such, the segregation of the NAA synthetic and degradative compartments was not restored by gene therapy. This inability to direct the vector to oligodendrocytes may limit the usefulness of ASPA gene therapy until vectors which target other cell types in the brain are developed. While gene therapy is a very promising technique, the technologies necessary for proper integration of selected genes into the correct cell populations in the brain remain to be developed, and additional basic research is required.
3.4. Potential Acetate Supplementation Therapy for Canavan Disease
Based on the idea that the loss of ability to metabolize NAA in the catabolic oligodendrocyte compartment of NAA metabolism results in intramyelinic osmotic damage, Baslow concluded that the etiology of Canavan disease does not involve an inability of oligodendrocytes to produce myelin (Baslow, 2000a). He stated his conclusion; “The demyelination in CD [Canavan disease] is probably not owing to the inability of oligodendrocytes to produce myelin, but to the continuous destruction of myelinating oligodendrocytes themselves by the NAA osmotic pressure generated in the sealed paranodal and the internodal regions” (page 67). However, based upon a growing body of evidence connecting NAA-derived acetate to aspects of myelin lipid formation (Chakraborty et al., 2001; D’Adamo, Jr. et al., 1968; D’Adamo, Jr. and Yatsu, 1966), we and others have proposed that the primary etiological mechanism in the pathogenesis of Canavan disease is reduced lipid synthesis due to a reduced supply of NAA-derived acetate in the brain during myelination (Hagenfeldt et al., 1987; Madhavarao et al., 2005; Mehta and Namboodiri, 1995) (see section 5. below).
ASPA acts to remove acetate from NAA in oligodendrocytes, and therefore ASPA activity against NAA would be expected to increase free acetate levels in these cells. A loss of ASPA enzyme function during postnatal myelination could then lead to an acetate deficiency in oligodendrocytes. Because acetate in the form of acetyl coenzyme A is a key building block of lipids, a lack of ASPA activity in oligodendrocytes could theoretically limit lipid synthesis in the myelinating cells of the brain when axonal myelination is maximal during early infancy. In this regard, we have observed an approximately 80% reduction in free acetate levels in the brains of ASPA −/− mice at the time of peak myelination (Madhavarao et al., 2005). For these reasons we have proposed dietary acetate supplementation therapy, for example with glyceryl triacetate (GTA), as a possible treatment for Canavan disease (Madhavarao et al., 2005; Mathew et al., 2005; Mehta and Namboodiri, 1995; Namboodiri et al., 2006b).
GTA is a triester with 3 acetate groups coupled to glycerol. Esterases present in most tissues of the body can cleave the acetate moieties, generating free acetate. Dosing experiments in mice demonstrated that GTA was 10 to 20 times more effective at delivering acetate to the brain than calcium acetate, and that it was well tolerated by developing mice (Mathew et al., 2005). If substantially elevated levels of acetate can be maintained in oligodendrocytes through dietary acetate supplementation during postnatal myelination, this should theoretically correct the acetate deficiency that occurs in Canavan disease. It is likely that acetate supplementation of Canavan patients would have to be initiated within the first few months after birth in order to ensure that critical neurodevelopmental stages requiring myelination are accomplished on schedule, including visual system development. Other potential problems would not be addressed by acetate supplementation including excess NAA concentrations in the brain, reduced aspartate levels in oligodendrocytes, and possibly reduced neuronal respiration on glutamate due to product inhibition of the mitochondrial enzymes Asp-NAT and aspartate aminotransferase (see section 6.3. below). If these turn out to contribute significantly to the pathogenesis of Canavan disease, then acetate supplementation therapy may only be partially effective as a treatment.
In a collaborative study between the Namboodiri and Matalon laboratories, preliminary results have been obtained with a relatively small number of ASPA −/− mice supplemented with GTA in the diet. The results indicate reduced vacuolization and reduced water content in the brain, improved motor function, increased lipid content and reduced mortality in homozygous ASPA −/− mice treated with GTA (unpublished observations). Because of the small numbers of treated mice, these results should be considered preliminary, and more conclusive data with a larger number of mice will be needed in order to find out whether GTA supplementation can prevent the severe phenotype associated with ASPA deficiency.
Breeding ASPA −/− mice and Tremor rats is difficult because heterozygote breeding pairs are required, which produce litters averaging only 25% ASPA −/− pups, and because the affected animals do not thrive and often die soon after birth. As such, many breeding cycles are required to obtain sufficient numbers of ASPA deficient pups. We are planning studies with GTA supplementation on a greater number of homozygous animals so the efficacy of acetate can be determined more conclusively as a therapeutic agent for the treatment of Canavan disease.
4. NAA and NAAG Biosynthesis
NAAG is the most concentrated neuropeptide in the human brain. It is synthesized enzymatically from NAA and glutamate (Arun et al., 2006; Boltshauser et al., 2004; Cangro et al., 1987; Gehl et al., 2004) and is localized in specific types of neurons throughout the CNS (Anderson et al., 1987; Moffett et al., 1993; Moffett and Namboodiri, 1995; Moffett and Namboodiri, 2006; Tieman et al., 1991; Tieman and Tieman, 1996). NAAG is released from synapses in a calcium dependent manner (Williamson et al., 1991), and acts through presynaptic metabotropic glutamate receptors to modulate the release of classical neurotransmitters (Xi et al., 2002; Zhao et al., 2001). NAAG biosynthesis has been studied for over three decades, but the biosynthetic and regulatory mechanisms involved remain poorly understood. To date, no NAAG biosynthetic enzyme has been isolated or characterized, and synthesis from radiolabeled precursors has only been demonstrated reliably in tissue explants and cell culture with intact cells. The primary reason that a NAAG biosynthetic enzyme has not been identified is due to the lack of ability to detect synthesis in tissue or cell homogenates, except for a few early reports that have not subsequently been corroborated (Reichelt et al., 1976a; Reichelt et al., 1976b; Reichelt and Kvamme, 1973).
The subsequent inability of other research groups to demonstrate enzymatic NAAG biosynthesis in brain tissue homogenates is a well known but under-reported fact in NAAG research. The loss of NAAG synthesis upon homogenization of tissues or cultured cells could have many possible explanations. These include the sequestration of a putative NAAG synthesizing enzyme or enzyme complex in a membrane compartment where optimal conditions for enzyme activity are maintained. Homogenization of cells or tissues leads to the mixing of extracellular and intracellular constituents, the release of proteases and peptidases, and the elimination of subcellular compartmentation and transmembrane gradients, all of which could adversely affect NAAG synthesis. Homogenization could expose a NAAG synthetase enzyme to inhibitory compounds that render it inactive. It is also possible that intact neuronal bioenergetic systems link NAAG synthesis to ATP hydrolysis, whereby homogenization interrupts an energy-dependent reaction. Another possibility is that NAAG synthetase is degraded very rapidly after synthesis, and has a very short functional half-life after homogenization. The inability to detect any NAAG synthesis from radiolabeled precursors in brain homogenates prevents further purification, identification and characterization of the enzyme or enzymes responsible. However, indirect means of examining some of the basic properties of a NAAG synthetase enzyme have been brought to bear in several different preparations as outlined below.
4.1. Evidence for a NAAG synthetase enzyme
The prevailing hypothesis is that NAAG is synthesized by the enzymatic ligation of NAA and glutamate, possibly by an energy-requiring process. The presence of a “NAAG synthetase” enzyme has been postulated based on indirect means in several studies (Arun et al., 2006; Cangro et al., 1987; Gehl et al., 2004; Urazaev et al., 2001; Williamson and Neale, 1988). Most recent studies which have successfully demonstrated NAAG biosynthesis were done in neural explants, including excised rat dorsal root ganglia (Cangro et al., 1987), excised frog retinas (Williamson and Neale, 1988), crayfish nerve cord (Urazaev et al., 2001) and hemisected rat spinal cord (Gehl et al., 2004). In several recent reports, NAAG biosynthesis has been observed in cell culture, including primary rat astrocyte cell culture (Gehl et al., 2004), and in a continuous human neuroblastoma cell line (Arun et al., 2004; Arun et al., 2006).
The first convincing studies on NAAG biosynthesis were conducted by Cangro and colleagues using excised rat dorsal root ganglia (DRG) in short-term culture (Cangro et al., 1987). They incubated DRG in tissue culture for 3 hr with radiolabeled glutamine, and found very low levels of incorporation into NAAG during that time frame. Interestingly, with this short time course 3H-glutamine was the best precursor, whereas glutamate was incorporated at less than half the rate. For reasons that are not understood, radiolabeled aspartate was not incorporated into NAAG during the 3 hr incubation period. The identity of the newly synthesized NAAG was confirmed by use of multiple chromatographic techniques in sequence, including cation exchange, anion exchange and reverse phase HPLC. Such corroboration is critical to ensure correct product identification, because a number of closely spaced radiolabeled peaks are observed on anion exchange HPLC chromatograms from NAAG labeling experiments.
Williamson and Neale studied NAAG biosynthesis in frog retinas in vivo by injecting 50 μCi of 3H-glutamate into the eye, waiting 18 hr, injecting again, and waiting another 6 hours before extracting the retinas (Williamson and Neale, 1988). With these long incubation times and high doses of radiolabel they found significant incorporation of glutamate into NAAG. These same investigators were not able to demonstrate the incorporation of radiolabeled precursors into NAAG in a chick retina preparation in vitro (Williamson and Neale, 1992). In those experiments radiolabeled NAAG was taken up robustly and released in a calcium dependent manner, but no significant incorporation into NAAG was seen with tritiated glutamine, glutamate or aspartate. However, it should be noted that for these experiments the investigators incubated retinal explants with only 5 μCi/ml radiolabeled precursors for 1 hr before determining the level of incorporation into NAAG, which may not have been long enough to synthesize detectable quantities of NAAG.
More recently, Neale and colleagues incubated hemisected spinal cords with 10 μCi of tritiated precursors (final concentrations: [3H]-Glu, 426 nM; [3H]-Gln, 400 nM; [3H]-Asp, 540 nM; or [3H]-NAA, 540 nM). Under these conditions, the earliest time point where NAAG synthesis could be detected was after a 90 min incubation with radiolabeled glutamine (Gehl et al., 2004). It would require at minimum that two enzymes convert glutamine and NAA into NAAG, including the conversion of glutamine to glutamate by glutaminase, and then the ligation of NAA to the newly synthesized glutamate. The reported time frame of 90 minutes is highly suggestive of a direct enzymatic route to NAAG biosynthesis. However, as with other studies using tissue explants, extremely low incorporation rates were obtained. The authors cited the fragility of the explanted spinal cord tissue as a reason for the low incorporation rates.
4.1.1. NAAG biosynthesis and protein synthesis inhibition
Inhibitors of protein synthesis have been reported to have no effect on the incorporation of radiolabeled precursors into NAAG in neural explants (Cangro et al., 1987; Gehl et al., 2004), suggesting that NAAG is not synthesized on ribosomes as a portion of a protein, and then cleaved to generate the active molecule as is the case with many other bioactive peptides. The first reliable report of NAAG biosynthesis in tissue explants demonstrated that incubation of excised rat dorsal root ganglia with cycloheximide or ansinomycin at 200 μM did not inhibit incorporation of radiolabel into the dipeptide (Cangro et al., 1987). In this report, the addition of protein synthesis inhibitors actually increased NAAG biosynthesis, and the authors attributed this to the increased availability of radiolabeled glutamate for NAAG biosynthesis, due to the reduced incorporation of glutamine and glutamate into proteins. In a later report, Neale and colleagues used a 30 min preincubation of spinal cord explants with 800 μM ansinomysin, before a 60 minute incubation with 3H-glutamine, and found no decrease in NAAG biosynthesis (Gehl et al., 2004). Again, the radiolabel incorporation rates were very low (198 ± 66 dpm/mg protein control vs. 219 ± 77 dpm/mg protein with ansiomycin). These results strongly favor enzymatic synthesis of NAAG from NAA and glutamate, rather than synthesis as part of a larger peptide which is then cleaved.
4.1.2. NAA concentration increases NAAG biosynthesis
The evidence to date indicates that NAAG is synthesized by peptide bond formation between NAA and glutamate, rather than by acetylation of aspartylglutamate. This suggests that increased substrate availability in the form of NAA (or glutamate) might lead to increased NAAG biosynthesis by mass action, thus implicating Asp-NAT as the first, and possibly regulatory enzyme in the biosynthetic pathway. Tritiated NAA injected intracerebroventricularly has been reported to be incorporated at low levels into NAAG (Sinichkin et al., 1977) suggesting that NAA acts as a direct precursor for NAAG synthesis. Gehl and colleagues reported robust NAAG synthesis in serum-deprived primary rat cortical astrocyte cell culture. These investigators also showed that cerebellar granule cells, but not cerebellar astrocytes, could incorporate tritiated aspartate into NAA, suggesting that if astrocytes can synthesize NAAG, they must obtain NAA as a precursor from another cellular source, such as uptake from the extracellular space. In fact, astrocytes, but not granule cells, were found to take NAA up from the culture medium. These investigators also showed that pre-incubation of serum-deprived astrocytes in culture with unlabeled NAA resulted in significantly increased NAAG biosynthesis from tritiated glutamate during the course of a 90 minute incubation period. These findings are highly suggestive of the existence of a NAAG synthetase enzyme which ligates NAA and glutamate, however they differ somewhat from the results in several other studies which have shown that astrocytes do not express detectable levels of NAAG (Moffett et al., 1993; Moffett and Namboodiri, 1995; Passani et al., 1998). It is possible that certain in vitro conditions, including serum deprivation, induce astrocytes in culture to produce high levels of NAAG (Cassidy and Neale, 1993a). The issue of NAAG synthesis by astrocytes and other glial cells in vivo is an area requiring further research.
Recently, Arun and coworkers have shown in a continuous human neuroblastoma cell line (SH-SY5Y) that the addition of unlabeled NAA to the culture media resulted in a dose-dependent increase in NAAG synthesis from radiolabeled precursors (Arun et al., 2006). The average endogenous NAA level in SH-SY5Y cells was about 824 ng/mg protein, and these levels increased to about 1869 ng/mg protein in cells exposed to 10 mM external NAA representing an approximately 2.2-fold increase in intracellular NAA. This doubling of intracellular NAA resulted in an approximately 30% increase in NAAG biosynthesis from radiolabeled glutamine over a 24 hour incubation period, clearly showing a relationship between NAA levels and NAAG biosynthesis.
5. NAA Breakdown and Myelin Lipid Synthesis
Canavan disease affects children throughout the world, but is most common in Ashkenazi Jewish and Saudi Arabian families (Adachi et al., 1973; Matalon et al., 1995). It is not known how many children are born each year with Canavan disease, and many cases certainly go undiagnosed. Currently there is no effective treatment available for restoring proper myelination and motor function. The pathogenic mechanisms operative in Canavan disease currently remain a matter of debate, but it has been proposed that the etiology involves a lack of NAA-derived acetate, which is required for some portion of myelin lipid synthesis during postnatal axonal myelination (Hagenfeldt et al., 1987; Kirmani et al., 2002; Madhavarao et al., 2004; Madhavarao et al., 2005; Mathew et al., 2005; Mehta and Namboodiri, 1995; Namboodiri et al., 2006b; Namboodiri et al., 2006a). Under this hypothesis, infants born with Canavan disease are normal at birth because ASPA activity is not critical until myelin synthesis is dramatically increased shortly after birth. The maturation of oligodendrocytes and the myelination of axons in the brain accelerates shortly after birth (Drobyshevsky et al., 2005). Therefore, the increase in expression of ASPA that parallels myelination in early development (D’Adamo, Jr. et al., 1973; Kirmani et al., 2003) becomes critical in order to liberate the acetate from NAA for the synthesis of acetyl CoA, which is necessary for the synthesis of certain myelin-associated lipids. A number of studies, including the recent demonstrations of the selective localization of ASPA in oligodendrocytes in the CNS (Bhakoo et al., 2001; Kirmani et al., 2002; Klugmann et al., 2003; Madhavarao et al., 2004), and the increasing NAA concentrations observed by MRS in human fetuses in utero during development (Girard et al., 2005) are consistent with the acetate-deficiency hypothesis of Canavan disease.
5.1. Incorporation of acetate moiety of NAA into brain lipids
It was shown in the 1950’s that the concentration of NAA in the rat brain is low at birth, and reaches adult levels at postnatal day 20 (Tallan, 1957). This developmental stage in the rat corresponds to the major period of postnatal myelination in the brain. In the 1960’s, D’Adamo and coworkers showed that the acetate moiety of NAA was incorporated most efficiently into brain lipids during CNS development, indicating that it was likely to be involved in the myelination of axons (D’Adamo et al., 1968; D’Adamo, Jr. and Yatsu, 1966). They noted that reduced but significant incorporation of NAA-derived acetate into brain lipids occurred in adult rats, suggesting that myelin turnover in adults involves the breakdown of NAA. In 1987, Hagenfeldt et al. proposed that the dysmyelination in Canavan disease was due to failure of N-acetylaspartate to serve as a carrier of acetyl groups from mitochondria to the cytosol for lipogenesis (Hagenfeldt et al., 1987). Several years later, Burri and colleagues showed that the acetate group from NAA was preferentially incorporated into brain lipids during brain development as compared with free acetate (Burri et al., 1991). They also found that NAA-derived acetate incorporation into brain lipids increased between 8 day old and 22 day old rat pups, whereas incorporation from free acetate during this postnatal myelination period was constant during development. In the mid 1990’s Mehta and Namboodiri corroborated that radiolabeled NAA and acetate were incorporated into acetyl CoA and brain lipids (Mehta and Namboodiri, 1995). In 2001, Ledeen and colleagues showed that radiolabeled NAA injected into the eye was transported down optic nerve axons, and that the acetate group was incorporated into the ensheathing myelin (Chakraborty et al., 2001).
5.2. NAA-derived acetate and Canavan disease
Recently, Namboodiri and coworkers demonstrated in the mouse model of Canavan disease (ASPA −/− knockout) that the rate of lipid synthesis in the brain was significantly reduced at the peak time of myelination (Madhavarao et al., 2005). They used the 3H2O method for determining the rate of myelination (Muse et al., 2001) in 17-day old wild type and ASPA −/− mice. Among nonpolar lipids in ASPA −/− mice, significant decreases were observed in glycerol 1-fatty acids (decreased by ~35%), cholesterol (decreased by 22%), cholesteryl fatty acids (decreased by ~35%), and glycerol trifatty acids (trimyristin, tripalmitin, trilaurin, and tristearin, decreased by ~21%). In contrast, glycerol 1,2-fatty acids (dimyristin, dipalmitin, dilaurin, and distearin) were not significantly reduced in the ASPA −/− mice. Among polar lipids, phospholipids and sulfatides (including phosphatidylinositol, phosphatidyl choline, phosphatidyl glycerol, phosphatidic acid, and cerebroside sulfate) were decreased by ~38% in ASPA −/− mice, whereas phosphatidyl ethanolamine, galactocerebroside, and hydroxy fatty acid ceramide, were decreased by ~35%. Ledeen and colleagues have also shown that myelin was decreased approximately 30% in ASPA −/− mice, wherein cerebroside 1 was decreased by approximately 65%, and cerebroside 2 was reduced by approximately 22% (Ledeen et al., 2006). Other myelin lipids, such as ethanolamine phosphoglycerides, were only modestly reduced. Many of these types of lipids, including cerebrosides and sulfatides, are critically important for proper myelination (Coetzee et al., 1998b; Dupree and Popko, 1999; Popko, 2000).
Another important observation from studies with ASPA −/− mice was that the free acetate levels in their brains were almost 5-fold lower than in wild-type mice at the time of peak postnatal myelination (Madhavarao et al., 2005). Because the only defect in these mice is the lack of ASPA activity, the finding of dramatically lowered brain acetate levels in developing ASPA −/− mice demonstrates that NAA is a critical source of free acetate in the brain during postnatal myelination. Exogenously applied acetate has been reported to be preferentially taken up and utilized by brain astrocytes (Lebon et al., 2002), but this was only tested between astrocytes and synaptosomes (Waniewski and Martin, 1998), not between other cell types from the CNS. Because neurons lack ASPA activity, it seems likely that NAA is transferred intact from neurons to oligodendrocytes, where it is then enzymatically degraded to free acetate and aspartate. In order for free acetate to participate in lipid synthesis it must first be converted to acetyl CoA (see section 5.5. below).
5.3. Other genetic disruptions in myelinogenesis
Leukodystrophies, including Canavan disease, are a class of genetic disorders that adversely affect axonal myelination or threaten oligodendrocyte survival, and which in turn result in either failed development or the destruction of white matter in the CNS (Maria et al., 2003). Numerous genetic mutations have been reported to interfere with myelination, either by affecting myelin lipid synthesis or breakdown, or by detrimentally affecting myelin-specific proteins (Duncan, 2005). Proper myelination of neuronal axons requires precise coordination of protein and lipid synthesis, their integration into oligodendrocyte membranes, as well as their subsequent breakdown and recycling. Genetic mutations that interfere with any point along the myelin lipid or protein synthetic and degradative pathways can lead to various severe leukodystrophies (see Figure 6). Because axonal loss is often a consequence of dysmyelinating diseases, MRS has revealed alterations in NAA levels in white matter in several leukodystrophies.
Figure 6.
Simplified schematic of NAA metabolism in neurons and oligodendrocytes, and the relationship to the malate-aspartate mitochondrial shuttle in neurons. The inner mitochondrial membrane in neurons is shown on the right, and the cytoplasm of oligodendrocytes on the left. Genetic mutations or deletions of the genes for the proteins designated by an (X) interrupt the flow of acetate groups from neurons to oligodendrocytes, preventing proper myelin lipid formation. The loss of aspartate-malate shuttle activity in aralar (−/−) mice blocks the ability of neurons to supply NAA to oligodendrocytes, thus compromising myelin lipid synthesis. NAA must be transported out of neurons and into oligodendrocytes at their areas of contact, so we can surmise that specific transporters are involved in NAA flux between them. The fate of NAA derived aspartate in oligodendrocytes is unknown, but it could enter the TCA cycle as oxaloacetate for energy production. Deactivation of ASPA in oligodendrocytes blocks acetate flux from neurons, in the form of NAA, from getting to the cytosol of oligodendrocytes, where myelin lipid synthesis occurs. Blocking downstream enzymes in galactolipid synthesis also compromises myelin lipid synthesis, and proper myelination in the CNS. Abbreviations: AAT, aspartate aminotransferase (EC 2.6.1.1); AcCoA, acetyl coenzyme A; ACS, acetyl CoA synthetase (EC 6.2.1.1); AKG-MT, alpha ketoglutarate-malate transporter; CGT, UDP-galactose:ceramide galactosyltransferase; MDH, malate dehydrogenase (EC 1.1.1.38).
Pelizaeus-Merzbacher disease is an X-linked recessive leukodystrophy involving mutations in or duplications of the gene for proteolipid protein (PLP), a critical myelin-associated protein in the CNS (Simons et al., 2002). The disease manifests itself when the gene carries specific mutations, or if multiple duplications result in 3 or more copies of the gene (Wolf et al., 2005). Pathogenesis involves lack of myelination and relatively late onset axonal degeneration, and the pace of disease progression depends on the specific mutation involved (Garbern, 2005). It is thought that mutations in PLP result in protein misfolding, and buildup in the endoplasmic reticulum. The resultant endoplasmic reticulum stress in oligodendrocytes is thought to lead to their death, and subsequent loss of myelin (Gow et al., 1998). Pelizaeus-Merzbacher disease has also been associated with alterations in NAA and NAAG levels in brain or cerebrospinal fluid. Increased levels of NAA in white matter have been reported by some researchers (Hanefeld et al., 2005; Takanashi et al., 2002), whereas other studies have reported decreased levels of NAA in white matter in this dysmyelinating disease (Pizzini et al., 2003). Increased NAAG in the cerebrospinal fluid has also been reported in patients with Pelizaeus-Merzbacher disease (Burlina et al., 2006a).
An extremely rare leukodystrophy is associated with very high levels of NAAG in the cerebrospinal fluid (Wolf et al., 2004). The disorder has been reported in two unrelated girls and is characterized by severe hypomyelination, absence of psychomotor development, and a near complete lack of myelination in cerebral MRI scans. In another remarkable case, a child with reduced myelination, psychomotor retardation, secondary microcephaly and seizures was reported to have brain NAA levels too low to detect by MRS, possibly due to a genetic mutation in the gene for the NAA synthetic enzyme Asp-NAT (Boltshauser et al., 2004; Burlina et al., 2006b). In this single known case of hypoacetylaspartia, NAA and NAAG were also undetectable in the patient’s cerebrospinal fluid by capillary electrophoresis. Interestingly, urinary levels of NAA and NAAG were found to be near normal in this patient, suggesting low levels of NAA and NAAG synthesis in the brain.
The specific biochemical or metabolic links between altered NAA and NAAG levels and dysmyelination in various leukodystrophies remain to be fully elucidated, but all of these findings point to NAA and NAAG as having roles in axonal physiology, axon-glial interactions, and the production and maintenance of the myelin sheath.
A recent study has shown that mice lacking the gene for aralar1, a mitochondrial aspartate-glutamate transporter, demonstrated pathologies similar to ASPA −/− mice (Jalil et al., 2005), including dysmyelination (Figure 6). The authors relate the dysmyelination observed in these mice to a lack of synthesis of aspartate, and resultant drastic reductions in NAA levels in the brain. The lack of NAA-derived acetate then leads to reduced myelin lipid synthesis and leukodystrophy, as is the case with ASPA −/− mice. An important observation concerning the dysmyelination in aralar1 deficient mice was that only select lipids were significantly reduced in the brain, including galactocerebrosides. These data provide additional evidence for NAA as being a major source of free acetate and acetyl CoA for lipid synthesis in the brain during development, and that NAA-derived acetate is critical for proper CNS myelination.
Other possible genetically based disruption points in myelinogenesis could include Asp-NAT, the alpha ketoglutarate-malate transporter, any mitochondrial or plasma membrane NAA transporters that are necessary to transfer NAA from neuronal mitochondria to the cytoplasm of oligodendrocytes, and finally, acetyl CoA synthetase in oligodendrocytes (see Figure 6). If redundant metabolic and transport pathways do not exist, mutations in genes for all of these proteins could theoretically lead to various leukodystrophies (Boltshauser et al., 2004).
CNS myelination is a demanding biological process that requires the precise control of gene expression, protein translation, protein folding, complex lipid synthesis, and the proper assembly of these components into the intricate architecture of the myelin sheath. Further, the maintenance of myelin requires a continuous and dynamic process of myelin component catabolism and recycling (Ando et al., 2003). Errors in any of these myelination-related functions can lead to dysmyelination and axonal degeneration. ASPA deficiency represents just one of the many vulnerabilities in the cellular apparatus required for myelin production via which leukodystrophies manifest white matter pathology. Further, perturbations in NAA and NAAG physiology appear to have important connections to myelination, providing fascinating avenues for further research into the production and maintenance of the myelin sheath.
5.4. Central vs. peripheral myelination
The processes of central and peripheral axonal myelination are accomplished by partially distinct mechanisms (Dupree et al., 1998; Fujita et al., 1998). The myelinating cells of the CNS and PNS are distinct, with each oligodendrocyte myelinating multiple axonal internodes in the CNS, but Schwann cells providing myelination for a single peripheral axonal internode. Many of the proteins expressed by oligodendrocytes and Schwann cells are distinct. For example, the major myelin-associated proteins in the CNS are PLP, its alternative splice variant DM20 and myelin basic protein (Garbern, 2005), whereas the predominant myelin-associated proteins in the PNS are protein zero (PO), peripheral myelin protein 22kD (PMP22) and myelin basic protein (Brown and Lemke, 1997; Jiang et al., 2000).
Mutations that affect myelination can have distinct effects on myelin formation in the CNS and PNS. For example, galactolipid-deficient mice have a CNS myelination disorder, but myelination in the PNS is far less affected in these animals (Coetzee et al., 1998a). Specifically, mice deficient in UDP-galactose:ceramide galactosyltransferase, a critical enzyme in the biosynthetic pathway to galactocerebroside and sulfatide, have reduced myelination, and exhibit myelin-related abnormalities in the CNS (see Figure 6). However, myelination in the PNS was found to be normal, except for a transient increase in myelin sheath-axonal detachments at postnatal day 10. It has also been shown that the gene for ASPA, which is expressed strongly in oligodendrocytes in the CNS, is not expressed significantly in the peripheral nervous system (Kirmani et al., 2003), suggesting a specific role for the NAA degrading enzyme in CNS myelination, but not in peripheral myelination mediated by Schwann cells. It is interesting in this regard that aralar1 −/− mice showed reduced levels of galactocerebrocides in the CNS, but not in the PNS, providing further support for a role of NAA-derived acetate in synthesis of specific lipids in the brain, but not in peripheral nerves (Jalil et al., 2005). Further, connexin Cx32 is a gap junction protein expressed in both oligodendrocytes and Schwann cells, and Cx32 mutations are associated with X-linked Charcot–Marie–Tooth disease, a peripheral demyelinating disease in which central myelination is unaffected (Menichella et al., 2003). All of these findings point to distinct mechanisms of myelin formation in the central and peripheral nervous systems.
5.5. Unique aspects of CNS lipid metabolism
In the 1960’s D’Adamo and colleagues showed that the incorporation of the acetate moiety of NAA into fatty acids in the brain during myelination was three times greater than from free acetate (D’Adamo, Jr. et al., 1968; D’Adamo, Jr. and Yatsu, 1966). They made note of the growing evidence suggesting that the control of fatty acid and sterol synthesis in the CNS is different from those in other tissues. Indeed, the evidence has since grown substantially demonstrating that brain lipid synthesis, like brain energy metabolism, is mechanistically distinct from lipid synthesis in many peripheral tissues. Both energy metabolism and lipid synthesis require a continuing supply of acetyl CoA, as does the synthesis of NAA (Figure 7). However, the generation of acetyl CoA is a complex, and poorly understood aspect of lipid synthesis (Nikolau et al., 2000). In many peripheral tissues, cells produce most or all of the acetyl CoA required for fatty acid synthesis by the action of the enzyme ATP citrate lyase (EC 2.3.3.8) (Goldberg and Brunengraber, 1980; Jones and Ashton, 1976). The primary source of carbon units for lipid synthesis through this lipogenic pathway is glucose (Bauer et al., 2005), and free acetate is not involved at any step. In the cytosol, ATP-citrate lyase catalyses the reaction of citrate and CoA to produce oxaloacetate and acetyl CoA, and the acetyl CoA produced can be used in turn for the synthesis of fatty acids and cholesterol.
Figure 7.
Three major fates for acetyl CoA produced in neurons. Acetyl CoA can be used in neurons for lipid synthesis via the ATP citrate lyase pathway, or energy production via the TCA cycle. When local energy and lipid synthesis demands have been met, a third route of acetyl CoA utilization present in neurons is the Asp-NAT mediated synthesis of NAA using aspartate as co-substrate, followed by export to oligodendrocytes for further metabolism. Pathologies that impair neuronal energetics would be expected to reduce NAA production as acetyl CoA is diverted to energy production and membrane lipid synthesis.
It seems likely that lipid metabolism differs in neurons and glia. For example, studies on the metabolism of N-acetylcarnitine in the brain have shown that glucose and N-acetylcarnitine have different lipogenic fates suggesting the existence of distinct lipogenic acetyl CoA pools in different cell types, or different subcellular compartments (Ricciolini et al., 1998). Evidence from studies of lipogenesis in intestinal colonocytes also points to cell-specific mechanisms of lipid synthesis. Colonocytes are rapidly dividing cells which synthesize the bulk of their own cholesterol and fatty acids from substrates such as acetate and butyrate derived from bacterial metabolism in the gut. When incubated with [14C]-labeled substrates including glucose, glutamine, proprionate, hydroxybutyrate, butyrate and acetate, it was found that acetate provided more carbon to lipids than the other substrates, indicating that TCA-generated acetyl-CoA was not significantly involved in de-novo lipid synthesis in colonocytes (Zambell et al., 2003). Further, hydroxycitrate did not inhibit acetate incorporation into lipids in these cells, suggesting that the citrate lyase reaction was not critical for lipid synthesis. So unlike liver cells, colonocytes do not use TCA-derived acetyl-CoA as the primary carbon source for lipid synthesis. Oligodendrocytes in the brain may also make use of a similar alternate pathway of acetyl CoA production involving free acetate. Indeed, a citrate lyase independent pathway of acetate incorporation into sterols has been reported in calf brain oligodendrocytes in culture (Pleasure et al., 1979).
In oligodendrocytes, it now appears that ASPA and NAA are required as an adjunct method of producing acetyl CoA for fatty acid synthesis, via the sequential actions of ASPA and acetyl CoA synthetase-1 (ACS1; EC 6.2.1.1), the other major enzyme involved in acetyl CoA synthesis (Figure 8). In preliminary experiments with recently generated antibodies to ACS1, we have observed expression of this enzyme in a sub-population of oligodendrocytes in the adult rat brain, which is consistent with a role in myelin lipid formation (unpublished observations).
Figure 8.
Two different methods of acetyl CoA synthesis leading to fatty acid synthesis. The ASPA/NAA system may only be critical for acetyl CoA (AcCoA) synthesis in certain cells types, such as oligodendrocytes (left), whereas the ATP citrate lyase system is present in most cell types (right). In cells other than oligodendrocytes, citrate provides the substrate for acetyl CoA synthesis. In oligodendrocytes, the ATP citrate lyase system is active, but in addition, NAA is a major substrate for the increased acetyl CoA synthesis required during postnatal myelination. In order to participate in lipid synthesis, the acetate derived from NAA in the cytoplasm of oligodendrocytes must be converted to acetyl CoA, possibly by the enzyme acetyl CoA synthetase-1. Abbreviations: AcCoA, acetyl coenzyme A; ACL, ATP citrate lyase (EC 2.3.3.8); ACS, acetyl CoA synthetase-1 (EC 6.2.1.1); ASPA, aspartoacylase (EC 3.5.1.15); CoA-SH, coenzyme A.
Radiolabeled carbon infusion and feeding experiments show much more rapid turnover of the acetate carbon in NAA than the aspartate moiety (Karelson et al., 2003), suggesting that the acetate from NAA is highly active in brain metabolism, particularly lipid synthesis during myelination. ATP citrate lyase is strongly expressed in the developing brain, and ATP citrate lyase-deficient mice die early in development, highlighting the essential nature of this reaction in many cell types, and the fact that ACS-1 can not compensate for the loss of the citrate lyase pathway (Beigneux et al., 2004). It appears likely that the brain uses both of these acetyl CoA synthesizing pathways together to meet the strong demand for lipid synthesis in the brain during postnatal myelination. The ASPA-ACS1 pathway in this view is a parallel pathway for acetyl CoA production and lipogenesis. Based upon observations that certain classes of lipids are deficient in the brains of ASPA −/− mice, while others are unaffected, it seems likely that the ASPA-ACS1 pathway is only critical for the formation of certain classes of lipids, in particular, steroids, cerebrosides and sulfatides. This raises the possibility that ACS1 knockout mice would not die before birth, because the ATP citrate lyase pathway in the brain would be sufficient for most or all prenatal lipid synthesis. However, much like ASPA −/− mice, ACS1 knockout mice may display postnatal symptoms similar to Canavan disease due to reduced acetyl CoA availability for myelin-related lipogenesis. It is noteworthy that aralar −/− mice, which lack aspartate-malate shuttle activity, show large reductions in galactocerebrocide levels in the CNS, while many other lipid classes appear unaffected (Jalil et al., 2005).
6. NAA in Neuronal Energy Metabolism
The earliest indication that NAA could be involved in energy metabolism was the second report by Harris Tallan in which he showed that NAA was present at high concentrations in the brains of birds and mammals, and that the distribution pattern closely paralleled the distribution of “respiratory activity” (Tallan, 1957). A subsequent report by Buniatian and coworkers in 1965 also indicated that NAA might be involved in brain energy metabolism (Buniatian et al.). Using rat brain cortical slices these investigators showed that glucose addition caused increased NAA levels and increased release of NAA into the medium, and that the absence of added glucose led to reduced NAA levels in the brain slices. In 1972 Nadler and Cooper injected radiolabeled NAA into rat brains and found two distinct metabolic compartments or pools which they associated with what appeared to be two distinct citric acid cycles in the brain, termed the “synthetic cycle” and the “energy cycle” (Nadler and Cooper, 1972a). Uptake and metabolism of NAA was rapid in the smaller synthetic cycle, and slower in the much larger energy cycle. It is likely that the distinct cycles were present in different cell types in the CNS, as discussed below.
6.1. ATP Synthesis and NAA Synthesis are Coupled
In 1979, Patel and Clark provided the first direct evidence showing a relationship between NAA synthesis and energy metabolism (Patel and Clark, 1979). They found that brain-derived mitochondrial preparations were distinct from those derived from other tissues in that they synthesized large amounts of NAA. Using isolated rat brain mitochondria, Patel and Clark demonstrated the synthesis and efflux of NAA from mitochondria incubated in the presence of glutamate and malate, plus either pyruvate or 3-hydroxybutyrate. The synthesis of NAA was stimulated by ADP (mitochondrial state 3), used glutamate as a transamination source for aspartate, and either pyruvate or 3-hydroxybutyrate as a source of acetyl CoA. An important finding of this study was that there was a reciprocal relationship between aspartate efflux and NAA efflux when rat brain mitochondria oxidized glutamate (10 mM) and malate (2.5 mM) in the presence of various concentrations of pyruvate. Without pyruvate as a source of acetyl-CoA, no NAA efflux was detected. Increasing pyruvate concentrations caused a decrease in aspartate efflux, and an increase in NAA efflux. However, NAA efflux did not replace aspartate efflux completely even at the highest concentration of pyruvate. Instead, the decrease in aspartate efflux reached a plateau at about 60% of the efflux levels observed in the absence of pyruvate. Similarly, NAA efflux also reached a maximum at about the same concentrations of pyruvate. Given that their preparations contained both neuronal and nonneuronal mitochondria, the above observation could reflect the inability of nonneuronal mitochondria to synthesize NAA.
In a subsequent study with rat brain mitochondria, Clarke and associates showed that NAA synthesis is decreased when oxygen consumption and ATP production are decreased using irreversible inhibitors of complexes III, IV and V of the mitochondrial respiratory chain (Bates et al., 1996). This study showed a biochemical coupling between NAA synthesis and energy production in brain mitochondria. This coupling relationship has been subsequently substantiated by additional reports noting decreases in NAA in a number of conditions of impaired energy metabolism in the brain, which are described below. For an overview of the data supporting a bioenergetic role for NAA in neurons, see (Clark, 1998).
6.2. Impairments in energy metabolism decrease NAA levels in brain
There are a number of experimental paradigms that have demonstrated decreases in NAA levels when brain energy metabolism is impaired by different mechanisms. Studies on traumatic brain injury have consistently shown decreased NAA levels in the brain using different model systems, and the decreases in NAA and ATP were temporally correlated, suggesting that the NAA reductions are related to energetic impairment (Gasparovic et al., 2001; Signoretti et al., 2001; Signoretti et al., 2004; Tavazzi et al., 2005; Vagnozzi et al., 2005). The reductions in NAA and ATP were positively correlated with the severity of the brain injury. In one study, cyclosporine showed neuroprotective properties by ameliorating mitochondrial damage, and helped to minimize the reductions in ATP and NAA (Signoretti et al., 2004). Based on research connecting NAA synthesis to ATP levels, measurement of NAA by magnetic resonance spectroscopy has been proposed as a non-invasive tool to monitor the recovery of brain energy metabolism following brain injury in a clinical setting.
Another experimental paradigm involves rat and primate models of progressive striatal neurodegeneration induced by the mitochondrial toxin 3-nitropropionic acid (3-NP). In rats treated with 3NP, NAA decrease was observed selectively in the striatum before any cell loss, but was associated with motor symptoms (Demougeot et al., 2001). Also, a similar selective and early striatal decrease in NAA was observed in 3-NP treated primates (Dautry et al., 2000). The early decrease in striatal NAA was partially reversed after 3 weeks of 3-NP withdrawal. These results suggest that NAA depletion reflects a reversible state of neuronal dysfunction and raise the possibility that quantitation of NAA by MRS might be a valuable tool to assess neuronal dysfunction and the effects of potential neuroprotective therapies. Interestingly, in another study, the NAA decrease in response to 3-NP was associated with an increase in acetate (Lee et al., 2000). However, the origin of this increased acetate remains unclear.
The mouse model for Huntington’s disease is another experimental paradigm wherein decreases in NAA levels are well documented. The drop in brain NAA levels commenced at about 6 weeks of age and coincided with the onset of symptoms (Jenkins et al., 2000). These decreases in NAA occurred in the absence of any neuronal cell death. Also, dietary creatine supplementation significantly improved survival, slowed development of brain atrophy and delayed decreases in NAA (Andreassen et al., 2001; Ferrante et al., 2000). These results also indicate that the levels of NAA in the brain reflect the health of neurons and are a reliable marker for monitoring neuronal energy impairment and dysfunction.
The overall weight of the evidence favors a link between NAA synthesis in neuronal mitochondria and energy metabolism, but studies have so far failed to make a direct connection between the synthesis of NAA and that of ATP. It is quite possible that the linkage between the syntheses of these two molecules is more indirect, for example, NAA synthesis can be considered an energy requiring process in the sense that it is dependent on the energy consuming synthesis of acetyl CoA.
6.3. A model of NAA synthesis linked to mitochondrial energetics in neurons
Studies of brain metabolism suffer from the fact that different cell types in the brain have mitochondria with different biochemical properties and pathways. Neurons and glia take up and metabolize different key metabolites, and express different complements of metabolic enzymes. The predominant astroglial localization of glutamine synthase permits the metabolism of glutamate to glutamine in astrocytes (Martinez-Hernandez et al., 1977; Norenberg and Martinez-Hernandez, 1979; Schousboe et al., 1997), which can then be excreted to the circulation, or recycled to neurons for conversion back to glutamate (Cooper, 2001). As discussed in Section 2.1., neuronal mitochondria express Asp-NAT, a unique enzyme which synthesizes NAA (Benuck and D’Adamo, Jr., 1968; Goldstein, 1969; Madhavarao et al., 2003; Truckenmiller et al., 1985). It had been proposed in the 1970’s that NAA synthesis in neuronal mitochondria might be a mechanism for transporting carbon from the mitochondrial matrix to the cytoplasm (Miller et al., 1996; Patel and Clark, 1979), much as citrate does in other cell types (Clark, 1998). However, this proposal was made before the discovery that the NAA degrading enzyme ASPA was not present in neurons, but instead was expressed predominantly in oligodendrocytes (Baslow et al., 1999; Kirmani et al., 2002; Madhavarao et al., 2004). The possibility that one function of NAA is to move acetate carbon units from neuronal mitochondria to the cytoplasm of oligodendrocytes was discussed above in Section 5.1.
Glucose is thought to be the primary energy source in brain, and there is controversy over the importance of other potential energy substrates, such as lactate (Aubert et al., 2005). Some studies have reported that the brain can use lactate for energy production (Tabernero et al., 1996; Tyson et al., 2003), but these findings have been disputed by other investigators (Chih and Roberts Jr, 2003; Fillenz, 2005). Amino acids have also been proposed as possible sources of energy in brain metabolism. Removal of glucose greatly accelerates glutamate transamination to aspartate in brain synaptosomes, suggesting that glutamate could be an energy source in the brain under some circumstances (Erecinska et al., 1988). The enzyme in mitochondria that accomplishes the task of glutamate transamination is aspartate aminotransferase (EC 2.6.1.1, AAT; also known as aspartate transaminase), which consumes oxaloacetate and glutamate to produce aspartate and alpha ketoglutarate (see Figures 6 and 8). Because alpha ketoglutarate can directly enter the TCA cycle for energy production, it is possible that glutamate could be used to meet the extra demand for ATP in neurons by oxidation of glutamate starting with the aspartate aminotransferase reaction.
The potentially important role of aspartate aminotransferase in facilitating respiration on glutamate in neuronal mitochondria was recognized by earlier investigators, and the name ‘mini citric acid cycle’ was coined to emphasize its role in neuronal energetics (Erecinska et al., 1988; Yudkoff et al., 1994). This truncated TCA cycle in neurons bypasses citrate and isocitrate formation as shown in Figure 9. Based on data obtained using radiolabeled aspartate and glutamine in rat brain synaptosomal preparations, Yudkoff, Erecinska and colleagues proposed that in the brain, amino acids could provide an alternative source of energy to help maintain ATP levels (Yudkoff et al., 1994). They made several important observations, including that in brain synaptosomes, the fastest reaction that supplies metabolites to the TCA cycle is aspartate aminotransferase, and the slowest step is from oxaloacetate to citrate. They also noted that bypassing the citrate synthase reaction, which consumes acetyl CoA and oxaloacetate to produce citrate, spares the mitochondrial pool of acetyl CoA, which then could be used for other reactions, including the synthesis of NAA (see Figure 9).
Figure 9.
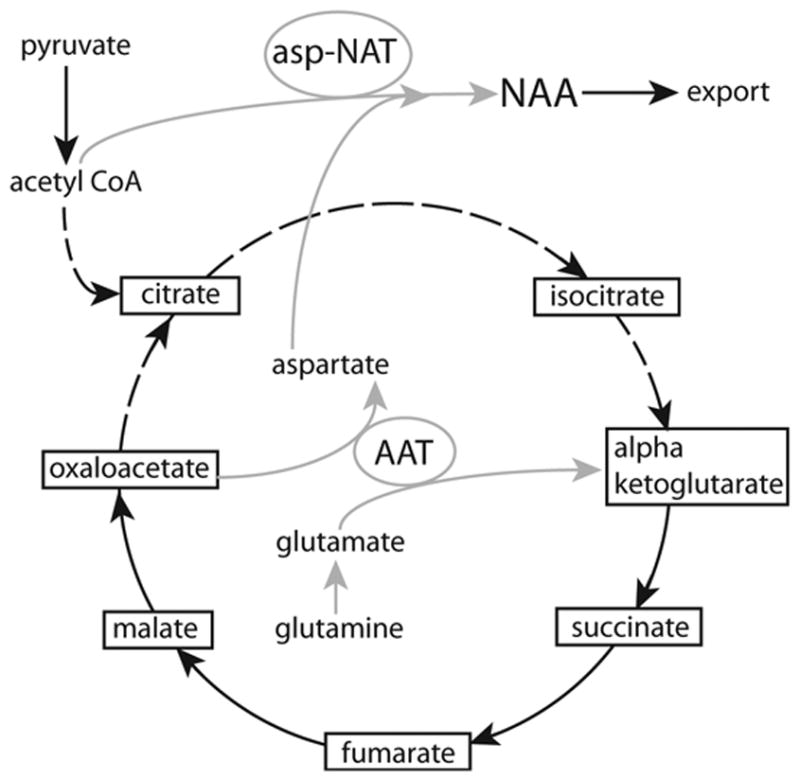
Schematic of the proposed “mini-TCA cycle” in neuronal mitochondria (Yudkoff et al., 1994), and the connection to NAA synthesis. Gray lines indicate the neuron-specific portion of the truncated TCA cycle, and the link to NAA synthesis, whereas dashed lines indicate the part of the TCA cycle that can be bypassed when oxidizing glutamate. As glutamate is converted to alpha ketoglutarate, the truncated TCA cycle produces excess aspartate, and the aspartate can be removed by acetylation through Asp-NAT (Madhavarao et al., 2005; Madhavarao and Namboodiri, 2006). The figure emphasizes the central role of aspartate aminotransferase (AAT) in the ability of neuronal mitochondria to bypass the slower citrate synthase reaction, and to oxidize glutamate through the truncated portion of the TCA cycle. There is no net cost in acetyl CoA utilization during the synthesis of NAA via Asp-NAT, because citrate production is circumvented. Reduced citrate production in neurons may reduce substrate availability for local lipid synthesis via the citrate lyase reaction (Figure 8), but extra NAA is generated which can then be exported and used for increased galactocerebroside and steroid synthesis in oligodendrocytes.
The truncated TCA cycle in neurons starts from glutamine, and proceeds through glutamate to alpha ketoglutarate without the action of glutamate dehydrogenase (EC 1.4.1.3), which would involve the production of ammonia. Glutamate dehydrogenase is present in neurons and glia (McKenna et al., 2000), and catalyzes the deamination of glutamate to alpha-ketoglutarate and ammonia using either NAD or NADP as cofactors (Mastorodemos et al., 2005). It is possible that neuronal mitochondria preferentially use the aspartate aminotransferase reaction instead of the glutamate dehydrogenase reaction to generate the key metabolite alpha ketoglutarate in order to avoid the problem of ammonia toxicity (Madhavarao et al., 2003; Madhavarao et al., 2005; Madhavarao and Namboodiri, 2006). This metabolic distinction between neurons and somatic cells such as hepatocytes may be due in part to the absence of an effective urea cycle system in the brain for nitrogen removal (see section 7. below).
In addition to acetyl CoA, NAA synthesis requires a continuous supply of aspartate in neuronal mitochondria. The aspartate-malate shuttle is a critical component of the inner membrane of neuronal mitochondria (Yudkoff et al., 1994), which moves reducing equivalents from the cytosol into mitochondria in the form of malate (Ramos et al., 2003). Intramitochondrial malate is then rapidly converted through oxaloacetate to aspartate via the aspartate aminotransferase reaction (Yudkoff et al., 1994). The aspartate-malate shuttle is essential for this process, and it involves several proteins and enzymes in mitochondria and the neuronal cytoplasm (see Figure 6). Aralar1 is a key glutamate-aspartate transporter involved in the aspartate-malate shuttle in the mitochondria of certain tissues, notably excitable tissues including skeletal muscle, heart and brain (Begum et al., 2002). Aralar1 moves aspartate out of mitochondria, and glutamate into mitochondria, and works in conjunction with the enzyme aspartate aminotransferase, which is another integral part of the malate-aspartate shuttle. Results with aralar1 −/− mice also provide support for the bioenergetic role for NAA linked to the aspartate-malate shuttle (Jalil et al., 2005). Aralar1 is the only mitochondrial aspartate-glutamate carrier in the brain, and aralar1 −/− mice lack malate-aspartate shuttle activity, have dramatically reduced brain NAA and aspartate levels, and show reduced neuronal respiration on glutamate plus malate (Jalil et al., 2005). It is possible that the acetylation of aspartate by Asp-NAT in neuronal mitochondria could combine with the action of aralar1 to remove product inhibition of the aspartate aminotransferase reaction. This would facilitate the conversion of glutamate to alpha ketoglutarate that can then enter the TCA cycle for energy production (Madhavarao et al., 2003; Madhavarao et al., 2005; Madhavarao and Namboodiri, 2006).
There have been a number of reports which do not support a bioenergetic role for NAA synthesis in the brain. Magnetic resonance spectroscopy studies on the indices of mitochondrial energetics in healthy individuals did not find a positive correlation between NAA levels and ATP (Pan and Takahashi, 2005). The lack of correlation between brain ATP and NAA levels was based entirely on in vivo MRS data. In addition, the determined concentration levels of free ADP and ATP varied by a factor of 1000. In terms of concentration however, NAA levels were comparable to those of ATP rather than ADP. It should also be noted that MRS techniques average ATP, ADP and NAA levels over a relatively large volume of tissue, which would contain all cell types present in the brain, thus averaging metabolite concentrations which may have large intercellular gradients. NAA synthesis in the brain has been shown to be coupled to glycogen and glucose metabolism (Choi and Gruetter, 2003; Moreno et al., 2001), but did not appear to be substantially involved in brain energy stores in the resting brain (Choi and Gruetter, 2004). Finally, the work of Lu and colleagues suggests that microsomal synthesis of NAA may be quantitatively more robust than mitochondrial synthesis (Lu et al., 2004), which would diminish any role for NAA in neuronal energy production. Much additional work will be required to determine the relative contributions of mitochondria and other compartments such as endoplasmic reticulum to NAA synthesis in neurons.
6.4. Fate of NAA in oligodendrocytes after postnatal myelination
A central question regarding any model of NAA metabolism concerns the role of the NAA-derived aspartate that is formed in oligodendrocytes as a result of ASPA- mediated catalysis. A study from Yudkoff and coworkers provides some clues concerning this important question (Miller et al., 1996). They found that intracranial injection of N-([2H3] acetyl)-L-[15N]-aspartate ([2H3,15N]NAA) led to its rapid decline which was associated with a rapid and quantitative appearance of [15N] glutamate. This finding indicated rapid transamination of the [15N] aspartate that was derived from the ASPA-mediated hydrolysis of [2H3,15N]NAA. Also, [15N] NAA was not detectable, indicating that [15N] aspartate was not reutilized for NAA synthesis. These results are consistent with the deacetylation of NAA taking place almost exclusively in oligodendrocytes, and the resulting aspartate being utilized for energy production in the citric acid cycle after conversion to oxaloacetate. Concerning the fate of NAA derived acetate in oligodendrocytes after postnatal myelination is complete, it should be noted that acetyl CoA is involved in numerous critical cellular reactions, and it can be degraded to CO2 through the TCA cycle for the derivatization of energy (Bluml et al., 2002). Therefore, NAA can supply both acetate and oxaloacetate to the TCA cycle for energy production (Nadler and Cooper, 1972a), at least in oligodendrocytes where ASPA is present. Glial cells preferentially utilize acetate for energy production through the TCA cycle, as opposed to neurons (Bluml et al., 2002; Hassel et al., 1995; Muir et al., 1986) suggesting that oligodendrocytes could make substantial use of NAA-derived acetate for energy production when myelination is complete. It has also been proposed that NAA could act as a reservoir of glutamate in the brain, wherein NAA is converted to aspartate in oligodendrocytes, which could then be converted to glutamate through the TCA cycle with favorable energetics (Clark et al., 2006).
7. Integrating Various NAA Functions in the CNS
Recently, Madhavarao and coworkers proposed a model whereby NAA has two primary roles in the nervous system; facilitation of energy metabolism in neuronal mitochondria, and a source of acetate for fatty acid and steroid synthesis in oligodendrocytes. In this model, Asp-NAT facilitates removal of excess aspartate from the matrix of neuronal mitochondria via acetylation, thus favoring alpha ketoglutarate formation from glutamate, and energy production via the citric acid cycle. Anaplerosis and cataplerosis are the complimentary metabolic processes which replenish TCA cycle intermediates, and remove excess intermediates respectively (Brunengraber and Roe, 2006). Principle anaplerotic molecules feeding into the TCA cycle include pyruvate, glutamate, and precursors of propionyl-CoA. Two important cataplerotic enzymes that help remove excess TCA intermediates are aspartate aminotransferase and citrate lyase (Owen et al., 2002). We propose that Asp-NAT also plays a cataplerotic role in neuronal mitochondria. NAA synthesis is associated with preventing product inhibition of aspartate aminotransferase by using the Asp-NAT reaction to remove excess aspartate from neuronal mitochondria. The malate-aspartate shuttle in neuronal mitochondria is required for aspartate synthesis by providing the precursor malate, and as such, shuttle activity is required for NAA synthesis in neuronal mitochondria. Converting the product aspartate into NAA helps to guide the aspartate aminotransferase reaction toward alpha ketoglutarate, thus facilitating energy production in neurons. Using this reaction would also spare acetyl CoA from the citrate synthetase reaction, so that it could participate in NAA synthesis instead.
In its lipogenic role, Asp-NAT activity works in conjunction with a putative mitochondrial NAA transporter to move acetate groups from the matrix of neuronal mitochondria to the cytoplasm in the form of NAA. Neuronal NAA must then be transferred to oligodendrocytes at their point of contact; the inner plasma membrane of the myelin sheath. It seems likely that specific dicarboxylate transport proteins or exchangers in both neurons and oligodendrocytes are involved in this process (Fujita et al., 2005). After axonal-glial transfer of NAA to oligodendrocytes, it provides acetate units for fatty acid and steroid synthesis in the cytosol via the actions of ASPA and cytoplasmic acetyl CoA synthetase (ACS1). The activity of ACS1 in the brain has been poorly characterized (Luong et al., 2000), but it may turn out to be another lipogenic enzyme present in oligodendrocytes that works in conjunction with ASPA. The proposed model stresses the trophic support between neurons and oligodendrocytes, where neurons supply oligodendrocytes with NAA-derived acetyl groups for fatty acid synthesis, and in turn, oligodendrocytes synthesize the critical myelin lipids, including sulfatide, galactocerebroside and cholesterol, which are essential for myelination and proper neuronal function (Figure 6). When lipid synthesis requirements have been met in oligodendrocytes, acetyl CoA and aspartate can be utilized for other functions, including protein synthesis and energy production.
7.1. Four cell-type model for NAA synthesis, utilization, breakdown and excretion
In order to place NAA metabolism in the CNS in a broader context, it is necessary to discuss intercellular trafficking of the relevant metabolites (Figure 10). The blood stream acts as both the source and sink for all brain metabolites, including glucose, amino acids and waste products. Endothelia and astrocytes provide a two-stage selective barrier to metabolite diffusion into and out of the brain in the form of the blood-brain barrier. Glucose is the brain’s primary energy source by providing the starting substrate for pyruvate production in neuronal cytoplasm, and utilization in mitochondria. Pyruvate is converted to acetyl CoA, which can enter the TCA cycle for oxidation. Additionally, acetyl CoA and aspartate can act as substrates for the synthesis of NAA, mediated by the enzyme Asp-NAT. Some portion of NAA in select neuronal populations is combined with glutamate to form NAAG, probably by a dipeptide synthetase. The bulk of the brain’s complement of NAA remains intraneuronal, and turns over relatively slowly. Some NAA is released in a controlled fashion from neurons as an osmolyte or nitrogen carrier (see section 7.1.1 below), or effluxes to extracellular space along its concentration gradient, where it is most likely taken up by astrocytes. NAAG released from neurons upon depolarization is either taken up by neurons, or is broken down to NAA and glutamate on the surface of astrocytes, which then take up both compounds. Some NAA is transported from neuronal axons to oligodendrocytes at their point of contact, and it is then broken down to acetate and aspartate within the oligodendrocytes. Acetate can be incorporated into acetyl CoA and then converted to lipids, or fully oxidized in the TCA cycle for energy. Aspartate can be used for protein synthesis, or can be converted to oxaloacetate for subsequent energy production. Ammonia produced in neurons by the enzyme glutaminase escapes to the extracellular space, and then into astrocytes where it participates in the glutamine synthetase reaction to produce glutamine. Glutamine can be passed back to neurons for conversion to glutamate and ammonia, or if the brain nitrogen load is high, the glutamine can be excreted from astrocytes to the circulation.
Figure 10.
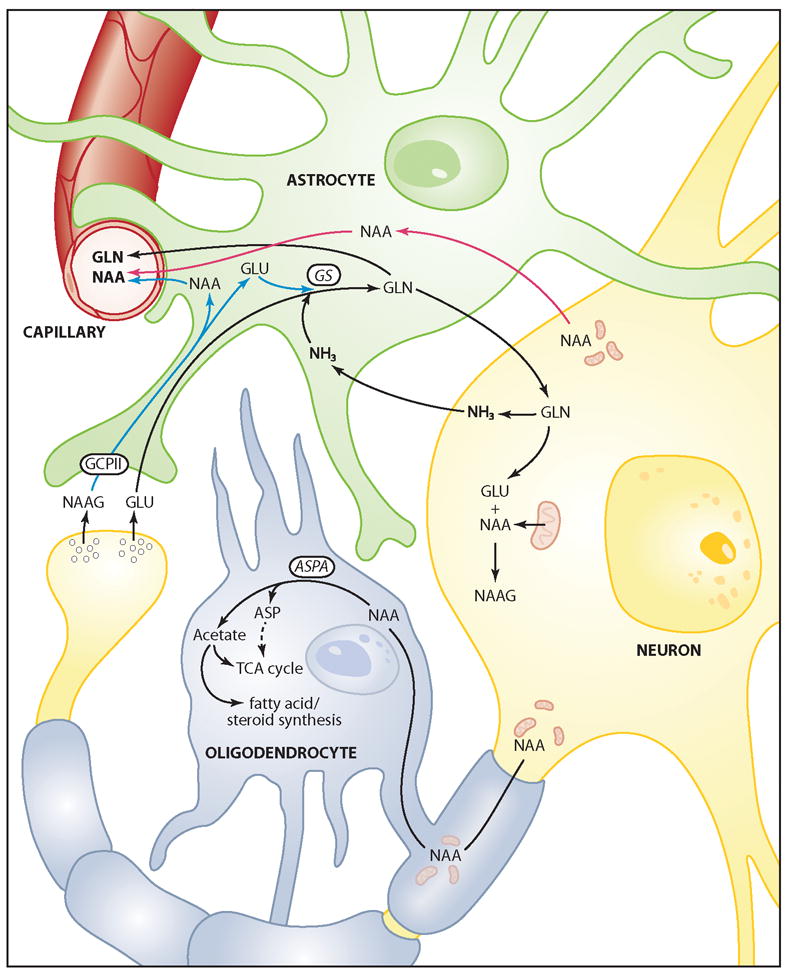
Hypothetical, simplified 4-cell model of NAA synthesis and metabolism in the brain. NAA is synthesized in neuronal mitochondria (see Figure 5), and can then either be transported to oligodendrocytes for fatty acid synthesis and energy production, or can be used for the synthesis of NAAG in neurons. NAAG is released from neuronal synapses along with other transmitters, such as glutamate, and the extracellular NAAG is hydrolyzed by GCPII on astrocytes, which then take up the breakdown products (blue lines). Glutamate and ammonia in astrocytes are converted to glutamine by glutamine synthase (GS), and the glutamine can be excreted to the circulation as a nitrogen removal system, or it can be transported back to neurons for reuse. When the brain nitrogen load is high, NAA excretion might act as a secondary nitrogen removal system, wherein NAA could hypothetically be released by neurons to the extracellular space, taken up by astrocytes, and then excreted to the circulation (red lines). Glutamate and glutamine cycling between neurons and astrocytes involves the production and detoxification of ammonia. Abbreviations: ASP, aspartate; ASPA, aspartoacylase; GCPII, glutamate carboxypeptidase II; GLN, glutamine; GLU, glutamate; GS, glutamine synthetase, NAA, N-acetylaspartate; NAAG, N-acetylaspartylglutamate; NH3, ammonia.
When glutamine and other amino acids are limiting in neurons, there would be less flux through the aspartate aminotransferase pathway of the truncated TCA cycle, and neuronal respiration would occur primarily through pyruvate conversion to acetyl CoA, and the synthesis of citrate from oxaloacetate and acetyl CoA. But when the nitrogen load in brain is increased, and excess glutamine is present, flux through aspartate aminotransferase would be accelerated, and both alpha ketoglutarate and aspartate would be formed. The alpha ketoglutarate could be oxidized through the TCA cycle, and the excess aspartate could be acetylated to NAA. A possible fate of NAA that has not been explored is that it could function as a secondary mechanism for nitrogen removal from the brain.
7.1.1. Possible Role for NAA in CNS Nitrogen Balance
Nitrogen balance involves the strict regulation of amino acid uptake, protein synthesis and degradation, amino acid excretion and amino acid oxidation. In the brain, as in other organs, the carbon skeletons of amino acids are oxidized for energy or stored as glycogen, but the excess amino nitrogen must be excreted. Ammonia metabolism in the brain is intimately associated with nitrogen recycling and removal. The primary route of ammonia metabolism and detoxification in brain is accomplished by the enzyme glutamine synthase in astrocytes (Cooper, 2001). This enzyme system not only efficiently removes ammonia from the blood as it enters the brain, but also detoxifies ammonia released from neurons. Neurons release glutamate and ammonia, and astrocytes take up both metabolites and produce glutamine through the glutamine synthase reaction, thus preventing ammonia toxicity. Excess glutamine in astrocytes is excreted to the bloodstream as required by the brain’s nitrogen load.
The brain lacks the urea cycle enzymes carbamoyl-phosphate synthase I and ornithine transcarbamylase, and therefore is unable to remove nitrogen in the form of urea. Instead, glutamine synthesis is the predominant route for nitrogen removal in the brain under both normal and hyperammonemic conditions (Cooper et al., 1979; Felipo and Butterworth, 2002). However, the glutamine synthase reaction occurs almost exclusively in astrocytes, and has a relatively low capacity for dealing with excess ammonia. We propose that a possible secondary nitrogen removal system in the brain could conceivably involve NAA synthesis and excretion (see Figure 10). Miller and colleagues used radiolabeled NAA infusion experiments to study NAA metabolism and found that after catalysis, the aspartate moiety was not reutilized for NAA synthesis (Miller et al., 1996). They suggested that these results indicated that one function of NAA was to transport amino group nitrogen from mitochondria to cytoplasm. Therefore it is possible that as the brain’s nitrogen load is increased, Asp-NAT could act to trap some aspartate as NAA, and neurons could increase their release of NAA to the extracellular space. It has been reported that NMDA application can stimulate a calcium-dependent release of NAA from neurons, but release was not stimulated by hyperosmotic conditions or potassium-induced depolarization (Tranberg et al., 2004). In another study, application of NMDA to brain slices resulted in the release of different metabolites than were released after potassium induced depolarization or glutamate application (Thatcher et al., 2002). NAA levels and energy state were decreased by NMDA application, and lactate levels were increased. Such findings suggest that NAA is associated with energy state, and that its release from neurons is a physiologically regulated response to NMDA receptor activity.
Thus, the lack of a degradative enzyme for NAA in neurons could serve in part as a secondary nitrogen removal system that works in conjunction with the astrocyte glutamine excretion system to eliminate excess nitrogen from the brain. Once aspartate is acetylated in neurons, NAA only has two known routes of further catabolism. First, it can be transferred to oligodendrocytes for degradation to acetate and aspartate, and subsequent lipid synthesis or energy metabolism. Second, NAA can be released from neurons to the extracellular space where it would be taken up by astrocytes via the NaDC3 transporter and then excreted to the circulation. NAA released to the circulation could be metabolized in other tissues, in particular kidney, which expresses ASPA at higher levels than brain (D’Adamo, Jr. et al., 1973). It is possible that one function of kidney ASPA involves scavenging of aspartate and acetate from the low levels of NAA in the circulation, which would be especially important when diet was poor (Wolfe, 2005). In this regard it is interesting to note that the NaDC3 dicarboxylate transporter has recently been localized to proximal kidney tubule cells (Bai et al., 2006), the same cell type in the kidney in which ASPA is present.
The greatly increased concentration of NAA that occurs in the urine of Canavan disease patients clearly indicates that normally, the majority of NAA produced in the brain is metabolized to acetate and aspartate, either in oligodendrocytes in the CNS, or after transport via the circulation, in kidney proximal tubules cells. It is interesting to note that in liver, which has a robust urea cycle, aspartate is conjugated to citrulline to form arginosuccinate as part of the urea cycle, and that the nitrogen atoms coming from aspartate and citrulline are eventually incorporated into urea and excreted. Therefore, the removal of nitrogen from the brain in the form of NAA may be a brain-specific mechanism for excretion of aspartate-associated nitrogen. Within the context of the hypothesis that NAA is involved in nitrogen removal from the brain, a lack of ASPA activity in brain and kidney explains the very large accumulation of NAA in urine during Canavan disease, because both catalytic compartments (oligodendrocytes and proximal tubule cells) are incapable of degrading NAA. The fact that brain levels of NAA only rise modestly in Canavan disease, whereas urine levels increase dramatically, indicates that excess NAA is efficiently cleared from the brain.
8. Summary and Future Directions
NAA remains an enigmatic molecule, but researchers are beginning to decipher the complex web of CNS biochemistry in which it is actively involved. NAA is one of the most prominent metabolites in magnetic resonance spectrograms of brain, and the measured levels are highly sensitive to brain injury or disease, providing an invaluable tool for diagnosis and evaluation in clinical settings. Increases in MRS sensitivity and reduced voxel size, in conjunction with emerging scanner technologies and computer techniques will dramatically improve the diagnostic capabilities of MRS in coming years. Whereas some proposed functions for NAA, such as an osmoregulatory role, remain tentative, other functions are now established, including a role in CNS fatty acid and steroid synthesis, particularly with respect to postnatal myelination. It is important to note that NAA provides a means for moving acetate and aspartate from neurons to oligodendrocytes, but the transport mechanisms underlying neuronal export and oligodendrocyte uptake of NAA remain to be determined. Many of the unique biochemical mechanisms of myelin lipid synthesis in the brain are poorly understood, and much additional research into the biochemistry of acetate utilization in the CNS is warranted, both as a building block for lipids, and as an energy source. The regulatory mechanisms which control the unique enzymes involved in CNS myelin lipid synthesis have only been cursorily examined, offering many opportunities for investigation. Further research into the precise etiology of Canavan disease is required to determine the best course of action in dealing with this fatal genetic disorder, including additional research into ASPA gene therapy and acetate supplementation. The proposed connections between NAA and neurotoxicity or disrupted osmoregulation associated with errors in NAA catabolism remain to be confirmed.
NAA appears to be a direct precursor for the enzymatic synthesis of the neuronal neuropeptide NAAG, which itself has important functions including the modulation of neurotransmitter release. How NAAG is synthesized from NAA remains a mystery, and further work will be required to identify the enzyme(s) responsible. Only limited research has been directed toward determining how the biosynthesis of either NAA or NAAG is regulated at the genetic or enzymatic levels, and purification of the enzymes involved, and identification of their respective genes will be crucial in furthering our understanding of these regulatory mechanisms. There is substantial evidence that NAA synthesis in neuronal mitochondria is tied to neuronal energy metabolism, but this topic is far from resolved, especially with respect to the relatively low turnover rate of NAA in the brain. The emerging bioenergetic functions of NAA in the nervous system are tantalizing targets for further research in part due to the established connection between neuronal health and NAA levels as revealed by magnetic resonance spectroscopy and other methods. The hypothesis that NAA excretion works in conjunction with glutamine excretion in regulating brain nitrogen balance is worthy of investigation. Because NAA is found at high concentrations only in the nervous system, the exceptionally strong expression of the NAA-specific hydrolyzing enzyme ASPA in tissues such as kidney is suggestive of possible distinct functions in peripheral tissues that may involve acetylated substrates other than NAA. Also in this regard, the localization of ASPA in the nuclei of oligodendrocytes and kidney proximal tubule cells is also suggestive of additional functional roles for this enzyme. These are potentially fruitful areas of research into the many complexities of NAA biochemistry and neurobiology in the central nervous system.
Acknowledgments
This work is dedicated to the memory of Dr. Suzannah Bliss Tieman. We would like to thank Dr. C. Demougeot for commenting on the manuscript. We apologize to any researchers whose relevant work we did not directly cite. This work was supported by a grant to MAAN from the NIH (Grant # RO1: NS39387), and by grants from the Samueli Institute for Information Biology and Jacob’s Cure. CNM was supported partially by a fellowship from the American Academy of Neurology Foundation, co-sponsored by the Canavan Foundation.
Abbreviations
- acetyl CoA
acetyl coenzyme A
- ACS1
acetyl CoA synthetase-1
- ASPA
aspartoacylase
- Asp-NAT
aspartate N-acetyltransferase
- CNS
central nervous system
- GTA
glyceryl triacetate
- HPLC
high performance liquid chromatography
- MRI
magnetic resonance imaging
- MRI
magnetic resonance imaging
- MRS
magnetic resonance spectroscopy
- NAA
N-acetylaspartate
- NAAG
N-acetylaspartylglutamate
- TCA cycle
tricarboxylic acid cycle
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abbott C, Bustillo J. What have we learned from proton magnetic resonance spectroscopy about schizophrenia? A critical update. Curr Opin Psychiatry. 2006;19:135–139. doi: 10.1097/01.yco.0000214337.29378.cd. [DOI] [PubMed] [Google Scholar]
- Adachi M, Schneck L, Cara J, Volk BW. Spongy degeneration of the central nervous system (van Bogaert and Bertrand type; Canavan’s disease) A review. Hum Pathol. 1973;4:331–347. doi: 10.1016/s0046-8177(73)80098-x. [DOI] [PubMed] [Google Scholar]
- Akimitsu T, Kurisu K, Hanaya R, Iida K, Kiura Y, Arita K, Matsubayashi H, Ishihara K, Kitada K, Serikawa T, Sasa M. Epileptic seizures induced by N-acetyl-L-aspartate in rats: in vivo and in vitro studies. Brain Res. 2000;861:143–150. doi: 10.1016/s0006-8993(00)02028-x. [DOI] [PubMed] [Google Scholar]
- Amstutz DR, Coons SW, Kerrigan JF, Rekate HL, Heiserman JE. Hypothalamic hamartomas: Correlation of MR imaging and spectroscopic findings with tumor glial content. AJNR Am J Neuroradiol. 2006;27:794–798. [PMC free article] [PubMed] [Google Scholar]
- Anderson KJ, Borja MA, Cotman CW, Moffett JR, Namboodiri MA, Neale JH. N-acetylaspartylglutamate identified in the rat retinal ganglion cells and their projections in the brain. Brain Res. 1987;411:172–177. doi: 10.1016/0006-8993(87)90696-2. [DOI] [PubMed] [Google Scholar]
- Ando S, Tanaka Y, Toyoda Y, Kon K. Turnover of myelin lipids in aging brain. Neurochem Res. 2003;28:5–13. doi: 10.1023/a:1021635826032. [DOI] [PubMed] [Google Scholar]
- Andreassen OA, Dedeoglu A, Ferrante RJ, Jenkins BG, Ferrante KL, Thomas M, Friedlich A, Browne SE, Schilling G, Borchelt DR, Hersch SM, Ross CA, Beal MF. Creatine increase survival and delays motor symptoms in a transgenic animal model of Huntington’s disease. Neurobiol Dis. 2001;8:479–491. doi: 10.1006/nbdi.2001.0406. [DOI] [PubMed] [Google Scholar]
- Arnold DL, Matthews PM, Francis G, Antel J. Proton magnetic resonance spectroscopy of human brain in vivo in the evaluation of multiple sclerosis: assessment of the load of disease. Magn Reson Med. 1990a;14:154–159. doi: 10.1002/mrm.1910140115. [DOI] [PubMed] [Google Scholar]
- Arnold DL, Shoubridge EA, Villemure JG, Feindel W. Proton and phosphorus magnetic resonance spectroscopy of human astrocytomas in vivo. Preliminary observations on tumor grading. NMR Biomed. 1990b;3:184–189. doi: 10.1002/nbm.1940030407. [DOI] [PubMed] [Google Scholar]
- Arun P, Madhavarao CN, Hershfield JR, Moffett JR, Namboodiri MA. SH-SY5Y neuroblastoma cells: a model system for studying biosynthesis of NAAG. Neuroreport. 2004;15:1167–1170. doi: 10.1097/00001756-200405190-00017. [DOI] [PubMed] [Google Scholar]
- Arun P, Madhavarao CN, Moffett JR, Namboodiri AM. Regulation of N-acetylaspartate and N-acetylaspartylglutamate biosynthesis by protein kinase activators. J Neurochem. 2006;98:2034–2042. doi: 10.1111/j.1471-4159.2006.04068.x. [DOI] [PubMed] [Google Scholar]
- Aubert A, Costalat R, Magistretti PJ, Pellerin L. Brain lactate kinetics: Modeling evidence for neuronal lactate uptake upon activation. Proc Natl Acad Sci U S A. 2005;102:16448–16453. doi: 10.1073/pnas.0505427102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai X, Chen X, Feng Z, Hou K, Zhang P, Fu B, Shi S. Identification of basolateral membrane targeting signal of human sodium-dependent dicarboxylate transporter 3. J Cell Physiol. 2006;206:821–830. doi: 10.1002/jcp.20553. [DOI] [PubMed] [Google Scholar]
- Bammer R, Skare S, Newbould R, Liu C, Thijs V, Ropele S, Clayton DB, Krueger G, Moseley ME, Glover GH. Foundations of advanced magnetic resonance imaging. NeuroRx. 2005;2:167–196. doi: 10.1602/neurorx.2.2.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barany M, Spigos DG, Mok E, Venkatasubramanian PN, Wilbur AC, Langer BG. High resolution proton magnetic resonance spectroscopy of human brain and liver. Magn Reson Imaging. 1987;5:393–398. doi: 10.1016/0730-725x(87)90128-7. [DOI] [PubMed] [Google Scholar]
- Barker PB, Bonekamp D, Riedy G, Smith M. Quantitation of NAA in the brain by magnetic resonance spectroscopy. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 183–197. [Google Scholar]
- Barker PB, Gillard JH. MR Spectroscopy in stroke. In: Gillard J, Waldman A, Barker PB, editors. Clinical MR Neuroimaging: Diffusion, Perfusion and Spectroscopy. Cambridge University Press; New York, NY: 2005. pp. 168–181. [Google Scholar]
- Barkovich AJ, Baranski K, Vigneron D, Partridge JC, Hallam DK, Hajnal BL, Ferriero DM. Proton MR spectroscopy for the evaluation of brain injury in asphyxiated, term neonates. AJNR Am J Neuroradiol. 1999;20:1399–1405. [PMC free article] [PubMed] [Google Scholar]
- Bartalini G, Margollicci M, Balestri P, Farnetani MA, Cioni M, Fois A. Biochemical diagnosis of Canavan disease. Childs Nerv Syst. 1992;8:468–470. doi: 10.1007/BF00274411. [DOI] [PubMed] [Google Scholar]
- Baslow MH. A review of phylogenetic and metabolic relationships between the acylamino acids, N-acetyl-L-aspartic acid and N-acetyl-L-histidine, in the vertebrate nervous system. J Neurochem. 1997;68:1335–1344. doi: 10.1046/j.1471-4159.1997.68041335.x. [DOI] [PubMed] [Google Scholar]
- Baslow MH. Function of the N-acetyl-L-histidine system in the vertebrate eye. Evidence in support of a role as a molecular water pump. J Mol Neurosci. 1998;10:193–208. doi: 10.1007/BF02761774. [DOI] [PubMed] [Google Scholar]
- Baslow MH. The existence of molecular water pumps in the nervous system: a review of the evidence. Neurochem Int. 1999;34:77–90. doi: 10.1016/s0197-0186(98)00073-4. [DOI] [PubMed] [Google Scholar]
- Baslow MH. Canavan’s spongiform leukodystrophy: a clinical anatomy of a genetic metabolic CNS disease. J Mol Neurosci. 2000a;15:61–69. doi: 10.1385/JMN:15:2:61. [DOI] [PubMed] [Google Scholar]
- Baslow MH. Functions of N-acetyl-L-aspartate and N-acetyl-L-aspartylglutamate in the vertebrate brain: role in glial cell-specific signaling. J Neurochem. 2000b;75:453–459. doi: 10.1046/j.1471-4159.2000.0750453.x. [DOI] [PubMed] [Google Scholar]
- Baslow MH. Evidence supporting a role for N-acetyl-L-aspartate as a molecular water pump in myelinated neurons in the central nervous system. An analytical review. Neurochem Int. 2002;40:295–300. doi: 10.1016/s0197-0186(01)00095-x. [DOI] [PubMed] [Google Scholar]
- Baslow MH. Brain N-acetylaspartate as a molecular water pump and its role in the etiology of Canavan disease: a mechanistic explanation. J Mol Neurosci. 2003a;21:185–190. doi: 10.1385/jmn:21:3:185. [DOI] [PubMed] [Google Scholar]
- Baslow MH. N-acetylaspartate in the vertebrate brain: metabolism and function. Neurochem Res. 2003b;28:941–953. doi: 10.1023/a:1023250721185. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Guilfoyle DN. Effect of N-acetylaspartic acid on the diffusion coefficient of water: a proton magnetic resonance phantom method for measurement of osmolyte-obligated water. Anal Biochem. 2002;311:133–138. doi: 10.1016/s0003-2697(02)00403-7. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Guilfoyle DN. Functions of N-acetylaspartate and N-acetylaspartylglutamate in brain: evidence of a role in maintenance of higher brain integrative activities of information processing and cognition. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 95–112. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Kitada K, Suckow RF, Hungund BL, Serikawa T. The effects of lithium chloride and other substances on levels of brain N-acetyl-L-aspartic acid in Canavan disease-like rats. Neurochem Res. 2002;27:403–406. doi: 10.1023/a:1015504031229. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Suckow RF, Hungund BL. Effects of ethanol and of alcohol dehydrogenase inhibitors on the reduction of N-acetylaspartate levels of brain in mice in vivo: a search for substances that may have therapeutic value in the treatment of Canavan disease. J Inherit Metab Dis. 2000;23:684–692. doi: 10.1023/a:1005618526988. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Suckow RF, Sapirstein V, Hungund BL. Expression of aspartoacylase activity in cultured rat macroglial cells is limited to oligodendrocytes. J Mol Neurosci. 1999;13:47–53. doi: 10.1385/JMN:13:1-2:47. [DOI] [PubMed] [Google Scholar]
- Baslow MH, Yamada S. Identification of N-acetylaspartate in the lens of the vertebrate eye: a new model for the investigation of the function of N-acetylated amino acids in vertebrates. Exp Eye Res. 1997;64:283–286. doi: 10.1006/exer.1996.0205. [DOI] [PubMed] [Google Scholar]
- Bates TE, Strangward M, Keelan J, Davey GP, Munro PM, Clark JB. Inhibition of N-acetylaspartate production: implications for 1H MRS studies in vivo. Neuroreport. 1996;7:1397–1400. [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, Thompson CB. ATP citrate lyase is an important component of cell growth and transformation. Oncogene. 2005 doi: 10.1038/sj.onc.1208773. [DOI] [PubMed] [Google Scholar]
- Begum L, Jalil MA, Kobayashi K, Iijima M, Li MX, Yasuda T, Horiuchi M, Del AA, Satrustegui J, Saheki T. Expression of three mitochondrial solute carriers, citrin, aralar1 and ornithine transporter, in relation to urea cycle in mice. Biochim Biophys Acta. 2002;1574:283–292. doi: 10.1016/s0167-4781(01)00376-1. [DOI] [PubMed] [Google Scholar]
- Behar KL, Boucher R, Fritch W, Manuelidis L. Changes in N-acetylaspartate and myo-inositol detected in the cerebral cortex of hamsters with Creutzfeldt-Jakob disease. Magn Reson Imaging. 1998;16:963–968. doi: 10.1016/s0730-725x(98)00109-x. [DOI] [PubMed] [Google Scholar]
- Beigneux AP, Kosinski C, Gavino B, Horton JD, Skarnes WC, Young SG. ATP-citrate lyase deficiency in the mouse. J Biol Chem. 2004;279:9557–9564. doi: 10.1074/jbc.M310512200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benuck M, D’Adamo AF., Jr Acetyl transport mechanisms. Metabolism of N-acetyl-L-aspartic acid in the non-nervous tissues of the rat. Biochim Biophys Acta. 1968;152:611–618. doi: 10.1016/0005-2760(68)90101-x. [DOI] [PubMed] [Google Scholar]
- Berger UV, Luthi-Carter R, Passani LA, Elkabes S, Black I, Konradi C, Coyle JT. Glutamate carboxypeptidase II is expressed by astrocytes in the adult rat nervous system. J Comp Neurol. 1999;415:52–64. doi: 10.1002/(sici)1096-9861(19991206)415:1<52::aid-cne4>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Breier A, Callicott JH, Adler C, Mattay VS, Shapiro M, Frank JA, Pickar D, Weinberger DR. The relationship between dorsolateral prefrontal neuronal N-acetylaspartate and evoked release of striatal dopamine in schizophrenia. Neuropsychopharmacology. 2000;22:125–132. doi: 10.1016/S0893-133X(99)00096-2. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Nawroz S, Mattay VS, Barnett AS, Duyn JH, Moonen CT, Frank JA, Tedeschi G, Weinberger DR. Regionally specific pattern of neurochemical pathology in schizophrenia as assessed by multislice proton magnetic resonance spectroscopic imaging. Am J Psychiatry. 1996;153:1554–1563. doi: 10.1176/ajp.153.12.1554. [DOI] [PubMed] [Google Scholar]
- Bertolino A, Weinberger DR. Proton magnetic resonance spectroscopy in schizophrenia. Eur J Radiol. 1999;30:132–141. doi: 10.1016/s0720-048x(99)00052-2. [DOI] [PubMed] [Google Scholar]
- Bhakoo KK, Craig TJ, Styles P. Developmental and regional distribution of aspartoacylase in rat brain tissue. J Neurochem. 2001;79:211–220. doi: 10.1046/j.1471-4159.2001.00561.x. [DOI] [PubMed] [Google Scholar]
- Bhakoo KK, Pearce D. In vitro expression of N-acetyl aspartate by oligodendrocytes: implications for proton magnetic resonance spectroscopy signal in vivo. J Neurochem. 2000;74:254–262. doi: 10.1046/j.1471-4159.2000.0740254.x. [DOI] [PubMed] [Google Scholar]
- Birnbaum SM. Amino acid acylases I and II from hog kidney. Methods Enzymol. 1955;2:115–119. [Google Scholar]
- Birnbaum SM, Levintow L, Kingsley RB, Greenstein JP. Specificity of amino acid acylases. J Biol Chem. 1952;194:455–470. [PubMed] [Google Scholar]
- Bjartmar C, Battistuta J, Terada N, Dupree E, Trapp BD. N-acetylaspartate is an axon-specific marker of mature white matter in vivo: a biochemical and immunohistochemical study on the rat optic nerve. Ann Neurol. 2002;51:51–58. doi: 10.1002/ana.10052. [DOI] [PubMed] [Google Scholar]
- Blakely RD, Robinson MB, Thompson RC, Coyle JT. Hydrolysis of the brain dipeptide N-acetyl-L-aspartyl-L-glutamate: subcellular and regional distribution, ontogeny, and the effect of lesions on N-acetylated-alpha-linked acidic dipeptidase activity. J Neurochem. 1988;50:1200–1209. doi: 10.1111/j.1471-4159.1988.tb10593.x. [DOI] [PubMed] [Google Scholar]
- Bluml S. In vivo quantitation of cerebral metabolite concentrations using natural abundance 13C MRS at 1.5 T. J Magn Reson. 1999;136:219–225. doi: 10.1006/jmre.1998.1618. [DOI] [PubMed] [Google Scholar]
- Bluml S, Moreno-Torres A, Shic F, Nguy CH, Ross BD. Tricarboxylic acid cycle of glia in the in vivo human brain. NMR Biomed. 2002;15:1–5. doi: 10.1002/nbm.725. [DOI] [PubMed] [Google Scholar]
- Boltshauser E, Schmitt B, Wevers RA, Engelke U, Burlina AB, Burlina AP. Follow-up of a child with hypoacetylaspartia. Neuropediatrics. 2004;35:255–258. doi: 10.1055/s-2004-821036. [DOI] [PubMed] [Google Scholar]
- Bonnet M, Ducournau D, Lumbroso P, Serpin G. [N-acetyl-aspartylglutamic acid eye drops in allergic-type conjuctivitis. Double-blind comparative clinical study] J Fr Ophtalmol. 1985;8:573–578. [PubMed] [Google Scholar]
- Bothwell JH, Rae C, Dixon RM, Styles P, Bhakoo KK. Hypo-osmotic swelling-activated release of organic osmolytes in brain slices: implications for brain oedema in vivo. J Neurochem. 2001;77:1632–1640. doi: 10.1046/j.1471-4159.2001.00403.x. [DOI] [PubMed] [Google Scholar]
- Bradford HF, Thomas AJ. Metabolism of glucose and glutamate by synaptosomes from mammalian cerebral cortex. J Neurochem. 1969;16:1495–1504. doi: 10.1111/j.1471-4159.1969.tb09904.x. [DOI] [PubMed] [Google Scholar]
- Briellmann RS, Wellard RM, Jackson GD. Seizure-associated abnormalities in epilepsy: evidence from MR imaging. Epilepsia. 2005;46:760–766. doi: 10.1111/j.1528-1167.2005.47604.x. [DOI] [PubMed] [Google Scholar]
- Brooks WM, Friedman SD, Gasparovic C. Magnetic resonance spectroscopy in traumatic brain injury. J Head Trauma Rehabil. 2001;16:149–164. doi: 10.1097/00001199-200104000-00005. [DOI] [PubMed] [Google Scholar]
- Brown AM, Lemke G. Multiple regulatory elements control transcription of the peripheral myelin protein zero gene. J Biol Chem. 1997;272:28939–28947. doi: 10.1074/jbc.272.46.28939. [DOI] [PubMed] [Google Scholar]
- Bruhn H, Frahm J, Gyngell ML, Merboldt KD, H:anicke W, Sauter R, Hamburger C. Noninvasive differentiation of tumors with use of localized H-1 MR spectroscopy in vivo: initial experience in patients with cerebral tumors [see comments] Radiology. 1989a;172:541–548. doi: 10.1148/radiology.172.2.2748837. [DOI] [PubMed] [Google Scholar]
- Bruhn H, Frahm J, Gyngell ML, Merboldt KD, Hanicke W, Sauter R. Cerebral metabolism in man after acute stroke: new observations using localized proton NMR spectroscopy. Magn Reson Med. 1989b;9:126–131. doi: 10.1002/mrm.1910090115. [DOI] [PubMed] [Google Scholar]
- Bruhn H, Frahm J, Merboldt KD, Hanicke W, Hanefeld F, Christen HJ, Kruse B, Bauer HJ. Multiple sclerosis in children: cerebral metabolic alterations monitored by localized proton magnetic resonance spectroscopy in vivo. Ann Neurol. 1992;32:140–150. doi: 10.1002/ana.410320205. [DOI] [PubMed] [Google Scholar]
- Brunengraber H, Roe CR. Anaplerotic molecules: Current and future. J Inherit Metab Dis. 2006;29:327–331. doi: 10.1007/s10545-006-0320-1. [DOI] [PubMed] [Google Scholar]
- Buniatian HC, Hovhannissian VS, Aprikian GV. The participation of N-acetyl-L-aspartic acid in brain metabolism. J Neurochem. 1965;12:695–703. doi: 10.1111/j.1471-4159.1965.tb06783.x. [DOI] [PubMed] [Google Scholar]
- Burgal M, Jorda A, Grisolia S. Effects of N-acetyl aspartate, aspartate, and glutamate on cAMP and cGMP levels in developing rat cerebral cortex. J Neurochem. 1982;38:1498–1500. doi: 10.1111/j.1471-4159.1982.tb07932.x. [DOI] [PubMed] [Google Scholar]
- Burlina AP, Corazza A, Ferrari V, Erhard P, Kunnecke B, Seelig J, Burlina AB. Detection of increased urinary N-acetylaspartylglutamate in Canavan disease [letter] Eur J Pediatr. 1994;153:538–539. doi: 10.1007/BF01957015. [DOI] [PubMed] [Google Scholar]
- Burlina AP, Ferrari V, Burlina AB, Ermani M, Boespflug-Tanguy O, Bertini E. N-acetylaspartylglutamate (NAAG) in pelizaeusmerzbacher disease. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006a. pp. 353–359. [Google Scholar]
- Burlina AP, Ferrari V, Facci L, Skaper SD, Burlina AB. Mast cells contain large quantities of secretagogue-sensitive N-acetylaspartate. J Neurochem. 1997;69:1314–1317. doi: 10.1046/j.1471-4159.1997.69031314.x. [DOI] [PubMed] [Google Scholar]
- Burlina AP, Schmitt B, Engelke U, Wevers RA, Burlina AB, Boltshauser E. Hypoacetylaspartia: clinical and biochemical follow-up of a patient. Adv Exp Med Biol. 2006b;576:283–287. doi: 10.1007/0-387-30172-0_20. [DOI] [PubMed] [Google Scholar]
- Burri R, Bigler P, Straehl P, Posse S, Colombo JP, Herschkowitz N. Brain development: 1H magnetic resonance spectroscopy of rat brain extracts compared with chromatographic methods. Neurochem Res. 1990;15:1009–1016. doi: 10.1007/BF00965747. [DOI] [PubMed] [Google Scholar]
- Burri R, Steffen C, Herschkowitz N. N-acetyl-L-aspartate is a major source of acetyl groups for lipid synthesis during rat brain development. Dev Neurosci. 1991;13:403–412. doi: 10.1159/000112191. [DOI] [PubMed] [Google Scholar]
- Bustillo J, Barrow R, Paz R, Tang J, Seraji-Bozorgzad N, Moore GJ, Bolognani F, Lauriello J, Perrone-Bizzozero N, Galloway MP. Long-Term Treatment of Rats with Haloperidol: Lack of an Effect on Brain N-Acetyl Aspartate Levels. Neuropsychopharmacology. 2005 doi: 10.1038/sj.npp.1300874. [DOI] [PubMed] [Google Scholar]
- Bustillo J, Wolff C, Gutierrez A, Dettmer TS, Cooper TB, Allan A, Lauriello J, Valenzuela CF. Treatment of rats with antipsychotic drugs: lack of an effect on brain N-acetyl aspartate levels. Schizophr Res. 2004;66:31–39. doi: 10.1016/s0920-9964(02)00528-5. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Bertolino A, Egan MF, Mattay VS, Langheim FJ, Weinberger DR. Selective relationship between prefrontal N-acetylaspartate measures and negative symptoms in schizophrenia. Am J Psychiatry. 2000a;157:1646–1651. doi: 10.1176/appi.ajp.157.10.1646. [DOI] [PubMed] [Google Scholar]
- Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, Goldberg TE, Weinberger DR. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex. 2000b;10:1078–1092. doi: 10.1093/cercor/10.11.1078. [DOI] [PubMed] [Google Scholar]
- Canavan MM. Schilder’s encephalitis perioxalis diffusa. Neurology. 1931;15:299–308. [Google Scholar]
- Cangro CB, Namboodiri MA, Sklar LA, Corigliano-Murphy A, Neale JH. Immunohistochemistry and biosynthesis of N-acetylaspartylglutamate in spinal sensory ganglia. J Neurochem. 1987;49:1579–1588. doi: 10.1111/j.1471-4159.1987.tb01030.x. [DOI] [PubMed] [Google Scholar]
- Cappellini M, Rapisardi G, Cioni ML, Fonda C. Acute hypoxic encephalopathy in the full-term newborn: correlation between Magnetic Resonance Spectroscopy and neurological evaluation at short and long term. Radiol Med (Torino) 2002;104:332–340. [PubMed] [Google Scholar]
- Caramanos Z, Narayanan S, Arnold DL. 1H-MRS quantification of tNA and tCr in patients with multiple sclerosis: a meta-analytic review. Brain. 2005;128:2483–2506. doi: 10.1093/brain/awh640. [DOI] [PubMed] [Google Scholar]
- Cartmell J, Schoepp DD. Regulation of neurotransmitter release by metabotropic glutamate receptors. J Neurochem. 2000;75:889–907. doi: 10.1046/j.1471-4159.2000.0750889.x. [DOI] [PubMed] [Google Scholar]
- Cassidy M, Neale JH. Localization and transport of N-acetylaspartylglutamate in cells of whole murine brain in primary culture. J Neurochem. 1993a;60:1631–1638. doi: 10.1111/j.1471-4159.1993.tb13385.x. [DOI] [PubMed] [Google Scholar]
- Cassidy M, Neale JH. N-acetylaspartylglutamate catabolism is achieved by an enzyme on the cell surface of neurons and glia. Neuropeptides. 1993b;24:271–278. doi: 10.1016/0143-4179(93)90015-3. [DOI] [PubMed] [Google Scholar]
- Chakraborty G, Mekala P, Yahya D, Wu G, Ledeen RW. Intraneuronal N-acetylaspartate supplies acetyl groups for myelin lipid synthesis: evidence for myelin-associated aspartoacylase. J Neurochem. 2001;78:736–745. doi: 10.1046/j.1471-4159.2001.00456.x. [DOI] [PubMed] [Google Scholar]
- Chang L, Ernst T, Tornatore C, Aronow H, Melchor R, Walot I, Singer E, Cornford M. Metabolite abnormalities in progressive multifocal leukoencephalopathy by proton magnetic resonance spectroscopy. Neurology. 1997;48:836–845. doi: 10.1212/wnl.48.4.836. [DOI] [PubMed] [Google Scholar]
- Chevance LG, Etievant M. [A peptide (a magnesium salt of N-acetyl (alpha, beta)-aspartyl-glutamic acid), Demonstration of the protection against local cellular destruction induced by in situ complement activation] J Pharmacol. 1986;17:676–685. [PubMed] [Google Scholar]
- Chih CP, Roberts EL., Jr Energy substrates for neurons during neural activity: a critical review of the astrocyte-neuron lactate shuttle hypothesis. J Cereb Blood Flow Metab. 2003;23:1263–1281. doi: 10.1097/01.WCB.0000081369.51727.6F. [DOI] [PubMed] [Google Scholar]
- Chinn RJ, Wilkinson ID, Hall-Craggs MA, Paley MN, Miller RF, Kendall BE, Newman SP, Harrison MJ. Toxoplasmosis and primary central nervous system lymphoma in HIV infection: diagnosis with MR spectroscopy. Radiology. 1995;197:649–654. doi: 10.1148/radiology.197.3.7480733. [DOI] [PubMed] [Google Scholar]
- Choi IY, Gruetter R. In vivo 13C NMR assessment of brain glycogen concentration and turnover in the awake rat. Neurochem Int. 2003;43:317–322. doi: 10.1016/s0197-0186(03)00018-4. [DOI] [PubMed] [Google Scholar]
- Choi IY, Gruetter R. Dynamic or inert metabolism? Turnover of N-acetyl aspartate and glutathione from D-[1-13C]glucose in the rat brain in vivo. J Neurochem. 2004;91:778–787. doi: 10.1111/j.1471-4159.2004.02716.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang MT, Lin WC, Tsai HY, Liu GC, Hu SW, Chiang IC. 3-T proton magnetic resonance spectroscopy of central neurocytoma: 3 case reports and review of the literature. J Comput Assist Tomogr. 2005;29:683–688. doi: 10.1097/01.rct.0000171240.95430.29. [DOI] [PubMed] [Google Scholar]
- Clark JB. N-acetyl aspartate: a marker for neuronal loss or mitochondrial dysfunction. Dev Neurosci. 1998;20:271–276. doi: 10.1159/000017321. [DOI] [PubMed] [Google Scholar]
- Clark JF, Doepke A, Filosa JA, Wardle RL, Lu A, Meeker TJ, Pyne-Geithman GJ. N-Acetylaspartate as a reservoir for glutamate. Med Hypotheses. 2006 doi: 10.1016/j.mehy.2006.02.047. [DOI] [PubMed] [Google Scholar]
- Clarke DD, Greenfield S, Dicker E, Tirri LJ, Ronan EJ. A relationship of N-acetylaspartate biosynthesis to neuronal protein synthesis. J Neurochem. 1975;24:479–485. doi: 10.1111/j.1471-4159.1975.tb07665.x. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Dupree JL, Popko B. Demyelination and altered expression of myelin-associated glycoprotein isoforms in the central nervous system of galactolipid-deficient mice. J Neurosci Res. 1998a;54:613–622. doi: 10.1002/(SICI)1097-4547(19981201)54:5<613::AID-JNR6>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Coetzee T, Suzuki K, Popko B. New perspectives on the function of myelin galactolipids. Trends Neurosci. 1998b;21:126–130. doi: 10.1016/s0166-2236(97)01178-8. [DOI] [PubMed] [Google Scholar]
- Coon AL, rias-Mendoza F, Colby GP, Cruz-Lobo J, Mocco J, Mack WJ, Komotar RJ, Brown TR, Connolly ES., Jr Correlation of cerebral metabolites with functional outcome in experimental primate stroke using in vivo 1H-magnetic resonance spectroscopy. AJNR Am J Neuroradiol. 2006;27:1053–1058. [PMC free article] [PubMed] [Google Scholar]
- Cooper AJ. Role of glutamine in cerebral nitrogen metabolism and ammonia neurotoxicity. Ment Retard Dev Disabil Res Rev. 2001;7:280–286. doi: 10.1002/mrdd.1039. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, McDonald JM, Gelbard AS, Gledhill RF, Duffy TE. The metabolic fate of 13N-labeled ammonia in rat brain. J Biol Chem. 1979;254:4982–4992. [PubMed] [Google Scholar]
- Criste GA, Trapp BD. N-acetyl-L-aspartate in multiple sclerosis. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 199–214. [Google Scholar]
- D’Adamo AF, Jr, Gidez LI, Yatsu FM. Acetyl transport mechanisms. Involvement of N-acetyl aspartic acid in de novo fatty acid biosynthesis in the developing rat brain. Exp Brain Res. 1968;5:267–273. doi: 10.1007/BF00235902. [DOI] [PubMed] [Google Scholar]
- D’Adamo AF, Gidez LI, Yatsu FM. Acetyl transport mechanisms. Involvement of N-acetyl aspartic acid in de novo fatty acid biosynthesis in the developing rat brain. Exp Brain Res. 1968;5:267–273. doi: 10.1007/BF00235902. [DOI] [PubMed] [Google Scholar]
- D’Adamo AF, Jr, Peisach J, Manner G, Weiler CT. N-acetyl-aspartate amidohydrolase: purification and properties. J Neurochem. 1977;28:739–744. doi: 10.1111/j.1471-4159.1977.tb10621.x. [DOI] [PubMed] [Google Scholar]
- D’Adamo AF, Jr, Smith JC, Woiler C. The occurrence of N-acetylaspartate amidohydrolase (aminoacylase II) in the developing rat. J Neurochem. 1973;20:1275–1278. doi: 10.1111/j.1471-4159.1973.tb00097.x. [DOI] [PubMed] [Google Scholar]
- D’Adamo AF, Jr, Yatsu FM. Acetate metabolism in the nervous system. N-acetyl-L-aspartic acid and the biosynthesis of brain lipids. J Neurochem. 1966;13:961–965. doi: 10.1111/j.1471-4159.1966.tb10292.x. [DOI] [PubMed] [Google Scholar]
- Danielsen ER, Ross B. Magnetic Resonance Spectroscopy Diagnosis of Neurological Diseases. Marcel Dekker; New York: 1999. [Google Scholar]
- Dautry C, Vaufrey F, Brouillet E, Bizat N, Henry PG, Conde F, Bloch G, Hantraye P. Early N-acetylaspartate depletion is a marker of neuronal dysfunction in rats and primates chronically treated with the mitochondrial toxin 3-nitropropionic acid. J Cereb Blood Flow Metab. 2000;20:789–799. doi: 10.1097/00004647-200005000-00005. [DOI] [PubMed] [Google Scholar]
- Davie CA, Hawkins CP, Barker GJ, Brennan A, Tofts PS, Miller DH, McDonald WI. Serial proton magnetic resonance spectroscopy in acute multiple sclerosis lesions. Brain. 1994;117:49–58. doi: 10.1093/brain/117.1.49. [DOI] [PubMed] [Google Scholar]
- Davies SE, Gotoh M, Richards DA, Obrenovitch TP. Hypoosmolarity induces an increase of extracellular N-acetylaspartate concentration in the rat striatum. Neurochem Res. 1998;23:1021–1025. doi: 10.1023/a:1020778832745. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Matthews PM, Arnold DL. Reversible decreases in N-acetylaspartate after acute brain injury. Magn Reson Med. 1995;34:721–727. doi: 10.1002/mrm.1910340511. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Narayanan S, Francis GS, Arnaoutelis R, Tartaglia MC, Antel JP, Matthews PM, Arnold DL. Evidence of axonal damage in the early stages of multiple sclerosis and its relevance to disability. Arch Neurol. 2001;58:65–70. doi: 10.1001/archneur.58.1.65. [DOI] [PubMed] [Google Scholar]
- De Stefano N, Narayanan S, Francis SJ, Smith S, Mortilla M, Tartaglia MC, Bartolozzi ML, Guidi L, Federico A, Arnold DL. Diffuse axonal and tissue injury in patients with multiple sclerosis with low cerebral lesion load and no disability. Arch Neurol. 2002;59:1565–1571. doi: 10.1001/archneur.59.10.1565. [DOI] [PubMed] [Google Scholar]
- Dedeoglu A, Choi JK, Cormier K, Kowall NW, Jenkins BG. Magnetic resonance spectroscopic analysis of Alzheimer’s disease mouse brain that express mutant human APP shows altered neurochemical profile. Brain Res. 2004;1012:60–65. doi: 10.1016/j.brainres.2004.02.079. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Eliaz Y, Feiwell R, Schuff N. Increased thalamic N-acetylaspartate in male patients with familial bipolar I disorder. Psychiatry Res. 2001;106:35–45. doi: 10.1016/s0925-4927(00)00083-4. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Johnson C, Eliaz Y, Schuff N. Reduced concentrations of thalamic N-acetylaspartate in male patients with schizophrenia. Am J Psychiatry. 2000a;157:644–647. doi: 10.1176/appi.ajp.157.4.644. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Johnson C, Pegues M. Proton magnetic resonance spectroscopy of the human brain in schizophrenia. Rev Neurosci. 2000b;11:147–158. doi: 10.1515/revneuro.2000.11.2-3.147. [DOI] [PubMed] [Google Scholar]
- Deicken RF, Zhou L, Schuff N, Weiner MW. Proton magnetic resonance spectroscopy of the anterior cingulate region in schizophrenia. Schizophr Res. 1997;27:65–71. doi: 10.1016/S0920-9964(97)00082-0. [DOI] [PubMed] [Google Scholar]
- Demougeot C, Bertrand N, Prigent-Tessier A, Garnier P, Mossiat C, Giroud M, Marie C, Beley A. Reversible loss of N-acetyl-aspartate in rats subjected to long-term focal cerebral ischemia. J Cereb Blood Flow Metab. 2003;23:482–489. doi: 10.1097/01.WCB.0000050066.57184.60. [DOI] [PubMed] [Google Scholar]
- Demougeot C, Garnier P, Mossiat C, Bertrand N, Giroud M, Beley A, Marie C. N-Acetylaspartate, a marker of both cellular dysfunction and neuronal loss: its relevance to studies of acute brain injury. J Neurochem. 2001;77:408–415. doi: 10.1046/j.1471-4159.2001.00285.x. [DOI] [PubMed] [Google Scholar]
- Demougeot C, Marie C, Giroud M, Beley A. N-acetylaspartate: a literature review of animal research on brain ischaemia. J Neurochem. 2004;90:776–783. doi: 10.1111/j.1471-4159.2004.02583.x. [DOI] [PubMed] [Google Scholar]
- Di Costanzo A, Trojsi F, Tosetti M, Giannatempo GM, Nemore F, Piccirillo M, Bonavita S, Tedeschi G, Scarabino T. High-field proton MRS of human brain. Eur J Radiol. 2003;48:146–153. doi: 10.1016/j.ejrad.2003.08.009. [DOI] [PubMed] [Google Scholar]
- Divry P, Mathieu M. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease [letter] Am J Med Genet. 1989;32:550–551. doi: 10.1002/ajmg.1320320425. [DOI] [PubMed] [Google Scholar]
- Drobyshevsky A, Song SK, Gamkrelidze G, Wyrwicz AM, Derrick M, Meng F, Li L, Ji X, Trommer B, Beardsley DJ, Luo NL, Back SA, Tan S. Developmental changes in diffusion anisotropy coincide with immature oligodendrocyte progression and maturation of compound action potential. J Neurosci. 2005;25:5988–5997. doi: 10.1523/JNEUROSCI.4983-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan ID. The PLP mutants from mouse to man. J Neurol Sci. 2005;228:204–205. doi: 10.1016/j.jns.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Coetzee T, Suzuki K, Popko B. Myelin abnormalities in mice deficient in galactocerebroside and sulfatide. J Neurocytol. 1998;27:649–659. doi: 10.1023/a:1006908013972. [DOI] [PubMed] [Google Scholar]
- Dupree JL, Popko B. Genetic dissection of myelin galactolipid function. J Neurocytol. 1999;28:271–279. doi: 10.1023/a:1007049310758. [DOI] [PubMed] [Google Scholar]
- Egan MF, Straub RE, Goldberg TE, Yakub I, Callicott JH, Hariri AR, Mattay VS, Bertolino A, Hyde TM, Shannon-Weickert C, Akil M, Crook J, Vakkalanka RK, Balkissoon R, Gibbs RA, Kleinman JE, Weinberger DR. Variation in GRM3 affects cognition, prefrontal glutamate, and risk for schizophrenia. Proc Natl Acad Sci U S A. 2004;101:12604–12609. doi: 10.1073/pnas.0405077101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ende G, Braus DF, Walter S, Weber-Fahr W, Henn FA. Multiregional 1H-MRSI of the hippocampus, thalamus, and basal ganglia in schizophrenia. Eur Arch Psychiatry Clin Neurosci. 2003;253:9–15. doi: 10.1007/s00406-003-0398-5. [DOI] [PubMed] [Google Scholar]
- Ende G, Hubrich P, Walter S, Weber-Fahr W, Kammerer N, Braus DF, Henn FA. Further evidence for altered cerebellar neuronal integrity in schizophrenia. Am J Psychiatry. 2005;162:790–792. doi: 10.1176/appi.ajp.162.4.790. [DOI] [PubMed] [Google Scholar]
- Erecinska M, Zaleska MM, Nissim I, Nelson D, Dagani F, Yudkoff M. Glucose and synaptosomal glutamate metabolism: studies with [15N]glutamate. J Neurochem. 1988;51:892–902. doi: 10.1111/j.1471-4159.1988.tb01826.x. [DOI] [PubMed] [Google Scholar]
- Estevez AY, O’Regan MH, Song D, Phillis JW. Hyposmotically induced amino acid release from the rat cerebral cortex: role of phospholipases and protein kinases. Brain Res. 1999;844:1–9. doi: 10.1016/s0006-8993(99)01801-6. [DOI] [PubMed] [Google Scholar]
- Fan G. Comments and controversies: magnetic resonance spectroscopy and gliomas. Cancer Imaging. 2006;6:113–115. doi: 10.1102/1470-7330.2006.0018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan TW, Higashi RM, Lane AN, Jardetzky O. Combined use of 1H-NMR and GC-MS for metabolite monitoring and in vivo 1H-NMR assignments. Biochim Biophys Acta. 1986;882:154–167. doi: 10.1016/0304-4165(86)90150-9. [DOI] [PubMed] [Google Scholar]
- Faull KF, Rafie R, Pascoe N, Marsh L, Pfefferbaum A. N-acetylaspartic acid (NAA) and N-acetylaspartylglutamic acid (NAAG) in human ventricular, subarachnoid, and lumbar cerebrospinal fluid. Neurochem Res. 1999;24:1249–1261. doi: 10.1023/a:1020973023059. [DOI] [PubMed] [Google Scholar]
- Felipo V, Butterworth RF. Neurobiology of ammonia. Prog Neurobiol. 2002;67:259–279. doi: 10.1016/s0301-0082(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Fenstermacher MJ, Narayana PA. Serial proton magnetic resonance spectroscopy of ischemic brain injury in humans. Invest Radiol. 1990;25:1034–1039. doi: 10.1097/00004424-199009000-00016. [DOI] [PubMed] [Google Scholar]
- Ferrante RJ, Andreassen OA, Jenkins BG, Dedeoglu A, Kuemmerle S, Kubilus JK, Kaddurah-Daouk R, Hersch SM, Beal MF. Neuroprotective effects of creatine in a transgenic mouse model of Huntington’s disease. J Neurosci. 2000;20:4389–4397. doi: 10.1523/JNEUROSCI.20-12-04389.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillenz M. The role of lactate in brain metabolism. Neurochem Int. 2005;47:413–417. doi: 10.1016/j.neuint.2005.05.011. [DOI] [PubMed] [Google Scholar]
- Franke C, Brinker G, Pillekamp F, Hoehn M. Probability of metabolic tissue recovery after thrombolytic treatment of experimental stroke: a magnetic resonance spectroscopic imaging study in rat brain. J Cereb Blood Flow Metab. 2000;20:583–591. doi: 10.1097/00004647-200003000-00016. [DOI] [PubMed] [Google Scholar]
- Fu L, Matthews PM, De Stefano N, Worsley KJ, Narayanan S, Francis GS, Antel JP, Wolfson C, Arnold DL. Imaging axonal damage of normal-appearing white matter in multiple sclerosis. Brain. 1998;121 ( Pt 1):103–113. doi: 10.1093/brain/121.1.103. [DOI] [PubMed] [Google Scholar]
- Fujita N, Kemper A, Dupree J, Nakayasu H, Bartsch U, Schachner M, Maeda N, Suzuki K, Popko B. The cytoplasmic domain of the large myelin-associated glycoprotein isoform is needed for proper CNS but not peripheral nervous system myelination. J Neurosci. 1998;18:1970–1978. doi: 10.1523/JNEUROSCI.18-06-01970.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita T, Katsukawa H, Yodoya E, Wada M, Shimada A, Okada N, Yamamoto A, Ganapathy V. Transport characteristics of N-acetyl-L-aspartate in rat astrocytes: involvement of sodium-coupled high-affinity carboxylate transporter NaC3/NaDC3-mediated transport system. J Neurochem. 2005;93:706–714. doi: 10.1111/j.1471-4159.2005.03067.x. [DOI] [PubMed] [Google Scholar]
- Fulham MJ, Bizzi A, Dietz MJ, Shih HH, Raman R, Sobering GS, Frank JA, Dwyer AJ, Alger JR, Di Chiro G. Mapping of brain tumor metabolites with proton MR spectroscopic imaging: clinical relevance. Radiology. 1992;185:675–686. doi: 10.1148/radiology.185.3.1438744. [DOI] [PubMed] [Google Scholar]
- Ganapathy V, Fujita T. Identity of the high-affinity sodium/carboxylate cotransporter NaC3 as the N-acetyl-L-aspartate transporter. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 67–76. [Google Scholar]
- Garbern JY. Pelizaeus-Merzbacher disease: pathogenic mechanisms and insights into the roles of proteolipid protein 1 in the nervous system. J Neurol Sci. 2005;228:201–203. doi: 10.1016/j.jns.2004.10.010. [DOI] [PubMed] [Google Scholar]
- Gasparovic C, Arfai N, Smid N, Feeney DM. Decrease and recovery of N-acetylaspartate/creatine in rat brain remote from focal injury. J Neurotrauma. 2001;18:241–246. doi: 10.1089/08977150151070856. [DOI] [PubMed] [Google Scholar]
- Gehl LM, Saab OH, Bzdega T, Wroblewska B, Neale JH. Biosynthesis of NAAG by an enzyme-mediated process in rat central nervous system neurons and glia. J Neurochem. 2004;90:989–997. doi: 10.1111/j.1471-4159.2004.02578.x. [DOI] [PubMed] [Google Scholar]
- George RL, Huang W, Naggar HA, Smith SB, Ganapathy V. Transport of N-acetylaspartate via murine sodium/dicarboxylate cotransporter NaDC3 and expression of this transporter and aspartoacylase II in ocular tissues in mouse. Biochim Biophys Acta. 2004;1690:63–69. doi: 10.1016/j.bbadis.2004.05.009. [DOI] [PubMed] [Google Scholar]
- Gideon P, Sperling B, rlien-Soborg P, Olsen TS, Henriksen O. Long-term follow-up of cerebral infarction patients with proton magnetic resonance spectroscopy. Stroke. 1994;25:967–973. doi: 10.1161/01.str.25.5.967. [DOI] [PubMed] [Google Scholar]
- Gillard JH, Waldman AD, Barker PB. Clinical MR Neuroimaging (Diffusion, Perfusion and Spectroscopy) Cambridge University Press; Cambridge, UK: 2005. [Google Scholar]
- Girard N, Fogliarini C, Viola A, Confort-Gouny S, Fur YL, Viout P, Chapon F, Levrier O, Cozzone P. MRS of normal and impaired fetal brain development. Eur J Radiol. 2005 doi: 10.1016/j.ejrad.2005.11.021. [DOI] [PubMed] [Google Scholar]
- Godbolt AK, Waldman AD, MacManus DG, Schott JM, Frost C, Cipolotti L, Fox NC, Rossor MN. MRS shows abnormalities before symptoms in familial Alzheimer disease. Neurology. 2006;66:718–722. doi: 10.1212/01.wnl.0000201237.05869.df. [DOI] [PubMed] [Google Scholar]
- Goldberg RP, Brunengraber H. Contributions of cytosolic and mitochondrial acetyl-CoA syntheses to the activation of lipogenic acetate in rat liver. Adv Exp Med Biol. 1980;132:413–418. doi: 10.1007/978-1-4757-1419-7_41. [DOI] [PubMed] [Google Scholar]
- Goldstein FB. Biosynthesis of N-acetyl-L-aspartic acid. Biochim Biophys Acta. 1959;33:583–584. doi: 10.1016/0006-3002(59)90161-1. [DOI] [PubMed] [Google Scholar]
- Goldstein FB. The enzymatic synthesis of N-acetyl-L-aspartic acid by subcellular preparations of rat brain. J Biol Chem. 1969;244:4257–4260. [PubMed] [Google Scholar]
- Gotoh M, Davies SE, Obrenovitch TP. Brain tissue acidosis: effects on the extracellular concentration of N-acetylaspartate. J Neurochem. 1997;69:655–661. doi: 10.1046/j.1471-4159.1997.69020655.x. [DOI] [PubMed] [Google Scholar]
- Gow A, Southwood CM, Lazzarini RA. Disrupted proteolipid protein trafficking results in oligodendrocyte apoptosis in an animal model of Pelizaeus-Merzbacher disease. J Cell Biol. 1998;140:925–934. doi: 10.1083/jcb.140.4.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham GD, Blamire AM, Rothman DL, Brass LM, Fayad PB, Petroff OA, Prichard JW. Early temporal variation of cerebral metabolites after human stroke. A proton magnetic resonance spectroscopy study. Stroke. 1993;24:1891–1896. doi: 10.1161/01.str.24.12.1891. [DOI] [PubMed] [Google Scholar]
- Graham SH, Meyerhoff DJ, Bayne L, Sharp FR, Weiner MW. Magnetic resonance spectroscopy of N-acetylaspartate in hypoxic- ischemic encephalopathy. Ann Neurol. 1994;35:490–494. doi: 10.1002/ana.410350420. [DOI] [PubMed] [Google Scholar]
- Gujar SK, Maheshwari S, Bjorkman-Burtscher I, Sundgren PC. Magnetic resonance spectroscopy. J Neuroophthalmol. 2005;25:217–226. doi: 10.1097/01.wno.0000177307.21081.81. [DOI] [PubMed] [Google Scholar]
- Gullans SR, Verbalis JG. Control of brain volume during hyperosmolar and hypoosmolar conditions. Annu Rev Med. 1993;44:289–301. doi: 10.1146/annurev.me.44.020193.001445. [DOI] [PubMed] [Google Scholar]
- Hagenfeldt L, Bollgren I, Venizelos N. N-acetylaspartic aciduria due to aspartoacylase deficiency- a new aetiology of childhood leukodystrophy. J Inherit Metab Dis. 1987;10:135–141. doi: 10.1007/BF01800038. [DOI] [PubMed] [Google Scholar]
- Hamosh A, Scott AF, Amberger JS, Bocchini CA, McKusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514–D517. doi: 10.1093/nar/gki033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanefeld FA, Brockmann K, Pouwels PJ, Wilken B, Frahm J, Dechent P. Quantitative proton MRS of Pelizaeus-Merzbacher disease: evidence of dys- and hypomyelination. Neurology. 2005;65:701–706. doi: 10.1212/01.wnl.0000174642.32187.20. [DOI] [PubMed] [Google Scholar]
- Harada M, Tanouchi M, Miyoshi H, Nishitani H, Kannuki S. Brain abscess observed by localized proton magnetic resonance spectroscopy. Magn Reson Imaging. 1994;12:1269–1274. doi: 10.1016/0730-725x(94)90092-6. [DOI] [PubMed] [Google Scholar]
- Harris K, Lin A, Bhattacharya P, Tran T, Wong W, Ross BD. Regulation of NAA-synthesis in the human brain in vivo: Canavan’s disease, Alzheimer’s disease and schizophrenia. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 263–273. [Google Scholar]
- Harte MK, Bachus SB, Reynolds GP. Increased N-acetylaspartate in rat striatum following long-term administration of haloperidol. Schizophr Res. 2005;75:303–308. doi: 10.1016/j.schres.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Haseler LJ, Arcinue E, Danielsen ER, Bluml S, Ross BD. Evidence from proton magnetic resonance spectroscopy for a metabolic cascade of neuronal damage in shaken baby syndrome. Pediatrics. 1997;99:4–14. doi: 10.1542/peds.99.1.4. [DOI] [PubMed] [Google Scholar]
- Hassel B, Sonnewald U, Fonnum F. Glial-neuronal interactions as studied by cerebral metabolism of [2-13C]acetate and [1-13C]glucose: an ex vivo 13C NMR spectroscopic study. J Neurochem. 1995;64:2773–2782. doi: 10.1046/j.1471-4159.1995.64062773.x. [DOI] [PubMed] [Google Scholar]
- Heilig CW, Stromski ME, Blumenfeld JD, Lee JP, Gullans SR. Characterization of the major brain osmolytes that accumulate in salt-loaded rats. Am J Physiol. 1989;257:F1108–F1116. doi: 10.1152/ajprenal.1989.257.6.F1108. [DOI] [PubMed] [Google Scholar]
- Hershfield J, Madhavarao CN, Moffett JR, Benjamins JA, Garbern JY, Namboodiri MA. Aspartoacylase is a regulated nuclear-cytoplasmic enzyme. FASEB J. 2006 doi: 10.1096/fj.05-5358fje. in press. [DOI] [PubMed] [Google Scholar]
- Higuchi T, Fernandez EJ, Maudsley AA, Shimizu H, Weiner MW, Weinstein PR. Mapping of lactate and N-acetyl-L-aspartate predicts infarction during acute focal ischemia: in vivo 1H magnetic resonance spectroscopy in rats. Neurosurgery. 1996;38:121–129. doi: 10.1097/00006123-199601000-00030. [DOI] [PubMed] [Google Scholar]
- Hoehn M, Nicolay K, Franke C, van der SB. Application of magnetic resonance to animal models of cerebral ischemia. J Magn Reson Imaging. 2001;14:491–509. doi: 10.1002/jmri.1213. [DOI] [PubMed] [Google Scholar]
- Hogan GR, Richardson EP., Jr Spongy degeneration of the nervous system (Canavan’s disease) Pediatrics. 1965;35:284–294. [PubMed] [Google Scholar]
- Hou J, Major EO. Progressive multifocal leukoencephalopathy: JC virus induced demyelination in the immune compromised host. J Neurovirol 6 Suppl. 2000;2:S98–S100. [PubMed] [Google Scholar]
- Huang W, Wang H, Kekuda R, Fei YJ, Friedrich A, Wang J, Conway SJ, Cameron RS, Leibach FH, Ganapathy V. Transport of N-acetylaspartate by the Na(+)-dependent high-affinity dicarboxylate transporter NaDC3 and its relevance to the expression of the transporter in the brain. J Pharmacol Exp Ther. 2000;295:392–403. [PubMed] [Google Scholar]
- Jack CR, Jr, Garwood M, Wengenack TM, Borowski B, Curran GL, Lin J, Adriany G, Grohn OH, Grimm R, Poduslo JF. In vivo visualization of Alzheimer’s amyloid plaques by magnetic resonance imaging in transgenic mice without a contrast agent. Magn Reson Med. 2004;52:1263–1271. doi: 10.1002/mrm.20266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr, Wengenack TM, Reyes DA, Garwood M, Curran GL, Borowski BJ, Lin J, Preboske GM, Holasek SS, Adriany G, Poduslo JF. In vivo magnetic resonance microimaging of individual amyloid plaques in Alzheimer’s transgenic mice. J Neurosci. 2005;25:10041–10048. doi: 10.1523/JNEUROSCI.2588-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagannathan NR, Desai NG, Raghunathan P. Brain metabolite changes in alcoholism: an in vivo proton magnetic resonance spectroscopy (MRS) study. Magn Reson Imaging. 1996;14:553–557. doi: 10.1016/0730-725x(96)00048-3. [DOI] [PubMed] [Google Scholar]
- Jakary A, Vinogradov S, Feiwell R, Deicken RF. N-acetylaspartate reductions in the mediodorsal and anterior thalamus in men with schizophrenia verified by tissue volume corrected proton MRSI. Schizophr Res. 2005;76:173–185. doi: 10.1016/j.schres.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, Li MX, Ramos M, Marmol P, Horiuchi M, Shimotsu K, Nakagawa S, Okubo A, Sameshima M, Isashiki Y, Del Arco A, Kobayashi K, Satrustegui J, Saheki T. Reduced N-acetylaspartate levels in mice lacking Aralar, a brain-and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem. 2005;280:31333–31339. doi: 10.1074/jbc.M505286200. [DOI] [PubMed] [Google Scholar]
- Jambou D, Lapalus P. Effect of N-acetyl-aspartyl-glutamate (Naaga) on in-vitro leukotriene synthesis by macrophage cell line P388D1. Int J Tissue React. 1990;12:273–280. [PubMed] [Google Scholar]
- Janson C, McPhee S, Bilaniuk L, Haselgrove J, Testaiuti M, Freese A, Wang DJ, Shera D, Hurh P, Rupin J, Saslow E, Goldfarb O, Goldberg M, Larijani G, Sharrar W, Liouterman L, Camp A, Kolodny E, Samulski J, Leone P. Clinical protocol. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum Gene Ther. 2002;13:1391–1412. doi: 10.1089/104303402760128612. [DOI] [PubMed] [Google Scholar]
- Jayasundar R, Shah T, Vaishya S, Singh VP, Sarkar C. In vivo and in vitro MR spectroscopic profile of central neurocytomas. J Magn Reson Imaging. 2003;17:256–260. doi: 10.1002/jmri.10252. [DOI] [PubMed] [Google Scholar]
- Jenkins BG, Klivenyi P, Kustermann E, Andreassen OA, Ferrante RJ, Rosen BR, Beal MF. Nonlinear decrease over time in N-acetyl aspartate levels in the absence of neuronal loss and increases in glutamine and glucose in transgenic Huntington’s disease mice. J Neurochem. 2000;74:2108–2119. doi: 10.1046/j.1471-4159.2000.0742108.x. [DOI] [PubMed] [Google Scholar]
- Jessen F, Scherk H, Traber F, Theyson S, Berning J, Tepest R, Falkai P, Schild HH, Maier W, Wagner M, Block W. Proton magnetic resonance spectroscopy in subjects at risk for schizophrenia. Schizophr Res. 2006 doi: 10.1016/j.schres.2006.06.011. [DOI] [PubMed] [Google Scholar]
- Jiang H, Duchala CS, Awatramani R, Shumas S, Carlock L, Kamholz J, Garbern J, Scherer SS, Shy ME, Macklin WB. Proteolipid protein mRNA stability is regulated by axonal contact in the rodent peripheral nervous system. J Neurobiol. 2000;44:7–19. doi: 10.1002/1097-4695(200007)44:1<7::aid-neu2>3.0.co;2-a. [DOI] [PubMed] [Google Scholar]
- Jones CT, Ashton IK. Lipid biosynthesis in liver slices of the foetal guinea pig. Biochem J. 1976;154:149–158. doi: 10.1042/bj1540149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung RE, Haier RJ, Yeo RA, Rowland LM, Petropoulos H, Levine AS, Sibbitt WL, Brooks WM. Sex differences in N-acetylaspartate correlates of general intelligence: an 1H-MRS study of normal human brain. Neuroimage. 2005;26:965–972. doi: 10.1016/j.neuroimage.2005.02.039. [DOI] [PubMed] [Google Scholar]
- Kadota O, Kohno K, Ohue S, Kumon Y, Sakaki S, Kikuchi K, Miki H. Discrimination of brain abscess and cystic tumor by in vivo proton magnetic resonance spectroscopy. Neurol Med Chir (Tokyo) 2001;41:121–126. doi: 10.2176/nmc.41.121. [DOI] [PubMed] [Google Scholar]
- Kalra S, Arnold DL. ALS surrogate markers MRS Amyotroph Lateral Scler Other. Motor Neuron Disord. 2004;5(Suppl 1):111–114. doi: 10.1080/17434470410019861. [DOI] [PubMed] [Google Scholar]
- Kalra S, Arnold DL. Magnetic resonance spectroscopy for monitoring neuronal integrity in amyotrophic lateral sclerosis. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 275–282. [DOI] [PubMed] [Google Scholar]
- Kalra S, Cashman NR, Genge A, Arnold DL. Recovery of N-acetylaspartate in corticomotor neurons of patients with ALS after riluzole therapy. Neuroreport. 1998;9:1757–1761. doi: 10.1097/00001756-199806010-00016. [DOI] [PubMed] [Google Scholar]
- Kantarci K, Jack CR., Jr Neuroimaging in Alzheimer disease: an evidence-based review. Neuroimaging Clin N Am. 2003;13:197–209. doi: 10.1016/s1052-5149(03)00025-x. [DOI] [PubMed] [Google Scholar]
- Kantarci K, Jack CR, Jr, Xu YC, Campeau NG, O’Brien PC, Smith GE, Ivnik RJ, Boeve BF, Kokmen E, Tangalos EG, Petersen RC. Regional metabolic patterns in mild cognitive impairment and Alzheimer’s disease: A 1H MRS study. Neurology. 2000;55:210–217. doi: 10.1212/wnl.55.2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Reynolds G, Petersen RC, Boeve BF, Knopman DS, Edland SD, Smith GE, Ivnik RJ, Tangalos EG, Jack CR., Jr Proton MR spectroscopy in mild cognitive impairment and Alzheimer disease: comparison of 1.5 and 3 T. AJNR Am J Neuroradiol. 2003;24:843–849. [PMC free article] [PubMed] [Google Scholar]
- Kantarci K, Xu Y, Shiung MM, O’Brien PC, Cha RH, Smith GE, Ivnik RJ, Boeve BF, Edland SD, Kokmen E, Tangalos EG, Petersen RC, Jack CR., Jr Comparative diagnostic utility of different MR modalities in mild cognitive impairment and Alzheimer’s disease. Dement Geriatr Cogn Disord. 2002;14:198–207. doi: 10.1159/000066021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karelson G, Ziegler A, Kunnecke B, Seelig J. Feeding versus infusion: a novel approach to study the NAA metabolism in rat brain. NMR Biomed. 2003;16:413–423. doi: 10.1002/nbm.845. [DOI] [PubMed] [Google Scholar]
- Kaul R, Balamurugan K, Gao GP, Matalon R. Canavan disease: genomic organization and localization of human ASPA to 17p13-ter and conservation of the ASPA gene during evolution. Genomics. 1994a;21:364–370. doi: 10.1006/geno.1994.1278. [DOI] [PubMed] [Google Scholar]
- Kaul R, Casanova J, Johnson AB, Tang P, Matalon R. Purification, characterization, and localization of aspartoacylase from bovine brain. J Neurochem. 1991;56:129–135. doi: 10.1111/j.1471-4159.1991.tb02571.x. [DOI] [PubMed] [Google Scholar]
- Kaul R, Gao GP, Aloya M, Balamurugan K, Petrosky A, Michals K, Matalon R. Canavan disease: mutations among Jewish and non-jewish patients. Am J Hum Genet. 1994b;55:34–41. [PMC free article] [PubMed] [Google Scholar]
- Kaul R, Gao GP, Balamurugan K, Matalon R. Cloning of the human aspartoacylase cDNA and a common missense mutation in Canavan disease. Nat Genet. 1993;5:118–123. doi: 10.1038/ng1093-118. [DOI] [PubMed] [Google Scholar]
- Kaul R, Gao GP, Matalon R, Aloya M, Su Q, Jin M, Johnson AB, Schutgens RB, Clarke JT. Identification and expression of eight novel mutations among non-Jewish patients with Canavan disease. Am J Hum Genet. 1996;59:95–102. [PMC free article] [PubMed] [Google Scholar]
- Kelley RI, Stamas JN. Quantification of N-acetyl-L-aspartic acid in urine by isotope dilution gas chromatography-mass spectrometry. J Inherit Metab Dis. 1992;15:97–104. doi: 10.1007/BF01800351. [DOI] [PubMed] [Google Scholar]
- Keshavan MS, Stanley JA, Pettegrew JW. Magnetic resonance spectroscopy in schizophrenia: methodological issues and findings--part II. Biol Psychiatry. 2000;48:369–380. doi: 10.1016/s0006-3223(00)00940-9. [DOI] [PubMed] [Google Scholar]
- Khan O, Shen Y, Caon C, Bao F, Ching W, Reznar M, Buccheister A, Hu J, Latif Z, Tselis A, Lisak R. Axonal metabolic recovery and potential neuroprotective effect of glatiramer acetate in relapsing-remitting multiple sclerosis. Mult Scler. 2005;11:646–651. doi: 10.1191/1352458505ms1234oa. [DOI] [PubMed] [Google Scholar]
- Kirmani BF, Jacobowitz DM, Kallarakal AT, Namboodiri MA. Aspartoacylase is restricted primarily to myelin synthesizing cells in the CNS: therapeutic implications for Canavan disease. Brain Res Mol Brain Res. 2002;107:176–182. doi: 10.1016/s0169-328x(02)00490-4. [DOI] [PubMed] [Google Scholar]
- Kirmani BF, Jacobowitz DM, Namboodiri MA. Developmental increase of aspartoacylase in oligodendrocytes parallels CNS myelination. Brain Res Dev Brain Res. 2003;140:105–115. doi: 10.1016/s0165-3806(02)00592-8. [DOI] [PubMed] [Google Scholar]
- Kitada K, Akimitsu T, Shigematsu Y, Kondo A, Maihara T, Yokoi N, Kuramoto T, Sasa M, Serikawa T. Accumulation of N-acetyl-L-aspartate in the brain of the tremor rat, a mutant exhibiting absence-like seizure and spongiform degeneration in the central nervous system. J Neurochem. 2000;74:2512–2519. doi: 10.1046/j.1471-4159.2000.0742512.x. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Leichtlein CB, Symes CW, Serikawa T, Young D, During MJ. Restoration of aspartoacylase activity in CNS neurons does not ameliorate motor deficits and demyelination in a model of Canavan disease. Mol Ther. 2005;11:745–753. doi: 10.1016/j.ymthe.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Klugmann M, Symes CW, Klaussner BK, Leichtlein CB, Serikawa T, Young D, During MJ. Identification and distribution of aspartoacylase in the postnatal rat brain. Neuroreport. 2003;14:1837–1840. doi: 10.1097/00001756-200310060-00016. [DOI] [PubMed] [Google Scholar]
- Knizley H., Jr The enzymatic synthesis of N-acetyl-L-aspartic acid by a water-insoluble preparation of a cat brain acetone powder. J Biol Chem. 1967;242:4619–4622. [PubMed] [Google Scholar]
- Kondo A, Nagara H, Akazawa K, Tateishi J, Serikawa T, Yamada J. CNS pathology in the neurological mutant rats zitter, tremor and zitter-tremor double mutant (spontaneously epileptic rat, SER). Exaggeration of clinical and neuropathological phenotypes in SER. Brain. 1991;114 ( Pt 2):979–999. doi: 10.1093/brain/114.2.979. [DOI] [PubMed] [Google Scholar]
- Kreis R, Arcinue E, Ernst T, Shonk TK, Flores R, Ross BD. Hypoxic encephalopathy after near-drowning studied by quantitative 1H-magnetic resonance spectroscopy. J Clin Invest. 1996;97:1142–1154. doi: 10.1172/JCI118528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kvittingen EA, Guldal G, Borsting S, Skalpe IO, Stokke O, Jellum E. N-acetylaspartic aciduria in a child with a progressive cerebral atrophy. Clin Chim Acta. 1986;158:217–227. doi: 10.1016/0009-8981(86)90285-8. [DOI] [PubMed] [Google Scholar]
- Lapalus P, Moulin G, Bayer V, Fredj-Reygrobellet D, Elena PP. Effects of a new anti-allergic agent: the magnesium salt of N-acetyl-aspartyl-glutamic acid on experimental allergic inflammation of the rabbit eye. Curr Eye Res. 1986;5:517–522. doi: 10.3109/02713688608996374. [DOI] [PubMed] [Google Scholar]
- Larsson HB, Christiansen P, Jensen M, Frederiksen J, Heltberg A, Olesen J, Henriksen O. Localized in vivo proton spectroscopy in the brain of patients with multiple sclerosis. Magn Reson Med. 1991;22:23–31. doi: 10.1002/mrm.1910220104. [DOI] [PubMed] [Google Scholar]
- Leary SM, Davie CA, Parker GJ, Stevenson VL, Wang L, Barker GJ, Miller DH, Thompson AJ. 1H magnetic resonance spectroscopy of normal appearing white matter in primary progressive multiple sclerosis. J Neurol. 1999;246:1023–1026. doi: 10.1007/s004150050507. [DOI] [PubMed] [Google Scholar]
- Lebon V, Petersen KF, Cline GW, Shen J, Mason GF, Dufour S, Behar KL, Shulman GI, Rothman DL. Astroglial contribution to brain energy metabolism in humans revealed by 13C nuclear magnetic resonance spectroscopy: elucidation of the dominant pathway for neurotransmitter glutamate repletion and measurement of astrocytic oxidative metabolism. J Neurosci. 2002;22:1523–1531. doi: 10.1523/JNEUROSCI.22-05-01523.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledeen RW, Wang J, Wu G, Lu ZH, Chakraborty G, Meyenhofer M, Tyring SK, Matalon R. Physiological role of N-acetylaspartate: contribution to myelinogenesis. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 131–143. [Google Scholar]
- Lee VM, Burdett NG, Carpenter A, Hall LD, Pambakian PS, Patel S, Wood NI, James MF. Evolution of photochemically induced focal cerebral ischemia in the rat. Magnetic resonance imaging and histology. Stroke. 1996;27:2110–2118. doi: 10.1161/01.str.27.11.2110. [DOI] [PubMed] [Google Scholar]
- Lee WT, Lee CS, Pan YL, Chang C. Temporal changes of cerebral metabolites and striatal lesions in acute 3-nitropropionic acid intoxication in the rat. Magn Reson Med. 2000;44:29–34. doi: 10.1002/1522-2594(200007)44:1<29::aid-mrm6>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Leone P, Janson CG, Bilaniuk L, Wang Z, Sorgi F, Huang L, Matalon R, Kaul R, Zeng Z, Freese A, McPhee SW, Mee E, During MJ, Bilianuk L. Aspartoacylase gene transfer to the mammalian central nervous system with therapeutic implications for Canavan disease. Ann Neurol. 2000;48:27–38. doi: 10.1002/1531-8249(200007)48:1<27::aid-ana6>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Leone P, Janson CG, McPhee SJ, During MJ. Global CNS gene transfer for a childhood neurogenetic enzyme deficiency: Canavan disease. Curr Opin Mol Ther. 1999;1:487–492. [PubMed] [Google Scholar]
- Lieberman EM, Achreja M, Urazaev AK. Synthesis of N-acetylaspartyl-glutamate (NAAG) and N-acetylaspartate (NAA) in axons and glia of the crayfish medial giant nerve fiber. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 303–315. [DOI] [PubMed] [Google Scholar]
- Lin A, Ross BD, Harris K, Wong W. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx. 2005;2:197–214. doi: 10.1602/neurorx.2.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin AP, Shic F, Enriquez C, Ross BD. Reduced glutamate neurotransmission in patients with Alzheimer’s disease -- an in vivo (13)C magnetic resonance spectroscopy study. MAGMA. 2003a;16:29–42. doi: 10.1007/s10334-003-0004-x. [DOI] [PubMed] [Google Scholar]
- Lin DD, Crawford TO, Barker PB. Proton MR spectroscopy in the diagnostic evaluation of suspected mitochondrial disease. AJNR Am J Neuroradiol. 2003b;24:33–41. [PMC free article] [PubMed] [Google Scholar]
- Lu ZH, Chakraborty G, Ledeen RW, Yahya D, Wu G. N-Acetylaspartate synthase is bimodally expressed in microsomes and mitochondria of brain. Brain Res Mol Brain Res. 2004;122:71–78. doi: 10.1016/j.molbrainres.2003.12.002. [DOI] [PubMed] [Google Scholar]
- Luong A, Hannah VC, Brown MS, Goldstein JL. Molecular characterization of human acetyl-CoA synthetase, an enzyme regulated by sterol regulatory element-binding proteins. J Biol Chem. 2000;275:26458–26466. doi: 10.1074/jbc.M004160200. [DOI] [PubMed] [Google Scholar]
- Luyten PR, den Hollander JA. Observation of metabolites in the human brain by MR spectroscopy. Radiology. 1986;161:795–798. doi: 10.1148/radiology.161.3.3786735. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Arun P, Moffett JR, Szucs S, Surendran S, Matalon R, Garbern J, Hristova D, Johnson A, Jiang W, Namboodiri MA. Defective N-acetylaspartate catabolism reduces brain acetate levels and myelin lipid synthesis in Canavan’s disease. Proc Natl Acad Sci U S A. 2005;102:5221–5226. doi: 10.1073/pnas.0409184102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madhavarao CN, Chinopoulos C, Chandrasekaran K, Namboodiri MA. Characterization of the N-acetylaspartate biosynthetic enzyme from rat brain. J Neurochem. 2003;86:824–835. doi: 10.1046/j.1471-4159.2003.01905.x. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Hammer JA, Quarles RH, Namboodiri MA. A radiometric assay for aspartoacylase activity in cultured oligodendrocytes. Anal Biochem. 2002;308:314–319. doi: 10.1016/s0003-2697(02)00225-7. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Moffett JR, Moore RA, Viola RE, Namboodiri MA, Jacobowitz DM. Immunohistochemical localization of aspartoacylase in the rat central nervous system. J Comp Neurol. 2004;472:318–329. doi: 10.1002/cne.20080. [DOI] [PubMed] [Google Scholar]
- Madhavarao CN, Namboodiri AM. NAA synthesis and functional roles. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 49–66. [Google Scholar]
- Marenco S, Bertolino A, Weinberger DR. MR proton spectroscopy. In: Lawrie SM, Johnstone EC, Weinberger DR, editors. Schizophrenia: from neuroimaging to neuroscience. Oxford University Press; Oxford, UK: 2004. pp. 73–92. [Google Scholar]
- Marenco S, Bertolino A, Weinberger DR. In vivo NMR measures of NAA and the neurobiology of schizophrenia. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006a. pp. 227–240. [Google Scholar]
- Marenco S, Steele SU, Egan MF, Goldberg TE, Straub RE, Sharrief AZ, Weinberger DR. Effect of metabotropic glutamate receptor 3 genotype on N-acetylaspartate measures in the dorsolateral prefrontal cortex. Am J Psychiatry. 2006b;163:740–742. doi: 10.1176/appi.ajp.163.4.740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis RU, Barkulis SS, Geiger A. A comparison between the incorporation of 14C from glucose into N-acetyly-L-aspartic acid and aspartic acid in brain perfusion experiments. J Neurochem. 1960;5:379–382. doi: 10.1111/j.1471-4159.1960.tb13377.x. [DOI] [PubMed] [Google Scholar]
- Maria BL, Deidrick KM, Moser H, Naidu S. Leukodystrophies: pathogenesis, diagnosis, strategies, therapies, and future research directions. J Child Neurol. 2003;18:578–590. doi: 10.1177/08830738030180090401. [DOI] [PubMed] [Google Scholar]
- Martinez-Hernandez A, Bell KP, Norenberg MD. Glutamine synthetase: glial localization in brain. Science. 1977;195:1356–1358. doi: 10.1126/science.14400. [DOI] [PubMed] [Google Scholar]
- Mastorodemos V, Zaganas I, Spanaki C, Bessa M, Plaitakis A. Molecular basis of human glutamate dehydrogenase regulation under changing energy demands. J Neurosci Res. 2005;79:65–73. doi: 10.1002/jnr.20353. [DOI] [PubMed] [Google Scholar]
- Matalon R, Michals K, Kaul R. Canavan disease: from spongy degeneration to molecular analysis. J Pediatr. 1995;127:511–517. doi: 10.1016/s0022-3476(95)70105-2. [DOI] [PubMed] [Google Scholar]
- Matalon R, Michals K, Sebesta D, Deanching M, Gashkoff P, Casanova J. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with canavan disease. Am J Med Genet. 1988;29:463–471. doi: 10.1002/ajmg.1320290234. [DOI] [PubMed] [Google Scholar]
- Matalon R, Michals-Matalon K. Molecular basis of Canavan disease. Eur J Paediatr Neurol. 1998;2:69–76. doi: 10.1016/s1090-3798(98)80044-5. [DOI] [PubMed] [Google Scholar]
- Matalon R, Rady PL, Platt KA, Skinner HB, Quast MJ, Campbell GA, Matalon K, Ceci JD, Tyring SK, Nehls M, Surendran S, Wei J, Ezell EL, Szucs S. Knock-out mouse for Canavan disease: a model for gene transfer to the central nervous system. J Gene Med. 2000;2:165–175. doi: 10.1002/(SICI)1521-2254(200005/06)2:3<165::AID-JGM107>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Matalon R, Surendran S, Rady PL, Quast MJ, Campbell GA, Matalon KM, Tyring SK, Wei J, Peden CS, Ezell EL, Muzyczka N, Mandel RJ. Adeno-associated virus-mediated aspartoacylase gene transfer to the brain of knockout mouse for canavan disease. Mol Ther. 2003;7:580–587. doi: 10.1016/s1525-0016(03)00066-2. [DOI] [PubMed] [Google Scholar]
- Mathew R, Arun P, Madhavarao C, Moffett J, Namboodiri A. Progress toward acetate supplementation therapy for Canavan disease: Glyceryl triacetate administration increases acetate, but not N-acetylaspartate levels in brain. J Pharmacol Exp Ther. 2005;315:297–303. doi: 10.1124/jpet.105.087536. [DOI] [PubMed] [Google Scholar]
- Mathiesen HK, Jonsson A, Tscherning T, Hanson LG, Andresen J, Blinkenberg M, Paulson OB, Sorensen PS. Correlation of global N-acetyl aspartate with cognitive impairment in multiple sclerosis. Arch Neurol. 2006a;63:533–536. doi: 10.1001/archneur.63.4.533. [DOI] [PubMed] [Google Scholar]
- Mathiesen HK, Jonsson A, Tscherning T, Hanson LG, Andresen J, Blinkenberg M, Paulson OB, Sorensen PS. Correlation of global N-acetyl aspartate with cognitive impairment in multiple sclerosis. Arch Neurol. 2006b;63:533–536. doi: 10.1001/archneur.63.4.533. [DOI] [PubMed] [Google Scholar]
- McIntosh JC, Cooper JR. Studies on the function of N-acetyl aspartic acid in brain. J Neurochem. 1965;12:825–835. doi: 10.1111/j.1471-4159.1965.tb10267.x. [DOI] [PubMed] [Google Scholar]
- McKenna MC, Stevenson JH, Huang X, Hopkins IB. Differential distribution of the enzymes glutamate dehydrogenase and aspartate aminotransferase in cortical synaptic mitochondria contributes to metabolic compartmentation in cortical synaptic terminals. Neurochem Int. 2000;37:229–241. doi: 10.1016/s0197-0186(00)00042-5. [DOI] [PubMed] [Google Scholar]
- McPhee SW, Francis J, Janson CG, Serikawa T, Hyland K, Ong EO, Raghavan SS, Freese A, Leone P. Effects of AAV-2-mediated aspartoacylase gene transfer in the tremor rat model of Canavan disease. Brain Res Mol Brain Res. 2005;135:112–121. doi: 10.1016/j.molbrainres.2004.12.007. [DOI] [PubMed] [Google Scholar]
- Mehta V, Namboodiri MA. N-acetylaspartate as an acetyl source in the nervous system. Brain Res Mol Brain Res. 1995;31:151–157. doi: 10.1016/0169-328x(95)00044-s. [DOI] [PubMed] [Google Scholar]
- Menichella DM, Goodenough DA, Sirkowski E, Scherer SS, Paul DL. Connexins are critical for normal myelination in the CNS. J Neurosci. 2003;23:5963–5973. doi: 10.1523/JNEUROSCI.23-13-05963.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menon DK, Sargentoni J, Peden CJ, Bell JD, Cox IJ, Coutts GA, Baudouin C, Newman CG. Proton MR spectroscopy in herpes simplex encephalitis: assessment of neuronal loss. J Comput Assist Tomogr. 1990;14:449–452. doi: 10.1097/00004728-199005000-00024. [DOI] [PubMed] [Google Scholar]
- Meyerhoff DJ, Blumenfeld R, Truran D, Lindgren J, Flenniken D, Cardenas V, Chao LL, Rothlind J, Studholme C, Weiner MW. Effects of heavy drinking, binge drinking, and family history of alcoholism on regional brain metabolites. Alcohol Clin Exp Res. 2004;28:650–661. doi: 10.1097/01.ALC.0000121805.12350.CA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyerhoff DJ, MacKay S, Bachman L, Poole N, Dillon WP, Weiner MW, Fein G. Reduced brain N-acetylaspartate suggests neuronal loss in cognitively impaired human immunodeficiency virus-seropositive individuals: in vivo 1H magnetic resonance spectroscopic imaging. Neurology. 1993;43:509–515. doi: 10.1212/wnl.43.3_part_1.509. [DOI] [PubMed] [Google Scholar]
- Miadonna A, Milazzo N, Salmaso C, Cottini M, Lorini M, Tedeschi A. N-acetyl-aspartyl-glutamic acid inhibits cellular recruitment and mediator release during the late allergen-induced nasal reaction. Eur J Clin Pharmacol. 1998;54:515–520. doi: 10.1007/s002280050506. [DOI] [PubMed] [Google Scholar]
- Miller SL, Daikhin Y, Yudkoff M. Metabolism of N-acetyl-L-aspartate in rat brain. Neurochem Res. 1996;21:615–618. doi: 10.1007/BF02527761. [DOI] [PubMed] [Google Scholar]
- Miyake M, Kakimoto Y, Sorimachi M. A gas chromatographic method for the determination of N-acetyl-L-aspartic acid, N-acetyl-aspartylglutamic acid and beta-citryl-L-glutamic acid and their distributions in the brain and other organs of various species of animals. J Neurochem. 1981;36:804–810. doi: 10.1111/j.1471-4159.1981.tb01665.x. [DOI] [PubMed] [Google Scholar]
- Miyake M, Morino H, Mizobuchi M, Kakimoto Y. N-acetyl-L-aspartic acid, N-acetyl-alpha-L-aspartyl-L-glutamic acid and beta-citryl-L-glutamic acid in human urine. Clin Chim Acta. 1982;120:119–126. doi: 10.1016/0009-8981(82)90082-1. [DOI] [PubMed] [Google Scholar]
- Moats RA, Ernst T, Shonk TK, Ross BD. Abnormal cerebral metabolite concentrations in patients with probable Alzheimer disease. Magn Reson Med. 1994;32:110–115. doi: 10.1002/mrm.1910320115. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Namboodiri MA. Differential distribution of N-acetylaspartylglutamate and N- acetylaspartate immunoreactivities in rat forebrain. J Neurocytol. 1995;24:409–433. doi: 10.1007/BF01181604. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Namboodiri MA. Expression of N-acetylaspartate and N-acetylaspartylglutamate in the nervous system. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 7–26. [Google Scholar]
- Moffett JR, Namboodiri MA, Cangro CB, Neale JH. Immunohistochemical localization of N-acetylaspartate in rat brain. Neuroreport. 1991;2:131–134. doi: 10.1097/00001756-199103000-00005. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Namboodiri MA, Neale JH. Enhanced carbodiimide fixation for immunohistochemistry: Application to the comparative distributions of N-acetylaspartylglutamate and N-acetylaspartate immunoreactivities in rat brain. J Histochem Cytochem. 1993;41:559–570. doi: 10.1177/41.4.8450195. [DOI] [PubMed] [Google Scholar]
- Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. [Google Scholar]
- Moghaddam B. Bringing order to the glutamate chaos in schizophrenia. Neuron. 2003;40:881–884. doi: 10.1016/s0896-6273(03)00757-8. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Jackson ME. Glutamatergic animal models of schizophrenia. Ann N Y Acad Sci. 2003;1003:131–137. doi: 10.1196/annals.1300.065. [DOI] [PubMed] [Google Scholar]
- Moore RA, Le Coq J, Faehnle CR, Viola RE. Purification and preliminary characterization of brain aspartoacylase. Arch Biochem Biophys. 2003;413:1–8. doi: 10.1016/s0003-9861(03)00055-9. [DOI] [PubMed] [Google Scholar]
- Moreno A, Ross BD, Bluml S. Direct determination of the N-acetyl-L-aspartate synthesis rate in the human brain by (13)C MRS and [1-(13)C]glucose infusion. J Neurochem. 2001;77:347–350. doi: 10.1046/j.1471-4159.2001.t01-1-00282.x. [DOI] [PubMed] [Google Scholar]
- Mostert JP, Sijens PE, Oudkerk M, De KJ. Fluoxetine increases cerebral white matter NAA/Cr ratio in patients with multiple sclerosis. Neurosci Lett. 2006;402:22–24. doi: 10.1016/j.neulet.2006.03.042. [DOI] [PubMed] [Google Scholar]
- Muir D, Berl S, Clarke DD. Acetate and fluoroacetate as possible markers for glial metabolism in vivo. Brain Res. 1986;380:336–340. doi: 10.1016/0006-8993(86)90231-3. [DOI] [PubMed] [Google Scholar]
- Muse ED, Jurevics H, Toews AD, Matsushima GK, Morell P. Parameters related to lipid metabolism as markers of myelination in mouse brain. J Neurochem. 2001;76:77–86. doi: 10.1046/j.1471-4159.2001.00015.x. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Cooper JR. Metabolism of the aspartyl moiety of N-acetyl-L-aspartic acid in the rat brain. J Neurochem. 1972a;19:2091–2105. doi: 10.1111/j.1471-4159.1972.tb05119.x. [DOI] [PubMed] [Google Scholar]
- Nadler JV, Cooper JR. N-acetyl-L-aspartic acid content of human neural tumours and bovine peripheral nervous tissues. J Neurochem. 1972b;19:313–319. doi: 10.1111/j.1471-4159.1972.tb01341.x. [DOI] [PubMed] [Google Scholar]
- Nakaiso M, Uno M, Harada M, Kageji T, Takimoto O, Nagahiro S. Brain abscess and glioblastoma identified by combined proton magnetic resonance spectroscopy and diffusion-weighted magnetic resonance imaging--two case reports. Neurol Med Chir (Tokyo) 2002;42:346–348. doi: 10.2176/nmc.42.346. [DOI] [PubMed] [Google Scholar]
- Namboodiri MA, Corigliano-Murphy A, Jiang G, Rollag M, Provencio I. Murine aspartoacylase: cloning, expression and comparison with the human enzyme. Brain Res Mol Brain Res. 2000;77:285–289. doi: 10.1016/s0169-328x(00)00068-1. [DOI] [PubMed] [Google Scholar]
- Namboodiri MA, Moffett JR, Arun P, Mathew R, Namboodiri S, Potti A, Hershfield J, Kirmani B, Jacobowitz DM, Madhavarao CN. Defective myelin lipid synthesis as a pathogenic mechanism of Canavan disease. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006a. pp. 145–163. [DOI] [PubMed] [Google Scholar]
- Namboodiri MA, Peethambaran A, Mathew R, Sambhu PA, Hershfield J, Moffett JR, Madhavarao CN. Canavan disease and the role of N-acetylaspartate in myelin synthesis. Mol Cell Endocrinol. 2006b;252:216–223. doi: 10.1016/j.mce.2006.03.016. [DOI] [PubMed] [Google Scholar]
- Narayanan S, De Stefano N, Francis GS, Arnaoutelis R, Caramanos Z, Collins DL, Pelletier D, Arnason BGW, Antel JP, Arnold DL. Axonal metabolic recovery in multiple sclerosis patients treated with interferon beta-1b. J Neurol. 2001;248:979–986. doi: 10.1007/s004150170052. [DOI] [PubMed] [Google Scholar]
- Nikolau BJ, Oliver DJ, Schnable PS, Wurtele ES. Molecular biology of acetyl-CoA metabolism. Biochem Soc Trans. 2000;28:591–593. [PubMed] [Google Scholar]
- Norenberg MD, Martinez-Hernandez A. Fine structural localization of glutamine synthetase in astrocytes of rat brain. Brain Res. 1979;161:303–310. doi: 10.1016/0006-8993(79)90071-4. [DOI] [PubMed] [Google Scholar]
- Odermatt B, Wellershaus K, Wallraff A, Seifert G, Degen J, Euwens C, Fuss B, Bussow H, Schilling K, Steinhauser C, Willecke K. Connexin 47 (Cx47)-deficient mice with enhanced green fluorescent protein reporter gene reveal predominant oligodendrocytic expression of Cx47 and display vacuolized myelin in the CNS. J Neurosci. 2003;23:4549–4559. doi: 10.1523/JNEUROSCI.23-11-04549.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohrmann P, Siegmund A, Suslow T, Pedersen A, Spitzberg K, Kersting A, Rothermundt M, Arolt V, Heindel W, Pfleiderer B. Cognitive impairment and in vivo metabolites in first-episode neuroleptic-naive and chronic medicated schizophrenic patients: A proton magnetic resonance spectroscopy study. J Psychiatr Res. 2006 doi: 10.1016/j.jpsychires.2006.07.002. [DOI] [PubMed] [Google Scholar]
- Ohrmann P, Siegmund A, Suslow T, Spitzberg K, Kersting A, Arolt V, Heindel W, Pfleiderer B. Evidence for glutamatergic neuronal dysfunction in the prefrontal cortex in chronic but not in first-episode patients with schizophrenia: a proton magnetic resonance spectroscopy study. Schizophr Res. 2005;73:153–157. doi: 10.1016/j.schres.2004.08.021. [DOI] [PubMed] [Google Scholar]
- Oppenheim C, Zuber M, Galanaud D, Detilleux M, Bolgert F, Mas JL, Chiras J, Meder JF. Spectroscopy and serial diffusion MR findings in hGH-Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2004;75:1066–1069. doi: 10.1136/jnnp.2003.020172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen OE, Kalhan SC, Hanson RW. The key role of anaplerosis and cataplerosis for citric acid cycle function. J Biol Chem. 2002;277:30409–30412. doi: 10.1074/jbc.R200006200. [DOI] [PubMed] [Google Scholar]
- Paley M, Cozzone PJ, Alonso J, Vion-Dury J, Confort-Gouny S, Wilkinson ID, Chong WK, Hall-Craggs MA, Harrison MJ, Gili J, Rovira A, Capellades J, Rio J, Ocana I, Nicoli F, Dhiver C, Gastaut JL, Gastaut JA, Wicklow K, Sauter R. A multicenter proton magnetic resonance spectroscopy study of neurological complications of AIDS. AIDS Res Hum Retroviruses. 1996;12:213–222. doi: 10.1089/aid.1996.12.213. [DOI] [PubMed] [Google Scholar]
- Pan JW, Takahashi K. Interdependence of N-acetyl aspartate and high-energy phosphates in healthy human brain. Ann Neurol. 2005;57:92–97. doi: 10.1002/ana.20317. [DOI] [PubMed] [Google Scholar]
- Parks MH, Dawant BM, Riddle WR, Hartmann SL, Dietrich MS, Nickel MK, Price RR, Martin PR. Longitudinal brain metabolic characterization of chronic alcoholics with proton magnetic resonance spectroscopy. Alcohol Clin Exp Res. 2002;26:1368–1380. doi: 10.1097/01.ALC.0000029598.07833.2D. [DOI] [PubMed] [Google Scholar]
- Parsons MW, Li T, Barber PA, Yang Q, Darby DG, Desmond PM, Gerraty RP, Tress BM, Davis SM. Combined (1)H MR spectroscopy and diffusion-weighted MRI improves the prediction of stroke outcome. Neurology. 2000;55:498–505. doi: 10.1212/wnl.55.4.498. [DOI] [PubMed] [Google Scholar]
- Passani L, Elkabes S, Coyle JT. Evidence for the presence of N-acetylaspartylglutamate in cultured oligodendrocytes and LPS activated microglia. Brain Res. 1998;794:143–145. doi: 10.1016/s0006-8993(98)00308-4. [DOI] [PubMed] [Google Scholar]
- Patel TB, Clark JB. Synthesis of N-acetyl-L-aspartate by rat brain mitochondria and its involvement in mitochondrial/cytosolic carbon transport. Biochem J. 1979;184:539–546. doi: 10.1042/bj1840539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TB, Clark JB. Mitochondrial/cytosolic carbon transfer in the developing rat brain. Biochim Biophys Acta. 1981;677:373–380. doi: 10.1016/0304-4165(81)90249-x. [DOI] [PubMed] [Google Scholar]
- Petroff OA, Ogino T, Alger JR. High-resolution proton magnetic resonance spectroscopy of rabbit brain: regional metabolite levels and postmortem changes. J Neurochem. 1988;51:163–171. doi: 10.1111/j.1471-4159.1988.tb04850.x. [DOI] [PubMed] [Google Scholar]
- Pizzini F, Fatemi AS, Barker PB, Nagae-Poetscher LM, Horska A, Zimmerman AW, Moser HW, Bibat G, Naidu S. Proton MR spectroscopic imaging in Pelizaeus-Merzbacher disease. AJNR Am J Neuroradiol. 2003;24:1683–1689. [PMC free article] [PubMed] [Google Scholar]
- Pleasure D, Lichtman C, Eastman S, Lieb M, Abramsky O, Silberberg D. Acetoacetate and D-(-)-beta-hydroxybutyrate as precursors for sterol synthesis by calf oligodendrocytes in suspension culture: extramitochondrial pathway for acetoacetate metabolism. J Neurochem. 1979;32:1447–1450. doi: 10.1111/j.1471-4159.1979.tb11083.x. [DOI] [PubMed] [Google Scholar]
- Pliss L, Balcar VJ, Bubenikova V, Pokorny J, FitzGibbon T, St’astny F. Morphology and ultrastructure of rat hippocampal formation after i.cv administration of N-acetyl-L-aspartyl-L-glutamate. Neurosci. 2003;122:93–101. doi: 10.1016/s0306-4522(03)00550-5. [DOI] [PubMed] [Google Scholar]
- Popko B. Myelin galactolipids: mediators of axon-glial interactions? Glia. 2000;29:149–153. doi: 10.1002/(sici)1098-1136(20000115)29:2<149::aid-glia8>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Port RE, Wolf W. Noninvasive methods to study drug distribution. Invest New Drugs. 2003;21:157–168. doi: 10.1023/a:1023569328717. [DOI] [PubMed] [Google Scholar]
- Pouwels PJ, Frahm J. Differential distribution of NAA and NAAG in human brain as determined by quantitative localized proton MRS. NMR Biomed. 1997;10:73–78. doi: 10.1002/(sici)1099-1492(199704)10:2<73::aid-nbm448>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Pouwels PJ, Frahm J. Regional metabolite concentrations in human brain as determined by quantitative localized proton MRS. Magn Reson Med. 1998;39:53–60. doi: 10.1002/mrm.1910390110. [DOI] [PubMed] [Google Scholar]
- Pratt H. Canavan’s disease and spongiform encephalopathy. Br Med J. 1972;4:427. doi: 10.1136/bmj.4.5837.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raichle ME, MacLeod AM, Snyder AZ, Powers WJ, Gusnard DA, Shulman GL. A default mode of brain function. Proc Natl Acad Sci U S A. 2001;98:676–682. doi: 10.1073/pnas.98.2.676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos M, Del Arco A, Pardo B, Martinez-Serrano A, Martinez-Morales JR, Kobayashi K, Yasuda T, Bogonez E, Bovolenta P, Saheki T, Satrustegui J. Developmental changes in the Ca2+-regulated mitochondrial aspartate-glutamate carrier aralar1 in brain and prominent expression in the spinal cord. Brain Res Dev Brain Res. 2003;143:33–46. doi: 10.1016/s0165-3806(03)00097-x. [DOI] [PubMed] [Google Scholar]
- Reichelt KL, Edminson PD, Kvamme E. The formation of peptido-amines from constituent amino acids and histamine in hypothalamic tissue. J Neurochem. 1976a;26:811–815. doi: 10.1111/j.1471-4159.1976.tb04456.x. [DOI] [PubMed] [Google Scholar]
- Reichelt KL, Edminson PD, Kvamme E. The formation of peptido-amines from constituent amino acids and histamine in hypothalamic tissue. J Neurochem. 1976b;26:811–815. doi: 10.1111/j.1471-4159.1976.tb04456.x. [DOI] [PubMed] [Google Scholar]
- Reichelt KL, Kvamme E. Histamine-dependent formation of N-acetyl-aspartyl peptides in mouse brain. J Neurochem. 1973;21:849–859. doi: 10.1111/j.1471-4159.1973.tb07529.x. [DOI] [PubMed] [Google Scholar]
- Ricciolini R, Scalibastri M, Kelleher JK, Carminati P, Calvani M, Arduini A. Role of acetyl-L-carnitine in rat brain lipogenesis: implications for polyunsaturated fatty acid biosynthesis. J Neurochem. 1998;71:2510–2517. doi: 10.1046/j.1471-4159.1998.71062510.x. [DOI] [PubMed] [Google Scholar]
- Rooney WD, Miller RG, Gelinas D, Schuff N, Maudsley AA, Weiner MW. Decreased N-acetylaspartate in motor cortex and corticospinal tract in ALS. Neurology. 1998;50:1800–1805. doi: 10.1212/wnl.50.6.1800. [DOI] [PubMed] [Google Scholar]
- Ross BD, Ernst T, Kreis R, Haseler LJ, Bayer S, Danielsen E, Bluml S, Shonk T, Mandigo JC, Caton W, Clark C, Jensen SW, Lehman NL, Arcinue E, Pudenz R, Shelden CH. 1H MRS in acute traumatic brain injury. J Magn Reson Imaging. 1998;8:829–840. doi: 10.1002/jmri.1880080412. [DOI] [PubMed] [Google Scholar]
- Rowland L, Bustillo JR, Lauriello J. Proton magnetic resonance spectroscopy (H-MRS) studies of schizophrenia. Semin Clin Neuropsychiatry. 2001;6:121–130. doi: 10.1053/scnp.2001.21838. [DOI] [PubMed] [Google Scholar]
- Sager TN, Hansen AJ, Laursen H. Correlation between N-acetylaspartate levels and histopathologic changes in cortical infarcts of mice after middle cerebral artery occlusion. J Cereb Blood Flow Metab. 2000;20:780–788. doi: 10.1097/00004647-200005000-00004. [DOI] [PubMed] [Google Scholar]
- Sager TN, Laursen H, Fink-Jensen A, Topp S, Stensgaard A, Hedehus M, Rosenbaum S, Valsborg JS, Hansen AJ. N-Acetylaspartate distribution in rat brain striatum during acute brain ischemia. J Cereb Blood Flow Metab. 1999a;19:164–172. doi: 10.1097/00004647-199902000-00008. [DOI] [PubMed] [Google Scholar]
- Sager TN, Laursen H, Hansen AJ. Changes in N-acetyl-aspartate content during focal and global brain ischemia of the rat. J Cereb Blood Flow Metab. 1995;15:639–646. doi: 10.1038/jcbfm.1995.79. [DOI] [PubMed] [Google Scholar]
- Sager TN, Thomsen C, Valsborg JS, Laursen H, Hansen AJ. Astroglia contain a specific transport mechanism for N-acetyl-L- aspartate. J Neurochem. 1999b;73:807–811. doi: 10.1046/j.1471-4159.1999.0730807.x. [DOI] [PubMed] [Google Scholar]
- Sager TN, Topp S, Torup L, Hanson LG, Egestad B, Moller A. Evaluation of CA1 damage using single-voxel 1H-MRS and un-biased stereology: Can non-invasive measures of N-acetyl-asparate following global ischemia be used as a reliable measure of neuronal damage? Brain Res. 2001;892:166–175. doi: 10.1016/s0006-8993(00)03274-1. [DOI] [PubMed] [Google Scholar]
- Schousboe A, Westergaard N, Waagepetersen HS, Larsson OM, Bakken IJ, Sonnewald U. Trafficking between glia and neurons of TCA cycle intermediates and related metabolites. Glia. 1997;21:99–105. [PubMed] [Google Scholar]
- Schuff N, Capizzano AA, Du AT, Amend DL, O’Neill J, Norman D, Kramer J, Jagust W, Miller B, Wolkowitz OM, Yaffe K, Weiner MW. Selective reduction of N-acetylaspartate in medial temporal and parietal lobes in AD. Neurology. 2002;58:928–935. doi: 10.1212/wnl.58.6.928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuff N, Meyerhoff DJ, Mueller S, Chao L, Sacrey DT, Laxer K, Weiner MW. N-acetylaspartate as a marker of neuronal injury in neurodegenerative disease. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 241–262. [Google Scholar]
- Seki T, Matsubayashi H, Amano T, Kitada K, Serikawa T, Sakai N, Sasa M. Adenoviral gene transfer of aspartoacylase into the tremor rat, a genetic model of epilepsy, as a trial of gene therapy for inherited epileptic disorder. Neurosci Lett. 2002;328:249–252. doi: 10.1016/s0304-3940(02)00522-0. [DOI] [PubMed] [Google Scholar]
- Seki T, Matsubayashi H, Amano T, Kitada K, Serikawa T, Sasa M, Sakai N. Adenoviral gene transfer of aspartoacylase ameliorates tonic convulsions of spontaneously epileptic rats. Neurochem Int. 2004;45:171–178. doi: 10.1016/j.neuint.2003.10.009. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Goldman-Rakic PS. The reduced neuropil hypothesis: a circuit based model of schizophrenia. Biol Psychiatry. 1999;45:17–25. doi: 10.1016/s0006-3223(98)00281-9. [DOI] [PubMed] [Google Scholar]
- Selemon LD, Kleinman JE, Herman MM, Goldman-Rakic PS. Smaller frontal gray matter volume in postmortem schizophrenic brains. Am J Psychiatry. 2002;159:1983–1991. doi: 10.1176/appi.ajp.159.12.1983. [DOI] [PubMed] [Google Scholar]
- Serikawa T, Ohno Y, Sasa M, Yamada J, Takaori S. A new model of petit mal epilepsy: spontaneous spike and wave discharges in tremor rats. Lab Anim. 1987;21:68–71. doi: 10.1258/002367787780740635. [DOI] [PubMed] [Google Scholar]
- Serles W, Li LM, Antel SB, Cendes F, Gotman J, Olivier A, Andermann F, Dubeau F, Arnold DL. Time course of postoperative recovery of N-acetyl-aspartate in temporal lobe epilepsy. Epilepsia. 2001;42:190–197. doi: 10.1046/j.1528-1157.2001.01300.x. [DOI] [PubMed] [Google Scholar]
- Shenton ME, Dickey CC, Frumin M, McCarley RW. A review of MRI findings in schizophrenia. Schizophr Res. 2001;49:1–52. doi: 10.1016/s0920-9964(01)00163-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigematsu H, Okamura N, Shimeno H, Kishimoto Y, Kan L, Fenselau C. Purification and characterization of the heat-stable factors essential for the conversion of lignoceric acid to cerebronic acid and glutamic acid: identification of N-acetyl-L-aspartic acid. J Neurochem. 1983;40:814–820. doi: 10.1111/j.1471-4159.1983.tb08052.x. [DOI] [PubMed] [Google Scholar]
- Shonk TK, Moats RA, Gifford P, Michaelis T, Mandigo JC, Izumi J, Ross BD. Probable Alzheimer disease: diagnosis with proton MR spectroscopy. Radiology. 1995;195:65–72. doi: 10.1148/radiology.195.1.7892497. [DOI] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Tavazzi B, Dunbar J, Amorini AM, Lazzarino G, Vagnozzi R. The protective effect of cyclosporin A upon N-acetylaspartate and mitochondrial dysfunction following experimental diffuse traumatic brain injury. J Neurotrauma. 2004;21:1154–1167. doi: 10.1089/neu.2004.21.1154. [DOI] [PubMed] [Google Scholar]
- Signoretti S, Marmarou A, Tavazzi B, Lazzarino G, Beaumont A, Vagnozzi R. N-Acetylaspartate reduction as a measure of injury severity and mitochondrial dysfunction following diffuse traumatic brain injury. J Neurotrauma. 2001;18:977–991. doi: 10.1089/08977150152693683. [DOI] [PubMed] [Google Scholar]
- Simmons ML, Frondoza CG, Coyle JT. Immunocytochemical localization of N-acetyl-aspartate with monoclonal antibodies. Neurosci. 1991;45:37–45. doi: 10.1016/0306-4522(91)90101-s. [DOI] [PubMed] [Google Scholar]
- Simons M, Kramer EM, Macchi P, Rathke-Hartlieb S, Trotter J, Nave KA, Schulz JB. Overexpression of the myelin proteolipid protein leads to accumulation of cholesterol and proteolipid protein in endosomes/lysosomes: implications for Pelizaeus-Merzbacher disease. J Cell Biol. 2002;157:327–336. doi: 10.1083/jcb.200110138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinichkin A, Sterri S, Edminson PD, Reichelt KL, Kvamme E. In vivo labelling of acetyl-aspartyl peptides in mouse brain from intracranially and intraperitoneally administered acetyl-L-[U-14C]aspartate. J Neurochem. 1977;29:425–431. doi: 10.1111/j.1471-4159.1977.tb10690.x. [DOI] [PubMed] [Google Scholar]
- Sistermans EA, de Coo RF, van Beerendonk HM, Poll-The BT, Kleijer WJ, van Oost BA. Mutation detection in the aspartoacylase gene in 17 patients with Canavan disease: four new mutations in the non-Jewish population. Eur J Hum Genet. 2000;8:557–560. doi: 10.1038/sj.ejhg.5200477. [DOI] [PubMed] [Google Scholar]
- Slusher BS, Tsai G, Yoo G, Coyle JT. Immunocytochemical localization of the N-acetyl-aspartyl- glutamate (NAAG) hydrolyzing enzyme N-acetylated alpha-linked acidic dipeptidase (NAALADase) J Comp Neurol. 1992;315:217–229. doi: 10.1002/cne.903150208. [DOI] [PubMed] [Google Scholar]
- Staffen W, Zauner H, Mair A, Kutzelnigg A, Kapeller P, Stangl H, Raffer E, Niederhofer H, Ladurner G. Magnetic resonance spectroscopy of memory and frontal brain region in early multiple sclerosis. J Neuropsychiatry Clin Neurosci. 2005;17:357–363. doi: 10.1176/jnp.17.3.357. [DOI] [PubMed] [Google Scholar]
- Stanley JA, Pettegrew JW, Keshavan MS. Magnetic resonance spectroscopy in schizophrenia: methodological issues and findings--part I. Biol Psychiatry. 2000;48:357–368. doi: 10.1016/s0006-3223(00)00949-5. [DOI] [PubMed] [Google Scholar]
- Steen RG, Hamer RM, Lieberman JA. Measurement of brain metabolites by 1H magnetic resonance spectroscopy in patients with schizophrenia: a systematic review and meta-analysis. Neuropsychopharmacology. 2005;30:1949–1962. doi: 10.1038/sj.npp.1300850. [DOI] [PubMed] [Google Scholar]
- Steen RG, Ogg RJ. Abnormally high levels of brain N-acetylaspartate in children with sickle cell disease. AJNR Am J Neuroradiol. 2005;26:463–468. [PMC free article] [PubMed] [Google Scholar]
- Tabernero A, Vicario C, Medina JM. Lactate spares glucose as a metabolic fuel in neurons and astrocytes from primary culture. Neurosci Res. 1996;26:369–376. doi: 10.1016/s0168-0102(96)01121-2. [DOI] [PubMed] [Google Scholar]
- Takanashi J, Inoue K, Tomita M, Kurihara A, Morita F, Ikehira H, Tanada S, Yoshitome E, Kohno Y. Brain N-acetylaspartate is elevated in Pelizaeus-Merzbacher disease with PLP1 duplication. Neurology. 2002;58:237–241. doi: 10.1212/wnl.58.2.237. [DOI] [PubMed] [Google Scholar]
- Tallan HH. Studies on the distribution of N-acetyl-L-aspartic acid in brain. J Biol Chem. 1957;224:41–45. [PubMed] [Google Scholar]
- Tallan HH, Moore S, Stein WH. N-Acetyl-L-aspartic acid in brain. J Biol Chem. 1956;219:257–264. [PubMed] [Google Scholar]
- Tanaka Y, Obata T, Sassa T, Yoshitome E, Asai Y, Ikehira H, Suhara T, Okubo Y, Nishikawa T. Quantitative magnetic resonance spectroscopy of schizophrenia: Relationship between decreased N-acetylaspartate and frontal lobe dysfunction. Psychiatry Clin Neurosci. 2006;60:365–372. doi: 10.1111/j.1440-1819.2006.01515.x. [DOI] [PubMed] [Google Scholar]
- Tavazzi B, Signoretti S, Lazzarino G, Amorini AM, Delfini R, Cimatti M, Marmarou A, Vagnozzi R. Cerebral oxidative stress and depression of energy metabolism correlate with severity of diffuse brain injury in rats. Neurosurgery. 2005;56:582–589. doi: 10.1227/01.neu.0000156715.04900.e6. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Davies SE, Obrenovitch TP, Doheny MH, Patsalos PN, Clark JB, Symon L. Investigation into the role of N-acetylaspartate in cerebral osmoregulation. J Neurochem. 1995;65:275–281. doi: 10.1046/j.1471-4159.1995.65010275.x. [DOI] [PubMed] [Google Scholar]
- Taylor DL, Davies SE, Obrenovitch TP, Urenjak J, Richards DA, Clark JB, Symon L. Extracellular N-acetylaspartate in the rat brain: in vivo determination of basal levels and changes evoked by high K+ J Neurochem. 1994;62:2349–2355. doi: 10.1046/j.1471-4159.1994.62062349.x. [DOI] [PubMed] [Google Scholar]
- Tedeschi G, Bonavita S, McFarland HF, Richert N, Duyn JH, Frank JA. Proton MR spectroscopic imaging in multiple sclerosis. Neuroradiology. 2002;44:37–42. doi: 10.1007/s002340100584. [DOI] [PubMed] [Google Scholar]
- Thatcher NM, Badar-Goffer RS, Ben-Yoseph O, McLean MA, Morris PG, Prior MJ, Taylor A, Bachelard HS. A comparison of some metabolic effects of N-methylaspartate stereoisomers, glutamate and depolarization: a multinuclear MRS study. Neurochem Res. 2002;27:51–58. doi: 10.1023/a:1014898421330. [DOI] [PubMed] [Google Scholar]
- Tieman SB, Neale JH, Tieman DG. N-acetylaspartylglutamate immunoreactivity in neurons of the monkey’s visual pathway. J Comp Neurol. 1991;313:45–64. doi: 10.1002/cne.903130105. [DOI] [PubMed] [Google Scholar]
- Tieman SB, Tieman DG. N-acetylaspartylglutamate immunoreactivity in human retina. Vision Res. 1996;36:941–947. doi: 10.1016/0042-6989(95)00185-9. [DOI] [PubMed] [Google Scholar]
- Tranberg M, Stridh MH, Guy Y, Jilderos B, Wigstrom H, Weber SG, Sandberg M. NMDA-receptor mediated efflux of N-acetylaspartate: physiological and/or pathological importance? Neurochem Int. 2004;45:1195–1204. doi: 10.1016/j.neuint.2004.06.005. [DOI] [PubMed] [Google Scholar]
- Trapp BD, Peterson J, Ransohoff RM, Rudick R, Mork S, Bo L. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- Truckenmiller ME, Namboodiri MA, Brownstein MJ, Neale JH. N-Acetylation of L-aspartate in the nervous system: differential distribution of a specific enzyme. J Neurochem. 1985;45:1658–1662. doi: 10.1111/j.1471-4159.1985.tb07240.x. [DOI] [PubMed] [Google Scholar]
- Tsai G, Coyle JT. N-acetylaspartate in neuropsychiatric disorders. Prog Neurobiol. 1995;46:531–540. doi: 10.1016/0301-0082(95)00014-m. [DOI] [PubMed] [Google Scholar]
- Tsai G, Passani LA, Slusher BS, Carter R, Baer L, Kleinman JE, Coyle JT. Abnormal excitatory neurotransmitter metabolism in schizophrenic brains. Arch Gen Psychiatry. 1995;52:829–836. doi: 10.1001/archpsyc.1995.03950220039008. [DOI] [PubMed] [Google Scholar]
- Tsai G, van Kammen DP, Chen S, Kelley ME, Grier A, Coyle JT. Glutamatergic neurotransmission involves structural and clinical deficits of schizophrenia. Biol Psychiatry. 1998;44:667–674. doi: 10.1016/s0006-3223(98)00151-6. [DOI] [PubMed] [Google Scholar]
- Tyson RL, Gallagher C, Sutherland GR. 13C-Labeled substrates and the cerebral metabolic compartmentalization of acetate and lactate. Brain Res. 2003;992:43–52. doi: 10.1016/j.brainres.2003.08.027. [DOI] [PubMed] [Google Scholar]
- Tyson RL, Sutherland GR. Labeling of N-acetylaspartate and N-acetylaspartylglutamate in rat neocortex, hippocampus and cerebellum from [1-13C]glucose. Neurosci Lett. 1998;251:181–184. doi: 10.1016/s0304-3940(98)00527-8. [DOI] [PubMed] [Google Scholar]
- Urazaev AK, Grossfeld RM, Fletcher PL, Speno H, Gafurov BS, Buttram JG, Lieberman EM. Synthesis and release of N-acetylaspartylglutamate (NAAG) by crayfish nerve fibers: implications for axon-glia signaling. Neurosci. 2001;106:237–247. doi: 10.1016/s0306-4522(01)00270-6. [DOI] [PubMed] [Google Scholar]
- Urenjak J, Williams SR, Gadian DG, Noble M. Specific expression of N-acetylaspartate in neurons, oligodendrocyte-type-2 astrocyte progenitors, and immature oligodendrocytes in vitro. J Neurochem. 1992;59:55–61. doi: 10.1111/j.1471-4159.1992.tb08875.x. [DOI] [PubMed] [Google Scholar]
- Vagnozzi R, Signoretti S, Tavazzi B, Cimatti M, Amorini AM, Donzelli S, Delfini R, Lazzarino G. Hypothesis of the postconcussive vulnerable brain: experimental evidence of its metabolic occurrence. Neurosurgery. 2005;57:164–171. doi: 10.1227/01.neu.0000163413.90259.85. [DOI] [PubMed] [Google Scholar]
- van Bogaert L, Bertrand I. Sur une idiotie familiale avec degerescence sponglieuse de neuraxe (note preliminaire) Acta Neurol. 1949;49:572–587. [Google Scholar]
- van Haren NE, Cahn W, Hulshoff Pol HE, Schnack HG, Caspers E, Lemstra A, Sitskoorn MM, Wiersma D, van den Bosch RJ, Dingemans PM, Schene AH, Kahn RS. Brain volumes as predictor of outcome in recent-onset schizophrenia: a multi-center MRI study. Schizophr Res. 2003;64:41–52. doi: 10.1016/s0920-9964(03)00018-5. [DOI] [PubMed] [Google Scholar]
- Varho T, Komu M, Sonninen P, Holopainen I, Nyman S, Manner T, Sillanpaa M, Aula P, Lundbom N. A new metabolite contributing to N-acetyl signal in 1H MRS of the brain in Salla disease. Neurology. 1999;52:1668–1672. doi: 10.1212/wnl.52.8.1668. [DOI] [PubMed] [Google Scholar]
- Verbalis JG. Control of Brain Volume During Hypoosmolality and Hyperosmolality. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 113–129. [Google Scholar]
- Vermathen P, Ende G, Laxer KD, Walker JA, Knowlton RC, Barbaro NM, Matson GB, Weiner MW. Temporal lobectomy for epilepsy: recovery of the contralateral hippocampus measured by (1)H MRS. Neurology. 2002;59:633–636. doi: 10.1212/wnl.59.4.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vion-Dury J, Confort-Gouny S, Nicoli F, Dhiver C, Gastaut JA, Gastaut JL, Cozzone PJ. Localized brain proton MRS metabolic patterns in HIV-related encephalopathies. C R Acad Sci III. 1994;317:833–840. [PubMed] [Google Scholar]
- Waldman AD, Rai GS, McConnell JR, Chaudry M, Grant D. Clinical brain proton magnetic resonance spectroscopy for management of Alzheimer’s and sub-cortical ischemic vascular dementia in older people. Arch Gerontol Geriatr. 2002;35:137–142. doi: 10.1016/s0167-4943(02)00014-6. [DOI] [PubMed] [Google Scholar]
- Walker PM, Ben SD, Giroud M, Brunotte F. Is NAA reduction in normal contralateral cerebral tissue in stroke patients dependent on underlying risk factors? J Neurol Neurosurg Psychiatry. 2006;77:596–600. doi: 10.1136/jnnp.2005.078238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker PM, Ben SD, Lalande A, Giroud M, Brunotte F. Time course of NAA T2 and ADC(w) in ischaemic stroke patients: 1H MRS imaging and diffusion-weighted MRI. J Neurol Sci. 2004;220:23–28. doi: 10.1016/j.jns.2004.01.012. [DOI] [PubMed] [Google Scholar]
- Wang X, Qian J, He R, Wei L, Liu N, Zhang Z, Huang Y, Lei H. Delayed changes in T1-weighted signal intensity in a rat model of 15-minute transient focal ischemia studied by magnetic resonance imaging/spectroscopy and synchrotron radiation X-ray fluorescence. Magn Reson Med. 2006;56:474–480. doi: 10.1002/mrm.20985. [DOI] [PubMed] [Google Scholar]
- Waniewski RA, Martin DL. Preferential utilization of acetate by astrocytes is attributable to transport. J Neurosci. 1998;18:5225–5233. doi: 10.1523/JNEUROSCI.18-14-05225.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wild JM, Wardlaw JM, Marshall I, Warlow CP. N-acetylaspartate distribution in proton spectroscopic images of ischemic stroke: relationship to infarct appearance on T2-weighted magnetic resonance imaging. Stroke. 2000;31:3008–3014. doi: 10.1161/01.str.31.12.3008. [DOI] [PubMed] [Google Scholar]
- Williamson LC, Eagles DA, Brady MJ, Moffett JR, Namboodiri MA, Neale JH. Localization and synaptic release of N-acetylaspartylglutamate in the chick retina and optic tectum. Eur J Neurosci. 1991;3:441–451. doi: 10.1111/j.1460-9568.1991.tb00831.x. [DOI] [PubMed] [Google Scholar]
- Williamson LC, Neale JH. Calcium-dependent release of N-acetylaspartylglutamate from retinal neurons upon depolarization. Brain Res. 1988;475:151–155. doi: 10.1016/0006-8993(88)90209-0. [DOI] [PubMed] [Google Scholar]
- Williamson LC, Neale JH. Uptake, metabolism, and release of N-[3H]acetylaspartylglutamate by the avian retina. J Neurochem. 1992;58:2191–2199. doi: 10.1111/j.1471-4159.1992.tb10963.x. [DOI] [PubMed] [Google Scholar]
- Wittsack HJ, Kugel H, Roth B, Heindel W. Quantitative measurements with localized 1H MR spectroscopy in children with Canavan’s disease. J Magn Reson Imaging. 1996;6:889–893. doi: 10.1002/jmri.1880060609. [DOI] [PubMed] [Google Scholar]
- Wolf NI, Sistermans EA, Cundall M, Hobson GM, vis-Williams AP, Palmer R, Stubbs P, Davies S, Endziniene M, Wu Y, Chong WK, Malcolm S, Surtees R, Garbern JY, Woodward KJ. Three or more copies of the proteolipid protein gene PLP1 cause severe Pelizaeus-Merzbacher disease. Brain. 2005;128:743–751. doi: 10.1093/brain/awh409. [DOI] [PubMed] [Google Scholar]
- Wolf NI, Willemsen MA, Engelke UF, van der Knaap MS, Pouwels PJ, Harting I, Zschocke J, Sistermans EA, Rating D, Wevers RA. Severe hypomyelination associated with increased levels of N-acetylaspartylglutamate in CSF. Neurology. 2004;62:1503–1508. doi: 10.1212/01.wnl.0000123094.13406.20. [DOI] [PubMed] [Google Scholar]
- Wolfe AJ. The acetate switch. Microbiol Mol Biol Rev. 2005;69:12–50. doi: 10.1128/MMBR.69.1.12-50.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolinsky JS, Narayana PA. Magnetic resonance spectroscopy in multiple sclerosis: window into the diseased brain. Curr Opin Neurol. 2002;15:247–251. doi: 10.1097/00019052-200206000-00004. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Berger GE, Lambert M, Conus P, Velakoulis D, Stuart GW, Desmond P, McGorry PD, Pantelis C. Prediction of functional outcome 18 months after a first psychotic episode: a proton magnetic resonance spectroscopy study. Arch Gen Psychiatry. 2006;63:969–976. doi: 10.1001/archpsyc.63.9.969. [DOI] [PubMed] [Google Scholar]
- Xi ZX, Baker DA, Shen H, Carson DS, Kalivas PW. Group II metabotropic glutamate receptors modulate extracellular glutamate in the nucleus accumbens. J Pharmacol Exp Ther. 2002;300:162–171. doi: 10.1124/jpet.300.1.162. [DOI] [PubMed] [Google Scholar]
- Yan HD, Ishihara K, Serikawa T, Sasa M. Activation by N-Acetyl-l-Aspartate of Acutely Dissociated Hippocampal Neurons in Rats via Metabotropic Glutamate Receptors. Epilepsia. 2003;44:1153–1159. doi: 10.1046/j.1528-1157.2003.49402.x. [DOI] [PubMed] [Google Scholar]
- Yaron Y, Schwartz T, Mey-Raz N, Amit A, Lessing JB, Malcov M. Preimplantation genetic diagnosis of Canavan disease. Fetal Diagn Ther. 2005;20:465–468. doi: 10.1159/000086834. [DOI] [PubMed] [Google Scholar]
- Yeo RA, Brooks WM, Jung RE. NAA and higher cognitive function in humans. In: Moffett JR, Tieman SB, Weinberger DR, Coyle JT, Namboodiri MA, editors. N-Acetylaspartate: A Unique Neuronal Molecule in the Central Nervous System. Springer Science + Business Media; New York, NY: 2006. pp. 215–226. [Google Scholar]
- Yi L, Zhang S, Zhang X. An experimental proton magnetic resonance spectroscopy analysis on early stage of acute focal cerebral ischemia. J Huazhong Univ Sci Technolog Med Sci. 2002;22:359–61. 366. doi: 10.1007/BF02896786. [DOI] [PubMed] [Google Scholar]
- Yudkoff M, Nelson D, Daikhin Y, Erecinska M. Tricarboxylic acid cycle in rat brain synaptosomes. Fluxes and interactions with aspartate aminotransferase and malate/aspartate shuttle. J Biol Chem. 1994;269:27414–27420. [PubMed] [Google Scholar]
- Zambell KL, Fitch MD, Fleming SE. Acetate and butyrate are the major substrates for de novo lipogenesis in rat colonic epithelial cells. J Nutr. 2003;133:3509–3515. doi: 10.1093/jn/133.11.3509. [DOI] [PubMed] [Google Scholar]
- Zeng BJ, Wang ZH, Ribeiro LA, Leone P, De Gasperi R, Kim SJ, Raghavan S, Ong E, Pastores GM, Kolodny EH. Identification and characterization of novel mutations of the aspartoacylase gene in non-Jewish patients with Canavan disease. J Inherit Metab Dis. 2002;25:557–570. doi: 10.1023/a:1022091223498. [DOI] [PubMed] [Google Scholar]
- Zhao J, Ramadan E, Cappiello M, Wroblewska B, Bzdega T, Neale JH. NAAG inhibits KCl-induced [(3)H]-GABA release via mGluR3, cAMP, PKA and L-type calcium conductance. Eur J Neurosci. 2001;13:340–346. [PubMed] [Google Scholar]