

Summary
MEF2 transcription factors are well-established regulators of muscle development. We have discovered an unanticipated role for MEF2C in the neural crest, where tissue-specific inactivation results in neonatal lethality due to severe craniofacial defects. We show that MEF2C is required for expression of the Dlx5, Dlx6, and Hand2 transcription factor genes in the branchial arches, and we identify a novel branchial arch-specific enhancer in the Dlx5/6 locus, which is activated synergistically by MEF2C and Dlx5, demonstrating that these factors interact to induce transcription. Mef2c and Dlx5/6 also interact genetically. Mice heterozygous for either Dlx5/6 or mef2c are normal at birth and survive to weaning. By contrast, heterozygosity for both mef2c and Dlx5/6 results in defective palate development and neonatal lethality. Taken together, the studies presented here define a novel, feed-forward transcriptional circuit between the MADS-box transcription factor MEF2C and the homeodomain transcription factors Dlx5 and Dlx6 in craniofacial development.
Introduction
Neural crest (NC) cells originate from the dorsal neural tube at the border between the neural plate and the lateral epidermis (Knecht and Bronner-Fraser, 2002; Trainor, 2005). When induced by contact mediated signals, NC precursor cells undergo an epithelial to mesenchymal transition, migrate throughout the developing embryo, and give rise to various tissues including the dorsal root and sympathetic ganglia, the glial cells of the peripheral and enteric nervous systems, melanocytes, and skeletal elements of the face and head (Knecht and Bronner-Fraser, 2002; Le Douarin et al., 2004; Trainor, 2005). Numerous congenital disorders are due to improper development of the NC and its derivatives, including craniofacial defects, which account for one third of all congenital anomalies (Farlie et al., 2004; Trainor, 2005).
Several transcription factors are known to be required for craniofacial development (Cobourne, 2000; Depew et al., 2005). Among these, the homeodomain transcription factors Dlx5 and Dlx6 are expressed in the NC-derived ectomesenchyme of the first and second branchial arches, and inactivation of Dlx5 and Dlx6 results in mice that have numerous craniofacial defects (Beverdam et al., 2002; Depew et al., 2002). Dlx6 is a direct transcriptional activator of the Hand2 gene, which encodes a basic helix-loop-helix (bHLH) transcription factor, in the first and second branchial arches via a conserved branchial arch enhancer element (Charite et al., 2001). Hand2 enhancer null mice exhibit cleft palate and hypoplasia of the mandible, among other craniofacial malformations (Yanagisawa et al., 2003). It is known that expression of both Dlx6 and Hand2 in the branchial arch mesenchyme requires endothelin signaling, but other components of this pathway, including the transcription factors required for activation of Dlx5 and Dlx6 are unknown (Charite et al., 2001; Clouthier et al., 2000; Ruest et al., 2004; Thomas et al., 1998).
The myocyte enhancer factor 2 (MEF2) family of transcription factors has four members in vertebrates, MEF2A–D (Black and Olson, 1998; McKinsey et al., 2002). MEF2 proteins bind to a consensus DNA-binding element known as a MEF2 site as homo- and heterodimers and interact with other transcription factors to function as both positive and negative regulators of gene expression in part through their association with class II histone deacetylases (HDACs)(McKinsey et al., 2001). Mef2c is the first member of the MEF2 family to be expressed during development, and mef2c null mice die at E9.5 with severe defects in cardiac and vascular development (Edmondson et al., 1994; Lin et al., 1998; Lin et al., 1997). Although MEF2C is widely appreciated for its role in the development of muscle lineages, its expression adjacent to the neural folds at E8.5 in early NC development has been reported previously (Edmondson et al., 1994), but its function in the NC has not been investigated.
In the present study, we used a conditional gene targeting approach in mice to inactivate mef2c in the NC, which results in lethality at birth due to upper airway obstruction. Mef2c NC knockout mice exhibit delayed ossification and hypoplasia or loss of the majority of the skeletal elements of the face and skull. We show that the transcription factor genes Dlx5, Dlx6, and Hand2 are not expressed in the NC component of the branchial arches in mef2c NC conditional knockout embryos, and we identify a MEF2-dependent transcriptional enhancer in the Dlx5/6 locus that is sufficient to direct expression to the branchial arches in vivo, establishing MEF2C as a direct transcriptional regulator of the Dlx6-Hand2 pathway in the NC. Furthermore, the Dlx5/6 enhancer identified in these studies is activated synergistically by MEF2C and Dlx5, indicating that these factors interact to regulate transcription. We also show that mef2c and Dlx5/6 interact genetically. Mice heterozygous for either Dlx5/6 or mef2c are normal, whereas mice heterozygous for both mef2c and Dlx5/6 do not survive and exhibit defective palate development. These studies highlight a previously unappreciated role for MEF2C in craniofacial development and identify a novel feed-forward transcriptional circuit between MEF2C and Dlx5/6 in craniofacial development.
Results
Mice lacking mef2c function in the neural crest die at birth due to upper airway obstruction
To determine the function of MEF2C in the NC, we conditionally inactivated a floxed allele of mef2c using Wnt1-Cre transgenic mice (Danielian et al., 1998; Vong et al., 2005). Animals lacking mef2c in the NC were born at expected Mendelian ratios and were animated and responsive to touch. However, mef2c conditional knockout mice quickly became cyanotic, and 100% of these animals died within an hour of birth (Fig. 1A).
Figure 1.
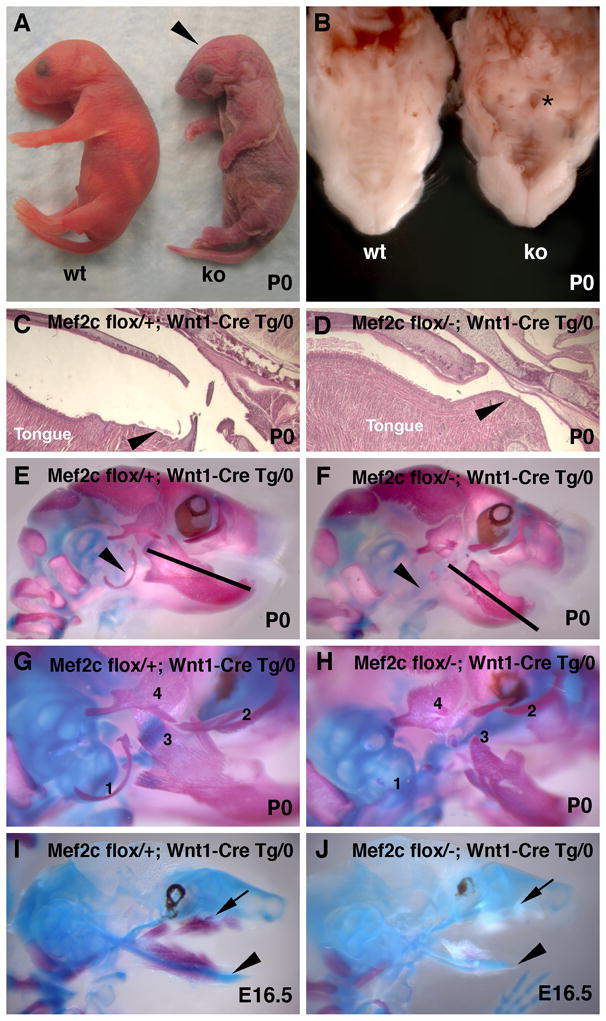
Mef2c is required for craniofacial development. (A) Mice lacking mef2c function in the neural crest have misshapen heads (arrowhead) and die from asphyxiation within an hour of birth. (B) Mef2c conditional knockout animals have a cleft of the posterior palate (asterisk). (C, D) Sagittal sections through the heads of mef2c neural crest conditional knockout (D) and littermate control (C) neonates show that the upper airway is constricted in the conditional knockout compared to control littermates (arrowheads). (E–J) Skeleton preparations of neonatal and fetal mice stained with alizarin red and alcian blue show that mef2c flox/-; Wnt1-Cre Tg/0 skulls (F, H, J) have multiple craniofacial defects compared to littermate controls (E, G, I). Arrowheads mark location of the tympanic ring in a control skull (E) and its absence in the mutant skull (H). Bars of equal size show that the mutant mandible is markedly shorter than the control (E, F). (G, H) Magnified view of the temporal region of control (G) and mutant (H) skulls showing the tympanic ring (1), zygomatic bone (2), mandibular processes (3), and temporal bone processes (4). Note that each of these structures is hypoplastic or missing in mef2c conditional knockout mice. (I, J) Delayed ossification in mef2c neural crest knockout mice at E16.5. Note the calcified bone in control (I) but not in mutant (J). In panels I and J, arrowheads mark Meckel’s cartilage, and arrows mark the developing maxilla.
The hearts, vasculature, and diaphragms of mef2c NC null mice showed no differences in gross morphology, histology, or marker gene expression compared to control littermates (data not shown). However, neonates lacking mef2c function in the NC exhibited a posterior cleft of the palate (Fig. 1B), a constricted airway, and defective position of the tongue near the back of the oral cavity compared to control littermates (Fig. 1C, D). These defects occurred with 100% penetrance in knockout animals (n = 63) and in none of the littermate controls. Knockout animals were tracheostomized with a 30G needle to bypass the upper airway, which resulted in recovery from cyanosis and restoration of viability prior to humane euthanasia (data not shown), demonstrating that mice lacking mef2c in the NC die at birth from asphyxiation caused by upper airway obstruction.
Mef2c function is required in the neural crest for craniofacial development
Skulls from mef2c NC conditional knockout neonates exhibited several defective or missing craniofacial structures compared to control littermates, including a hypoplastic mandible, zygomatic arch, and temporal bone (Fig. 1, compare panels E, G to F, H). Additionally, the coronoid, condular, and angular processes of the mutant mandibles were severely hypoplastic when compared to wild type littermate controls, and the tympanic ring was absent in mice lacking mef2c function in the NC (Fig. 1G, H). At E16.5, wild type skulls displayed normal Meckel’s cartilage and extensive ossification in the mandible and maxilla (Fig. 1I). By contrast, mef2c NC knockout embryos exhibited hypoplastic Meckel’s cartilage and a lack of ossification as well as hypoplasia of other future skeletal elements in the skull (Fig. 1J). The differences in skeletal development could be observed as early as E13.5, a time prior to the onset of ossification, when Meckel’s cartilage was already hypoplastic in conditional knockout animals (data not shown). Taken together, these results clearly establish that MEF2C function is required in the NC for proper craniofacial development.
Branchial arch expression of Dlx5, Dlx6, and Hand2 requires MEF2C
In order to place MEF2C into a pathway for craniofacial development, we examined the expression of several transcription factor genes required for craniofacial development at E9.5, which is the time when mef2c is expressed in the NC component of the branchial arches and developing craniofacial mesenchyme (Fig. 2A, B). Notably, Dlx5, Dlx6, and Hand2 expression in the first and second branchial arches was almost completely absent in mef2c NC knockout mice compared to control littermates at E9.5 (Fig. 2C–H; also Supplemental Fig. S1, panels A–D). Prx1 null mice also have a similar craniofacial phenotype to mef2c NC conditional mice (Martin et al., 1995). However, no differences were observed in Prx1 expression in mef2c NC knockout and control embryos (Supplemental Fig. S1, panels E, F), indicating that branchial arch development in general was not defective at this stage and that Prx1 is not a downstream target of MEF2C. No differences were observed in the Wnt1-Cre fate map between NC knockout and control embryos in a ROSA26R lacZ reporter background (Soriano, 1999), indicating that contribution of NC cells to the branchial arches and viscerocranium was not grossly defective (Fig. 2I, J). Likewise, no obvious changes were observed in proliferation or apoptosis between knockout and control embryos at E9.5 or E10.5 (Supplemental Fig. S1, panels G–J). Taken together, these results suggest that the requirement for mef2c is in post-migratory NC and that MEF2C is an upstream regulator of the Dlx6-Hand2 pathway in craniofacial development.
Figure 2.
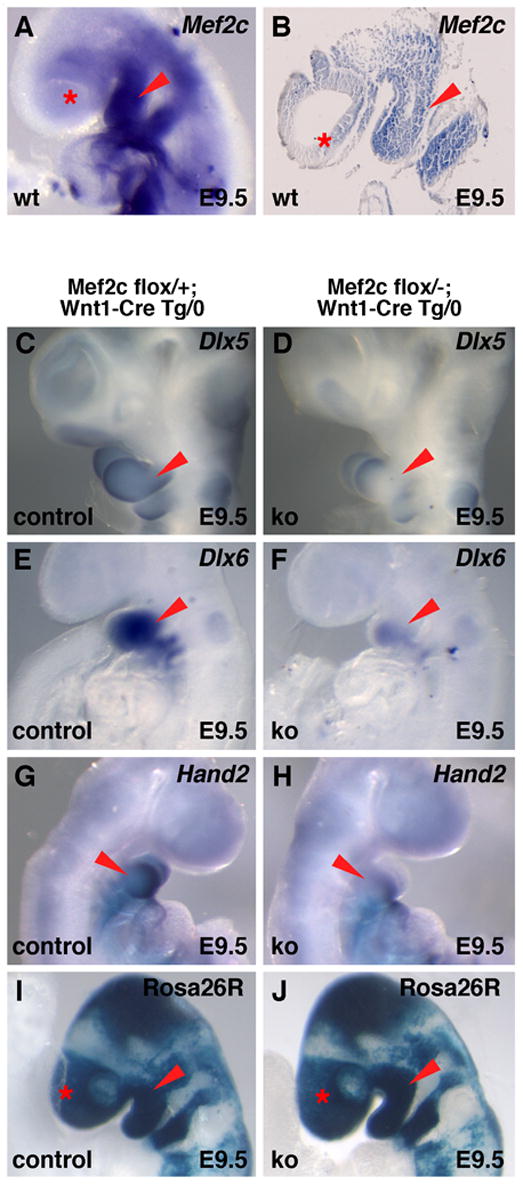
Dlx5, Dlx6, and Hand2 expression in the branchial arches requires mef2c. (A, B) Whole mount (A) and sagittal section (B) in situ hybridization showing endogenous mef2c expression in wild type embryos at E9.5. Note the robust expression of mef2c in the first and second branchial arches. (C–H) Dlx5, Dlx6, and Hand2 expression are nearly absent in the branchial arches of mef2c neural crest knockout embryos at E9.5. Note that Dlx6 expression is still faintly detected in the first arch of mef2c neural crest knockout embryos (F) and that Dlx5 and Hand2 expression appear to be absent except in a small region at the distal tip of the first arch (D, H). (I, J) X-gal staining of E9.5 embryos generated by crossing a ROSA26R lacZ reporter allele into the mef2c conditional knockout background showed no obvious defects in neural crest contribution to the branchial arches or craniofacial mesenchyme compared to littermate controls. Red arrowheads mark the first branchial arch in all panels; asterisks mark craniofacial ectomesenchymal expression.
A novel, branchial arch enhancer from the Dlx5/6 locus is directly activated by MEF2C
Dlx6 is closely linked to Dlx5 in the mouse genome (Zerucha et al., 2000), and deletion of both genes results in very severe craniofacial defects. Inactivation of Dlx5/6 in mice results in loss of Hand2 expression in the branchial arches at E9.5, and Dlx6 has been shown to be a direct transcriptional regulator of Hand2 through a conserved enhancer in the Hand2 locus (Beverdam et al., 2002; Charite et al., 2001; Depew et al., 2002; Ruest et al., 2004). Since Dlx5, Dlx6, and Hand2 were down regulated in the absence of mef2c function in the NC, we hypothesized that MEF2C might directly regulate these genes. To identify potential transcriptional enhancers from these genes, we analyzed all of the conserved, noncoding sequences in the Dlx5/6 and Hand2 loci for MEF2 sites, and we identified a deeply conserved element immediately upstream of the Dlx6 coding sequence (Fig. 3A) that contained four highly conserved MEF2 sites and multiple potential homeodomain protein (Hox) binding sites (Fig. 3B). We tested this region of the Dlx5/6 locus for enhancer activity by cloning it into the Hsp68-lacZ reporter plasmid and using this construct to generate transgenic embryos. This novel region of the Dlx5/6 locus directed expression to the first and second branchial arches at E9.5 (Fig. 3C) in a pattern very similar to the endogenous Dlx5 and Dlx6 expression patterns in the arches (Fig. 2C, E).
Figure 3.

Identification of a MEF2-dependent transcriptional enhancer from the Dlx5/6 locus. (A) Schematic representation of the Dlx5/6 locus. Red boxes denote the Dlx5 and Dlx6 transcribed regions; arrows show the direction of transcription. The blue box denotes the location of the enhancer identified in this study. (B) ClustalW alignment of the mouse, human, and chicken sequences in the conserved region of the Dlx6 branchial arch enhancer. Conserved MEF2 consensus elements are highlighted by light blue boxes; conserved Hox/Dlx binding sites are highlighted by yellow-green boxes; asterisks denote conservation across all three species. (C) The Dlx5/6 enhancer directs lacZ expression to the first and second branchial arches in transgenic embryos at E9.5 as shown by whole mount X-gal staining. The arrowhead indicates branchial arch expression of the Dlx5/6-lacZ transgene. (D) MEF2C trans-activates the Dlx5/6 branchial arch enhancer. Cotransfection of a MEF2C-VP16 expression plasmid with the Dlx5/6-TK-lacZ results in 12-fold activation of the reporter (lane 4). This activation is dependent upon the presence of the MEF2 sites since mutation of those elements almost completely abolished MEF2 trans-activation (lane 6). No trans-activation of the parent reporter by MEF2C-VP16 was observed (lane 2). Data are presented as the mean plus standard error for six independent experiments. (E) MEF2C and Dlx5 synergistically activate the Dlx5/6 enhancer. Cotransfection of a MEF2C or Dlx5 expression plasmid results in 3-fold and 50-fold activation of the Dlx5/6 enhancer, respectively (lanes 4, 5). Cotransfection of both expression plasmids results in strong synergistic activation of over 400-fold (lane 6). No trans-activation of the TK parent reporter by MEF2C plus Dlx5 was observed (lane 2). Data are presented as the mean plus standard error for three independent experiments.
All four of the candidate MEF2 sites in the Dlx5/6 locus were bound by MEF2C binding by EMSA, although only the MEF2–3 and MEF2–4 sites exhibited robust binding in vitro (data not shown). To determine if the Dlx6 enhancer was responsive to MEF2 activation via its conserved MEF2 sites, we tested a Dlx6-TK-lacZ reporter construct for MEF2C trans-activation in 3T3 cells (Fig. 3D). Cotransfection with a MEF2C-VP16 expression plasmid resulted in activation of the Dlx5/6 reporter more than 10-fold over control cotransfections (Fig. 3D, lane 4) and this activation was dependent on the presence of intact MEF2 sites since mutation of the four sites resulted in a complete loss of activation of the enhancer element by MEF2C (Fig. 3D, lane 6). Taken together, these results demonstrate that the MEF2 sites in this novel Dlx5/6 branchial arch enhancer are functional and further support the notion that Dlx5 and Dlx6 are direct transcriptional targets of MEF2C via this enhancer element.
Cooperative activation of the Dlx5/6 branchial arch enhancer by MEF2C and Dlx5
The presence of multiple conserved Hox sites, combined with the presence of functional MEF2 sites in the Dlx5/6 branchial arch enhancer suggested that this enhancer might also be activated by Dlx proteins themselves. We tested this possibility and the possibility that MEF2C might participate in a feed-forward transcriptional circuit with Dlx5/6 by activating their expression and then cooperating with them to amplify Dlx5/6 expression (Fig. 3E). Indeed, Dlx5 activated expression of the Dlx5/6 enhancer more than 50-fold (Fig. 3E, lane 5). Furthermore, under conditions in which wild type MEF2C activated the enhancer only 3-fold (Fig. 3E, lane 4), cotransfection of Dlx5 and MEF2C resulted in clear synergy by activating reporter expression more than 400-fold (Fig. 3E, lane 6).
Genetic interaction between Mef2c and Dlx5/6 results in neonatal lethality in compound heterozygous mice
The transcriptional synergy between MEF2C and Dlx5 suggested that these genes might interact. Therefore, we crossed Dlx5/6+/− mice with mef2c+/− mice to test for genetic interaction. Because Dlx5 and Dlx6 are closely linked, the two genes have been deleted together in a single gene targeting event (Depew et al., 2002). Heterozygosity for either mef2c or Dlx5/6 alone did not significantly affect neonatal viability (Fig. 4A). By contrast, 100% of double heterozygous offspring were obviously sick at birth and died on P0 (Fig. 4A), indicating a clear genetic interaction between the Dlx5/6 and mef2c loci. Compared to wild type littermates, Dlx5/6+/−; mef2c+/− mice exhibited a small, incomplete palate and a displaced tongue (Fig. 4B, C), which is consistent with the palate and displaced tongue phenotype in mef2c NC conditional knockout mice (Fig. 1D) and further supports the conclusion that MEF2C functions as a transcriptional activator and partner of Dlx5 and Dlx6 in craniofacial development (Fig. 4D).
Figure 4.
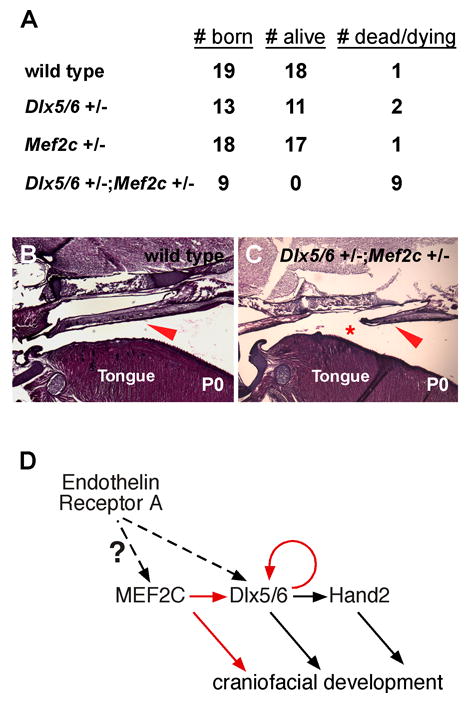
Mef2c and Dlx5/6 interact genetically. (A) Mef2c+/− mice were crossed to Dlx5/6+/− mice and the total number of offspring was scored on P0 (# born). Among the total of each genotype born, animals were scored as either alive and viable (# alive) or as dead or clearly cyanotic and dying (# dead/dying). Nearly all wild type, Mef2c+/−, and Dlx5/6+/− mice were viable at birth. By contrast, 100% (9/9) Dlx5/6+/−;Mef2c+/− mice died or were clearly cyanotic on P0, indicating a strong genetic interaction. (B, C) Sagittal sections of the heads of wild type (B) or Dlx5/6+/−;Mef2c+/− (C) mice collected on P0. Compared to wild type littermates, Dlx5/6+/−;Mef2c+/− mice exhibit a misshapen (arrowheads) and incomplete (asterisk) palate and an improper position of the tongue at the rear of the oral cavity. (D) A model transcriptional pathway for craniofacial development in which MEF2C functions downstream of endothelin receptor A signaling to activate Dlx5 and Dlx6, which in turn, activates Hand2 and also reinforces their own expression in a feed-forward fashion. Solid arrows denote direct regulation; dashed arrows denote direct or indirect regulation.
Discussion
A MEF2-dependent transcriptional pathway for craniofacial development
We have uncovered a novel role for MEF2C function in the NC as an essential regulator of craniofacial development. The defects in mef2c NC knockout mice are similar to those found in other transcription factor knockouts, including Prx1, Dlx5/6, and Hand2 knockout animals (Beverdam et al., 2002; Depew et al., 2002; Martin et al., 1995; Yanagisawa et al., 2003). We show that Dlx5, Dlx6, and Hand2 expression are dramatically reduced in mef2c NC knockout embryos, and we present evidence that MEF2C is a direct transcriptional regulator of Dlx5/6 via a novel branchial arch enhancer. Additionally, we show that mef2c and Dlx5/6 genetically interact and that MEF2C functions as a transcriptional partner with Dlx5 in a feed-forward transcriptional circuit by synergistically activating a novel branchial enhancer in the Dlx5/6 locus (Fig. 4D).
Modular control of branchial arch gene regulation
It has been noted previously that deletion of the Hand2 branchial arch enhancer does not completely abolish Hand2 expression in the branchial arches as a small region of distal expression within the arches appears to be governed by a hypothetical, unidentified regulatory element (Charite et al., 2001; Ruest et al., 2004; Yanagisawa et al., 2003). Interestingly, the loss of Hand2 expression seen in the absence of mef2c function in the NC is nearly complete except at the distal end of the first arch, which appears to correspond to the Dlx6-independent Hand2 expression domain (Fig. 2H; Yanagisawa et al., 2003). Thus, Hand2 regulation, even within the branchial arches, appears to be controlled by multiple enhancer modules (Charite et al., 2001; Ruest et al., 2004; Yanagisawa et al., 2003). Similarly, a previously identified intergenic Dlx5/6 enhancer directs expression to the forebrain and to a subset of the endogenous Dlx5 and Dlx6 expression patterns in the branchial arches (Ruest et al., 2003; Zerucha et al., 2000). Here, we identify an additional regulatory module immediately upstream of the proposed Dlx6 transcriptional start site that regulates expression in the branchial arches (Fig. 3). This type of compartmentalized regulation provides the opportunity for independent control of gene expression in discrete lineages or for multiple different upstream pathways to intersect in the same lineage.
MEF2C as a potential downstream effector of endothelin signaling
Dlx6 and Hand2 expression in branchial arch NC requires endothelin signaling (Charite et al., 2001; Clouthier et al., 2000; Ruest et al., 2004; Thomas et al., 1998). Endothelin signaling regulates a diverse array of normal developmental and physiological processes, including cardiovascular and craniofacial development in the embryo and vascular tone postnatally (Clouthier et al., 1998; Kedzierski and Yanagisawa, 2001; Kurihara et al., 1994). Endothelin signaling is also critical in a number of pathological processes as well, including hypertension, atherosclerosis, and pathological cardiac hypertrophy and heart failure (Kedzierski and Yanagisawa, 2001). Targeted inactivation of several endothelin pathway components, including Edn1, Ednra, and Ece1 each result in lethality at birth due to craniofacial defects and mechanical asphyxiation similar to the phenotypes exhibited by mef2c NC knockout mice, including loss of Dlx6 and Hand2 expression (Charite et al., 2001; Clouthier et al., 1998; Clouthier et al., 2000; Kurihara et al., 1994; Ruest et al., 2004; Thomas et al., 1998; Yanagisawa et al., 1998). Based on these observations, it will be interesting to determine if MEF2C functions as a downstream effector of endothelin signaling during craniofacial development (Fig. 4D).
Implications for MEF2C involvement in congenital craniofacial defects
Craniofacial defects are among the most common serious congenital anomalies in humans, affecting as many as 1 in every 300 births (Stanier and Moore, 2004). However, the affected genes and developmental mechanisms underlying the majority of craniofacial disorders remain unknown (Stanier and Moore, 2004). This is likely due to the fact that many craniofacial anomalies with a genetic component are highly variable in penetrance or severity, several present as part of broader syndromes, and some craniofacial defects are thought to result from combinations of environmental and genetic influences (Farlie et al., 2004; Stanier and Moore, 2004). For example, Pierre Robin sequence (PRS) is characterized by hypoplastic mandible, cleft palate, and obstruction of the upper airway by the tongue (Dinwiddie, 2004; Farlie et al., 2004). The etiology of PRS is thought to be primarily environmental in nature, although there is evidence for a genetic component to this complex and to broader syndromes that include the PRS phenotypes (Dinwiddie, 2004; Farlie et al., 2004). Mice lacking mef2c function in the NC appear to have a similar set of defects in the oral cavity, including a small lower jaw, cleft palate, and a displaced position of the tongue, suggesting the possibility that developmental processes regulated by MEF2C may contribute to this or other similar craniofacial defects in humans.
Experimental Procedures
Cloning and Mutagenesis
The Dlx6 enhancer fragment described in these studies was amplified from mouse genomic DNA using the primers Dlx6-F 5′-ccaccacacaagcttgctaccccacac-3′ and Dlx6R 5′-tgtgttcagaagcaggggccctag-3′ and then cloned into plasmids Hsp68-lacZ and pTK-β-gal for transgenic and transfection analyses, respectively. Mutagenesis was performed as described previously (Dodou et al., 2003). The sequences of the mutagenic oligonucleotides and other oligonucleotides used for cloning are available on request.
Transgenic Mice
Transgenic mice were generated by oocyte microinjection using standard methods (Hogan et al., 1994) as described previously (Dodou et al., 2003). Mef2c mutant mice carrying the Mef2cflox allele and the conventional knockout allele, mef2cTm1, have each been described (Lin et al., 1997; Vong et al., 2005). Wnt1-Cre, Dlx5/6+/−, and Rosa26R lacZ reporter mice have also each been described (Danielian et al., 1998; Depew et al., 2002; Soriano, 1999). Transgenic and knockout alleles were detected by Southern blot. All experiments using animals complied with federal and institutional guidelines and were reviewed and approved by the UCSF Institutional Animal Care and Use Committee.
X-gal staining, in situ hybridization, and skeleton preparations
X-gal staining to detect β-galactosidase expression was performed as described previously (Dodou et al., 2003). In situ hybridization was performed according to standard methods using digoxigenin-labeled antisense probes as described previously (Rojas et al., 2005). Skeleton and cartilage preparations were performed according to standard procedures (Hogan et al., 1994).
Cell Culture and Transfections
3T3 cells were maintained in DMEM supplemented with 10% FBS. Transfections were performed with FuGene 6 (Roche), according to the manufacturer’s recommendations in 35 mm dishes. A total of 1 μg each of reporter and expression plasmid was used in each transfection so that the quantity of DNA was always held constant at 2 μg per sample. Cells were cultured for 48 h after transfection, harvested, and assayed using the Luminescent β-gal Detection System (Clontech), as described previously (Dodou et al., 2003). Plasmid pRK5-MEF2C contains the mouse MEF2C cDNA in the pRK-5 mammalian expression vector (BD Pharmingen). Plasmid pRK5-MEF2C-VP16 is the same except that the Herpesvirus VP16 activation domain has been fused in-frame to the C-terminus of the MEF2C coding sequence.
Supplementary Material
Acknowledgments
We thank Gail Martin, Deepak Srivastava, Jim Martin, John Rubenstein, Eric Olson and Rhonda Bassel-Duby for providing probes. We are grateful to Juhee Jeong, John Rubenstein, and David Clouthier for helpful discussions, and John Rubenstein and Lou Reichhardt for providing mice. MPV and PA were supported by predoctoral fellowships from the Howard Hughes Medical Institute and the Canadian Institutes of Health Research, respectively. This work was supported by grants from the NIH to BLB.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Beverdam A, Merlo GR, Paleari L, Mantero S, Genova F, Barbieri O, Janvier P, Levi G. Jaw transformation with gain of symmetry after Dlx5/Dlx6 inactivation: mirror of the past? Genesis. 2002;34:221–227. doi: 10.1002/gene.10156. [DOI] [PubMed] [Google Scholar]
- Black BL, Olson EN. Transcriptional control of muscle development by myocyte enhancer factor-2 (MEF2) proteins. Annu Rev Cell Dev Biol. 1998;14:167–196. doi: 10.1146/annurev.cellbio.14.1.167. [DOI] [PubMed] [Google Scholar]
- Charite J, McFadden DG, Merlo G, Levi G, Clouthier DE, Yanagisawa M, Richardson JA, Olson EN. Role of Dlx6 in regulation of an endothelin-1-dependent, dHAND branchial arch enhancer. Genes Dev. 2001;15:3039–3049. doi: 10.1101/gad.931701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clouthier DE, Hosoda K, Richardson JA, Williams SC, Yanagisawa H, Kuwaki T, Kumada M, Hammer RE, Yanagisawa M. Cranial and cardiac neural crest defects in endothelin-A receptor-deficient mice. Development. 1998;125:813–824. doi: 10.1242/dev.125.5.813. [DOI] [PubMed] [Google Scholar]
- Clouthier DE, Williams SC, Yanagisawa H, Wieduwilt M, Richardson JA, Yanagisawa M. Signaling pathways crucial for craniofacial development revealed by endothelin-A receptor-deficient mice. Dev Biol. 2000;217:10–24. doi: 10.1006/dbio.1999.9527. [DOI] [PubMed] [Google Scholar]
- Cobourne MT. Construction for the modern head: current concepts in craniofacial development. J Orthod. 2000;27:307–314. doi: 10.1093/ortho/27.4.307. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- Depew MJ, Lufkin T, Rubenstein JL. Specification of jaw subdivisions by Dlx genes. Science. 2002;298:381–385. doi: 10.1126/science.1075703. [DOI] [PubMed] [Google Scholar]
- Depew MJ, Simpson CA, Morasso M, Rubenstein JL. Reassessing the Dlx code: the genetic regulation of branchial arch skeletal pattern and development. J Anat. 2005;207:501–561. doi: 10.1111/j.1469-7580.2005.00487.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinwiddie R. Congenital upper airway obstruction. Paediatr Respir Rev. 2004;5:17–24. doi: 10.1016/j.prrv.2003.10.001. [DOI] [PubMed] [Google Scholar]
- Dodou E, Xu SM, Black BL. mef2c is activated directly by myogenic basic helix-loop-helix proteins during skeletal muscle development in vivo. Mech Dev. 2003;120:1021–1032. doi: 10.1016/s0925-4773(03)00178-3. [DOI] [PubMed] [Google Scholar]
- Edmondson DG, Lyons GE, Martin JF, Olson EN. Mef2 gene expression marks the cardiac and skeletal muscle lineages during mouse embryogenesis. Development. 1994;120:1251–1263. doi: 10.1242/dev.120.5.1251. [DOI] [PubMed] [Google Scholar]
- Farlie PG, McKeown SJ, Newgreen DF. The neural crest: basic biology and clinical relationships in the craniofacial and enteric nervous systems. Birth Defects Res C Embryo Today. 2004;72:173–189. doi: 10.1002/bdrc.20013. [DOI] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the mouse embryo. 2. Plainview, NY: Cold Spring Harbor Laboratory Press; 1994. [Google Scholar]
- Kedzierski RM, Yanagisawa M. Endothelin system: the double-edged sword in health and disease. Annu Rev Pharmacol Toxicol. 2001;41:851–876. doi: 10.1146/annurev.pharmtox.41.1.851. [DOI] [PubMed] [Google Scholar]
- Knecht AK, Bronner-Fraser M. Induction of the neural crest: a multigene process. Nat Rev Genet. 2002;3:453–461. doi: 10.1038/nrg819. [DOI] [PubMed] [Google Scholar]
- Kurihara Y, Kurihara H, Suzuki H, Kodama T, Maemura K, Nagai R, Oda H, Kuwaki T, Cao WH, Kamada N, et al. Elevated blood pressure and craniofacial abnormalities in mice deficient in endothelin-1. Nature. 1994;368:703–710. doi: 10.1038/368703a0. [DOI] [PubMed] [Google Scholar]
- Le Douarin NM, Creuzet S, Couly G, Dupin E. Neural crest cell plasticity and its limits. Development. 2004;131:4637–4650. doi: 10.1242/dev.01350. [DOI] [PubMed] [Google Scholar]
- Lin Q, Lu J, Yanagisawa H, Webb R, Lyons GE, Richardson JA, Olson EN. Requirement of the MADS-box transcription factor MEF2C for vascular development. Development. 1998;125:4565–4574. doi: 10.1242/dev.125.22.4565. [DOI] [PubMed] [Google Scholar]
- Lin Q, Schwarz J, Bucana C, Olson EN. Control of mouse cardiac morphogenesis and myogenesis by transcription factor MEF2C. Science. 1997;276:1404–1407. doi: 10.1126/science.276.5317.1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JF, Bradley A, Olson EN. The paired-like homeo box gene MHox is required for early events of skeletogenesis in multiple lineages. Genes Dev. 1995;9:1237–1249. doi: 10.1101/gad.9.10.1237. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. Control of muscle development by dueling HATs and HDACs. Curr Opin Genet Dev. 2001;11:497–504. doi: 10.1016/s0959-437x(00)00224-0. [DOI] [PubMed] [Google Scholar]
- McKinsey TA, Zhang CL, Olson EN. MEF2: a calcium-dependent regulator of cell division, differentiation and death. Trends Biochem Sci. 2002;27:40–47. doi: 10.1016/s0968-0004(01)02031-x. [DOI] [PubMed] [Google Scholar]
- Rojas A, De Val S, Heidt AB, Xu SM, Bristow J, Black BL. Gata4 expression in lateral mesoderm is downstream of BMP4 and is activated directly by Forkhead and GATA transcription factors through a distal enhancer element. Development. 2005;132:3405–3417. doi: 10.1242/dev.01913. [DOI] [PubMed] [Google Scholar]
- Ruest LB, Hammer RE, Yanagisawa M, Clouthier DE. Dlx5/6-enhancer directed expression of Cre recombinase in the pharyngeal arches and brain. Genesis. 2003;37:188–194. doi: 10.1002/gene.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruest LB, Xiang X, Lim KC, Levi G, Clouthier DE. Endothelin-A receptor-dependent and -independent signaling pathways in establishing mandibular identity. Development. 2004;131:4413–4423. doi: 10.1242/dev.01291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Stanier P, Moore GE. Genetics of cleft lip and palate: syndromic genes contribute to the incidence of non-syndromic clefts. Hum Mol Genet . 2004;13 Spec No 1:R73–81. doi: 10.1093/hmg/ddh052. [DOI] [PubMed] [Google Scholar]
- Thomas T, Kurihara H, Yamagishi H, Kurihara Y, Yazaki Y, Olson EN, Srivastava D. A signaling cascade involving endothelin-1, dHAND and msx1 regulates development of neural-crest-derived branchial arch mesenchyme. Development. 1998;125:3005–3014. doi: 10.1242/dev.125.16.3005. [DOI] [PubMed] [Google Scholar]
- Trainor PA. Specification of neural crest cell formation and migration in mouse embryos. Semin Cell Dev Biol. 2005;16:683–693. doi: 10.1016/j.semcdb.2005.06.007. [DOI] [PubMed] [Google Scholar]
- Vong LH, Ragusa MJ, Schwarz JJ. Generation of conditional Mef2cloxP/loxP mice for temporal- and tissue-specific analyses. Genesis. 2005;43:43–48. doi: 10.1002/gene.20152. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Clouthier DE, Richardson JA, Charite J, Olson EN. Targeted deletion of a branchial arch-specific enhancer reveals a role of dHAND in craniofacial development. Development. 2003;130:1069–1078. doi: 10.1242/dev.00337. [DOI] [PubMed] [Google Scholar]
- Yanagisawa H, Yanagisawa M, Kapur RP, Richardson JA, Williams SC, Clouthier DE, de Wit D, Emoto N, Hammer RE. Dual genetic pathways of endothelin-mediated intercellular signaling revealed by targeted disruption of endothelin converting enzyme-1 gene. Development. 1998;125:825–836. doi: 10.1242/dev.125.5.825. [DOI] [PubMed] [Google Scholar]
- Zerucha T, Stuhmer T, Hatch G, Park BK, Long Q, Yu G, Gambarotta A, Schultz JR, Rubenstein JL, Ekker M. A highly conserved enhancer in the Dlx5/Dlx6 intergenic region is the site of cross-regulatory interactions between Dlx genes in the embryonic forebrain. J Neurosci. 2000;20:709–721. doi: 10.1523/JNEUROSCI.20-02-00709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.