

The most important contribution to the understanding and management of paraneoplastic neurologic disorders (PND) is the discovery that many of these disorders are immune mediated. It is believed that cytotoxic T-cell responses and antibodies that target neuronal proteins usually expressed by the underlying tumor cause the neurologic symptoms [1]. The detection of these antibodies has provided diagnostic tests that allow recognition of the disorder as paraneoplastic and direct the search of the tumor to selected organs [2].
Despite the diagnostic usefulness of the paraneoplastic antibodies, their detection does not supersede the importance of the clinical assessment, because some antibodies can be found in patients who have cancer without PND [3] and, conversely, approximately 40% of patients who have PND do not have detectable antibodies [4]. Furthermore, rigorous correlations indicate that in the appropriate clinical setting some antibodies are specific markers of PND (ie, anti-Hu, anti-Yo, anti-CV2, anti-Ma2) [4], whereas others (ANNA3, PCA2) are less specific markers of PND [5].
A better understanding of the function of the paraneoplastic neuronal (or onconeuronal) antigens along with modelling PND in animals results in improved treatment strategies. For the clinician who currently confronts these patients, however, the best chance to affect the neurologic outcome depends on: (1) the prompt diagnosis of the disorder, (2) the early discovery and treatment of the tumor, and (3) the use of immunotherapy. Likewise, any clinical features or tests suggesting that the patient's syndrome is not a PND are also important to prevent delays incurred by unnecessary oncologic evaluations.
In 60% of patients who have PND the neurologic symptoms develop before the presence of cancer is known, so these patients are usually seen first by general practitioners or neurologists [6]. In an attempt to improve the recognition of these syndromes, the authors recently proposed a logical approach to the management of limbic encephalitis and postulated that many patients without well-characterized antibodies harbor novel immune responses [6,7]. This approach takes into consideration the type of syndrome, the neuroimaging and cerebrospinal fluid (CSF) findings, and whether the autoantigens are intracellular or are located in the cell membrane. Disorders associated with intracellular autoantigens usually associate with cytotoxic T-cell mechanisms and are less likely to improve than are disorders that associate with autoantigens in the cell membrane. This review summarizes the authors' findings of limbic encephalitis and postulate that a similar approach can be used for syndromes involving other areas of the nervous system.
HISTORICAL REMARKS
Limbic encephalitis causes impressive deficits that are characteristically dominated by rapid and severe loss of short-term memory, but recognition of this syndrome did not occur until the 1960s, when most other PNDs were already known. It was Brierley and colleagues [8] who initially reported three patients who had “subacute encephalitis of later adult life, mainly affecting the limbic areas”; two of the patients had evidence of cancer (one confirmed at autopsy), but the investigators considered “most unlikely that this finding was in any way related to the encephalitis although its occurrence should be noted.” In 1968 Corsellis and colleagues [9] coined the term “limbic encephalitis” to describe one patient who had severe short-term memory loss and two patients who had memory loss and dementia in association with bronchial carcinoma; the three patients had inflammatory and degenerative changes concentrated in the temporal parts of the limbic gray matter. The same investigators reviewed eight previously reported cases and established for the first time a relationship between limbic encephalitis and systemic cancer.
Once the relationship between cancer and the limbic dysfunction was established, three pathogenic hypotheses were proposed: (1) a degeneration (not further defined) of the nervous system in which inflammatory infiltrates were a secondary “reaction to the tissue breakdown,” (2) a viral infection, and (3) an immune-mediated response against the nervous system that is the currently accepted hypothesis.
The first immune response identified in association with limbic encephalitis was the anti-Hu antibody [10]. This antibody associates with small cell lung cancer (SCLC) and paraneoplastic limbic encephalitis that usually affects other areas of the nervous system (encephalomyelitis). Since then, other immune responses have been identified, some of them with more syndrome specificity for limbic dysfunction than the anti-Hu immune response (Table 1) [11-13].
Table 1.
Paraneoplastic antibodies that may associate with limbic encephalitis
| Antibody | Syndrome | Cancer |
|---|---|---|
| Hu | Limbic encephalitis, encephalomyelitis | SCLC, other |
| Ma proteins | Limbic, hypothalamic,a and brainstem encephalitis | Testis, lung, other |
| CV2/CRMP5 | Limbic, striatal encephalitis (chorea), cerebellar ataxia, peripheral neuropathy, uveitis | SCLC, thymoma |
| Amphiphysin | Limbic encephalitis, stiff-person syndrome | Breast, SCLC |
Hypothalamic pituitary hormonal deficits; decreased CSF hypocretin (associated with hypersomnia, narcolepsy).
RECOGNIZING THE SYNDROME
Paraneoplastic limbic encephalitis usually presents with irritability, depression, insomnia or hypersomnia, seizures, hallucinations, and short-term memory loss that may progress to frank dementia [14]. Psychomotor or temporal lobe seizures predominate over generalized seizures [15]. Most patients have EEG abnormalities that may include foci of epileptic activity in one or both temporal lobes or focal or generalized slow activity [16]. In patients who have seizures, the memory deficits can be difficult to examine, and the absent history of cancer may complicate even more the recognition of the disorder as paraneoplastic. Conversely, in patients known to have cancer (∼30%–40% of cases) the development of similar symptoms may suggest several complications of cancer or its treatment and diverse etiologies that can also occur in noncancer patients (Table 2). The clinical history and identification of inflammatory abnormalities in the CSF rules out some of these etiologies and increases the index of suspicion for paraneoplastic limbic encephalitis, although similar findings can be found in viral infections and autoimmune disorders. Approximately 70% of patients who have limbic encephalitis develop MRI abnormalities in the medial temporal lobes that are best seen with fluid-attenuated inversion recovery (FLAIR) sequences [14,16]. The abnormalities can be asymmetric and occasionally may show contrast enhancement. Brain fluorodeoxyglucose-PET (FDG-PET) is particularly useful in patients without seizures and normal MRI; it usually shows FDG hyperactivity in temporal lobes and may reveal other areas of hypermetabolism, suggesting additional foci of encephalitis (Fig. 1) [6,17,18].
Table 2.
Differential diagnosis of limbic encephalopathy
| Disorder | Distinctive features or tests |
|---|---|
| Disorders that selectively involve the limbic system | |
| Herpes simplex encephalitis | HSV DNA in CSF (sensitivity 94%, specificity 98%) |
| Paraneoplastic limbic encephalitis | Paraneoplastic antibodies detectable in serum and CSF of 60% of patients (see subtypes in Table 1) |
| Autoimmune non-paraneoplastic limbic encephalitis | VGKC (may also occur as paraneoplastic manifestation of thymoma, SCLC) |
| Disorders that predominantly involve the limbic system | |
| Degenerative disorders (Alzheimer disease, fronto-temporal dementia, mild cognitive impairment) | Amnestic syndrome may predominate at early stages |
| Severe hypoxia | History of cardiac arrest, carbon monoxide poisoning, or drug overdose |
| Transient global amnesia | Bitemporal hypoperfusion as shown by SPECT or abnormal diffusion-weighted MRI |
| Temporal lobe seizures | Abnormal FLAIR and diffusion-weighted MRI in temporal lobes following status epilepticus; hippocampal atrophy in mesial temporal sclerosis |
| Endocrine dysfunction (Cushing disease, corticosteroid treatment, post-traumatic stress disorder) | Decreased hippocampal volume may be found in chronic hypercortisolism |
| Wernicke-Korsakoff encephalopathy | Deficit of B1: poor nutrition, consumption by tumor |
| Doxifluridine | |
| Disorders that may involve the limbic system | |
| Head trauma | Contusion affecting inferomedial or anterior temporal lobes (“contre-coup” lesion) |
| Systemic autoimmune disorders | |
| Lupus erythematosus | Ribosomal-P antibodies |
| Hashimoto encephalitis | Thyroperoxidase and thyroglobulin antibodies |
| Sjögren syndrome | Anti-Ro(SSA)/La(SSB); salivary gland biopsy |
| Infections | HHV6 (usually after stem cell transplantation) |
| Neurosyphilis | |
| Tumors (gliomatosis cerebri) | Diagnostic brain biopsy |
| Stroke with bilateral posterior cerebral artery involvement | Amnestic syndrome often coexists with cortical blindness, prosopagnosia, apraxia of ocular movements, and other |
Abbreviations: Anti-Ro(SSA)/La(SSB), Sjögren's syndrome serology; CSF, cerebrospinal fluid; HSV, herpes simplex virus; VGKC, voltage-gated potassium channels.
Fig. 1.
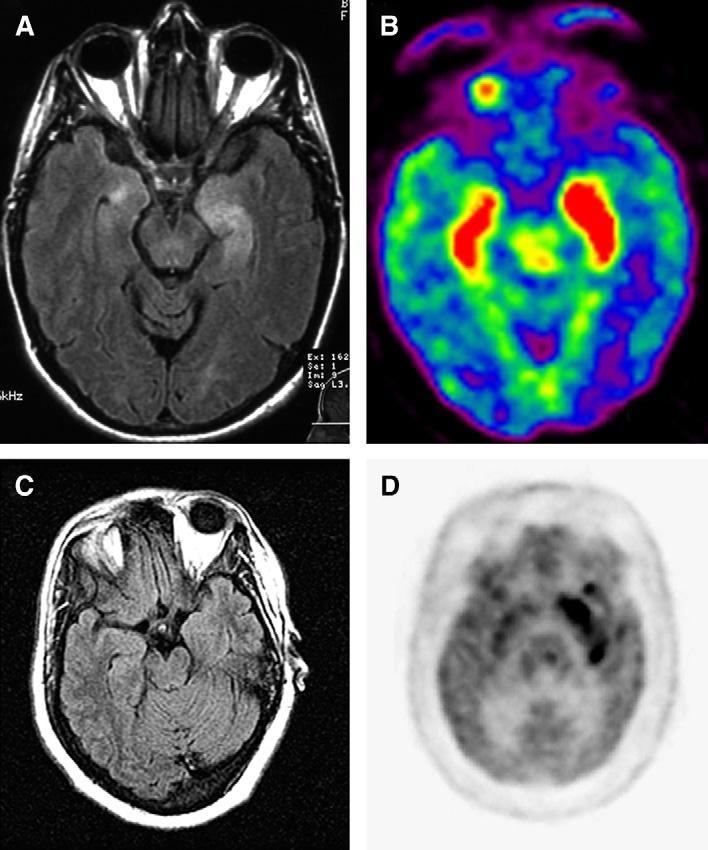
MRI and FDG-PET in patients who have paraneoplastic limbic encephalitis (A) (brain MRI) and (B) (FDG-PET) of a patient who had SCLC and anti-Hu associated paraneoplastic limbic encephalitis; note that there is bilateral medial temporal lobe FLAIR hyperintensity that correlates with FDG hyperactivity (red) in the PET study. (C) Brain MRI and (D) FDG-PET of a patient who had paraneoplastic encephalitis associated with carcinoma of the thymus; in this patient the PET study revealed an extensive area of FDG hyperactivity (dark signal in the medial left temporal lobe) that was not noted in the MRI.
Overall the information provided by the clinical and electrophysiologic findings, routine CSF studies, and MRI and metabolic neuroimaging serves to establish the diagnosis of limbic encephalitis in 70% to 80% of patients. None of these studies, however, defines a paraneoplastic etiology that should always be considered in patients who have a subacute limbic syndrome.
RECOGNIZING THE PARANEOPLASTIC ETIOLOGY
There are several disorders unrelated to cancer that may cause limbic dysfunction (Table 2). A paraneoplastic etiology can only be established with the demonstration of paraneoplastic antibodies in serum or CSF or with the demonstration of a tumor [4]. Neuropathologic studies do not establish a paraneoplastic etiology, because similar inflammatory infiltrates, neuronal loss, microglial activation, and gliosis can be observed in non-paraneoplastic disorders.
The antibodies that are reliable markers of a paraneoplastic etiology are shown in Table 1. Antibodies to voltage-gated potassium channels (VGKC) are considered markers of non-paraneoplastic limbic encephalitis, but in fact may occur in patients who have cancer, and therefore their detection does not exclude a paraneoplastic etiology [19].
In a study of 50 patients and a review of 176 previously reported cases, the tumors more frequently associated with limbic encephalitis were SCLC, testicular tumors, teratoma of the ovary, Hodgkin lymphoma, and breast cancer [14].
DIAGNOSTIC DILEMMAS
In 40% to 50% of patients who have a clinical syndrome compatible with limbic encephalitis, no paraneoplastic antibodies are identified [14,20]. Additionally there are other patients whose symptoms of limbic dysfunction are atypical or develop in association with clinical and neuroimaging findings, suggesting involvement outside the limbic system [21]. Because the CSF of these patients often demonstrates pleocytosis and intrathecal IgG synthesis, and because symptoms may respond to immunotherapy, a possible immunopathogenesis was considered [22]. The working hypothesis was that many of these patients develop antibody-associated immune responses that were missed using current diagnostic techniques. To test this hypothesis the authors examined patients' sera and CSF with several tissue processing and immunohistochemical techniques and with cultures of rat hippocampal neurons; these studies resulted in the identification of several novel autoantibodies.
NOVEL NEURONAL ANTIBODIES
Encephalitis Associated with Antibodies Against the Neuropil and Dendrites of Hippocampus
Using modified immunohistochemical techniques, Ances and colleagues [6] found that patients who had encephalitis predominantly involving the temporal lobes harbored serum or CSF antibodies to antigens expressed in the cell membrane of neurons and dendritic processes of the neuropil of the hippocampus. These antibodies are detected using paraformaldehyde fixed tissue from non-perfused animals and are missed by immunoblot, immunoprecipitation with dendrotoxin (used to detect VGKC antibodies), and immunohistochemistry with methanol-acetone or acetone-fixed tissue. Because some of these novel antibodies are highly specific for hippocampal proteins, they are also missed by commercial laboratories that only use cerebellum as the tissue substrate for immunologic studies. Two additional features that may complicate the detection of these antibodies are the predominant detection in the CSF rather than serum and a tendency to disappear with the neurologic improvement.
Preliminary characterization of the autoantigens indicated that these are diverse and concentrated in the hippocampus. Some autoantigens partially colocalized with synaptophysin and spinophilin, suggesting an immune-mediated attack on hippocampal dendrites (Fig. 2) [23].
Fig. 2.
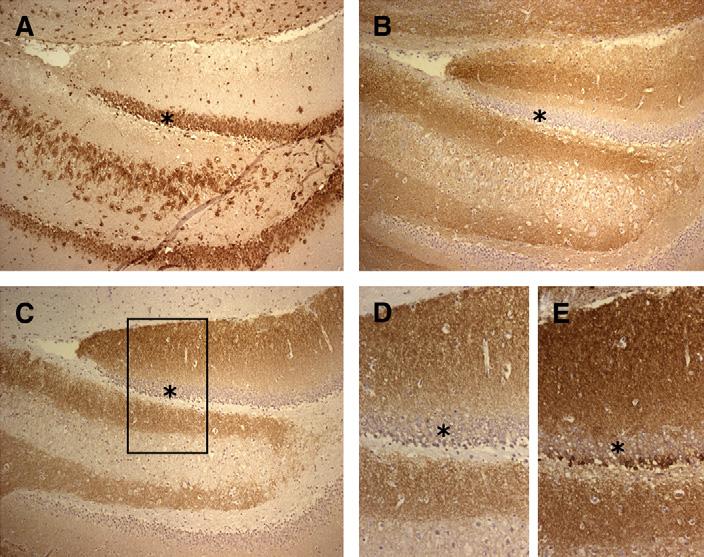
Antibodies to intracellular and cell-membrane antigens in patients who had limbic encephalitis. (A) Coronal section of rat hippocampus immunolabeled with anti-Hu antibodies. (B) and (C) Consecutive sections of hippocampus immunolabeled with Kv 1.2 VGKC antibodies (B) and a novel neuropil antibody (C). (D) The same antibody reactivity demonstrated in (C) at a higher magnification (rectangular region). (E) Reactivity of another neuropil antibody in a consecutive section of the same region. Note the difference between (A) and the rest of the panels; the anti-Hu antibody (A) reacts with intracellular antigens (Hu), whereas the other antibodies react with areas of the neuropil that are rich in dendrites and synapses but spare the neuronal cell bodies. In all panels (*) are placed in the same region (neurons of the dentate gyrus) to allow comparison between reactivities. (A, B, and C) ×100; (D and E) ×200. Sections counterstained with hematoxylin.
Attempts to characterize each of the neuropil autoantigens and corresponding sub-syndromes have resulted in the identification of a specific immune-mediated phenotype in patients who have ovarian teratoma (see later discussion) [7]. These patients' sera or CSF frequently show immunolabeling of antigens that are expressed at the cytoplasmic membrane of hippocampal neurons and processes (Fig. 3A,B). The immunolabeling also occurs in cultures of live hippocampal neurons to which minimal amounts of antibodies are added briefly to the media, suggesting that the antigens are on the cell surface (Fig. 3C). Immunoprobing of a hippocampal-expression library with patients' antibodies resulted in the isolation of EFA6A, a protein that interacts with a member of the two-pore-domain potassium channel family and is involved in the regulation of the dendritic development of hippocampal neurons. Some members of this ion channel family (ie, TASK-1 and TASK-3) are known to play roles in controlling breathing and arousal [24,25].
Fig. 3.
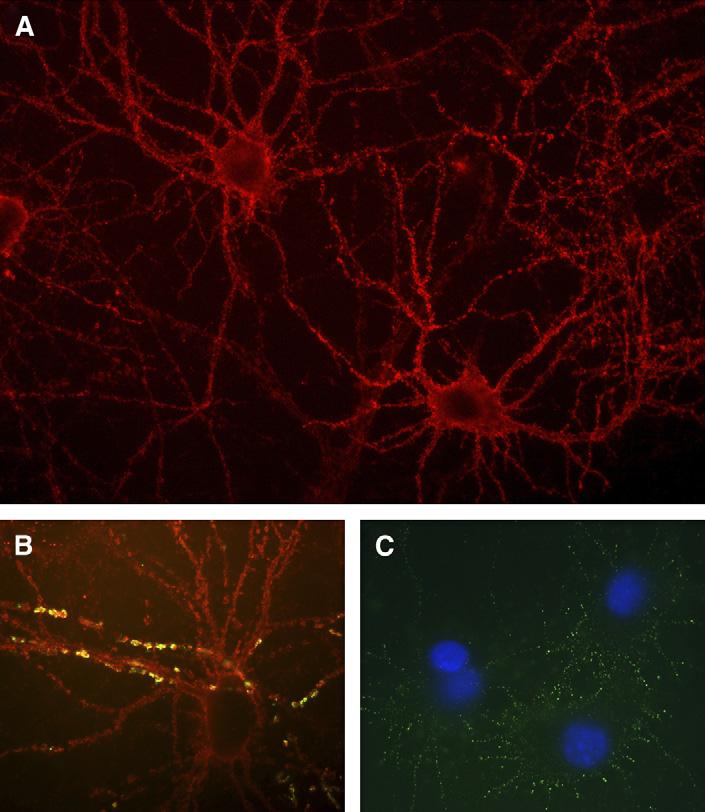
Immunolabeling of cultured hippocampal neurons. (A) Immunolabeling of neurons with CSF of a patient who had carcinoma of the thymus and paraneoplastic limbic encephalitis associated with antibodies to the neuropil of the hippocampus (antigen unknown). Note the intense immunolabeling of the neuronal cell membrane. (B) Immunolabeling of neurons with antibodies (red) from serum of a patient who had limbic encephalitis without a known cancer. The segments of dendrites in yellow demonstrate co-localization of the autoantigen (unknown) with spinophilin, a marker of dendritic spines. (C) Immunolabeling of the surface of live hippocampal neurons with antibodies from a patient who had encephalitis associated with ovarian teratoma; the autoantigen co-localizes with EFA6A. (A) ×400; (B and C) ×800 with oil.
CLINICAL-IMMUNOLOGIC PHENOTYPES OF PARANEOPLASTIC LIMBIC ENCEPHALITIS
Based on these studies the authors propose three groups of immune-mediated limbic encephalitis, each likely including several sub-phenotypes as outlined here and in Table 3.
Table 3.
Clinical features, response to treatment, and prognosis related to type of antibody and location of antigens
| Neuronal antibodies: Hu, Ma2, CV2/CRMP5, amphiphysin, atypical (intracellular antigens) |
VGKC antibodies (cell membrane antigens) |
Novel neuropil antibodies (cell membrane antigens) |
|
|---|---|---|---|
| Hippocampal specificity of antibodies | No; antibodies react with neurons of any part of the neuraxis | Mild; all patients with similar pattern of antibody reactivity | Intense; different patterns (some with pure limbic reactivity) |
| CSF inflammatory abnormalitiesa | Frequent | Infrequent (normal CSF or with mild abnormalities) | Frequent |
| Intrathecal synthesis of antibodies | Frequent | Infrequent/absent | Frequent |
| Hyponatremia | No (except some patients with SCLC) | Frequent | No |
| Clinical phenotypes other than limbic encephalitis | Several according to type of antibody [66] | Neuromyotonia; Morvan syndrome [38] | Prominent behavioral and psychiatric symptoms and seizures; central hypoventilation may occur [6,7] |
| Brain MRI | Frequent medial temporal lobe FLAIR/T2 hyperintensities (classic findings) | Frequent classic findings | Infrequent classic findings but frequent temporal lobe involvement |
| Tumor association | SCLC, non-SCLC, testicular tumors, thymoma, other | Infrequent: SCLC, thymoma | Frequent: teratoma, thymoma |
| Response to treatment (tumor and/or immunosuppression) | Rare; except for patients with testicular tumors and Ma2 encephalitis | Frequent (corticosteroids, IVIg, plasma exchange) | Frequent (tumor and/or corticosteroids, IVIg, plasma exchange) |
| Clinical course | Progressive until stabilization or death (Hu, CV2/CRMP5); relapses are rare | Relapses may occur and are treatable | Relapses may occur and are treatable |
| Antibody titers | Usually detectable for months or years | Decrease or disappear in months | Decrease or disappear in months |
Pleocytosis, increased protein concentration, elevated IgG index, oligoclonal bands.
Limbic Encephalitis and Antibodies to Well-Characterized Intracellular Onconeuronal Antigens (Hu, Ma, CV2/CRMP5, and Amphiphysin)
Clinical features that preferentially associate with each autoantigen are shown in Table 1 [20,26-28]. Irrespective of the type of immunity, the CSF of these patients usually shows moderate pleocytosis and increased protein concentration, elevated IgG synthesis, and oligoclonal bands. The corresponding paraneoplastic antibodies are found in the CSF and almost always in the serum. The antibodies are usually detectable in serum for months or years after treatment of the tumor or immunotherapy [29]. Autopsy and biopsy studies suggest that cytotoxic T-cell mechanisms play a prominent pathogenic role [26,30,31]. There are no animal models for any of these immunities except for amphiphysin-associated stiff-person syndrome [32]. The management should focus on the prompt diagnosis and treatment of the tumor. At early stages of the disease, when there is evidence of inflammation suggested by CSF analysis or PET neuroimaging, immunotherapies directed at antibodies and cytotoxic T-cells can be considered [33,34]. The use of corticosteroids, IVIg, or plasma exchange has limited value if the tumor is not treated. Cyclophosphamide has been used in individual cases with mild to moderate effects [35,36].
Some patients who have antibodies to Ma2 proteins have better neurologic outcome than patients who have other antibodies (ie, Hu, CV2/CRMP5). The reason for the different outcomes is unclear but may be related in part to the fact immunity to Ma2 often associates with testicular tumors that are easier to remove and are more responsive to chemotherapy than other neoplasms (ie, SCLC) [26].
Limbic Encephalitis and Antibodies to VGKC (Kv1.1, Kv1.2, and Kv 1.6)
In addition to limbic encephalitis, patients who have these antibodies may develop peripheral nerve hyperexcitability, autonomic dysfunction, hyperhydrosis, seizures (without evidence of limbic encephalopathy), rapid eye movement sleep behavior abnormalities, or a combination of peripheral and central nervous system dysfunction called Morvan syndrome [37-39].
The limbic encephalitis of patients who have VGKC antibodies frequently associates with hyponatremia and infrequently with cancer. Hyponatremia cannot be used as a distinctive feature of this immunity, however, because it also occurs in 11% of patients who have SCLC as part of the paraneoplastic syndrome of inappropriate secretion of antidiuretic hormone (SIADH) [40]. The brain MRI findings of patients who have VGKC antibodies are similar to other types of immune-mediated limbic encephalitis [41], although some paraneoplastic immunities (ie, anti-Ma2) often show additional abnormalities in the hypothalamus and upper brainstem that may enhance with contrast [42].
When compared with any other type of limbic encephalitis, patients who have VGKC antibodies are less likely to develop CSF pleocytosis or intrathecal synthesis of IgG and usually do not have intrathecal synthesis of VGKC antibodies [6,38,41].
There is no single distinctive feature of limbic encephalitis associated with VGKC antibodies; this disorder therefore should be considered in patients who have no smoking history and develop subacute symptoms of limbic dysfunction in association with hyponatremia with or without autonomic dysfunction or peripheral nerve excitability. Supportive laboratory studies include symmetric or asymmetric medial temporal lobe MRI FLAIR hyperintensities and normal or mild CSF abnormalities. For this group of patients, in particular those who are young, a thymoma may be present [43]. Similar findings can occur in older individuals and smokers, and in these patients a paraneoplastic manifestation of SCLC should be strongly considered [19,37].
The treatment of limbic encephalitis with VGKC antibodies is based on IgG-depleting strategies, such as plasma exchange or IVIg, in combination with corticosteroids. In the authors' experience, tapering prednisone over 3 to 4 weeks after intravenous methylprednisolone may result in neurologic relapse. It is therefore reasonable to use daily prednisone for 3 to 4 weeks and then convert to an every-other-day regimen for several months. Significant neurologic improvement or recovery occurs in 70% of the patients [38,41]. At early stages of the disorder some patients develop life-threatening hyponatremia and status epilepticus; the long-term outcome is usually good, although short-term memory deficits may persist.
Limbic Encephalitis of Patients Who Have Antibodies to Novel Neuronal Membrane Antigens
This is a heterogeneous group of disorders that may encompass a larger number of patients than those who have antibodies to VGKC (Kv1.1, Kv1.2, and Kv 1.6). The common clinical phenotype includes predominant behavioral and psychiatric symptoms (that may obscure short-term memory deficits), seizures, and brain MRI abnormalities that are less frequently restricted to the hippocampus than in classic limbic encephalitis. FDG-PET may reveal foci of hypermetabolism in the frontotemporal lobes, brainstem, or cerebellum. Combining MRI and FDG-PET studies, the temporal lobes are preferentially affected. Patients harboring these novel antibodies are more likely to have intrathecal IgG synthesis, CSF pleocytosis, and systemic tumors (thymoma, teratoma) than those who have VGKC antibodies and do not develop hyponatremia [6].
In contrast to patients who have paraneoplastic antibodies to intracellular antigens, the encephalitis of patients who have any of these collectively termed “neuropil antibodies” improves with immunotherapy and, if present, treatment of the associated tumor. This clinical improvement usually associates with improvement of MRI and FDG-PET abnormalities and a decrease of antibody titers [6].
The Encephalitic Syndrome Associated with Ovarian Teratoma and Antibodies to Antigens That Co-Localize with EFA6A
A subgroup of patients who have antibodies to neuronal cell membrane antigens have ovarian teratoma. These patients harbor antibodies to antigens that co-localize with EFA6A, which is a hippocampal protein upregulated by N-Methyl-D-Aspartate (NMDA) receptors. The clinical and immunologic similarities of these patients suggest that they are affected by a distinct syndrome. This includes subacute psychiatric symptoms, short-term memory deficits, seizures, rapid decrease of level of consciousness, and frequent central hypoventilation [44,45]. The clinical picture, young age of the patients, and absent history of cancer may lead to a diagnosis of acute psychosis, malingering, or drug abuse. A similar encephalitic syndrome had been previously reported in several patients who have ovarian teratoma; in most instances an immunopathogenesis was suspected but not identified [22,46-49].
The CSF of these patients usually shows pleocytosis and elevated protein concentration and IgG index. In some patients the EFA6A-like reactive antibodies can only be demonstrated in the CSF.
Because EFA6A interacts with a member of the two-pore domain potassium channel, and some members of this potassium channel family seem to be important for control of breathing and arousal, it is tempting to speculate that the patients' antibodies disrupt the function of these channels [24,25].
Despite the severity of the clinical features, most patients who have teratoma-associated encephalitis improve with treatment of the tumor, plasma exchange, IVIg, and corticosteroids [7]. The limited number of cases prevents firm treatment recommendations, but removal of the teratoma seems important. Some of these patients require intensive and prolonged care because of ventilatory problems, and the recovery is usually slow (weeks or months). In a recent review of nine patients who had encephalitis associated with ovarian teratoma, one of the patients died as a result of neurologic complications and the other patients significantly improved or totally recovered. Since then, the authors have identified four additional patients who have the same syndrome and recovered after combining these treatments; one patient did not improve after tumor removal but had a dramatic response to corticosteroids and cyclophosphamide.
Limbic Encephalitis: A Model for Other Paraneoplastic Disorders
A question raised by these studies is whether a similar diversity of phenotypes and immune responses against intracellular and cell membrane antigens occurs in other paraneoplastic disorders. For syndromes involving the peripheral nervous system, several immune mechanisms mediated by humoral or cytotoxic T-cell responses have been demonstrated. For example, antibodies to ion channels or receptors are involved in the dysfunction of the neuromuscular junction and nerves, resulting in the Lambert-Eaton myasthenic syndrome (LEMS), neuromyotonia, and myasthenia gravis [50,51]. Humoral immune mechanisms also seem to play critical roles in dermatomyositis and several neuropathies associated with monoclonal gammopathies [52-54], whereas predominant cytotoxic T-cell responses are involved in polymyositis, vasculitis of the nerve and muscle, and several subacute predominantly axonal neuropathies [55-58].
In the paraneoplastic disorders of the CNS, the study of novel immune responses is limited by the difficulty in obtaining tissue and in demonstrating antibodies against cell membrane antigens. In limbic encephalitis, an observation that led the authors to reassess the presence of immune responses was that some patients improve with immunosuppressants. For other paraneoplastic disorders, however, such as cerebellar or brainstem encephalopathies, the potential for recovery or improvement is more limited. This has been noted in patients who have disorders mediated by antibodies, such as LEMS and cerebellar degeneration in association with antibodies to voltage-gated calcium channels (VGCC). Although immunomodulation and immunosuppression improve the symptoms of LEMS, the concomitant cerebellar deficits are much less responsive to treatment [59-61]. Despite this low potential for recovery, there are reports of patients who have paraneoplastic cerebellar dysfunction whose symptoms improved after treatment of the tumor or immunosuppression [62-65]. In some of these patients, as in approximately 40% of patients who have cancer and subacute cerebellar degeneration without paraneoplastic antibodies, the CSF showed inflammatory abnormalities identical to those found in patients who have paraneoplastic antibodies, suggesting an immune-mediated pathogenesis. These findings and recent evidence that patients may harbor antibodies that spare neuronal cell bodies but react intensively with the neuropil and dendrites of the molecular layer of the cerebellum (Fig. 4) (Josep Dalmau, MD, unpublished data, 2005) suggests that the antigen diversity described in limbic encephalitis may also occur in other syndromes. The authors anticipate that analysis of the CSF at symptom presentation with techniques that allow detection of antibodies to cell membrane antigens will reveal novel immune responses. Characterization of these immune responses hastens diagnosis, improves treatment of the associated disorders, indicates the relative risk for an underlying tumor (paraneoplasia), and suggests the type of tumor more frequently involved.
Fig. 4.
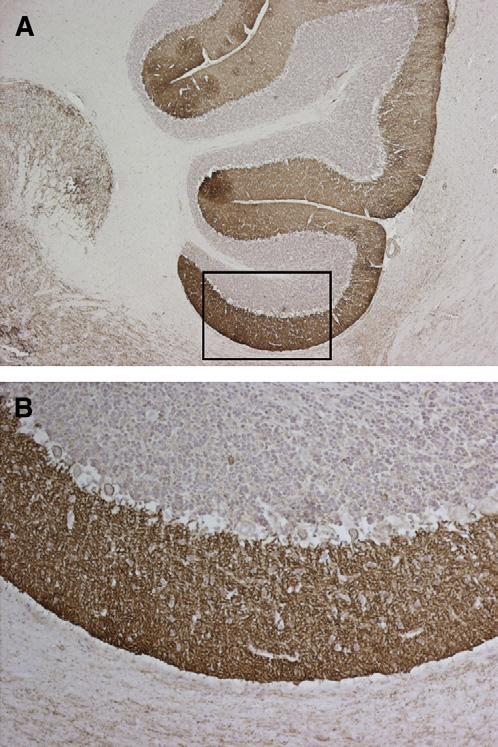
Patient who had subacute cerebellar dysfunction and CSF antibodies against the molecular layer of the cerebellum. (A) A section of rat cerebellum immunostained with the antibodies from the CSF of a patient who had predominant subacute cerebellar degeneration and mild memory deficits without a known cancer. (B) Higher magnification of the area indicated in (A). Note that there is selective and intense immunolabeling of the neuronal processes running in the molecular layer of the cerebellum; the neuronal cell bodies of the granular layer, Purkinje cells, and cells of the molecular layer are spared. The dendrotoxin immunoprecipitation assay was negative, indicating that the autoantigen (unknown) is not a Kv1.1, 1,2, or 1,6 VGKC. (A) ×200; (B) ×400. Sections mildly counterstained with hematoxylin.
SUMMARY
The authors recently proposed a logical approach to the management of limbic encephalitis and postulated that many patients who do not have well-characterized antineuronal antibodies harbor novel immune responses. This approach takes into consideration the type of syndrome, the neuroimaging and CSF findings, and whether the autoantigens are intracellular or located in the cell membrane. Disorders related with the first class of autoantigens usually associate with antibodies and cytotoxic T-cell mechanisms and are less likely to improve than are disorders associated with the second class of autoantigens. The latter comprises a diverse group of encephalitides with emerging sub-phenotypes in which humoral mechanisms seem to play a pathogenic role. This article summarizes the authors' experience with limbic encephalitis and postulate that a similar approach can be used for other paraneoplastic disorders.
Acknowledgment
This work has been supported in part by RO1CA89054 and RO1CA107192 (JD). We thank Dr. Myrna R. Rosenfeld for critical review of the manuscript.
References
- 1.Dalmau J, Gultekin HS, Posner JB. Paraneoplastic neurologic syndromes: pathogenesis and physiopathology. Brain Pathol. 1999;9:275–84. doi: 10.1111/j.1750-3639.1999.tb00226.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Posner JB. Neurologic complications of cancer. F.A. Davis Co.; Philadelphia: 1995. [Google Scholar]
- 3.Dalmau J, Furneaux HM, Gralla RJ, et al. Detection of the anti-Hu antibody in the serum of patients with small cell lung cancer—a quantitative western blot analysis. Ann Neurol. 1990;27:544–52. doi: 10.1002/ana.410270515. [DOI] [PubMed] [Google Scholar]
- 4.Graus F, Delattre JY, Antoine JC, et al. Recommended diagnostic criteria for paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 2004;75:1135–40. doi: 10.1136/jnnp.2003.034447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pittock SJ, Kryzer TJ, Lennon VA. Paraneoplastic antibodies coexist and predict cancer, not neurological syndrome. Ann Neurol. 2004;56:715–9. doi: 10.1002/ana.20269. [DOI] [PubMed] [Google Scholar]
- 6.Ances BM, Vitaliani R, Taylor RA, et al. Treatment-responsive limbic encephalitis identified by neuropil antibodies: MRI and PET correlates. Brain. 2005;128:1764–77. doi: 10.1093/brain/awh526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vitaliani R, Mason W, Ances B, et al. Paraneoplastic encephalitis, psychiatric symptoms, and hypoventilation in ovarian teratoma. Ann Neurol. 2005;58:594–604. doi: 10.1002/ana.20614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brierley JB, Corsellis JAN, Hierons R, et al. Subacute encephalitis of later adult life. Mainly affecting the limbic areas. Brain. 1960;83:357–68. [Google Scholar]
- 9.Corsellis JA, Goldberg GJ, Norton AR. “Limbic encephalitis” and its association with carcinoma. Brain. 1968;91:481–96. doi: 10.1093/brain/91.3.481. [DOI] [PubMed] [Google Scholar]
- 10.Dalmau J, Graus F, Rosenblum MK, et al. Anti-Hu–associated paraneoplastic encephalomyelitis/sensory neuronopathy. A clinical study of 71 patients. Medicine (Balt) 1992;71:59–72. doi: 10.1097/00005792-199203000-00001. [DOI] [PubMed] [Google Scholar]
- 11.Voltz R, Gultekin SH, Rosenfeld MR, et al. A serologic marker of paraneoplastic limbic and brain-stem encephalitis in patients with testicular cancer. N Engl J Med. 1999;340:1788–95. doi: 10.1056/NEJM199906103402303. [DOI] [PubMed] [Google Scholar]
- 12.Honnorat J, Antoine JC, Derrington E, et al. Antibodies to a subpopulation of glial cells and a 66 kDa developmental protein in patients with paraneoplastic neurological syndromes. J Neurol Neurosurg Psychiatry. 1996;61:270–8. doi: 10.1136/jnnp.61.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antoine JC, Absi L, Honnorat J, et al. Antiamphiphysin antibodies are associated with various paraneoplastic neurological syndromes and tumors. Arch Neurol. 1999;56:172–7. doi: 10.1001/archneur.56.2.172. [DOI] [PubMed] [Google Scholar]
- 14.Gultekin SH, Rosenfeld MR, Voltz R, et al. Paraneoplastic limbic encephalitis: neurological symptoms, immunological findings and tumour association in 50 patients. Brain. 2000;123:1481–94. doi: 10.1093/brain/123.7.1481. [DOI] [PubMed] [Google Scholar]
- 15.Fadul CE, Stommel EW, Dragnev KH, et al. Focal paraneoplastic limbic encephalitis presenting as orgasmic epilepsy. J Neurooncol. 2005;72:195–8. doi: 10.1007/s11060-004-2242-9. [DOI] [PubMed] [Google Scholar]
- 16.Lawn ND, Westmoreland BF, Kiely MJ, et al. Clinical, magnetic resonance imaging, and electroencephalographic findings in paraneoplastic limbic encephalitis. Mayo Clin Proc. 2003;78:1363–8. doi: 10.4065/78.11.1363. [DOI] [PubMed] [Google Scholar]
- 17.Kassubek J, Juengling FD, Nitzsche EU, et al. Limbic encephalitis investigated by 18FDG-PET and 3D MRI. J Neuroimaging. 2001;11:55–9. doi: 10.1111/j.1552-6569.2001.tb00011.x. [DOI] [PubMed] [Google Scholar]
- 18.Scheid R, Lincke T, Voltz R, et al. Serial 18F-fluoro-2-deoxy-D-glucose positron emission tomography and magnetic resonance imaging of paraneoplastic limbic encephalitis. Arch Neurol. 2004;61:1785–9. doi: 10.1001/archneur.61.11.1785. [DOI] [PubMed] [Google Scholar]
- 19.Pozo-Rosich P, Clover L, Saiz A, et al. Voltage-gated potassium channel antibodies in limbic encephalitis. Ann Neurol. 2003;54:530–3. doi: 10.1002/ana.10713. [DOI] [PubMed] [Google Scholar]
- 20.Alamowitch S, Graus F, Uchuya M, et al. Limbic encephalitis and small cell lung cancer. Clinical and immunological features. Brain. 1997;120:923–8. doi: 10.1093/brain/120.6.923. [DOI] [PubMed] [Google Scholar]
- 21.Mihara M, Sugase S, Konaka K, et al. The “pulvinar sign” in a case of paraneoplastic limbic encephalitis associated with non-Hodgkin's lymphoma. J Neurol Neurosurg Psychiatry. 2005;76:882–4. doi: 10.1136/jnnp.2004.049783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nokura K, Yamamoto H, Okawara Y, et al. Reversible limbic encephalitis caused by ovarian teratoma. Acta Neurol Scand. 1997;95:367–73. doi: 10.1111/j.1600-0404.1997.tb00227.x. [DOI] [PubMed] [Google Scholar]
- 23.Law AJ, Weickert CS, Hyde TM, et al. Reduced spinophilin but not microtubule-associated protein 2 expression in the hippocampal formation in schizophrenia and mood disorders: molecular evidence for a pathology of dendritic spines. Am J Psychiatry. 2004;161:1848–55. doi: 10.1176/ajp.161.10.1848. [DOI] [PubMed] [Google Scholar]
- 24.Washburn CP, Bayliss DA, Guyenet PG. Cardiorespiratory neurons of the rat ventrolateral medulla contain TASK-1 and TASK-3 channel mRNA. Respir Physiol Neurobiol. 2003;138:19–35. doi: 10.1016/s1569-9048(03)00185-x. [DOI] [PubMed] [Google Scholar]
- 25.Bayliss DA, Talley EM, Sirois JE, et al. TASK-1 is a highly modulated pH-sensitive ‘leak’ K(+) channel expressed in brainstem respiratory neurons. Respir Physiol. 2001;129:159–74. doi: 10.1016/s0034-5687(01)00288-2. [DOI] [PubMed] [Google Scholar]
- 26.Dalmau J, Graus F, Villarejo A, et al. Clinical analysis of anti-Ma2-associated encephalitis. Brain. 2004;127:1831–44. doi: 10.1093/brain/awh203. [DOI] [PubMed] [Google Scholar]
- 27.Yu Z, Kryzer TJ, Griesmann GE, et al. CRMP-5 neuronal autoantibody: marker of lung cancer and thymoma-related autoimmunity. Ann Neurol. 2001;49:146–54. [PubMed] [Google Scholar]
- 28.Dorresteijn LD, Kappelle AC, Renier WO, et al. Anti-amphiphysin associated limbic encephalitis: a paraneoplastic presentation of small-cell lung carcinoma. J Neurol. 2002;249:1307–8. doi: 10.1007/s00415-002-0677-5. [DOI] [PubMed] [Google Scholar]
- 29.Llado A, Mannucci P, Carpentier AF, et al. Value of Hu antibody determinations in the follow-up of paraneoplastic neurologic syndromes. Neurology. 2004;63:1947–9. doi: 10.1212/01.wnl.0000144340.03364.bf. [DOI] [PubMed] [Google Scholar]
- 30.Jean WC, Dalmau J, Ho A, et al. Analysis of the IgG subclass distribution and inflammatory infiltrates in patients with anti-Hu-associated paraneoplastic encephalomyelitis. Neurology. 1994;44:140–7. doi: 10.1212/wnl.44.1.140. [DOI] [PubMed] [Google Scholar]
- 31.Bernal F, Graus F, Pifarre A, et al. Immunohistochemical analysis of anti-Hu-associated paraneoplastic encephalomyelitis. Acta Neuropathol (Berl) 2002;103:509–15. doi: 10.1007/s00401-001-0498-0. [DOI] [PubMed] [Google Scholar]
- 32.Sommer C, Weishaupt A, Brinkhoff J, et al. Paraneoplastic stiff-person syndrome: passive transfer to rats by means of IgG antibodies to amphiphysin. Lancet. 2005;365:1406–11. doi: 10.1016/S0140-6736(05)66376-3. [DOI] [PubMed] [Google Scholar]
- 33.Sillevis SP, Grefkens J, De Leeuw B, et al. Survival and outcome in 73 anti-Hu positive patients with paraneoplastic encephalomyelitis/sensory neuronopathy. J Neurol. 2002;249:745–53. doi: 10.1007/s00415-002-0706-4. [DOI] [PubMed] [Google Scholar]
- 34.Graus F, Keime-Guibert F, Rene R, et al. Anti-Hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients. Brain. 2001;124:1138–48. doi: 10.1093/brain/124.6.1138. [DOI] [PubMed] [Google Scholar]
- 35.Keime-Guibert F, Graus F, Fleury A, et al. Treatment of paraneoplastic neurological syndromes with antineuronal antibodies (Anti-Hu, anti-Yo) with a combination of immunoglobulins, cyclophosphamide, and methylprednisolone. J Neurol Neurosurg Psychiatry. 2000;68:479–82. doi: 10.1136/jnnp.68.4.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Vernino S, O'Neill BP, Marks RS, et al. Immunomodulatory treatment trial for paraneoplastic neurological disorders. Neuro-oncol. 2004;6:55–62. doi: 10.1215/S1152851703000395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liguori R, Vincent A, Clover L, et al. Morvan's syndrome: peripheral and central nervous system and cardiac involvement with antibodies to voltage-gated potassium channels. Brain. 2001;124:2417–26. doi: 10.1093/brain/124.12.2417. [DOI] [PubMed] [Google Scholar]
- 38.Vincent A, Buckley C, Schott JM, et al. Potassium channel antibody-associated encephalopathy: a potentially immunotherapy-responsive form of limbic encephalitis. Brain. 2004;127:701–12. doi: 10.1093/brain/awh077. [DOI] [PubMed] [Google Scholar]
- 39.Iranzo A, Graus F, Clover L, et al. Rapid eye movement sleep behavior disorder and potassium channel antibody-associated limbic encephalitis. Ann Neurol. 2006;59:178–81. doi: 10.1002/ana.20693. [DOI] [PubMed] [Google Scholar]
- 40.List AF, Hainsworth JD, Davis BW, et al. The syndrome of inappropriate secretion of antidiuretic hormone (SIADH) in small-cell lung cancer. J Clin Oncol. 1986;4:1191–8. doi: 10.1200/JCO.1986.4.8.1191. [DOI] [PubMed] [Google Scholar]
- 41.Thieben MJ, Lennon VA, Boeve BF, et al. Potentially reversible autoimmune limbic encephalitis with neuronal potassium channel antibody. Neurology. 2004;62:1177–82. doi: 10.1212/01.wnl.0000122648.19196.02. [DOI] [PubMed] [Google Scholar]
- 42.Rosenfeld MR, Eichen JG, Wade DF, et al. Molecular and clinical diversity in paraneoplastic immunity to Ma proteins. Ann Neurol. 2001;50:339–48. [PubMed] [Google Scholar]
- 43.Buckley C, Oger J, Clover L, et al. Potassium channel antibodies in two patients with reversible limbic encephalitis. Ann Neurol. 2001;50:73–8. doi: 10.1002/ana.1097. [DOI] [PubMed] [Google Scholar]
- 44.Muni RH, Wennberg R, Mikulis DJ, et al. Bilateral horizontal gaze palsy in presumed paraneoplastic brainstem encephalitis associated with a benign ovarian teratoma. J Neuroophthalmol. 2004;24:114–8. doi: 10.1097/00041327-200406000-00004. [DOI] [PubMed] [Google Scholar]
- 45.Stein-Wexler R, Wootton-Gorges SL, Greco CM, et al. Paraneoplastic limbic encephalitis in a teenage girl with an immature ovarian teratoma. Pediatr Radiol. 2005;35:694–7. doi: 10.1007/s00247-005-1402-1. [DOI] [PubMed] [Google Scholar]
- 46.Okamura H, Oomori N, Uchitomi Y. An acutely confused 15-year-old girl. Lancet. 1997;350:488. doi: 10.1016/S0140-6736(97)06208-9. see comments. [DOI] [PubMed] [Google Scholar]
- 47.Aydiner A, Gurvit H, Baral I. Paraneoplastic limbic encephalitis with immature ovarian teratoma—a case report. J Neurooncol. 1998;37:63–6. doi: 10.1023/a:1005875822605. [DOI] [PubMed] [Google Scholar]
- 48.Fadare O, Hart HJ. Anti-Ri antibodies associated with short-term memory deficits and a mature cystic teratoma of the ovary. Int Semin Surg Oncol. 2004;1:11. doi: 10.1186/1477-7800-1-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee AC, Ou Y, Lee WK, et al. Paraneoplastic limbic encephalitis masquerading as chronic behavioural disturbance in an adolescent girl. Acta Paediatr. 2003;92:506–9. doi: 10.1111/j.1651-2227.2003.tb00588.x. [DOI] [PubMed] [Google Scholar]
- 50.Newsom-Davis J. Therapy in myasthenia gravis and Lambert-Eaton myasthenic syndrome. Semin Neurol. 2003;23:191–8. doi: 10.1055/s-2003-41135. [DOI] [PubMed] [Google Scholar]
- 51.Hart IK, Maddison P, Newsom-Davis J, et al. Phenotypic variants of autoimmune peripheral nerve hyperexcitability. Brain. 2002;125:1887–95. doi: 10.1093/brain/awf178. [DOI] [PubMed] [Google Scholar]
- 52.Kissel JT, Mendell JR, Rammohan KW. Microvascular deposition of complement membrane attack complex in dermatomyositis. N Engl J Med. 1986;314:329–34. doi: 10.1056/NEJM198602063140601. [DOI] [PubMed] [Google Scholar]
- 53.Rudnicki SA, Harik SI, Dhodapkar M, et al. Nervous system dysfunction in Waldenström's macroglobulinemia: response to treatment. Neurology. 1998;51:1210–3. doi: 10.1212/wnl.51.4.1210. [DOI] [PubMed] [Google Scholar]
- 54.Vital A, Favereaux A, Martin-Dupont P, et al. Anti-myelin-associated glycoprotein antibodies and endoneurial cryoglobulin deposits responsible for a severe neuropathy. Acta Neuropathol (Berl) 2001;102:409–12. doi: 10.1007/s004010100381. [DOI] [PubMed] [Google Scholar]
- 55.Amato AA, Barohn RJ. Idiopathic inflammatory myopathies. Neurol Clin. 1997;15:615–48. doi: 10.1016/s0733-8619(05)70337-6. [DOI] [PubMed] [Google Scholar]
- 56.Younger DS, Dalmau J, Inghirami G, et al. Anti-Hu-associated peripheral nerve and muscle microvasculitis. Neurology. 1994;44:181–3. doi: 10.1212/wnl.44.1.181-a. [DOI] [PubMed] [Google Scholar]
- 57.Blumenthal D, Schochet S, Jr, Gutmann L, et al. Small-cell carcinoma of the lung presenting with paraneoplastic peripheral nerve microvasculitis and optic neuropathy. Muscle Nerve. 1998;21:1358–9. doi: 10.1002/(sici)1097-4598(199810)21:10<1358::aid-mus25>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 58.Antoine JC, Mosnier JF, Absi L, et al. Carcinoma associated paraneoplastic peripheral neuropathies in patients with and without anti-onconeural antibodies. J Neurol Neurosurg Psychiatry. 1999;67:7–14. doi: 10.1136/jnnp.67.1.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Clouston PD, Saper CB, Arbizu T, et al. Paraneoplastic cerebellar degeneration. III. Cerebellar degeneration, cancer, and the Lambert-Eaton myasthenic syndrome. Neurology. 1992;42:1944–50. doi: 10.1212/wnl.42.10.1944. [DOI] [PubMed] [Google Scholar]
- 60.Blumenfeld AM, Recht LD, Chad DA, et al. Coexistence of Lambert-Eaton myasthenic syndrome and subacute cerebellar degeneration: differential effects of treatment. Neurology. 1991;41:1682–5. doi: 10.1212/wnl.41.10.1682. [DOI] [PubMed] [Google Scholar]
- 61.Goldstein JM, Waxman SG, Vollmer TL, et al. Subacute cerebellar degeneration and Lambert-Eaton myasthenic syndrome associated with antibodies to voltage-gated calcium channels: differential effect of immunosuppressive therapy on central and peripheral defects. J Neurol Neurosurg Psychiatry. 1994;57:1138–9. doi: 10.1136/jnnp.57.9.1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Paone JF, Jeyasingham K. Remission of cerebellar dysfunction after pneumonectomy for bronchogenic carcinoma. N Engl J Med. 1980;302:156. doi: 10.1056/NEJM198001173020305. [DOI] [PubMed] [Google Scholar]
- 63.Counsell CE, McLeod M, Grant R. Reversal of subacute paraneoplastic cerebellar syndrome with intravenous immunoglobulin. Neurology. 1994;44:1184–5. doi: 10.1212/wnl.44.6.1184. [DOI] [PubMed] [Google Scholar]
- 64.Stark E, Wurster U, Patzold U, et al. Immunological and clinical response to immunosuppressive treatment in paraneoplastic cerebellar degeneration. Arch Neurol. 1995;52:814–8. doi: 10.1001/archneur.1995.00540320098016. [DOI] [PubMed] [Google Scholar]
- 65.David YB, Warner E, Levitan M, et al. Autoimmune paraneoplastic cerebellar degeneration in ovarian carcinoma patients treated with plasmapheresis and immunoglobulin. A case report. Cancer. 1996;78:2153–6. [PubMed] [Google Scholar]
- 66.Bataller L, Dalmau JO. Paraneoplastic disorders of the central nervous system: update on diagnostic criteria and treatment. Semin Neurol. 2004;24:461–71. doi: 10.1055/s-2004-861540. [DOI] [PubMed] [Google Scholar]