

Abstract
Molecular oxygen is involved in hydroxylation and subsequent degradation of HIF-1α, a subunit of HIF-1 transcription factor, therefore oxygen shortage (hypoxia) stabilizes this protein. However, HIF-1α can also be stabilized by transition metal ions in the presence of oxygen, suggesting that a different mechanism is involved in metal-induced hypoxic stress. Recently, we showed that the depletion of intracellular ascorbate by metals may lead to the inhibition of hydroxylases. Since nickel(II) has similarity to iron(II), an alternative hypothesis suggests that iron substitution for nickel in the enzyme inhibits hydroxylase activity. Here we investigated the induction of HIF-1 by another metal, chromium, which can not replace iron in the enzyme. We show that chromium(VI), but not chromium(III) can oxidize ascorbate both in cells and in cell-free system. In agreement with these data chromium(VI) stabilizes HIF-1α protein in cells only until it is reduced to chromium(III). In contrast, nickel(II) was found to be a catalyst, which facilitate continuous oxidation of ascorbate by ambient oxygen. These data correlate with extended stabilization of HIF-1α following acute exposure to nickel(II). The HIF-1-dependent reporter assays revealed that 20–24 h was required to fully develop HIF-1 transcriptional response and the acute exposure to nickel(II), but not chromium(VI), meets this requirement. However, repeated (chronic) exposure to chromium(VI) can also lead to extended stabilization of HIF-1α. Thus, obtained data emphasize important role of ascorbate in regulation of HIF-1 transcriptional activity in metal-exposed human lung cells.
Keywords: Ascorbate, Chromium(VI), Nickel(II), HIF-1α, HIF-dependent transcription
Introduction
Modulation of gene expression by metals in the respiratory system is an important factor contributing to the disease development and progression [1]. Ni(II) exposure activates a number of transcription factors in lung epithelial cells including HIF-1, which plays a critical role in the induction of genes involved in inflammation and apoptosis [2–4]. A HIF-1 complex contains an α and a β subunit [5]. Under normoxia, the HIF-1α subunit is rapidly degraded via the von Hippel-Lindau tumor suppressor gene product (pVHL)-mediated ubiquitin-proteasome pathway [6, 7]. The interaction of pVHL and HIF-1α proteins is triggered by the hydroxylation of two prolines in HIF-1α’s oxygen dependent degradation domain. Shortage of oxygen (hypoxia) prevents hydroxylation and subsequent pVHL interaction. This stabilizes HIF-1α protein and results in formation of HIF-1 transcription complex.
Unlike under hypoxia, HIF-1α is stabilized by metals in the presence of oxygen, suggesting that different mechanisms are involved in metal-induced HIF-1 activation. Two metals Co(II) and Ni(II) were shown to be good inducers of HIF-1 [8]. Since they are similar to Fe(II), it was suggested that Ni(II) could either replace iron in the hydroxylases or interfere with iron uptake [9, 10]. Alternatively it was suggested that reactive oxygen species (ROS) may play an important role in HIF-1α stabilization and metals, which are known to generate free radicals in cells, may stabilize HIF-1α through this mechanism [11]. However, there are sufficient data to indicate that Ni(II) and perhaps other metals do not require free ROS generation for HIF-1α stabilization [12, 13]. Recently, we showed that the depletion of intracellular ascorbate by Ni(II) or Co(II) may lead to the inhibition of prolyl hydroxylases [8, 14], which, along with asparaginyl hydroxylase or factor inhibiting HIF (FIH), act as intracellular oxygen sensors [15]. These enzymes are members of the Fe(II)-, 2-oxoglutarate (2OG)-, and ascorbate-dependent family of dioxygenases. The role of ascorbate is to reduce enzyme-bound iron, which is important for maintaining hydroxylases activity.
The active role of ascorbate in the hydroxylase reactions may explain the controversy about the role of ROS in HIF-1 activation and reconcile previously obtained data. Indeed, ROS may deplete through oxidation a variety of reducing molecules. However, since only ascorbate is capable of maintaining iron in the reduced state its presence is critical for hydroxylase activity. Transition metals like Ni(II) and Co(II) can deplete intracellular ascorbate via oxidation and/or by inhibiting ascorbate uptake by cells [14, 16]. If ascorbate depletion is sufficient for HIF-1 induction by Ni(II) or Co(II), which can also substitute for Fe(II) in prolyl hydroxylases, then exposure to other transition metal ions, that can oxidize ascorbate, but would not be able to replace iron, should also stabilize HIF-1α protein. Cr(VI), known to oxidize ascorbate directly, is one of such metal ions [17]. Previous studies showed that Cr(VI) is a principal ascorbate oxidant in rat lung, liver and kidneys [18, 19]. Here we compared the accumulation of HIF-1α protein in human lung epithelial cells following Ni(II) or Cr(VI) exposure. The results show the difference in the time course of this accumulation. Thus, HIF-1α protein is induced only by Cr(VI) and disappears after the latter is reduced to Cr(III). Cell-free experiments show rapid phase of ascorbate oxidation by Cr(VI), which ends after the completion of Cr(VI) reduction to Cr(III). In contrast, Ni(II) appeared to be a catalyst of such oxidation. This leads to a complete depletion of cellular ascorbate and prolonged HIF-1α stabilization in Ni(II)-exposed cells. Since HIF-dependent transcriptional response requires time, prolonged HIF-1α stabilization by Ni(II), but not Cr(VI) results in strong activation of HIF-dependent genes.
Materials and Methods
Reagents
NiSO4 · 6H2O was obtained from Alfa Aesar (Ward Hill, MA). Protease inhibitors cocktail was purchased from Roche (Indianapolis, IN). K2CrO4, CrCl3 · 6H2O, L-ascorbic acid and other chemicals were obtained from Sigma-Aldrich (St. Louis, MO).
Cell Lines and Culture Conditions
The 1HAEo- cell line was obtained from Dr. D.C. Gruenert [20]. Cells were grown on plastic coated with a mixture of BSA (Invitrogen Corporation, Carlsbad, CA) and collagen (Cohesion, Palo Alto, CA) in Minimum Essential Medium with Earle’s modified salts (Invitrogen) containing 10% FCS, 2 mM L-glutamine, 100 μg/ml streptomycin, and 100 U/ml penicillin. A549 cells were grown on F12K medium (Invitrogen) supplemented with 10% FCS, 100 μg/ml streptomycin, and 100 U/ml penicillin. For exposure, cells were plated at a density 25–30×103 cells/cm2 in complete medium under 95% air, 5% CO2, at 37ºC. Cells were exposed to metals 30–36 hours later.
HPLC Determination of Ascorbate Oxidation
Chromatographic separations were done on a Waters Alliance 2695 HPLC system (Milford, MA, USA) with Waters 996 Photodiode array detector using a reverse-phase column Luna C18, 150×4.6 mm (Phenomenex, Torrance, CA, USA) equipped with C18 guard column (4.0×2.0 mm, Phenomenex). The column was heated to 30°C, and the flow rate was 0.6 ml/min. The composition of the mobile phase was modified from Lykkesfeldt [21]. Briefly, the eluent consisted of 15% of methanol, 36.6 μM tetraoctylammonium bromide (Sigma-Aldrich), 0.189 mM n-dodecyltrimethylammonium chloride (Sigma-Aldrich), 1.0 mM Na2-EDTA, (Sigma-Aldrich), 100 mM sodium acetate (EMD Chemicals Inc., Darmstadt, Germany), and 100 mM sodium phosphate monobasic. The pH of the mobile phase was adjusted to pH 5.4 with concentrated ortho-phosphoric acid (Sigma-Aldrich). The samples were incubated in the internal thermostate of the HPLC at 37°C. They consisted of 50 μM ascorbic acid with 0, 100, or 250 μM Ni(II), 0 or 25 μM Cr(III), or 0, 5, or 25 μM Cr(VI) in 50 mM Hepes buffer pH 7.4. Traces of other metal ions were removed from Hepes buffer by pre-treatment with Chelex 100 Resin (Bio-Rad Laboratories) for 24 h. Fifty-μL aliquots of samples were analyzed every 2 h. All measurements were done in triplicate. Cellular samples for ascorbate measurements were prepared as described by Lykkesfeldt [21]. In this case ascorbate was measured using coulometric detector ESA Coulochem II (ESA Inc., Bedford, MA) equipped with a Model 5010A analytical cell operated at +300 mV. All measurements were done in duplicates.
RNA Preparation and Microarray Analysis
Total RNA was extracted using an RNeasy kit (Qiagen, Valencia, CA) from 1HAEo- cells treated with 5 μM Cr(VI) or 0.5 mM Ni(II) for 4 h or 24 h. Cell density and conditions of treatment were identical to conditions of nuclear extracts isolation. Synthesis of cDNA and cRNA, hybridization of labeled cRNA to the human 133A 2.0 chips (Affymetrix Inc., San Diego, CA), and measurement of signal intensities were carried out according to manufacturer’s protocols. Two chips were used for each treatment and the experiment was repeated twice. The results were averaged and the data were stored and analyzed using MS SQL Server Database (Microsoft) and Data Mining Tool software (Affymetrix Inc., San Diego, CA).
Western Blotting
Total cell protein extracts were obtained after lysing cells in the lysis buffer (Cell Signaling, Beverly, MA) for 15 min at 4°C. Equal loading of protein was assured by prior quantitation using the Bradford assay. Forty-μg protein extracts were separated by gel electrophoresis in 4–12% NuPage bis-tris minigels (Invitrogen) in MOPS buffer, pH 7.7 and transferred onto a PVDF membranes (Invitrogen). Nuclei for nuclear extracts preparation were obtained as described earlier [14]. Nuclei were resuspended in 50–150 μl of extraction buffer (0.42 M NaCl, 25% glycerol, 2 mM MgCl2, 0.2 mM Na2-EDTA, 1 mM DTT, 20 mM Hepes, pH 7.9 plus 0.5 mM PMSF, 10 μg/ml Antipain, 1.5 μg/ml Aprotinin, 3 μg/ml Leupeptin, 3 μg/ml Pepstatin, and phosphatase inhibitors) and incubated on ice for 15 min, followed by maximum speed centrifugation in the Eppendorf centrifuge for 15 min at 4°C. Supernatants were aliquoted and stored at −70°C. Fifteen-μg of protein were separated by 4–12% gradient NuPage SDS Bis-Tris gels (Invitrogen, Carlsbad, CA) in MOPS-SDS running buffer (Invitrogen), under reducing conditions. Proteins were transferred onto PVDF membranes. The membranes were blocked using 5% solution of dry milk (Blotting Grade Blocker Non-Fat Dry Milk, Bio-Rad Laboratories, Hercules, CA) in the presence of 5% goat serum (Sigma, Sigma-Aldrich, St. Louis, MO, USA). The protein bands were visualized in membranes using anti-HIF-1α (BD Biosciences Pharmingen, San Jose, CA, USA), anti-HIF-1β, or anti-β-actin antibodies (Novus Biologicals, Littleton, CO, USA). The secondary antibodies were goat anti-rabbit HRP-conjugated antibodies, or goat anti-mouse HRP-conjugated antibodies (Cell Signaling, Danvers, MA, USA). Chemiluminescent signal was detected using Super Signal kit (Pierce Biotechnology Inc., Rockford, Il, USA). The anti-CA9 and NDRG1/Cap43 antibodies were used as previously described [16].
MTS Assay
The MTS assay (Promega, Madison, WI) was utilized for evaluation of cellular reducing capacity. MTS is bioreduced by cells into a formazan product that is soluble outside of cells in the tissue culture medium. The quantity of formazan product as measured by the amount of 490 nm absorbance is directly proportional to the reducing potential of cells. The assay was performed in 96 well plates according to the manufacturer’s instructions in triplicate. Cell number was counted in parallel wells. The results shown are the mean ± SD of one representative experiment.
MTT Assay
The cytotoxicity following metals exposure was assessed by MTT assay (Promega, Madison, WI) according to the manufacturer’s protocol.
Uptake of 51Cr(VI) by Human Lung Cells
For 51Cr(VI) uptake analysis 1HAEo- and A549, cells were plated into 96- well plates at a density of 25×103 cells/well in complete medium. Aliquots of Na251CrO4 (Perkin Elmer, Boston, MA) corresponding to 5, 10 and 20 μM Cr(VI) were added to cell cultures for the time periods shown in the Figure. The measurements were taken in triplicates. After incubation, cells were washed 3 times with 200 μl cold Ca+2 and Mg+2 - free PBS, containing 1 mM EDTA. Aliquots of 100 μl of 0.5% triton X-100 lysis buffer were added to each well and the content of wells was taken for radioactivity counting. The experiment was repeated twice. Cell number was counted in parallel wells. The results shown are the mean values ± SD of one representative experiment.
HRE-Luc Reporter Assay
The transient transfection experiments were carried out as previously described [14]. The transfection experiments were repeated at least twice and each condition has been tested in quadruplicate. The results shown are the mean values ± SD.
Results
HIF-1α Stabilization by Cr(VI)
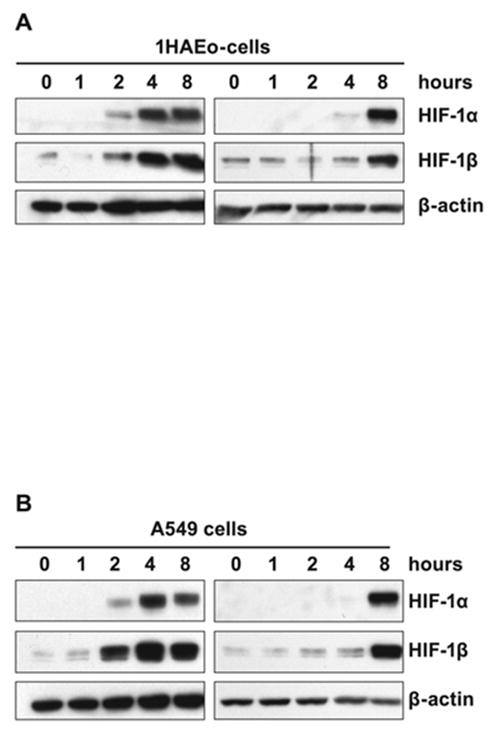
The time-course of accumulation of both HIF-1 α and β subunits in the nuclear fraction of human lung 1HAEo- cells following Ni(II) or Cr(VI) exposure is shown in Fig. 1A, C. The analysis of dose-dependent induction of HIF-1α protein by Cr(VI) revealed no differences between 1 and 10 μM, but enhancement by 25 μM Cr(VI) (Fig. 1B). Since 25 μM Cr(VI) produced significant toxic effect, 5 μM, a concentration of Cr(VI) which caused only 5–10% cell death measured by MTT assay, was chosen for the study. The 500 μM concentration of Ni(II) tested in the present study produced similar toxicity. Cr(VI) as the anion CrO4-2 quickly enters cells where it is reduced by ascorbate to Cr(III) [19, 22]. Thus, it was expected that Cr(VI) would deplete intracellular ascorbate faster than Ni(II). Indeed, the comparison of time-courses of Ni(II)- and Cr(VI)-induced HIF-1α nuclear accumulation shows earlier accumulation following Cr(VI) exposure (Fig. 1A, C). The HIF-1β subunit was not induced by metal’s exposure (not shown), but was accumulated in the nucleus together with HIF-1α (Fig. 1A, C). The maximal accumulation of both subunits in 1HAEo- cells exposed to 5 μM Cr(VI) was observed between 4 to 8 h. After that the amount of both proteins in the nucleus declined.
Figure 1.
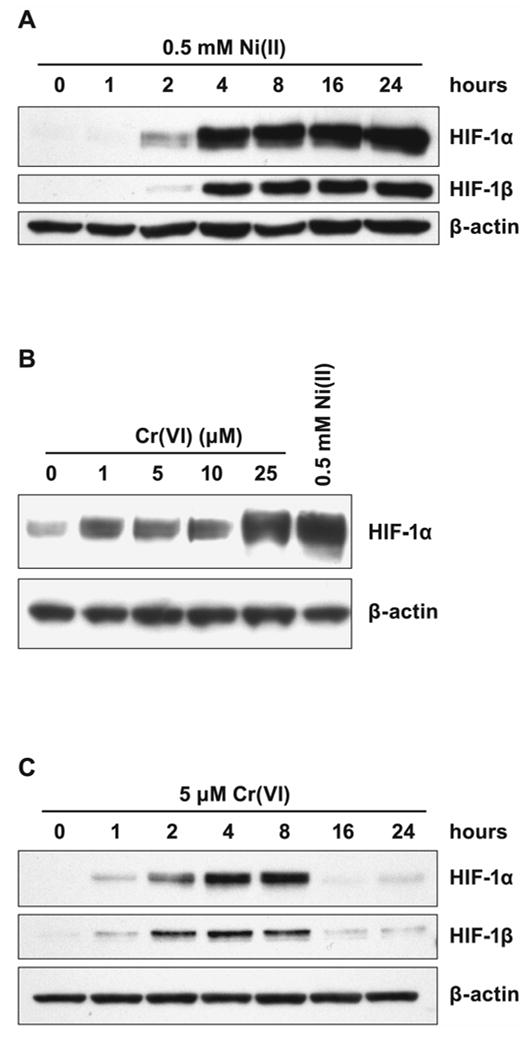
Effects of Ni(II) or Cr(VI) exposure on HIF-1α, and HIF-1β levels in 1HAEo- cells. A, 1HAEo- cells were exposed to 0.5 mM NiSO4 for the time periods shown in the Figure. Nuclear protein extracts (15 μg) were prepared for immunoblotting as described in Materials and Methods and probed with antibodies against HIF-1α, or HIF-1β, and were also probed with antibodies against β-actin to provide loading control. B, 1HAEo- cells were exposed for 4 h to concentrations of K2CrO4 shown in the Figure. The effect of 0.5 mM NiSO4 is shown for comparison. The assay was carried out as described in A. C, 1HAEo- cells were exposed to 5 μM K2CrO4 for the time periods shown in the Figure. The assay was carried out as described in A.
In contrast, in Ni(II) exposed 1HAEo- cells nuclear accumulation of both HIF-1 subunits was markedly increased by 4 h and remained high up to 24 h. To examine whether the drop in HIF-1α protein level in Cr(VI) exposed cells was due to the reduction of all available Cr(VI) and not due to other inhibitory mechanisms which could down-regulate HIF-1α protein in exposed cells, we performed a combined exposure to Ni(II) and Cr(VI). No suppression of HIF-1α protein (Fig. 2A) or HIF-dependent transcription (Fig. 2B) was observed following 20 h of combined exposure to Ni(II) and Cr(VI). Moreover, exposure to both Ni(II) and Cr(VI) resulted in additive effect on both HIF-1α protein level and HIF-dependent transcription. To further confirm that the decline in the amount of HIF-1α protein after 8 h was due to the reduction of available Cr(VI), a fresh aliquot of Cr(VI) was added to cells exposed to Cr(VI) for 16 h. The addition of 5 or 10 μM K2CrO4 resulted in reappearance of HIF-1α protein after an additional 4 h exposure (Fig. 2C).
Figure 2.
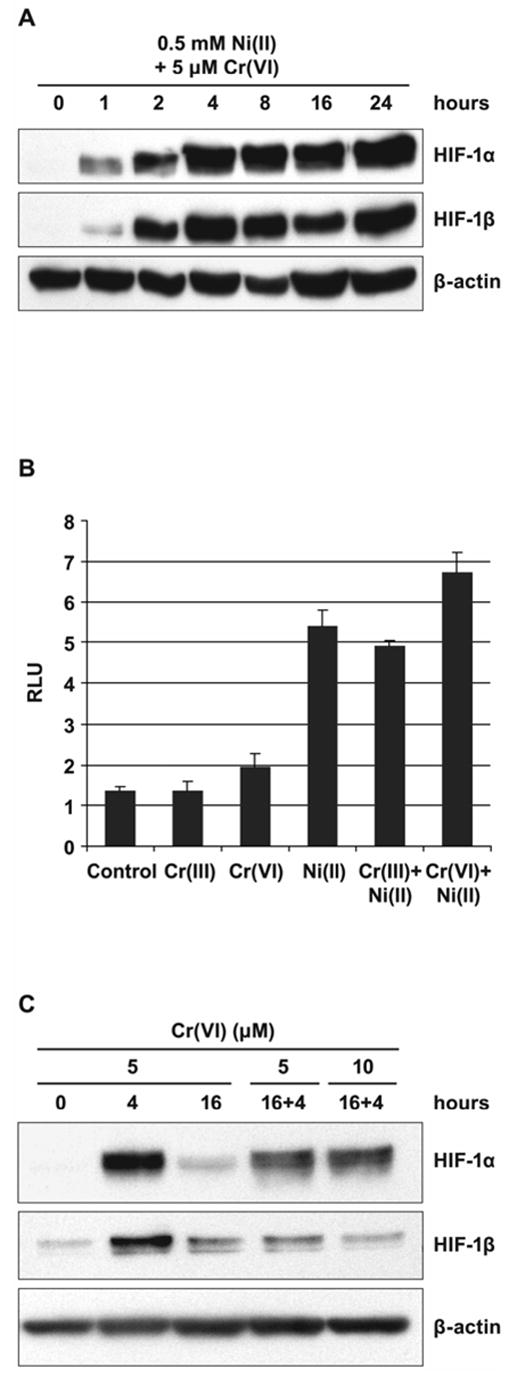
Induction of HIF-1 transcription factor in 1HAEo- cells by combined exposure to Ni(II) and Cr(VI). A, 1HAEo- cells were exposed to 0.5 mM NiSO4 and 5 μM K2CrO4 for the time periods shown in the Figure. Nuclear fractions were prepared for immunoblotting as described in Materials and Methods and probed with antibodies against HIF-1α and HIF-1β, they were also probed with antibodies against β-actin to provide loading control. B, 1HAEo- cells were transfected with HRE-Luc reporter plasmid and exposed to 5 μM CrCl3, 5 μM K2CrO4, or 0.5 mM NiSO4 alone or in combination as shown in the Figure for 20 h. Luciferase activity is expressed as relative luciferase units (RLU). The data are presented as means ± SD. C, 1HAEo- cells were exposed to 5 μM K2CrO4 for 4 or 16 h; after 16 h of exposure, fresh aliquots of 5 and 10 μM K2CrO4 were added to the media for an additional 4-h period. The assay was carried out as described in A.
To further test whether the reduction of Cr(VI) is responsible for the decline of HIF-1α protein, another lung epithelial cell line, A549 was utilized. Unlike 1HAEo- cells these are tumor cells which have significantly higher glycolytic activity than 1HAEo- and therefore a higher reducing capacity as detected by MTS assay (not shown). The higher extracellular reducing capacity of A549 cells causes lower uptake of labeled 51Cr(VI) into A549 cells as compared with 1HAEo- cells (Fig. 3). Thus exposure of A549 cells to 5 μM 51Cr(VI) for 20 h resulted in the accumulation of only one-third the amount of intracellular chromium when compared with 1HAEo- cells. The addition of 50 μM ascorbate to cell medium resulted in quick reduction of all extracellular Cr(VI) into Cr(III), which could not enter the cells (not shown).
Figure 3. Comparison of Cr(VI) uptake into A549 and 1HAEo- cells.
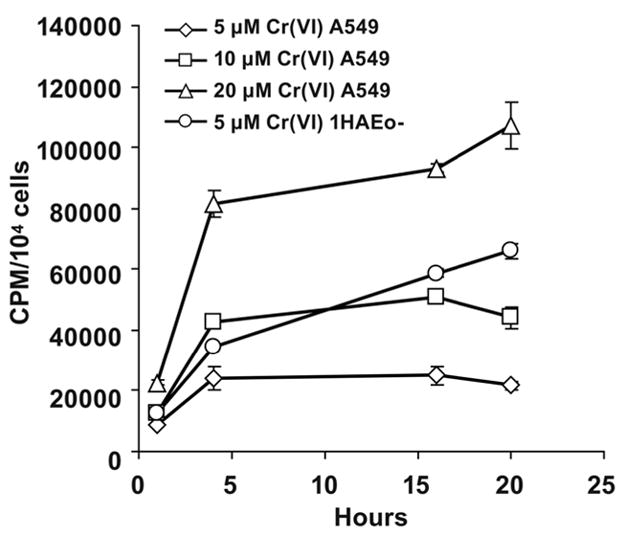
Uptake of Cr(VI) into A549 and 1HAEo- cells was assessed by monitoring accumulation of labeled 51Cr(VI) at time intervals shown in the Figure. Each measurement has been done in triplicates. The data are presented as means ± SD.
The stabilization of HIF-1α protein following Ni(II) exposure in A549 cells was similar to that in 1HAEo- cells (Fig. 4A). However, exposure to 5 μM Cr(VI) did not result in the HIF-1α protein accumulation in these cells (Fig. 4B). As expected from the experiments with labeled 51Cr(VI), more Cr(VI) was required for A549 cells to have intracellular Cr(VI) comparable with the amounts delivered into 1HAEo- cells (Fig. 3). Increasing Cr(VI) concentration up to 20–25 μM resulted in a significant accumulation of HIF-1α protein in A549 cells (Fig. 4B). However, in these cells HIF-1α protein also disappeared after 8 h of exposure suggesting complete reduction of Cr(VI) during this time by A549 cells (Fig. 4C). As in 1HAEo- cells, the addition of fresh Cr(VI) to the medium caused reappearance of HIF-1α protein (Fig. 4D).
Figure 4.
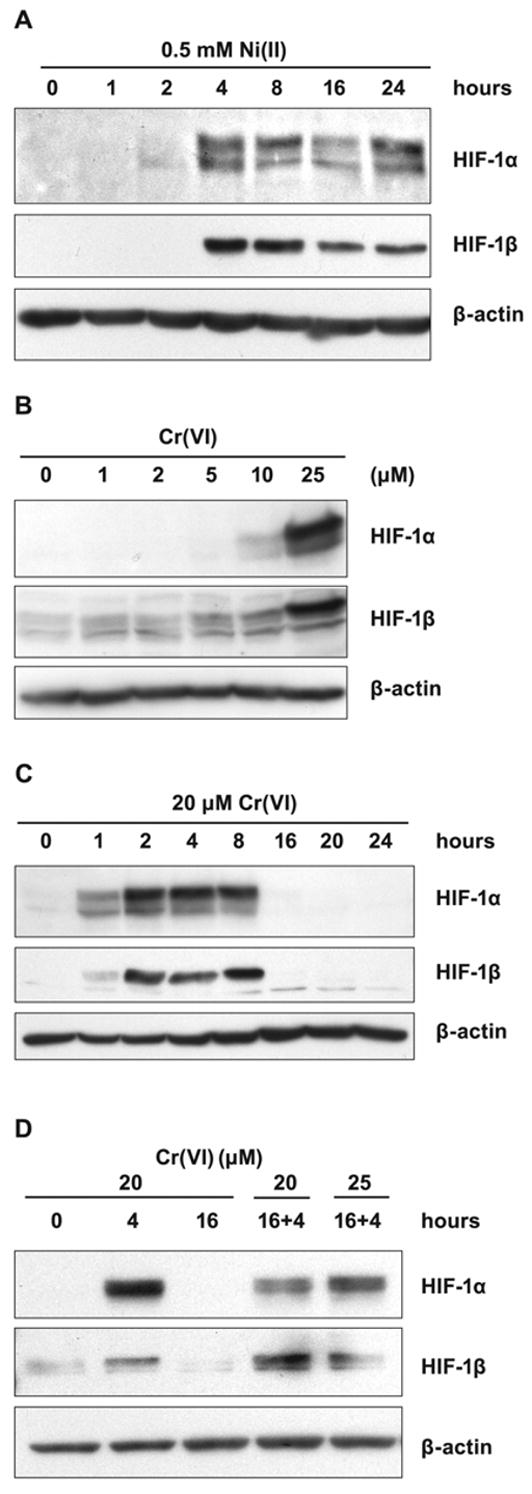
Effects of Ni(II) exposure on HIF-1α and HIF-1β levels in A549 cells. A, A549 cells were exposed to 0.5 mM NiSO4 for the time periods shown in the Figure. Nuclear protein extracts (15 μg) were prepared for immunoblotting as described in Materials and Methods and probed with antibodies directed against HIF-1α and HIF-1β. The membrane was also probed with antibodies against β-actin to provide loading control. B, A549 cells were exposed for 4 h to various concentrations of K2CrO4 shown in the Figure. The assay was carried out as described in A. C, A549 cells were exposed to 20 μM K2CrO4 for the time periods shown in the Figure. The assay was carried out as described in A. D, A549 cells were exposed to 20 μM K2CrO4 for 4 or 16 h, after 16 h fresh aliquots of K2CrO4 were added to make 20 and 25 μM for an additional 4 h. The assay was carried out as described in A.
Effect of Metal Ions on Ascorbate Oxidation in Cell-Free System
In order to compare the effects of the investigated metal ions on the course of ascorbate oxidation, we incubated 50 μM ascorbate in HEPES buffer, pH 7.4, in the absence or presence of 5 or 25 μM Cr(VI), 25 μM Cr(III), or 100 μM Ni(II), under air, at 37ºC (Fig. 5A, B). The concentration of reagents were similar to those used in cell cultures; the 50 μM ascorbic acid concentration corresponded to that found in cells in our previous experiments [14]. The HPLC measurements revealed that in the absence of metals, the concentration of ascorbic acid decreased gradually to 25% of the original level in 16 h of incubation (Fig. 5A). The addition of 5 or 25 μM Cr(VI) resulted in a marked acceleration of ascorbate oxidation (Fig. 5A). However, as soon as all Cr(VI) was reduced (approximately 3 h) ascorbate oxidation slowed down to the rate similar to that observed in the presence of Cr(III) (Fig. 5A). Interestingly, oxidation of ascorbate with ambient oxygen in the presence of Cr(III) was significantly slower than without the metal, suggesting that Cr(III) may even stabilize ascorbate via unknown mechanism (Fig. 5A).
Figure 5.
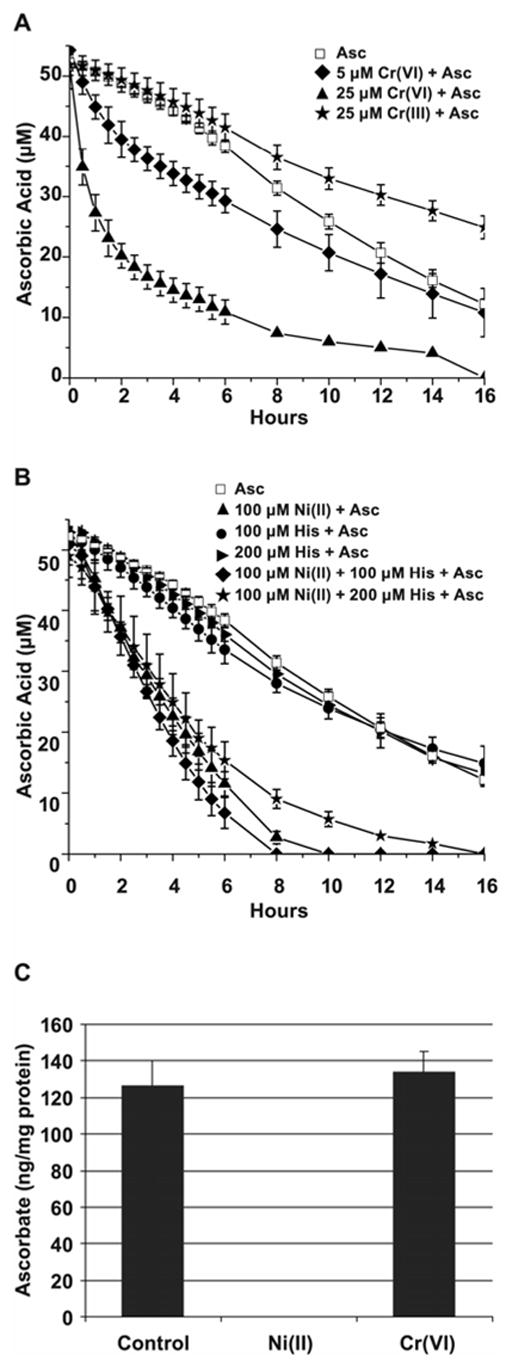
Kinetics of ascorbate oxidation in the presence of metal ions. A, Ascorbate oxidation by Cr(VI) or Cr(III) in cell-free system. 50 μM of ascorbate in Hepes buffer was incubated at 37ºC alone or in the presence of 25 μM Cr(III), or 5 μM, or 25 μM Cr(VI) for the time periods shown in the Figure. The data are presented as mean values ± the SD. B, Ascorbate oxidation by Ni(II) in cell-free system. 50 μM of ascorbate in Hepes buffer was incubated at 37ºC alone or in the presence of 100 μM Ni(II), 100 μM or 200 μM histidine, or all together for the time periods shown in the Figure. The data presened are mean values ± the SD. C, Ascorbate oxidation by metals in 1HAEo- cells. The cells were preloaded with 50 μM ascorbate for 2 h followed by incubation with 0.5 mM NiSO4 or 5 μM K2CrO4 for additional 16 h. Levels of reduced ascorbate were measured at the end of incubation using HPLC as described in Materials and Methods. The data are presented as means ± SD.
In contrast, the nickel-mediated oxidation of ascorbate, though initially slower, was eventually more efficient and resulted in a complete oxidation of ascorbate in 10 h of incubation (Fig. 5B). In the medium or inside the cell Ni(II) can be bound to histidine or histidine-containing peptides. The complex of nickel-histidine 1:1 facilitated ascorbate oxidation more efficiently than nickel alone resulting in complete oxidation of ascorbate in 8 h of incubation (Fig. 5B). Although the 1:2 nickel-histidine complex tended to slow the ascorbate oxidation down, it was still more efficient than that of ascorbate alone. Histidine alone had no effect. Thus, these experiments predicted that Ni(II), or histidine-bound Ni(II) should deplete intracellular ascorbate more efficiently than Cr(VI), which may be quickly and irreversibly converted into inert (or even protective) Cr(III).
Effect of Metal Ions on Ascorbate Oxidation in Cells
The above data suggested that exposure to Ni(II) or Cr(VI) could affect intracellular ascorbate differently. To assess these effects in 1HAEo- cells, cells preloaded with 50 μM ascorbate for 2 h were exposed to 0.5 mM Ni(II) or 5 μM Cr(VI) for 16 h. After this time, the intracellular levels of ascorbate in untreated or Cr(VI)-exposed cells appeared to be similar, whereas in Ni(II)-exposed cells ascorbate was not detectable (Fig. 5C). Taken together, these results provided the first indication that ascorbate was irreversibly destroyed in cells by Ni(II) exposure, but only transiently by Cr(VI)-exposure.
To investigate further whether depletion of ascorbate causes inhibition of hydroxylase activity and HIF-1α protein stabilization, two types of experiments were carried out. First, 1HAEo- and A549 cells were preloaded with 25 μM of ascorbate for 2 h, washed and exposed to Ni(II) for 1, 2, 4 and 8 h. The time of HIF-1α appearance was shifted by 4 h when compared with the time-course without ascorbate preloading (Fig. 6A, B). These data suggested that additional 4 h were needed to destroy higher levels intracellular ascorbate by Ni(II). Preloading cells with 50 μM of Fe(II), followed by Ni(II) exposure did not affect HIF-1α stabilization (not shown). To further examine the involvement of intracellular ascorbate in HIF-1 α degradation the protein was stabilized by exposure to Ni(II) or Cr(VI) for 8 h, then cells were washed and incubated in fresh complete medium with or without supplementation with 50 μM ascorbate for additional 2 and 4 h (Fig. 7A, B). In 1HAEo- cells, change of culture medium following Ni(II) exposure did not decrease HIF-1α levels, however, supplementation with 50 μM ascorbate resulted in the decline of HIF-1α levels after 2 h and disappearance after 4 h (Fig. 7A). Change of medium following Cr(VI) exposure resulted in the disappearance of HIF-1α protein 1 h later (Fig. 7A). In A549 cells, change of medium following Ni(II) or Cr(VI) exposure resulted in a decline of HIF-1α levels already after 2 h (Fig. 7B). In order to understand the different response of 1HAEo- and A549 cells to a medium change, we compared ascorbate accumulation in 1HAEo- and A549 cells following metals exposure. Significantly more ascorbate was transported into A549 cells as compared with 1HAEo- cells during 2 h (Fig. 7C). The comparison of expression levels of the SVCT2 ascorbate transporter revealed 2.5 higher levels of expression in A549 cells (not shown), suggesting that these cells can more efficiently acquire ascorbate. These experiments indicate that the addition of fresh medium which contains low amounts of ascorbate was sufficient for A549 cells, whereas 1HAEo- cells required supplementation of medium with 50 μM ascorbate. In both cases this resulted in activation of hydroxylase activity and destruction of metals-induced HIF-1α protein.
Figure 6.
Preloading of cells with ascorbate hinders HIF-1α protein stabilization by Ni(II) exposure. A. 1HAEo- cells, or B, A549 cells were preloaded with 25 μM ascorbate for 2 h, washed to remove extracellular ascorbate and exposed to 0.5 mM NiSO4 for various time periods shown in the Figure. Right panel HIF-1α and HIF-1β induction in ascorbate preloaded cells. Left panel cells without ascorbate preloading shown for comparison. Nuclear protein extracts (15 μg) were prepared for immunoblotting as described in Materials and Methods and probed with antibodies against HIF-1α, HIF-1β. Antibodies against β-actin were used to provide loading control.
Figure 7.
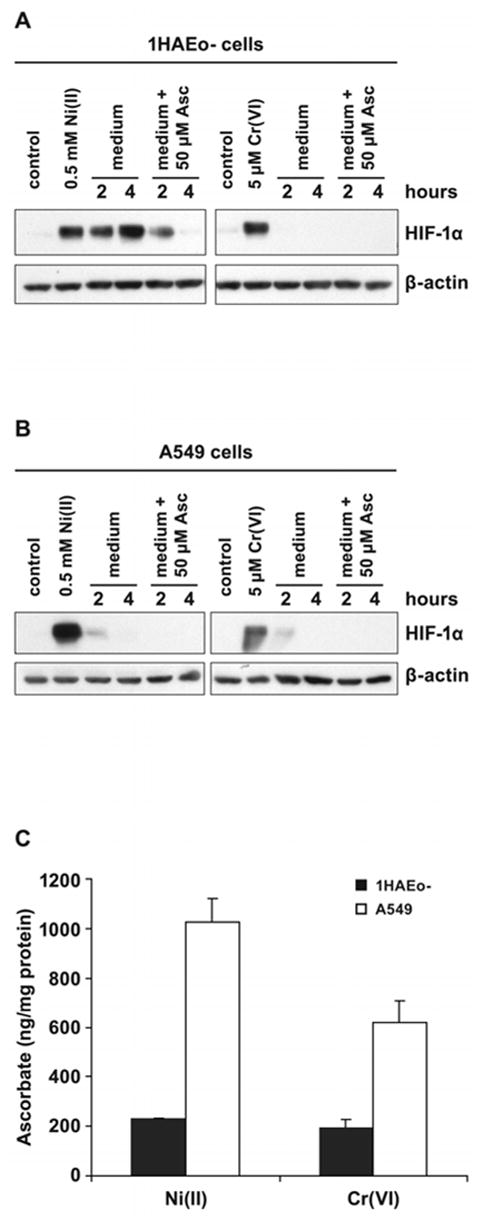
The addition of ascorbate destabilizes metal-induced HIF-1α protein. A, 1HAEo- cells were exposed to 0.5 mM NiSO4 or 5 μM K2CrO4 for 6 h.. B, A549 were exposed to 0.5 mM NiSO4 and 20 μM K2CrO4 for 6 h. The medium was changed to the fresh medium with or without 50 μM ascorbate for the time periods shown in the Figure. Nuclear protein extracts (15 μg) were prepared for immunoblotting as described in Materials and Methods and probed with antibodies against HIF-1α and also probed with antibodies against β-actin to provide loading control. C, The intracellular accumulation of reduced ascorbate following Ni(II) or Cr(VI) exposure. 1HAEo- cells (dotted bars) were exposed to 0.5 mM NiSO4 or 5 μM K2CrO4 for 6 h, or A549 cells (striped bars) were exposed to 0.5 mM NiSO4 or 20 μM K2CrO4 for 6 h, then washed and loaded with 50 μM ascorbate. The intracellular ascorbate level was determined at 2 h of incubation with ascorbate using HPLC as described in Materials and Methods. The data are presented as means ± SD.
Time Requirement for HIF-1-dependent Transcription
Exposure to Ni(II) or Cr(VI) revealed strong nuclear accumulation of both α and β HIF-1 subunits in both 1HAEo- and A549 cells after 4 h of exposure (Fig. 1B and 4C). However, in the nuclear fraction of Cr(VI)-exposed cells these subunits were no longer detectable after 24 h. In order to assess the level of HIF-1-dependent transcription at 4 h and to compare it to the level of transcription activity at 24 h, we first utilized the HRE-Luc reporter assay. In 1HAEo- cells Ni(II)-induced HIF-1-dependent transcription activity was not changed at 4 h of exposure, but was strongly induced in 24 h. In contrast, Cr(VI)-exposed cells demonstrated poor induction of HIF-dependent transcription from 4 to 24 h of exposure (Fig. 8A). The comparison of expression of two hypoxia-inducible proteins CA9 and NDRG1/Cap43 in Cr(VI)- or Ni(II)- exposed cells also demonstrated strong induction by Ni(II) at 20 h, but not by Cr(VI) (Fig. 8B). This data suggested that the stabilization of HIF-1α protein and nuclear accumulation of both subunits of HIF-1 transcription factor in metal-exposed cells did not immediately initiate HIF-dependent transcription.
Figure 8.
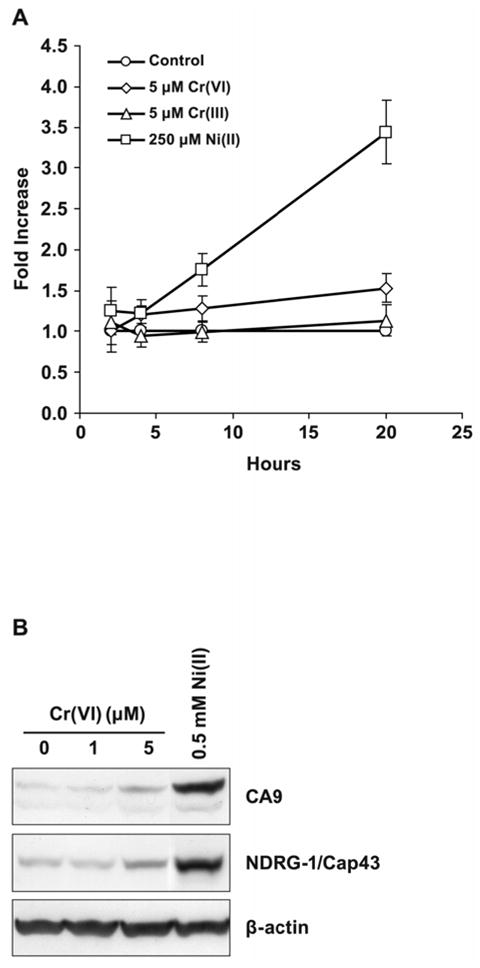
Exposure of cells to Ni(II), but not Cr(VI), stimulates HIF-dependent transcription. A, Time-course of HIF-dependent transcription induced by Ni(II) or Cr(VI) in 1HAEo- cells. 1HAEo- cells were transfected with HRE-Luc reporter plasmid and incubated with 5 μM CrCl3, 5 μM K2CrO4 or 0.25 mM NiSO4 for various time periods shown in the Figure. Relative luciferase activity was detected and expressed as the fold increase over control. Data are presented as mean values ± SD. B, Induction of NDRG1/Cap43 or CA9 proteins by various concentrations of Cr(VI) or 0.5 mM NiSO4. 1HAEo- cells were exposed to metals for 20 h. Total cell extracts were prepared for immunoblotting and probed with antibodies directed against NDRG1/Cap43 or CA9 proteins. The same blot was probed with antibodies against β-actin to provide loading control.
In order to gain a global view on HIF-dependent gene response of 1HAEo- cells exposed to Ni(II) or Cr(VI) for 4 or 24 h, we performed GeneChip analysis. Gene expression profiling was performed by using RNA samples isolated from cells exposed to both metals at 4 and 24 h in two independent experiments. We used Affymetrix human genome U133 plus 2.0 arrays which contained probe sets allowing for the detection of over 50,000 transcripts representing 38,000 genes. Various computational and statistical algorithms were applied to identify genes whose overall expression level in two independent batches of 1HAEo- cells was altered by metals exposure at 4 and 24 h at least 2-fold compared with controls. When the results of two experiments were averaged, 22,277 genes showed “present signal” in all groups. Following Ni(II) exposure 1,093 genes were up-regulated at least two fold in 4-h and 790 were up-regulated in 24-h. The exposure to Cr(VI) resulted in up-regulation of 1,354 genes in 4-h and 1,088 in 24-h. Next we analyzed top 40 genes which were up-regulated by Ni(II) or Cr(VI) exposure at 4 and 24 h. The analysis of the top 40 genes induced by 4 or 24 h of Cr(VI) or 4 h of Ni(II) exposure did not reveal presence of hypoxia-inducible genes (not shown). At the same time, the analysis of the top 40 genes up-regulated by 24 h of Ni(II) exposure revealed that at least half of the genes were typical hypoxia-inducible genes. The top 40 genes are shown on Table 1. It is possible that the other genes may also be hypoxia-inducible. Among them are genes coding for proteins involved in glucose metabolism, apoptosis, extracellular matrix metabolism, as well as angiogenic growth factors and cytokines, transporters, signal transducers, and transcriptional regulators.
Table 1.
List of top 40 genes induced by Ni(II) or Cr(VI) at 4 and 24 h
| FOLD CHANGE | |||||
|---|---|---|---|---|---|
| 0.5 mM Ni(II) | 5 μM Cr(VI) | Hypoxia up regulateda | |||
| Unigene symbol | 4 h | 24 h | 4 h | 24 h | |
| LOX | 0.4 | 66.7 | 0.4 | 0.7 | Y9 |
| STC1 | 1.1 | 36.6 | 7.1 | 0.7 | Y46 |
| C10orf10 | 2.1 | 27.1 | 1.5 | 0.6 | |
| TNFRSF25 | 5 | 22.2 | 3 | 0.8 | |
| ANGPTL4 | 2.7 | 20.9 | 1 | 0.8 | Y9 |
| ALDOC | 0.6 | 15.8 | 0.5 | 0.5 | Y9 |
| ENO2 | 0.6 | 15.8 | 2.4 | 0.3 | Y26 |
| GPM6B | 2.6 | 11 | 7 | 1 | |
| NDRG1 | 0.9 | 10.8 | 0.6 | 0.7 | Y29 |
| HIST1H3B | 0.5 | 8.7 | 1.1 | 1.3 | |
| C21orf7 | 0.4 | 8.5 | 3.5 | 0.6 | |
| ZBTB10 | 0 | 7.7 | 4 | 0.7 | |
| CXCR4 | 0.7 | 7.5 | 2.3 | 0.7 | Y35 |
| HIG2 | 0.7 | 6.8 | 0.5 | 0.4 | Y9 |
| GAL3ST4 | 0.4 | 6.8 | 0.9 | 0.6 | |
| TFR2 | 1 | 6.6 | 2.6 | 1.2 | Y43 |
| P4HA2 | 0.7 | 5.8 | 0.6 | 1.1 | Y31 |
| FABP3 | 0.8 | 5.8 | 2.4 | 0.7 | |
| CDKN2D | 0.4 | 5.6 | 2.5 | 0.6 | |
| BNIP3 | 0.4 | 5.5 | 0.9 | 1.2 | Y31 |
| NAV3 | 2.2 | 5.4 | 1.2 | 1.2 | |
| MTSS1 | 0.6 | 5.3 | 2.4 | 0.7 | |
| PPP2R5B | 0.6 | 5.2 | 1.1 | 0.7 | |
| HK2 | 0.7 | 5.1 | 1 | 0.7 | Y31 |
| SLC2A3 | 1.1 | 5.1 | 1.3 | 0.9 | Y9 |
| NPAS2 | 0.6 | 5 | 1.3 | 0.7 | |
| PDE3A | 1.8 | 5 | 4.8 | 1.1 | Y27 |
| BCL9 | 0.2 | 5 | 8.8 | 0.7 | |
| PGK1 | 0.8 | 4.9 | 0.8 | 0.9 | Y31 |
| PDK1 | 0.6 | 4.8 | 0.3 | 0.7 | Y26 |
| INSIG2 | 0.4 | 4.5 | 1.3 | 0.6 | Y26 |
| RNASE4 | 0.4 | 4.5 | 0.8 | 0.7 | Y9 |
| CSF3R | 0.8 | 4.3 | 1.3 | 1.1 | |
| ETV2 | 0.4 | 4.2 | 0.3 | 0.7 | Y9 |
| PILRA | 0.5 | 4.2 | 3.2 | 0.6 | |
| JUN | 0.8 | 4.1 | 0.8 | 1.1 | Y31 |
| PFKFB3 | 1.6 | 4.1 | 1.3 | 1.2 | Y31 |
| E2IG5 | 0.5 | 4.1 | 0.8 | 0.9 | Y26 |
| BCL2L11 | 0.3 | 4.1 | 2.9 | 0.6 | |
| ETS1 | 0.8 | 4 | 1.9 | 0.8 | Y9 |
Reference number, Y = Yes
Strong up-regulation of these genes by 24 h but not by 4 h suggested that the induction by metals of the majority of hypoxic genes responding to hypoxia requires time. The exposure to Ni(II) permanently destroys ascorbate and keeps HIF-1α at high levels throughout the time of exposure. In contrast, single exposure to Cr(VI) rapidly, but transiently depletes ascorbate, causing stabilization of HIF-1α protein only for a short period of time.
Discussion
One of the mechanisms by which transition metals may induce inflammatory response and apoptosis causing lung injury [2, 3] is stabilization of HIF-1α protein and activation of HIF-1 transcription factor [5, 23]. The HIF-1α stabilization by Ni(II) or Co(II) has been the subject of recent studies [8]. The hydroxylation of two prolines in HIF-1α protein enables its binding to the von Hippel-Lindau protein (pVHL), a component of the ubiquitin ligase complex, followed by ubiquitylation and subsequent proteasomal degradation. A family of HIF-specific hydroxylases (Fe(II)- and 2OG-dependent dioxygenases) is involved in the hydroxylation process [8, 24]. Since Ni(II) ion is similar to Fe(II) ion it has been suggested that nickel may replace iron in the hydroxylases and thus lower or abolish their activity [9, 10]. Alternatively, we have suggested that the depletion of intracellular ascorbate is the key event in the HIF-1 induction by metals [14]. This is because ascorbate is responsible for maintenance of enzyme-bound iron in the reduced state [14, 25, 26]. If ascorbate depletion is the primary event in HIF-1 induction by Ni(II) and Co(II) then exposure to other oxidatively active metals, which can oxidize ascorbate, but can not substitute for Fe(II), should also induce HIF-1 transcription factor. Indeed, the induction of HIF-1α protein by Cr(VI) was shown in prostate cancer DU145 cells [27]. That paper indicated the involvement of oxidative stress in HIF-1α stabilization by Cr(VI). It is known that Cr(VI) quickly enters cells in vitro and in vivo where it can be reduced by ascorbate or glutathione [17–19, 22]. Thus, Cr(VI) will oxidize and deplete intracellular ascorbate stores. This may cause oxidative stress observed by Gao et al., [27] in prostate cancer cells. Other oxidatively active metal ions like arsenite or vanadate were also shown to stabilize the HIF-1α protein [28, 29].
In this work, for the first time, we used two cell lines representing human lung epithelial cells, which are relevant target of exposure to Ni(II) or Cr(VI). Both cell lines, when equalized for Cr(VI) and ascorbate uptake, showed essentially similar results. The exposure of these cell lines to both metals resulted in stabilization of the HIF-1α and HIF-2α proteins. When the time-course of HIFα’s stabilization by two metals was compared, we observed remarkable differences. Thus, in Ni(II)-exposed cells HIF-1α protein was strongly induced by 4 h and remained high during the 24 h exposure. In contrast, in Cr(VI)-exposed cells, HIF-1α protein degraded after 8–16 h. This disappearance of HIF-1α protein at later time points in Cr(VI)-treated cells provided first indication that reduced Cr(III) could not stabilize HIF-1α protein. The down-regulation of HIF-1α protein during the course of Cr(VI) treatment was not due to any chromium-specific acceleration of the destruction of this protein, since co-exposure with Ni(II) did not manifest any destabilization of Ni(II)-induced HIF-1α protein. Instead, the co-exposure produced a slight increase of HIF-1α protein and HIF-dependent transcription. Moreover, the repeated dosing of Cr(VI) caused reappearance of HIF-1α protein. These data indicated that only Cr(VI) before it is reduced to Cr(III) can cause HIF-1α protein stabilization. Since Cr(VI) can not substitute for Fe(II) in the enzyme and only Cr(VI) is able to stabilize HIF-1α protein the most likely explanation of the observed events is that the metals act by targeting intracellular ascorbic acid.
Further evidence emphasizing the role of ascorbate in HIF-1α regulation was obtained in experiments with preloading the cells with either ascorbate or iron. Only the preloading of ascorbate significantly delayed the HIF-1α appearance suggesting that additional time was needed for the metals to deplete intracellular ascorbate below levels sustaining activity of the hydroxylases. Moreover, addition of ascorbate to the medium after metal exposure caused a rapid destabilization of HIF-1α protein. Preloading cells with iron had no effect on HIF-1α protein stabilization. Thus, some redistribution of iron following Ni(II) exposure observed by other [9] may be a result of iron oxidation in exposed cells. This is a subject of additional investigations. In conjunction with our previous data that metals suppress ascorbate uptake and inhibit intracellular hydroxylase activity [14, 16] our data imply that modulations of ascorbate levels by wide variety of transition metals, not necessarily iron-replacing metals, may cause stabilization and activation of HIF-1 transcription factor.
To get more insights into the mechanism of ascorbate oxidation by Ni(II) or Cr(VI), we investigated effects of these metals on the rate of ascorbate oxidation in a cell-free system. Indeed, we found that Cr(VI) oxidized ascorbate quickly, but only until all Cr(VI) was reduced to Cr(III). The later, was found to even delay the spontaneous ascorbate oxidation by air. Since Cr(III) was inert or even had protective effect on ascorbate, it was conceivable that, after all Cr(VI) was reduced, the dehydroascorbate (DHA) could be recycled to ascorbate. The recycling of dehydroascorbate is a well known part of ascorbate metabolism in cells [30]. The data obtained in cells were in agreement with cell-free experiments. The HPLC analyses of ascorbate levels showed that it can be recovered in Cr(VI)- but not in Ni(II)-exposed cells. Thus, from these experiments we concluded that ascorbate oxidation caused by Cr(VI) was transient and led to only a temporary inhibition of hydroxylases and stabilization of HIF-1α protein. When all Cr(VI) was reduced, DHA reduction by cells might lead to the reactivation of hydroxylases and disappearance of HIF-1α protein. This scenario was in line with the results of repeated addition of Cr(VI), which demonstrated further stabilization of HIF-1α protein. Interestingly, ascorbate recycling may be strongly stimulated by ebselen [31] and it was previously shown that ebselen abolished transcriptional activation of Co(II)-induced hypoxic genes [11]. Taken together, these observations emphasize active role of ascorbate recycling in HIF-1α protein destabilization.
In contrast, Ni(II) appeared to be a catalyst, which facilitates ascorbate oxidation by ambient oxygen and histidine-Ni(II) complexes demonstrated even stronger catalytic activity than Ni(II) alone. It is important to note, that, found in this study, Ni(II) catalytic activity is specifically exerted towards ascorbate. Other investigators did not find Ni(II) to affect another abundant cellular antioxidant glutathione under similar experimental conditions [32]. Thus, the exposure to Ni(II) has the capacity strongly and permanently deplete intracellular ascorbate that leads to activation of hypoxic genes [14, 16]. The proposed mechanism for the role of ascorbate in the regulation of the HIF activity is presented schematically in Figure 9. Important feature of Ni(II) activity is irreversible destruction of ascorbate, whereas chromium is only active as Cr(VI), but inactive in Cr(III) form.
Figure 9.
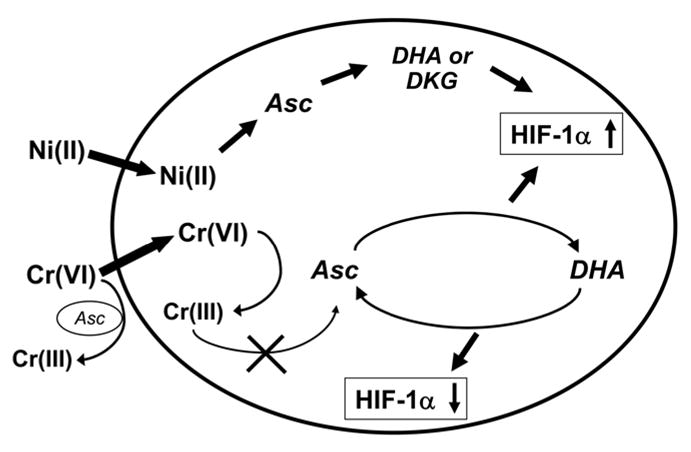
Proposed model for the role of ascorbate in the modulation of HIF activity by metals. Ascorbate (Asc) concentration inside the cell is significantly higher than outside. When Ni(II) enters cells it continuously and irreversibly oxidizes Asc to dehydroascorbate (DHA), diketogulonate (DKG) and other degradation products. This results in prolonged hydroxylases inactivation and HIF up-regulation. When Cr(VI) enters cells it can be reduced by Asc to inactive Cr(III). This results in Asc oxidation to DHA and HIF up-regulation. After all Cr(VI) is reduced, DHA can be recycled back to Asc leading to HIF down-regulation. Ascorbate also can reduce Cr(VI) to cell-impermeable Cr(III) outside of cells.
The time-course of metal-induced HIF-dependent transcription is being reported for the first time. The HIF-1 transcription complex is composed from two subunits, an α and a β. The exposure metals resulted in translocation of both subunits to the nucleus. In agreement with other observations we found that HIF-1β subunit was constitutively expressed and its protein level was unchanged following Ni(II)- or Cr(VI)-exposure [27]; but it is also translocated to the nucleus, where it dimerizes with regulatory subunits. It is interesting to note simultaneous appearance and disappearance of both HIF-1 subunits in the nucleus following metals exposure. Since Ni(II) exposure stabilized HIF-1α protein permanently and Cr(VI) exposure caused only a transient up-regulation of this protein, we compared the effect of both metals on HIF-dependent transcription. It was conceivable that HIF-1-dependent transcription could follow the pattern of HIF-1α stabilization, i.e. should be high at 4 h exposure to both metals, but declined at 24 h of exposure in Cr(VI)-treated cells. In Ni(II)-exposed cells, the transcription should remain unchanged or even increase with time. Moreover, since exposure to Cr(VI) causes nuclear accumulation of both subunits earlier than exposure to Ni(II), it was expected that Cr(VI) could activate HIF-1-dependent transcription earlier. However, the HRE-Luc reporter assay revealed that the HIF-dependent transcription response was only weakly activated at 4 h or even 8 h following exposure to both metals. Further testing has been done by the analysis of gene expression using the Affymetrix GeneChip assay. Previously, we have identified a number of genes that were induced by both hypoxia and Ni(II) [4, 33, 34]. Among them were genes coding for glycolytic enzymes and glucose transporters [33]. In this study the strongest induction by Ni(II) at 24 h was found for lysyl oxidase (LOX). LOX is a known HIF-dependent gene [35, 36]. In contrast, LOX was not induced by Cr(VI) at 4 or 24 h. Stanniocalcin (STC1), also a known HIF-dependent gene [37], was strongly induced by Ni(II) at 24 h, and only moderately induced by Cr(VI) at 4, but not at 24 h. Other hypoxia-inducible genes that are involved in glycolysis such as ALDOC, ENO2, HK2, PGK1, PDK1, and glucose transport (SLC2A3) [36, 38] were induced by Ni(II) at 24 h, but not at 4 h. They were not induced by Cr(VI) at both time points. Interestingly, CSF3R, the receptor for granulocyte colony-stimulating factor, which is involved in stimulation of glucose and dehydroascorbate transport [39], was induced by Ni(II) at 24, but not induced by Cr(VI) at 4 or 24 h. The hypoxia-inducible genes, NDRG1/Cap43 and BNIP3, which we showed previously to be induced by 20 h of Ni(II) exposure in mouse fibroblasts [4, 34], were also induced by Ni(II) at 24 h in human lung cells. They were, however, not induced by Ni(II) at 4 h, or by Cr(VI) at 4 or 24 h.
Only a few hypoxia-inducible genes were induced by Cr(VI) after 4 h of exposure. Among them were STC1, ENO2, CXCR4, TFR2, and PDE3A [40–42]. The transcription of all these genes returned to normal or was even slightly downregulated at the 24th h of Cr(VI) exposure.
Our study indicates that although HIF-1 is undoubtedly important for activation of hypoxia-inducible genes, the formation of active transcription complex takes longer than 4–8 h and may involve many more signal transduction pathways which must be combined to allow for the expression of hypoxia-inducible genes.
The expression analysis of hypoxia-inducible genes emphasize the ability of Cr(VI)-exposed lung cells to overcome consequences of initial loss of ascorbate without the activation of HIF-1-dependent transcription. However, we found that repeated exposure of human lung cells to Cr(VI) resulted in reappearance of HIF-1, suggesting that chronic exposure to Cr(VI) may cause prolonged activation of HIF-1 transcription factor and subsequent activation of hypoxia-inducible genes. Prolonged exposure to metal ions is important for their damaging effects. For example in vivo soluble Ni(II), given to an animal, is rapidly cleared from exposed tissues and therefore single exposure is less toxic and carcinogenic than exposure to sparingly-soluble particulate nickel compound such as Ni3S2, which can be slowly solubilized in tissues thereby maintaining steady levels of Ni(II) over longer period of time. However, there is evidence that chronic exposure to soluble Ni(II) may also be carcinogenic [43].
The induction of HIF-1-dependent transcription by nickel may be important for the nickel-induced carcinogenic process [33, 44]. It triggers cellular reactions typical of hypoxia, including the up-regulation of expression of genes involved in glucose transport, glycolysis, neovascularization, and survival. This enables cells to overcome nutritive deprivation, stimulates their escape from the hostile metabolic microenvironment and favors unrestricted growth [45]. Activation of HIF-1 has been implicated in the pathophysiologic alterations of both smooth muscle and endothelial cell biology in patients with pulmonary hypertension [46]. These events, when combined with activation of proinflammatory cytokines in lungs can significantly contribute to the development and progression of respiratory system disorders found in nickel-exposed individuals. It has been shown that higher dietary intake and serum concentrations of ascorbate maintain pulmonary function and protect against the development of chronic respiratory symptoms [47]. The present work suggests that high levels of ascorbate in lungs are important for alleviating harmfull effects of metals, particularly those mediated through up-regulation of HIF-dependent genes.
Acknowledgments
We thank R. Bare and L. North for excellent technical assistance, Dr. E. Goncharova for invaluable help in the analysis of Affymetrix GeneChip data. Critical comments of Dr. J. Phang are greatly appreciated. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government.
Abbreviations
- Asc
ascorbate
- CA9
carbonic anhydrase 9
- Cr(VI)
chromium (VI)
- DHA
dehydroascorbate
- HIF
hypoxia inducible transcription factor
- HRE
HIF response element
- Ni(II)
nickel(II)
- 2-OG
2-oxoglutarate
- ROS
reactive oxygen species
- RLU
relative luciferase units
- pVHL
von Hippel-Lindau protein
Footnotes
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Barchowsky A, O’Hara KA. Metal-induced cell signaling and gene activation in lung diseases. Free Radic Biol Med. 2003;34:1130–1135. doi: 10.1016/s0891-5849(03)00059-5. [DOI] [PubMed] [Google Scholar]
- 2.Krick S, Eul BG, Hanze J, Savai R, Grimminger F, Seeger W, Rose F. Role of hypoxia-inducible factor-1alpha in hypoxia-induced apoptosis of primary alveolar epithelial type II cells. Am J Respir Cell Mol Biol. 2005;32:395–403. doi: 10.1165/rcmb.2004-0314OC. [DOI] [PubMed] [Google Scholar]
- 3.Madjdpour C, Jewell UR, Kneller S, Ziegler U, Schwendener R, Booy C, Klausli L, Pasch T, Schimmer RC, Beck-Schimmer B. Decreased alveolar oxygen induces lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2003;284:L360–367. doi: 10.1152/ajplung.00158.2002. [DOI] [PubMed] [Google Scholar]
- 4.Salnikow K, Blagosklonny MV, Ryan H, Johnson R, Costa M. Carcinogenic nickel induces genes involved with hypoxic stress. Cancer Res. 2000;60:38–41. [PubMed] [Google Scholar]
- 5.Wang GL, Semenza GL. Purification and characterization of hypoxia-inducible factor 1. J Biol Chem. 1995;270:1230–1237. doi: 10.1074/jbc.270.3.1230. [DOI] [PubMed] [Google Scholar]
- 6.Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292:468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 7.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 8.Maxwell P, Salnikow K. HIF-1: an oxygen and metal responsive transcription factor. Cancer Biol Ther. 2004;3:29–35. doi: 10.4161/cbt.3.1.547. [DOI] [PubMed] [Google Scholar]
- 9.Davidson T, Chen H, Garrick MD, D’Angelo G, Costa M. Soluble nickel interferes with cellular iron homeostasis. Mol Cell Biochem. 2005;279:157–162. doi: 10.1007/s11010-005-8288-y. [DOI] [PubMed] [Google Scholar]
- 10.Davidson TL, Chen H, Di Toro DM, D’Angelo G, Costa M. Soluble nickel inhibits HIF-prolyl-hydroxylases creating persistent hypoxic signaling in A549 cells. Mol Carcinog. 2006;45:479–489. doi: 10.1002/mc.20176. [DOI] [PubMed] [Google Scholar]
- 11.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci U S A. 1998;95:11715–11720. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Andrew AS, Klei LR, Barchowsky A. Nickel requires hypoxia-inducible factor-1 alpha, not redox signaling, to induce plasminogen activator inhibitor-1. Am J Physiol Lung Cell Mol Physiol. 2001;281:L607–615. doi: 10.1152/ajplung.2001.281.3.L607. [DOI] [PubMed] [Google Scholar]
- 13.Salnikow K, Su W, Blagosklonny MV, Costa M. Carcinogenic metals induce hypoxia-inducible factor-stimulated transcription by reactive oxygen species-independent mechanism. Cancer Res. 2000;60:3375–3378. [PubMed] [Google Scholar]
- 14.Salnikow K, Donald SP, Bruick RK, Zhitkovich A, Phang JM, Kasprzak KS. Depletion of intracellular ascorbate by the carcinogenic metals nickel and cobalt results in the induction of hypoxic stress. J Biol Chem. 2004;279:40337–40344. doi: 10.1074/jbc.M403057200. [DOI] [PubMed] [Google Scholar]
- 15.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karaczyn A, Ivanov S, Reynolds M, Zhitkovich A, Kasprzak KS, Salnikow K. Ascorbate depletion mediates up-regulation of hypoxia-associated proteins by cell density and nickel. J Cell Biochem. 2006;97:1025–1035. doi: 10.1002/jcb.20705. [DOI] [PubMed] [Google Scholar]
- 17.Zhitkovich A. Importance of Chromium-DNA Adducts in Mutagenicity and Toxicity of Chromium(VI) Chem Res Toxicol. 2005;18:3–11. doi: 10.1021/tx049774+. [DOI] [PubMed] [Google Scholar]
- 18.Standeven AM, Wetterhahn KE. Ascorbate is the principal reductant of chromium(VI) in rat lung ultrafiltrates and cytosols, and mediates chromium-DNA binding in vitro. Carcinogenesis. 1992;13:1319–1324. doi: 10.1093/carcin/13.8.1319. [DOI] [PubMed] [Google Scholar]
- 19.Standeven AM, Wetterhahn KE. Ascorbate is the principal reductant of chromium (VI) in rat liver and kidney ultrafiltrates. Carcinogenesis. 1991;12:1733–1737. doi: 10.1093/carcin/12.9.1733. [DOI] [PubMed] [Google Scholar]
- 20.Gruenert DC, Finkbeiner WE, Widdicombe JH. Culture and transformation of human airway epithelial cells. Am J Physiol. 1995;268:L347–360. doi: 10.1152/ajplung.1995.268.3.L347. [DOI] [PubMed] [Google Scholar]
- 21.Lykkesfeldt J. Determination of ascorbic acid and dehydroascorbic acid in biological samples by high-performance liquid chromatography using subtraction methods: reliable reduction with tris[2-carboxyethyl]phosphine hydrochloride. Anal Biochem. 2000;282:89–93. doi: 10.1006/abio.2000.4592. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki Y, Fukuda K. Reduction of hexavalent chromium by ascorbic acid and glutathione with special reference to the rat lung. Arch Toxicol. 1990;64:169–176. doi: 10.1007/BF02010721. [DOI] [PubMed] [Google Scholar]
- 23.Salnikow K, An WG, Melillo G, Blagosklonny MV, Costa M. Nickel-induced transformation shifts the balance between HIF-1 and p53 transcription factors. Carcinogenesis. 1999;20:1819–1823. doi: 10.1093/carcin/20.9.1819. [DOI] [PubMed] [Google Scholar]
- 24.Dann CE, 3rd, Bruick RK. Dioxygenases as O2-dependent regulators of the hypoxic response pathway. Biochem Biophys Res Commun. 2005;338:639–647. doi: 10.1016/j.bbrc.2005.08.140. [DOI] [PubMed] [Google Scholar]
- 25.de Jong L, Albracht SP, Kemp A. Prolyl 4-hydroxylase activity in relation to the oxidation state of enzyme-bound iron. The role of ascorbate in peptidyl proline hydroxylation. Biochim Biophys Acta. 1982;704:326–332. doi: 10.1016/0167-4838(82)90162-5. [DOI] [PubMed] [Google Scholar]
- 26.Majamaa K, Gunzler V, Hanauske-Abel HM, Myllyla R, Kivirikko KI. Partial identity of the 2-oxoglutarate and ascorbate binding sites of prolyl 4-hydroxylase. J Biol Chem. 1986;261:7819–7823. [PubMed] [Google Scholar]
- 27.Gao N, Jiang BH, Leonard SS, Corum L, Zhang Z, Roberts JR, Antonini J, Zheng JZ, Flynn DC, Castranova V, Shi X. p38 Signaling-mediated hypoxia-inducible factor 1alpha and vascular endothelial growth factor induction by Cr(VI) in DU145 human prostate carcinoma cells. J Biol Chem. 2002;277:45041–45048. doi: 10.1074/jbc.M202775200. [DOI] [PubMed] [Google Scholar]
- 28.Gao N, Shen L, Zhang Z, Leonard SS, He H, Zhang XG, Shi X, Jiang BH. Arsenite induces HIF-1alpha and VEGF through PI3K, Akt and reactive oxygen species in DU145 human prostate carcinoma cells. Mol Cell Biochem. 2004;255:33–45. doi: 10.1023/b:mcbi.0000007259.65742.16. [DOI] [PubMed] [Google Scholar]
- 29.Hwang JT, Lee M, Jung SN, Lee HJ, Kang I, Kim SS, Ha J. AMP-activated protein kinase activity is required for vanadate-induced hypoxia-inducible factor 1alpha expression in DU145 cells. Carcinogenesis. 2004;25:2497–2507. doi: 10.1093/carcin/bgh253. [DOI] [PubMed] [Google Scholar]
- 30.May JM, Qu ZC, Whitesell RR, Cobb CE. Ascorbate recycling in human erythrocytes: role of GSH in reducing dehydroascorbate. Free Radic Biol Med. 1996;20:543–551. doi: 10.1016/0891-5849(95)02130-2. [DOI] [PubMed] [Google Scholar]
- 31.Zhao R, Holmgren A. Ebselen is a dehydroascorbate reductase mimic, facilitating the recycling of ascorbate via mammalian thioredoxin systems. Antioxid Redox Signal. 2004;6:99–104. doi: 10.1089/152308604771978390. [DOI] [PubMed] [Google Scholar]
- 32.Krezel A, Szczepanik W, Sokolowska M, Jezowska-Bojczuk M, Bal W. Correlations between complexation modes and redox activities of Ni(II)-GSH complexes. Chem Res Toxicol. 2003;16:855–864. doi: 10.1021/tx034012k. [DOI] [PubMed] [Google Scholar]
- 33.Salnikow K, Davidson T, Zhang Q, Chen LC, Su W, Costa M. The involvement of hypoxia-inducible transcription factor-1-dependent pathway in nickel carcinogenesis. Cancer Res. 2003;63:3524–3530. [PubMed] [Google Scholar]
- 34.Salnikow K, Davidson T, Costa M. The role of hypoxia-inducible signaling pathway in nickel carcinogenesis. Environ Health Perspect. 2002;110(Suppl 5):831–834. doi: 10.1289/ehp.02110s5831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JT, Jeffrey SS, Giaccia AJ. Lysyl oxidase is essential for hypoxia-induced metastasis. Nature. 2006;440:1222–1226. doi: 10.1038/nature04695. [DOI] [PubMed] [Google Scholar]
- 36.Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, Giaccia AJ. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene. 2003;22:5907–5914. doi: 10.1038/sj.onc.1206703. [DOI] [PubMed] [Google Scholar]
- 37.Yeung HY, Lai KP, Chan HY, Mak NK, Wagner GF, Wong CK. Hypoxia-inducible factor-1-mediated activation of stanniocalcin-1 in human cancer cells. Endocrinology. 2005;146:4951–4960. doi: 10.1210/en.2005-0365. [DOI] [PubMed] [Google Scholar]
- 38.Mense SM, Sengupta A, Zhou M, Lan C, Bentsman G, Volsky DJ, Zhang L. Gene expression profiling reveals the profound upregulation of hypoxia-responsive genes in primary human astrocytes. Physiol Genomics. 2006;25:435–449. doi: 10.1152/physiolgenomics.00315.2005. [DOI] [PubMed] [Google Scholar]
- 39.Vera JC, Rivas CI, Zhang RH, Golde DW. Colony-stimulating factors signal for increased transport of vitamin C in human host defense cells. Blood. 1998;91:2536–2546. [PubMed] [Google Scholar]
- 40.Tacchini L, Fusar Poli D, Bernelli-Zazzera A, Cairo G. Transferrin receptor gene expression and transferrin-bound iron uptake are increased during postischemic rat liver reperfusion. Hepatology. 2002;36:103–111. doi: 10.1053/jhep.2002.33997. [DOI] [PubMed] [Google Scholar]
- 41.Murray F, MacLean MR, Pyne NJ. Increased expression of the cGMP-inhibited cAMP-specific (PDE3) and cGMP binding cGMP-specific (PDE5) phosphodiesterases in models of pulmonary hypertension. Br J Pharmacol. 2002;137:1187–1194. doi: 10.1038/sj.bjp.0704984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schioppa T, Uranchimeg B, Saccani A, Biswas SK, Doni A, Rapisarda A, Bernasconi S, Saccani S, Nebuloni M, Vago L, Mantovani A, Melillo G, Sica A. Regulation of the chemokine receptor CXCR4 by hypoxia. J Exp Med. 2003;198:1391–1402. doi: 10.1084/jem.20030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kasprzak KS, Sunderman FW, Salnikow K. Nickel carcinogenesis. Mutat Res. 2003;533:67–97. doi: 10.1016/j.mrfmmm.2003.08.021. [DOI] [PubMed] [Google Scholar]
- 44.Salnikow K, Kasprzak K. S. Ascorbate depletion: a critical step in nickel carcinogenesis? Environ Health Perspect. 2005;113:577–584. doi: 10.1289/ehp.7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Semenza GL. Targeting HIF-1 for cancer therapy. Nat Rev Cancer. 2003;3:721–732. doi: 10.1038/nrc1187. [DOI] [PubMed] [Google Scholar]
- 46.Semenza GL. Involvement of hypoxia-inducible factor 1 in pulmonary pathophysiology. Chest. 2005;128:592S–594S. doi: 10.1378/chest.128.6_suppl.592S. [DOI] [PubMed] [Google Scholar]
- 47.Schwartz J, Weiss ST. Relationship between dietary vitamin C intake and pulmonary function in the First National Health and Nutrition Examination Survey (NHANES I) Am J Clin Nutr. 1994;59:110–114. doi: 10.1093/ajcn/59.1.110. [DOI] [PubMed] [Google Scholar]