

Abstract
Tumor necrosis factor (TNF-α) in various cell types induces either cell death or mitogenesis through different signaling pathways. In the present study, we determined in human corneal epithelial cells how TNF-α also promotes cell survival. Human corneal epithelial (HCE) cells were cultured in DMEM/F-12 medium containing 10% FBS. TNF-α stimulation induced activation of a voltage-gated K+ channel detected by measuring single channel activity using patch clamp techniques. The effect of TNF-α on downstream events included NFκB nuclear translocation and increases in DNA binding activities, but did not elicit ERK, JNK, or p38 limb signaling activation. TNF-α induced increases in p21 expression resulting in partial cell cycle attenuation in the G1 phase. Cell cycle progression was also mapped by flow cytometer analysis. Blockade of TNF-α-induced K+ channel activity effectively prevented NFκB nuclear translocation and binding to DNA, diminishing the cell-survival protective effect of TNF-α. In conclusion, TNF-α promotes survival of HCE cells through sequential stimulation of K+ channel and NFκB activities. This response to TNF-α is dependent on stimulating K+ channel activity because following suppression of K+ channel activity TNF-α failed to activate NFκB nuclear translocation and binding to nuclear DNA.
Keywords: Patch clamp, K+ channel blockers, Cell death, Cell cycle mapping
Introduction
TNF-α has pleiotropic effects in many cell types that include activation of apoptosis, inflammation, and immune responses. These responses underlie the pathogenesis of various diseases, including diabetes and cancer. Conversely, TNF-α can also induce through different signaling pathway module proliferation and differentiation [1]. TNF-α is one of a number of cytokines found in tears and its expression is elevated in allergic conjunctivitis [2]. TNF-α is also secreted by corneal epithelial cells in response to inflammatory stimulation caused by bacterial and viral infections [3–6]. However, a cell survival-promoting role for TNF-α has not been described in this tissue.
The TNF-α-linked cell signaling pathways that elicit apoptosis have been extensively characterized. Upon binding of TNF-α to Tumor Necrosis Factor Receptor 1 (TNF-R1), a type I membrane protein in the TNFR superfamily in mammalian cells, TNF-R1 monomers cluster resulting in death signal initiation [7–10]. There is a common structure among these receptors known as the “death domain” (DD) on the cytoplasmic side of the membrane [10]. Activation of a DD-containing receptor by TNF-α can lead to recruitment of intracellular DD-containing adaptors such as Fas associated death domain (FADD) and TNFR associated death domain (TRADD). TNF-α activates TNF-R1 coupled receptor DDs and they recruit several signaling molecules (adaptor proteins) to the activated receptor including: FADD, TRAFs, and receptor-interaction protein (RIP) leading to activation of caspase 8, NFκB, and c-Jun N-terminal kinase (JNK) signaling pathways [11,12]. NFκB protein is a member of the cytoplasmic heterodimeric transcription factor family associated with cell survival [13,14]. Upon phosphorylation by IKK, NFκB dissociates from IκB and IκB is then degraded by proteosome complexes [15]. Unbound NFκBs dimerize and become active transcription factors. They then translocate into the nucleus to initiate gene transcription in response to TNF-α.
Even though TNF-α induces proliferation in some cell types, a cell signaling role for TNF-α-induced K+ channel activation in eliciting this response has not been described. Their involvement is tenable because K+ channels are a component of signaling pathways that mediate mitogenic responses to serum-containing growth factors in many cell types including corneal epithelial cells [16–19]. K+ channel activity belonging to the Kv3.4 family in these cells is essential for EGF-induced proliferation [16,20]. Inhibition of K+ channel activity with specific channel blockers results in attenuation of the cell cycle in the G1 phase [16–19]. On the other hand, UV irradiation also induces robust increases in channel activity in these cells, which in turn triggers apoptosis through activation of JNK signal cascades [20–22]. TNF-α-stimulated increases in K+ channel activities are important for TNF-α-induced cellular effects in cortical neurons, kidney epithelial, and liver cells [23–25]. TNF-α also induces mRNA expression of various K+ channel types during a systemic inflammatory response as well as tumor cell proliferation in brain and cancer cells [26–28]. However, TNF-α suppresses particular types of K+ channel activity in other cell types, indicating that the effect of TNF-α on K+ channel activity is dependent on the channel type and cell origins [26,29]. In the present study, we investigated the role of TNF-α-induced changes in K+ channel activity in determining human corneal epithelial cell fate. Our results reveal that such changes are requisite for TNF-α-induced NFκB nuclear translocation and activation, which in turn improves corneal epithelial cell survival. On the other hand, TNF-α-induced cellular responses in these cells do not involve activation of any of the MAP kinase limb pathways. It is currently unclear why these MAP kinase pathways are not involved in mediating improved cell survival in response to TNF-α exposure.
Methods
Cell culture of corneal epithelial cells
SV40-immortalized human corneal epithelial (HCE) cells were grown in DMEM/F-12 culture medium containing 10% fetal bovine serum, 5 μg/ml insulin, and 10,000 units/ml penicillin and 10,000 μg/ml streptomycin, and maintained in an incubator supplied with 95% air and 5% CO2 at 37°C. Before experimentation, cells were synchronized in the G1 phase of the cell cycle by serum deprivation for at least 24 h. The medium was replaced every 2 days and the cells were passed by treatment with 0.05% trypsin-EDTA. For apoptosis induction, cells were placed 6 cm away from a safe light containing UV-C in a tissue culture hood and irradiated with 40 μJ/cm2. After irradiation, they were collected and rinsed with phosphate-buffered saline (PBS). To suppress K+ channel activity, the cells were exposed to a K+ channel blocker, 4-aminopyridine (4-AP) and preincubated for 20 min prior to exposure to TNF-α dependent on the time requirement of different experiments. All chemicals unless otherwise specified were obtained from Sigma Chemicals.
Nuclear staining with ethidium bromide and acridine orange
Cell viability and apoptosis were determined by nuclear staining to detect nuclear DNA condensation. A dye mixture containing ethidium bromide and acridine orange, each of which was present at 100 μg/ml (EB/AO), was added to each cell culture dish at a final density of 3 × 105/ml. Cell populations were scored according to their staining color and intensity using a UV-fluorescence microscope (Nikon). Nuclei staining green have not lost membrane integrity. In contrast, corneal epithelial cells in which the nuclei stained orange have lost membrane integrity. Apoptotic cells can be distinguished from viable cells on the basis of the absence or presence of nuclear condensation/fragmentation.
Immunoblot analysis
The cells (2 × 105) were rinsed twice with ice-cold PBS and solubilized in sodium dodecyl sulfate polyacrylamide (SDS) sample buffer containing 62.5 mM Tris–HCl pH 6.8, 2% W/V SDS, 10% glycerol, 50 mM DTT, 0.01% w/v bromophenol blue, or phenol red. The resulting suspensions were denatured by boiling for 5 min. After fractionation of cell lysates with 12% polyacrylamide gel (PAGE), proteins were electrotransferred to PVDF membranes. Membranes were exposed to blocking buffer containing 5% nonfat milk in TBS-0.1% Tween 20 (TBS-T) for 1 h at room temperature (RT, 21–23°C), and then incubated overnight or for 1 h with antibodies of interest at 4°C. All antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA). After three washes with TBS-T buffer, membranes were incubated with alkaline phosphatase (AP)-linked secondary antibody for 1 h at RT. The proteins were detected with a Phototope-Star Western Blot Detection kit (Cell Signaling Technology, Beverly, MA). In addition, caspase 3 activity was detected to determine cell apoptosis by using an antibody against cleaved caspase 3. This antibody recognizes endogenous levels of the large fragment (17/19 kDa) of activated caspase-3 resulting from cleavage adjacent to Asp175 (Cell Signaling Technology, Beverly, MA).
Electrophoretic mobility shift assay (EMSA)
Nuclear proteins were extracted by centrifugation at 25 K rpm for 20 min. The consensus oligonucleotide for NFκB transcription factor (5′-AGTTGAGGGGACTTTCCCAGGC-3′) was 5′-end-labeled with γ-32P by using T4 poly-nucleotide kinase. After labeling with 32P, DNA probes were used to hybridize with extracted nuclear proteins. Competitive reactions were conducted by adding 1 pM nonlabeled DNA probe into each sample. DNA–protein complexes were displayed by electrophoresis on a 6% nondenaturing polyacrylamide gel. For the supershift assay, DNA–protein complexes were further incubated with a specific antibody against NFκB-p65. Specific competition experiments were performed by using unlabeled oligonucleotide to compete with the labeled oligonucleotide for NFκB. Nonspecific competition experiments were performed by using unlabeled Sp1 oligonucleotides to interact with the labeled NFκB oligonucleotides.
Cell-attached and whole-cell patch clamps
Glass pipettes with a resistance of 3–4 MΩ were manufactured with a two-stage puller (PP-83, Narishige). For the cell-attached single channel patch clamp, electrode pipettes and bath chamber solutions contained (mM): 140 KCl, 5 NaCl, 1 CaCl2, 1 MgCl2, 1 EGTA, and 10 HEPES (pH 7.4). Single channel currents were recorded with an Axonpatch 200A amplifier (Axon Instruments, Foster City, CA) and filtered with a 4-pole low-pass filter at 1 kHz and digitized at 22 kHz with a pulse-code modulator (A.R. Vetter, Rebersburg, PA). The nystatin-perforated patch clamp technique was used for whole cell K+ current recording. Pipettes with a resistance of 3–4 MΩ when filled with 150 mM KCl solution were manufactured with a two-stage puller (PP-83, Narishige) and fire-polished before use. The pipette tip was filled with a solution containing (in mM) 140 KCl, 2 MgCl2, 0.5 CaCl2, 2 ATP, 0.05 GTP, 1 EGTA, and 10 HEPES (titrated with KOH to pH 7.2). The remainder of the pipette was back-filled with the same pipette solution supplemented with 200 μg/ml nystatin. Cell membrane potential was held at −60 mV and cells were stimulated by a voltage pulse applied from −80 mV to +60 mV at 20 mV increments. The duration of each pulse was 1000 ms. The bath solution was composed of (in mM) 140 NaCl, 2 KCl, 1 CaCl2, 10 HEPES (pH 7.4). A pCLAMP program (Axon Instruments, Foster City, CA) was used to analyze single and whole-cell patch clamp channel data. Channel activity was determined as NPo, where N represents the number of channels in the patch and Po represents the open channel probability. Patched cells were held for 2 to 3 min after the seal is stabilized before adding TNF-α. All experiments were performed at room temperature (21–23°C). Channel blockers, including 4-AP and blood-derived substance-I (BDS-I), were applied into the patch pipettes to inhibit channel activity.
Cell cycle and statistical analysis
Cell cycle analysis was performed using flow cytometry. After cells were trypsinized and fixed with 70% ethanol and 50 mM glycine, they were resuspended in PBS containing RNase A (100 μg/ml) and propidium iodide (PI, 25 μg/ml). Cell cycle phases were mapped with a FACScan and analyzed with Cell Quest software (Becton Dickinson, Mountain View, CA). For statistical analysis of Western blots, autoradiographic films were scanned and relative densities of each signal band were analyzed using an Image program. The data are represented as mean ± SE and statistical significance was determined with the paired Student’s t test at P < 0.05.
Results
TNF-α-induced attenuation of cell cycle progression
In corneal epithelial cells, serum-containing factors transiently stimulate ERK and p38 resulting in stimulation of proliferation and migration [30,31]. On the other hand, exposure to UV-C induces JNK and p38 activation resulting in apoptosis [21]. Time- and concentration-dependent effects of TNF-α on MAP kinase signaling pathways were studied because it has been shown that TNF-α induces JNK signaling pathway in many cell types. Exposure to TNF-α stimulation at different concentrations up to 50 ng/ml had no effect on either JNK, Erk, or p38 MAP kinase activities, respectively. Similarly, none of these pathways were activated following exposure to TNF-α for up to 60 min (Figs. 1A and B). Even after 16 h of exposure to TNF-α, TNF-α stimulation failed to phosphorylate JNK (Fig. 1C). On the other hand as described, exposure to UV-C irradiation caused rapid activation of both JNK isoforms after 10 min (Fig. 1A), which was sustained for a subsequent 16 h (Fig. 1C). These results were highly reproducible from 3–4 independent experiments. To assess whether TNF-α could affect another parameter associated with cell fate, we next determined if it alters an aspect of cell cycle progression induced by exposure to FBS. This was done by measuring with Western analysis the effects of TNF-α on the level of protein expression of a cell cycle inhibitory protein, p21. Fig. 2A compares the time course of p21 expression in TNF-α-induced cells with their untreated counterpart. The data are shown graphically in Fig. 2B, indicating that TNF-α increased p21 expression up to 16 h. Progression of the cell cycle in response to TNF-α stimulation was detected by cell cycle mapping using a flow cytometer (Fig. 2C). The cells were synchronized by serum-depletion for at least 24 h and then stimulated with 10% FBS in the absence and presence of TNF-α. In TNF-α- and FBS-stimulated cells, the cell population of serum-starved cells in the G1 phase decreased from 56% to 50% whereas in those only exposed to FBS, the decline reached 44% (Fig. 2D). On the other hand, the S phase population following FBS starvation contained 23% of the total whereas FBS exposure caused the number to rise from 23% to 34%. With those exposed to FBS and TNF-α, the S phase population rose to only 29%. In addition, sub-G0 stage of the cell cycle was analyzed by comparing the differences among staved, FBS-stimulated, and TNF-α-induced cells (Fig. 2E). These changes suggest that TNF-α attenuates cell cycle progression. This interpretation is consistent with the decline in population of TNF-α treated cells in the S phase as well as the rise in p21 protein expression induced by this cytokine.
Fig. 1.
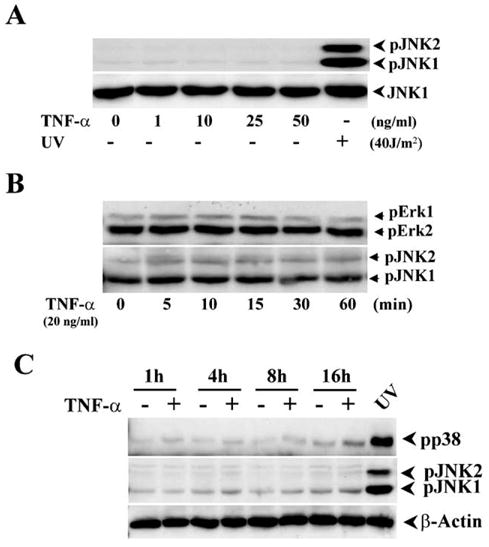
Effect of TNF-α on MAK kinase activation in HCE cells. (A) Dose-dependent effects of TNF-α stimulation on JNK activity. TNF-α was added into HCE cells at different concentrations. UV irradiation was used to activate JNK as a positive control. JNK activation was analyzed by Western blots using Western analysis with antibodies against phospho-JNK 30 min after the stimulation. (B) Effect of TNF-α stimulation on Erk activity. Effects of TNF-α stimulation on Erk and JNK activation were measured at indicated time points using Western analysis with antibodies against phospho-JNK and phospho-Erk. (C) Effect of TNF-α stimulation on p38 activity. Effects of TNF-α stimulation on p38 and JNK activation with/without UV irradiation were detected following a time course. HCE cells were incubated with 20 mg/ml TNF-α for different incubation periods. Whole cell lysates from UV exposed HCE cells were used as a positive control of phospho-JNK and phospho-p38.
Fig. 2.
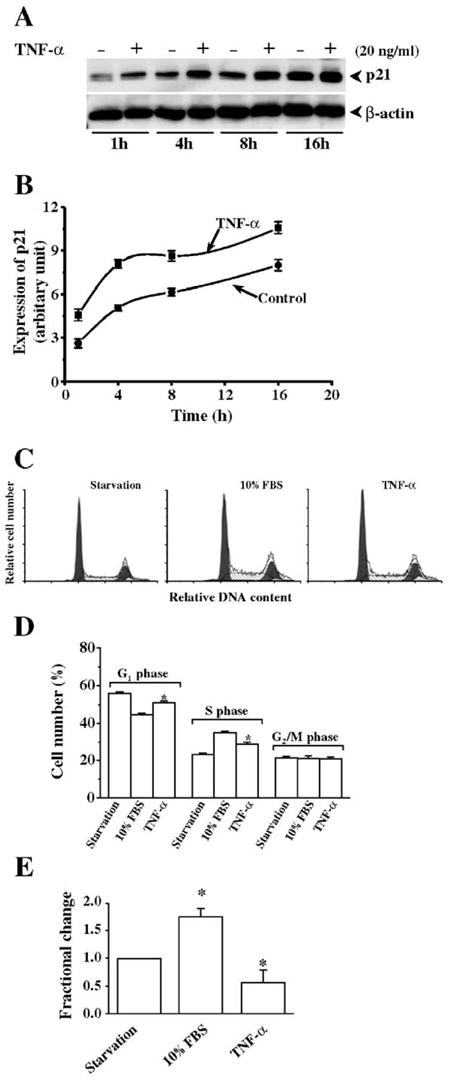
Effects of TNF-α stimulation on p21 expression and cell cycle progression. (A) Effect of TNF-α stimulation on p21 expression in HCE cells. TNF-α (20 ng/ml) was applied to activate p21 expression detected by Western blots following a time course up to 16 h. Expression of β-actin was detected as a loading control. (B) TNF-α stimulation-induced p21 expression plotted as a function of time. Statistical analysis revealed significant increases in p21 expression in response to TNF-α stimulation (n = 3, P < 0.05). (C) Effect of TNF-α stimulation on cell cycle progression. HCE cells were synchronize in G1 phase by serum-starvation for at least 24 h and then grown in normal DMEM/F12 medium containing 10% FBS in the absence or presence of 20 ng/ml TNF-α. HCE cells were harvested 18 h later for cell cycle mapping experiment using a flow cytometer. (D) Statistical analysis of cell cycle progression. (E) Statistical analysis of change in sub-G0 stage of the cell cycle. Symbol “*” indicates the significant difference between TNF-α-stimulated and unstimulated cells (n = 4, P < 0.05).
TNF-α-induced K+ channel activity in HCE cells
In corneal epithelial cells, exposure to either mitogens or UV light induces increases in K+ channel activity followed by increases in proliferation or apoptosis [16,20]. Such activation is requisite for these responses to occur because they are attenuated if K+ channel activity is suppressed by 4-AP or other K+ channel blockers. As TNF-α also affects cell fate, we determined whether changes in K+ channel activity are a component of a signaling pathway mediating such a response. Accordingly, the nystatin-perforated whole-cell patch clamp was used to study the effect of TNF-α on K+ channel activity by recording the whole-cell current resulted from depolarization of the membrane potential from a holding potential of −80 to +60 mV in 20 mV increments. Upon exposure of RCE cells to 40 ng/ml TNF-α following a time course up to 25 min, the amplitude of the K+ current was markedly increased within 10 min and then gradually decreased (Figs. 3A and B). The long-term effect of TNF-α on K+ channel activity was determined by patching these cells 18 h after exposure of cells to 20 ng/ml TNF-α. TNF-α-induced increases in K+ current amplitude appeared after 18 h (Fig. 3C). K+ currents in response to 20 ng/ml TNF-α stimulation were plotted as a function of membrane potentials (I–V curve) with/without TNF-α stimulation (Fig. 3D). To further confirm the effect of TNF-α on K+ channel activity, K+ channel activity was then investigated by using the cell-attached patch clamp. Serum-starved HCE cells were used in control experiments to record baseline K+ channel activity. Application of 20 ng/ml of TNF-α onto the patched cells induced robust K+ channel activity at a membrane potential of −60 mV (Fig. 3E). TNF-α-induced K+ channel activities were recorded up to 30 min and there was no running down in channel activity. The channel opening properties were analyzed as long as the recording period. TNF-α-induced K+ channel activity significantly increased to 30.2 ± 3.1% (n = 8), compared to K+ channel activity in control cells (9.1 ± 1.6%, n = 14). However, TNF-α-induced K+ channel activities were effectively suppressed by 4-AP (1 mM) and BDS-I (400 nM) in the patch pipette. Application of 4-AP and BDS-I markedly suppressed TNF-α-induced K+ channel activity resulting in the channel opening to decline to 8.4 ± 0.8% and 7.6 ± 0.9%, respectively (Fig. 3F). Such inhibition is consistent with our previous study that showed that there is 4-AP and BDS-I sensitive K+ channel activity in these cells in response to mitogen and stress stimulations.
Fig. 3.
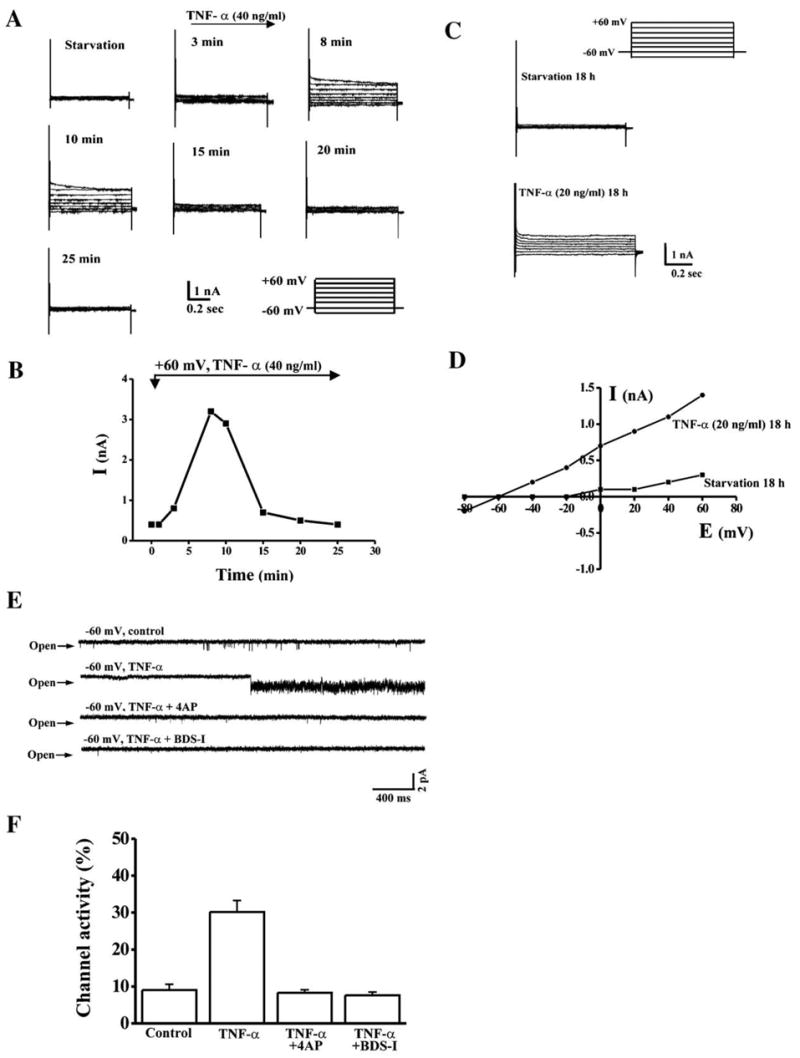
Effect of TNF-α on increases in K+ channel activity. (A) Whole-cell current recorded from TNF-α-stimulated cells. Nystatin-perforated whole-cell patch clamp was performed to record K+ currents in response to TNF-α stimulation. (B) Time course recorded from a TNF-α-stimulated cell. Amplitudes of K+ current were recorded after 40 ng/ml TNF-α was applied in the patch chamber. (C) Long-term effect of TNF-α on K+ currents. Whole-cell currents were recorded 18 h after TNF-α stimulation. (D) Current–voltage (I–V) relationship of TNF-α-stimulated K+ currents. I–V curves of K+ currents were obtained in cells 18 h after serum starvation and TNF-α stimulation. (E) Single channel activity recorded in TNF-α-stimulated cells. Single channel activity was recorded by the cell-attached patch clamping in HCE cells in the absence and presence of 20 ng/ml TNF-α. The membrane potential was held at −60 mV and 4-AP (1 mM) or BDS-I (400 nM) was used to suppress K+ channel activity. (F) Comparison of single channel activity in the absence and presence of TNF-α. Single channel open probability was calculated using a pCLAMP program. Symbol “*” indicates the significant difference between TNF-α-stimulated and unstimulated cells (n = 4, P < 0.05).
Dependence of TNF-α-enhanced cell survival on K+ channel activity
As TNF-α induced attenuation of cell cycle progression by resulting a rise in the cell population in the G1 phase, we determined if this effect is associated with an increase in cell viability. Accordingly, the dependence was evaluated by TNF-α-induced cell cycle attenuation on K+ channel activity. This was done by evaluating viability of cells that were stimulated with 20 ng/ml TNF-α in the absence and presence of various dosages of 4-AP (Fig. 4). Cell death was determined 36 h after TNF-α induction by detection of chromatin condensation using nuclear staining with ethidium bromide (EB) and acridine orange (AO). Treatment of cells with just TNF-α (20 ng/ml) or 4-AP (1 mM) did not affect HCE cell viability. However, suppression of K+ channel activity with 4-AP in TNF-α-induced cells triggered 4-AP dose-dependent programmed cell death. In addition, cell apoptotic response was also measured by detection of caspase 3 activity by detection of cleaved caspase 3 Tsp175 fragment (Fig. 4B). Caspase 3 activity was increased in TNF-α-induced and K+ channel-inhibited cells. These data suggest that TNF-α promotion of cell survival is dependent on its ability to induce increases in K+ channel activity.
Fig. 4.
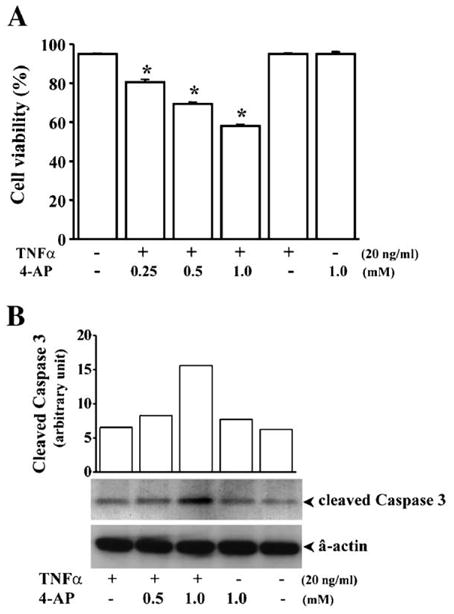
Effect of suppressing K+ channel activity on TNF-α induced alterations of cell viability. HCE cells were treated with different dosages of 4-AP 30 min prior to TNF-α stimulation (20 ng/ml). (A) HCE cell apoptosis detected by nuclear staining with ethidium bromide and acidine orange (EB/AO). HCE cell apoptosis was determined 36 h after TNF-α stimulation by EB/AO to detect chromatin condensation/fragmentation. Cell population was scored according to the color using a UV-fluorescence microscope (Nikon). Symbol “*” indicates the significant difference between cells with and without TNF-α stimulation and 4-AP treatment (P < 0.05). (B) HCE cell apoptosis detected by measuring caspase 3 activity. HCE cell apoptosis was determined by Western blot analysis of extracts from TNF-α-stimulated and K+ channel-inhibited HCE cells using Cleaved Caspase-3 (Asp175) (5A1) Rabbit mAb.
TNF-α-induced NFκB nuclear translocation and activation
To determine signaling downstream events linked to TNF-α-induced K+ channel activation events in HCE cells, we first probed for NFκB nuclear translocation and activation. This was done by separating nuclear proteins from their cytosolic counterparts and by evaluating with Western analysis NFκB density in these two fractions. Nuclear translocation of NFκB was markedly increased in a dose-dependent manner from 20 to 100 ng/ml of TNF-α concentration (Fig. 5A). With 20 ng/ml of TNF-α, NFκB nuclear translocation became evident during the first 15 min of a 60 min measuring period (Fig. 5B). These results indicate a variation of TNF-R1-linked signaling pathway because TNF-α stimulation induces nuclear translocation of NFκB without activating JNK cascades.
Fig. 5.
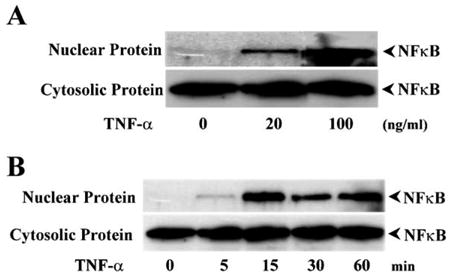
Effect of TNF-α on NFκB translocation in HCE cells. (A) Dose-dependent response of TNF-α-induced NFκB nuclear translocation. Nuclear and cytosolic proteins were isolated from TNF-α-stimulated cells and expressions of NFκB were detected by Western blots. (B) Time course of TNF-α induced NFκB translocation in the nuclei. HCE cells were stimulated with 20 ng/ml TNF-α and collected for Western analysis at indicated time points.
Effect of altered K+ channel activity on TNF-α-induced NFκB activation
Activation of NFκB in TNF-α-induced cells was further assessed by measuring its capability to undergo nuclear translocation and selectively bind to DNA fragments. Time-dependent activation of NFκB in response to TNF-α induction was determined using electrophoresis gel shifting assay (EMSA). Nuclear proteins were extracted from HCE cells before and after TNF-α stimulation following a time course. NFκB-specific and 32P-labeled DNA probes were hybridized with gel-displayed nuclear NFκB proteins (Fig. 6A). TNF-α induced two strong DNA binding bands from 15 to 60 min and their formation was competitively blocked by unlabeled NFκB-specific probes, but not with unrelated DNAs. A supershift band was observed in the lane where the sample was preincubated with anti-NFκB antibody, suggesting that these bands are indeed NFκB-specific bands. The dependence of TNF-α-induced NFκB nuclear translocation on K+ channel activity was determined by treating HCE cells with various dosages of 4-AP. TNF-α-induced DNA binding activity of NFκB was markedly inhibited by suppression of K+ channel activity with 4-AP in a dose-dependent manner (Fig. 6B). Suppression of K+ channel activity with 4-AP also inhibited TNF-α-induced NFκB nuclear translocation analyzed by Western blot in nuclear and cytosolic separated proteins (Fig. 6C). These results provide evidence that TNF-α-induced NFκB nuclear translocation is dependent on upstream increases in K+ channel activity.
Fig. 6.
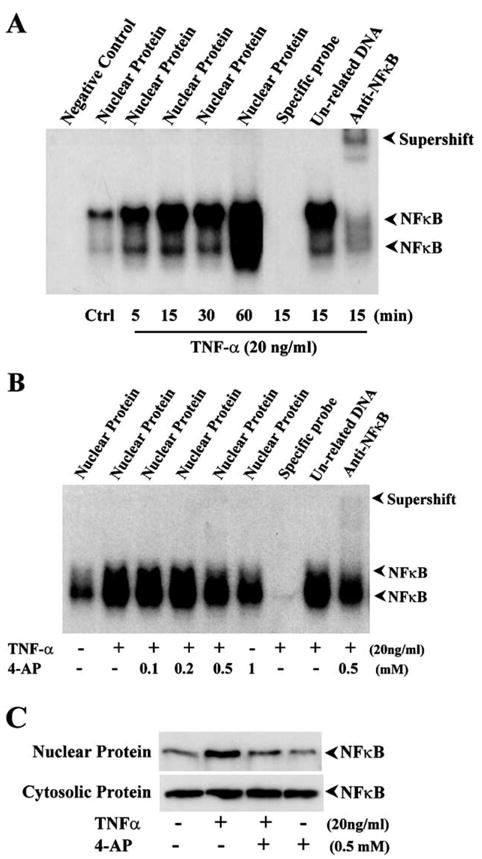
Effects of TNF-α and suppression of K+ channel activity on NFκB activation. (A) Time-dependent activation of NFκB in response to TNF-α stimulation. HCE cells were simulated with 20 ng/ml TNF-α and then lysed at indicated time points. (B) Dose-dependent effect of suppressing K+ channel activity on TNF-α-induced NFκB activation. Nuclear extracts were displayed in EMSA as described in method. Unlabeled NFκB consensus oligonucleotides (specific competitor), unlabeled SP1 oligonucleotides (nonspecific competitor), anti-NFκB (p65) antibody (for supershift lane), and various concentrations of 4-AP were added to reaction, respectively. (C) Inhibition of TNF-α induced NFκB transnuclear activity by suppressing K+ channel activity. HCE cells were treated with 1 mM 4-AP for 30 min prior to the stimulation of 20 ng/ml TNF-α for 15 min. The cell nuclear and cytosolic extracts were displayed in PAGE.
Discussion
The present study explores mechanisms mediating TNF-α control of corneal epithelial cell survival and apoptosis. It has important physiological relevance to corneal epithelial wound healing because TNF-α is increased in tears during infections and inflammations of the cornea [3,4,32]. Interestingly, TNF-α may have a dual effect of inducing cell death, but its predominant effect is to affect an increase in cell viability through K+ channel activation [6]. Recent studies show that TNF-α is able to activate NFκB in many different cell types and this response is important to determine cell fate. The ability of NFκB to suppress apoptosis can be overridden by protein synthesis inhibitors such as cycloheximide resulting in sensitizing the cells to TNF-induced apoptosis [33–35]. We found in corneal epithelial cells that TNF-α-induced NFκB activation is ligand dose-dependent and time-dependent (Figs. 5 and 6). NFκB activation by TNF-α was identified based on NFκB nuclear translocation and increases in the ability of NFκB to bind to its target DNA. Some members of the TNF superfamily induce apoptosis, whereas others act as survival factors or induce cell proliferation. In corneal epithelial cells, TNF-α did not induce apoptosis, but instead increases in expression of p21 and cell cycle attenuation in the G1 phase (Fig. 2).
TNF-stimulation activates MAP kinase cascades in some cell types, especially, the JNK signaling pathway [36–38]. There are crosstalks between JNK and NFκB in the TNF-α-induced signaling network. TNF-α-induced JNK activation may not be involved in induction of apoptosis when NFκB protects against apoptosis [39]. In the absence of NFκB activity, TNF-α-induced apoptosis increased, whereas direct activation of NFκB protects cell against apoptosis. Similarly, in different cell types, there is an inverse relationship between TNF-α-induced activation of the JNK module and NFκB. In cases where there is JNK module activation, NFκB stimulation is suppressed whereas in other cases increased NFκB stimulation is associated with less JNK activation [40–43]. In the present study, we found that Erk, JNK, and p38 MAP kinase pathways were not activated by application of TNF-α. Lack of TNF-α-induced activation of JNK in HCE cells may enhance the antiapoptotic effect of NFκB. This notion is supported by recent studies in hepatocytes showing that the JNK inhibitor SP600125 minimizes TNF-α-induced apoptosis through suppression of caspase 3 activation and DNA laddering [43].
K+ channels are universally present in the cell membrane and important in maintaining growth factor-stimulated cell growth and UV irradiation-induced apoptosis. K+ channel activity has been found to be a key determinant for cell progression through the G1 checkpoint of the cell cycle [16,18,44,45]. In K+ channel activity-suppressed cells, retinoblastoma protein (pRB) is dephosphorylated and effectively inhibits the cell from progressing through the G1/S transition [18]. These results suggest that changes in K+ channel activity can modulate the magnitude of a mitogenic response to a growth factor or cytokine. On the other hand, increases in K+ channel activity are likely involved in mediating programmed cell death (apoptosis). In corneal epithelial cells, UV irradiation can induce hyper-activation of K+ channels with a magnitude of 2-fold greater than that of mitogen-induced channel activity [20]. The hyper-activation of K+ channels at this magnitude may cause an excessive loss of intracellular K+ ions, resulting in a rapid cell shrinkage, activation of JNK cascades, and other catastrophic cellular effects [20,22]. Their diverse control suggests that there exists a crosstalk at various levels between K+ channel activity in the cell membrane and growth factor receptor-linked signaling cascades or stress-induced signaling pathways [16,17,19,22,46]. In earlier studies, our lab and others have shown that the K+ channel activity is the major conductance in corneal epithelial cells. In the present study, we found in HCE cells that TNF-α induces increases in activity of a 4-AP sensitive K+ channel (Fig. 3). Such stimulation elicits activation through possibly a myriad of changes that include initially volume shrinkage leading to changes in cytoskeletal configuration and scaffolding protein conformation. In addition, TNF-α may also activate other pathways besides K+ channel pathways. However, the exact details cannot be dealt with here because it is a topic for future investigation.
In previous studies, we have identified subtypes of K+ channel in corneal epithelial cells using a specific panel of K+ channel antibodies. The TNF-α-induced K+ channel is similar to those K+ channels in the Kv3.4 family because it is sensitive to 4-AP and BDS-I and has a similarity in the single channel conductance [47]. The K+ channel subtype Kv3.4 is highly expressed in whole cell lysates and in cell membrane preparations of these cells. TNF-α-induced increases in K+ channel activity are upstream from activation and nuclear translocation of NFκB because exposure to 4-AP prevented TNF-α-induced NFκB binding to DNA and its nuclear translocation (Fig. 6). In our previous studies, 4-AP inhibited K+ channel at μM levels (IC50 = 106 μM), indicating that there is a good agreement between the inhibitory effects of 4-AP on K+ channel activity and those on NFκB activation [20,47]. Furthermore, such suppression obviated the protective effect of TNF-α on cell death. Overriding this protective effect results in an increase in apoptosis of these cells, while application of TNF-α or 4-AP alone to cells does not induce apoptosis (Fig. 4). Our results further indicate that this cell membrane channel activity is responsible for initiation of TNF-α-induced activation of NFκB signaling that is essential for cell survival in response to cytokine and stress stimulations. In conclusion, our results reveal for the first time that TNF-α-induced stimulation of a 4-AP-sensitive K+ channel is required for NFκB nuclear translocation and DNA binding activity, which in turn promotes cell survival.
Acknowledgments
This study was supported by grants from the National Institutes of Health (NIH) EY04795 to PR, and EY12953 and EY15282 to LL.
References
- 1.Baud V, Karin M. Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol. 2001;11:372–377. doi: 10.1016/s0962-8924(01)02064-5. [DOI] [PubMed] [Google Scholar]
- 2.Cook EB, Stahl JL, Lowe L, Chen R, Morgan E, Wilson J, Varro R, Chan A, Graziano FM, Barney NP. Simultaneous measurement of six cytokines in a single sample of human tears using microparticle-based flow cytometry: allergics vs. non-allergics. J Immunol Methods. 2001;254:109–118. doi: 10.1016/s0022-1759(01)00407-0. [DOI] [PubMed] [Google Scholar]
- 3.Bitko V, Garmon NE, Cao T, Estrada B, Oakes JE, Lausch RN, Barik S. Activation of cytokines and NF-kappa B in corneal epithelial cells infected by respiratory syncytial virus: potential relevance in ocular inflammation and respiratory infection. BMC Microbiol. 2004;4:28. doi: 10.1186/1471-2180-4-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kumar A, Zhang J, Yu FS. Innate immune response of corneal epithelial cells to Staphylococcus aureus infection: role of peptidoglycan in stimulating proinflammatory cytokine secretion. Invest Ophthalmol Visual Sci. 2004;45:3513–3522. doi: 10.1167/iovs.04-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Minami M, Kita M, Yan XQ, Yamamoto T, Iida T, Sekikawa K, Iwakura Y, Imanishi J. Role of IFN-gamma and tumor necrosis factor-alpha in herpes simplex virus type 1 infection. J Interferon Cytokine Res. 2002;22:671–676. doi: 10.1089/10799900260100150. [DOI] [PubMed] [Google Scholar]
- 6.Kurpakus-Wheater M, Sexton R, McDermott ML, Mrock LK, Sosne G. Caspase-9 activation in hypoxic human corneal epithelial cells. Apoptosis. 2003;8:681–688. doi: 10.1023/A:1026164332473. [DOI] [PubMed] [Google Scholar]
- 7.Ashkenazi A, Marsters SA, Capon DJ, Chamow SM, Figari IS, Pennica D, Goeddel DV, Palladino MA, Smith DH. Protection against endotoxic shock by a tumor necrosis factor receptor immunoadhesin. Proc Natl Acad Sci U S A. 1991;88:10535–10539. doi: 10.1073/pnas.88.23.10535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gruss HJ, Dower SK. Tumor necrosis factor ligand superfamily: involvement in the pathology of malignant lymphomas. Blood. 1995;85:3378–3404. [PubMed] [Google Scholar]
- 9.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith CA, Gruss HJ, Davis T, Anderson D, Farrah T, Baker E, Sutherland GR, Brannan CI, Copeland NG, Jenkins NA, et al. CD30 antigen, a marker for Hodgkin’s lymphoma, is a receptor whose ligand defines an emerging family of cytokines with homology to TNF. Cell. 1993;73:1349–1360. doi: 10.1016/0092-8674(93)90361-s. [DOI] [PubMed] [Google Scholar]
- 11.Ichijo H. Differentiation of the chick retinotectal topographic map by remodeling in specificity and refinement in accuracy. Brain Res Dev Brain Res. 1999;117:199–211. doi: 10.1016/s0165-3806(99)00126-1. [DOI] [PubMed] [Google Scholar]
- 12.Dempsey PW, Doyle SE, He JQ, Cheng G. The signaling adaptors and pathways activated by TNF superfamily. Cytokine Growth Factor Rev. 2003;14:193–209. doi: 10.1016/s1359-6101(03)00021-2. [DOI] [PubMed] [Google Scholar]
- 13.Beg AA, Baltimore D. An essential role for NF-kappaB in preventing TNF-alpha-induced cell death. Science. 1996;274:782–784. doi: 10.1126/science.274.5288.782. [DOI] [PubMed] [Google Scholar]
- 14.Gilmore TD, Koedood M, Piffat KA, White DW. Rel/NF-kappaB/IkappaB proteins and cancer. Oncogene. 1996;13:1367–1378. [PubMed] [Google Scholar]
- 15.Zandi E, Rothwarf DM, Delhase M, Hayakawa M, Karin M. The IkappaB kinase complex (IKK) contains two kinase subunits, IKKalpha and IKKbeta, necessary for IkappaB phosphorylation and NF-kappaB activation. Cell. 1997;91:243–252. doi: 10.1016/s0092-8674(00)80406-7. [DOI] [PubMed] [Google Scholar]
- 16.Roderick C, Reinach PS, Wang L, Lu L. Modulation of rabbit corneal epithelial cell proliferation by growth factor-regulated K(+) channel activity. J Membr Biol. 2003;196:41–50. doi: 10.1007/s00232-003-0623-1. [DOI] [PubMed] [Google Scholar]
- 17.Xu D, Wang L, Dai W, Lu L. A requirement for K+-channel activity in growth factor-mediated extracellular signal-regulated kinase activation in human myeloblastic leukemia ML-1 cells. Blood. 1999;94:139–145. [PubMed] [Google Scholar]
- 18.Xu B, Wilson BA, Lu L. Induction of human myeloblastic ML-1 cell G1 arrest by suppression of K+ channel activity. Am J Physiol. 1996;271:C2037–C2044. doi: 10.1152/ajpcell.1996.271.6.C2037. [DOI] [PubMed] [Google Scholar]
- 19.Wang L, Xu B, White ER, Lu L. Growth factor-mediated K+ channel activity associated with human myeloblastic ML-1 cell proliferation. Am J Physiol. 1997;273:C1657–C1665. doi: 10.1152/ajpcell.1997.273.5.C1657. [DOI] [PubMed] [Google Scholar]
- 20.Wang L, Li T, Lu L. UV-induced corneal epithelial cell death by activation of potassium channels. Invest Ophthalmol Visual Sci. 2003;44:5095–5101. doi: 10.1167/iovs.03-0590. [DOI] [PubMed] [Google Scholar]
- 21.Lu L, Wang L, Shell B. UV-induced signaling pathways associated with corneal epithelial cell apoptosis. Invest Ophthalmol Visual Sci. 2003;44:5102–5109. doi: 10.1167/iovs.03-0591. [DOI] [PubMed] [Google Scholar]
- 22.Wang L, Xu D, Dai W, Lu L. An ultraviolet-activated K+ channel mediates apoptosis of myeloblastic leukemia cells. J Biol Chem. 1999;274:3678–3685. doi: 10.1074/jbc.274.6.3678. [DOI] [PubMed] [Google Scholar]
- 23.Houzen H, Kikuchi S, Kanno M, Shinpo K, Tashiro K. Tumor necrosis factor enhancement of transient outward potassium currents in cultured rat cortical neurons. J Neurosci Res. 1997;50:990–999. doi: 10.1002/(SICI)1097-4547(19971215)50:6<990::AID-JNR9>3.0.CO;2-8. [DOI] [PubMed] [Google Scholar]
- 24.Nietsch HH, Roe MW, Fiekers JF, Moore AL, Lidofsky SD. Activation of potassium and chloride channels by tumor necrosis factor alpha. Role in liver cell death. J Biol Chem. 2000;275:20556–20561. doi: 10.1074/jbc.M002535200. [DOI] [PubMed] [Google Scholar]
- 25.Wei Y, Babilonia E, Pedraza PL, Ferreri NR, Wang WH. Acute application of TNF stimulates apical 70-pS K+ channels in the thick ascending limb of rat kidney. Am J Physiol Renal Physiol. 2003;285:F491–F497. doi: 10.1152/ajprenal.00104.2003. [DOI] [PubMed] [Google Scholar]
- 26.Vicente R, Coma M, Busquets S, Moore-Carrasco R, Lopez-Soriano FJ, Argiles JM, Felipe A. The systemic inflammatory response is involved in the regulation of K(+) channel expression in brain via TNF-alpha-dependent and -independent pathways. FEBS Lett. 2004;572:189–194. doi: 10.1016/j.febslet.2004.07.030. [DOI] [PubMed] [Google Scholar]
- 27.Vicente R, Escalada A, Coma M, Fuster G, Sanchez-Tillo E, Lopez-Iglesias C, Soler C, Solsona C, Celada A, Felipe A. Differential voltage-dependent K+ channel responses during proliferation and activation in macrophages. J Biol Chem. 2003;278:46307–46320. doi: 10.1074/jbc.M304388200. [DOI] [PubMed] [Google Scholar]
- 28.Wang H, Zhang Y, Cao L, Han H, Wang J, Yang B, Nattel S, Wang Z. HERG K+ channel, a regulator of tumor cell apoptosis and proliferation. Cancer Res. 2002;62:4843–4848. [PubMed] [Google Scholar]
- 29.Wang J, Wang H, Zhang Y, Gao H, Nattel S, Wang Z. Impairment of HERG K(+) channel function by tumor necrosis factor-alpha: role of reactive oxygen species as a mediator. J Biol Chem. 2004;279:13289–13292. doi: 10.1074/jbc.C400025200. [DOI] [PubMed] [Google Scholar]
- 30.Sharma GD, He J, Bazan HE. p38 and ERK1/2 coordinate cellular migration and proliferation in epithelial wound healing: evidence of cross-talk activation between MAP kinase cascades. J Biol Chem. 2003;278:21989–21997. doi: 10.1074/jbc.M302650200. [DOI] [PubMed] [Google Scholar]
- 31.Kang SS, Li T, Xu D, Reinach PS, Lu L. Inhibitory effect of PGE2 on EGF-induced MAP kinase activity and rabbit corneal epithelial proliferation. Invest Ophthalmol Visual Sci. 2000;41:2164–2169. [PubMed] [Google Scholar]
- 32.Leonardi A, Brun P, Abatangelo G, Plebani M, Secchi AG. Tear levels and activity of matrix metalloproteinase (MMP)-1 and MMP-9 in vernal keratoconjunctivitis. Invest Ophthalmol Visual Sci. 2003;44:3052–3058. doi: 10.1167/iovs.02-0766. [DOI] [PubMed] [Google Scholar]
- 33.Gaur U, Aggarwal BB. Regulation of proliferation, survival and apoptosis by members of the TNF superfamily. Biochem Pharmacol. 2003;66:1403–1408. doi: 10.1016/s0006-2952(03)00490-8. [DOI] [PubMed] [Google Scholar]
- 34.Manna SK, Aggarwal BB. Lipopolysaccharide inhibits TNF-induced apoptosis: role of nuclear factor-kappaB activation and reactive oxygen intermediates. J Immunol. 1999;162:1510–1518. [PubMed] [Google Scholar]
- 35.Manna SK, Mukhopadhyay A, Van NT, Aggarwal BB. Silymarin suppresses TNF-induced activation of NF-kappa B, c-Jun N-terminal kinase, and apoptosis. J Immunol. 1999;163:6800–6809. [PubMed] [Google Scholar]
- 36.Murata-Ohsawa M, Tohda S, Kogoshi H, Nara N. The Notch ligand, Delta-1, reduces TNF-alpha-induced growth suppression and apoptosis by decreasing activation of caspases in U937 cells. Int J Mol Med. 2004;14:861–866. [PubMed] [Google Scholar]
- 37.Rosette C, Karin M. Ultraviolet light and osmotic stress: activation of the JNK cascade through multiple growth factor and cytokine receptors. Science. 1996;274:1194–1197. doi: 10.1126/science.274.5290.1194. [DOI] [PubMed] [Google Scholar]
- 38.Sathyanarayana P, Barthwal MK, Kundu CN, Lane ME, Bergmann A, Tzivion G, Rana A. Activation of the Drosophila MLK by ceramide reveals TNF-alpha and ceramide as agonists of mammalian MLK3. Mol Cell. 2002;10:1527–1533. doi: 10.1016/s1097-2765(02)00734-7. [DOI] [PubMed] [Google Scholar]
- 39.Liu ZG, Hsu H, Goeddel DV, Karin M. Dissection of TNF receptor 1 effector functions: JNK activation is not linked to apoptosis while NF-kappaB activation prevents cell death. Cell. 1996;87:565–576. doi: 10.1016/s0092-8674(00)81375-6. [DOI] [PubMed] [Google Scholar]
- 40.Shishodia S, Koul D, Aggarwal BB. Cyclooxygenase (COX)-2 inhibitor celecoxib abrogates TNF-induced NF-kappa B activation through inhibition of activation of I kappa B alpha kinase and Akt in human non-small cell lung carcinoma: correlation with suppression of COX-2 synthesis. J Immunol. 2004;173:2011–2022. doi: 10.4049/jimmunol.173.3.2011. [DOI] [PubMed] [Google Scholar]
- 41.Papa S, Zazzeroni F, Pham CG, Bubici C, Franzoso G. Linking JNK signaling to NF-{kappa}B: a key to survival. J Cell Sci. 2004;117:5197–5208. doi: 10.1242/jcs.01483. [DOI] [PubMed] [Google Scholar]
- 42.Liu H, Ma Y, Pagliari LJ, Perlman H, Yu C, Lin A, Pope RM. TNF-alpha-induced apoptosis of macrophages following inhibition of NF-kappa B: a central role for disruption of mitochondria. J Immunol. 2004;172:1907–1915. doi: 10.4049/jimmunol.172.3.1907. [DOI] [PubMed] [Google Scholar]
- 43.Schwabe RF, Uchinami H, Qian T, Bennett BL, Lemasters JJ, Brenner DA. Differential requirement for c-Jun NH2-terminal kinase in TNFalpha- and Fas-mediated apoptosis in hepatocytes. FASEB J. 2004;18:720–722. doi: 10.1096/fj.03-0771fje. [DOI] [PubMed] [Google Scholar]
- 44.Dubois JM, Rouzaire-Dubois B. Role of potassium channels in mitogenesis. Prog Biophys Mol Biol. 1993;59:1–21. doi: 10.1016/0079-6107(93)90005-5. (Review, 149 refs) [DOI] [PubMed] [Google Scholar]
- 45.Lewis RS, Cahalan MD. Ion channels and signal transduction in lymphocytes. Annu Rev Physiol. 1990;52:415–430. doi: 10.1146/annurev.ph.52.030190.002215. [DOI] [PubMed] [Google Scholar]
- 46.Lu L, Reinach PS, Kao WW. Corneal epithelial wound healing. Exp Biol Med (Maywood) 2001;226:653–664. doi: 10.1177/153537020222600711. [DOI] [PubMed] [Google Scholar]
- 47.Wang L, Fyffe RE, Lu L. Identification of a Kv3.4 channel in corneal epithelial cells. Invest Ophthalmol Visual Sci. 2004;45:1796–1803. doi: 10.1167/iovs.03-1056. [DOI] [PubMed] [Google Scholar]