

Abstract
Neurophysiologic processes underlie the uncontrolled, compulsive behaviors defining the addicted state. These “hard-wired” changes in the brain are considered critical for the transition from casual to addictive drug use. This review of preclinical and clinical (primarily neuroimaging) studies will describe how the delineation between pleasure, reward, and addiction has evolved as our understanding of the biologic mechanisms underlying these processes has progressed. Although the mesolimbic dopaminergic efflux associated with drug reward was previously considered the biologic equivalent of pleasure, dopaminergic activation occurs in the presence of unexpected and novel stimuli (either pleasurable or aversive) and appears to determine the motivational state of wanting or expectation. The persistent release of dopamine during chronic drug use progressively recruits limbic brain regions and the prefrontal cortex, embedding drug cues into the amygdala (through glutaminergic mechanisms) and involving the amygdala, anterior cingulate, orbitofrontal cortex, and dorsolateral prefrontal cortex in the obsessive craving for drugs. The abstinent, addicted brain is subsequently primed to return to drug use when triggered by a single use of drug, contextual drug cues, craving, or stress, with each process defined by a relatively distinct brain region or neural pathway. The compulsive drive toward drug use is complemented by deficits in impulse control and decision making, which are also mediated by the orbitofrontal cortex and anterior cingulate. Within this framework, future targets for pharmacologic treatment are suggested.
Keywords: amygdala, cocaine, cues, dopamine, impulsive behavior, nucleus accumbens, substance-related disorders, ventral tegmental area
Rewards are pleasurable. Addictions hurt. Reward is experienced in response to discrete stimuli, providing enjoyment and arousal. Addictions involve persistent, compulsive, and uncontrolled behaviors that are both maladaptive and destructive. Despite these obvious differences, reward and addiction were previously thought to share common neurobiologic underpinnings. This unified model has undergone a dramatic revision, and reward and addiction are now conceptualized as distinct neurochemical processes involving different, albeit overlapping, neuroanatomical circuits. In this review, I will begin with a historical perspective of mesolimbic dopamine and reward, followed by two theories of addiction (i.e., dopamine depletion and sensitization) that have guided medication development for several years. Newer interpretations of dopamine’s role in reward and addiction are then presented, reflecting our present understanding of dopamine as mediator of expectation, wanting, and novelty. Two general mechanisms involved in drug relapse will then be discussed: compulsive drive states, considered as four brain regions/pathways, each mediating a distinct relapse trigger (i.e., priming, drug cues, craving, and stress); and the inhibitory dyscontrol that can exacerbate the compulsive drug drive. The role of mesocorticolimbic dopaminergic and glutaminergic pathways, intracellular mechanisms, and relevant brain regions in compulsive drug drive and inhibitory dyscontrol will be the focus of these sections on drug relapse.
A few caveats are warranted. First, I will often use the term “addiction”1 (using terminology recommended by the American Academy of Pain Medicine, American Pain Society, and American Society of Addiction Medicine) rather than “dependence”2 to refer to the complex of behaviors that include withdrawal, tolerance, loss of control, compulsive use, and continued use despite adverse consequences. Second, addiction will be discussed specifically as it relates to substances of abuse, primarily cocaine. However, the neurobiology, brain structures, and behavioral and cognitive processes involved in the addictive use of substances most likely relate to nonsubstance addictions, such as those involving sex and gambling. Third, any review of neurobiological processes in addiction must, by necessity, be selective in its approach and regretfully omit important advances in the field. Last, this review will focus upon the biologic processes involved in the return to substance use after a period of abstinence—processes that pose, I believe, the most interesting and perplexing questions in understanding the nature of addiction. Thus, theories of addiction involving “opponent” or withdrawal processes will not be discussed.3-5
REWARD AND MESOLIMBIC DOPAMINE
Olds and Milner6 initiated our modern understanding of brain reward mechanisms in 1954. In this seminal work, rodents were given the opportunity to administer electrical stimulation to various brain regions. Specific brain areas were found to elicit persistent self-stimulation, often to the exclusion of any other behaviors. Confirming in humans the results of Olds and Milner, Heath7,8 demonstrated that subjects would similarly self-administer electrical stimuli to specific “pleasure” areas of the brain (for ethical commentary, see Baumeister’s 2000 article).9 Over the subsequent decades, the brain structures, neuronal pathways, and relevant neurotransmitters implicated in the experience of reward and reinforcement were further refined (see review by Gardner).10 The mesolimbic pathway, in particular, was identified as the key component in reward assessment. This pathway originates with dopaminergic cell bodies in the ventral tegmental area (VTA), a dopamine-rich nucleus located in the ventral portion (or tegmentum, meaning “covering”) of the midbrain. These dopaminergic axons project and primarily terminate in the nucleus accumbens (NAc) in the ventral striatum, but also extend into the amygdala, bed nucleus of stria terminalis (BNST), lateral septal area, and lateral hypothalamus. The VTA is in, close proximity to the substantia nigra, another dopamine-rich nucleus. Whereas the substantia nigra projects primarily to the dorsal striatum (via the mesostriatal pathway) and mediates motor activity, the mesolimbic pathway mediates reward.11
The experience of reward is accompanied by activation of the mesolimbic dopaminergic pathway. Natural rewards, such as food or sex—as well as most substances that are abused by humans, including alcohol, amphetamine, caffeine, cocaine, marijuana, nicotine, opiates, and phencyclidine10,12-17—increase extracellular concentrations of mesolimbic dopamine (DA). The stimulants cocaine and amphetamine directly amplify the mesolimbic dopaminergic signal at the postsynaptic DA receptor through different synaptic mechanisms. Cocaine increases synaptic dopaminergic concentrations by blocking the presynaptic dopamine transporter (DAT).18 The DAT is responsible for reabsorbing synaptic DA back into the presynaptic neuron, and occupancy of the DAT by cocaine prevents DA re-uptake. The amphetamines increase synaptic DA primarily by increasing DA release from the synaptic vesicles.19 Both cocaine and the, amphetamines increase both the absolute concentration of DA in the synapse and the time interval that DA remains at the postsynaptic receptor site. Due to their direct effect upon dopaminergic activity, the stimulants (particularly cocaine) are considered prototypic drugs of reward and have thus become the focus of biological and treatment studies (see following sections). Although nonstimulant drugs indirectly interact with the mesolimbic pathways through a variety of receptor systems, these compounds share the common pharmacologic property of stimulating mesolimbic DA—primarily in the NAc.10 These nonstimulant drugs bind with either G protein-coupled or ligand-gated ion channel receptors. Drugs that bind with G protein-coupled receptors (and their respective binding site(s)), include tetrahydrocannabinol (THC) (an agonist at cannabinoid receptors); opiates, such as heroin or morphine (agonists at opioid receptors, activating dopamine via VTA GABAergic disinhibition);20 and caffeine (an antagonist at striatal adenosine A2 receptors). Drugs that bind with ligand-gated ion channel receptors include alcohol (an allostatic facilitator at the GABAergic receptors and inhibitor of N-methyl-D-aspartate [NMDA] glutamate receptors); phencyclidine (PCP) (blocks NMDA glutamate receptors); and nicotine products, such as cigarettes (agonists at nicotinic cholinergic receptors).
The anatomical distinction made between drug reward and drug withdrawal further confirmed the importance of the mesolimbic pathway in drug self-administration. In a classic study by Bozarth and Wise,21 morphine was administered into either the VTA or periventricular gray (PVG) region of rats for 72 hours (the PVG is a brain stem region containing a high concentration of opioid receptors). Following the administration of the opioid antagonist naloxone, PVG morphine-infused rats showed signs of withdrawal, whereas VTA morphine-infused rats did not. Furthermore, rats could be trained to self-administer morphine into the VTA but not the PVG. This study demonstrated a clear separation between the biological processes involved in withdrawal versus reward, further supporting the role of the VTA in drug reward.
A wealth of preclinical studies has directly examined the effects of increasing or decreasing DA in the NAc upon drug self-administration.22-24 Using an in vivo “microdialysis” technique, minute changes in extracellular neurotransmitter concentrations can be assessed in real time during experimental manipulations. With this technique, Pettit and Justice25 found a correlation between the amount of cocaine self-administered by rodents and the extracellular DA released in the NAc. Conversely, an attenuation of the self-administration of stimulants follows either the administration of DA antagonists (e.g., haloperidol)26,27 or the neurotoxic lesioning of the dopaminergic cells in the NAc.28 Functional neuroimaging studies have further confirmed the relevance of the mesolimbic pathway and DA release to the cocaine-induced high. Using functional magnetic resonance imaging (fMRI), Breiter and colleagues29 administered cocaine to cocaine-addicted subjects while their brains were being scanned. Over the next several minutes, subjects were able to distinguish the “rush” that they reported during the first minute or two following cocaine use from the “craving” experienced following dissipation of the rush. Since fMRI techniques allow measures of regional brain activation to be assessed every few seconds, distinct images of brain activity were obtained during both the “rush” and “craving” experiences. During the rush, but not craving, increased brain activation was observed in the VTA, consistent with activation of the mesolimbic pathway. In a study using positron emission tomography (PET), Volkow and colleagues30 administered cocaine to subjects during imaging, and reported that the level of DAT occupancy or DAT blockade was significantly associated with the magnitude of self-reported high. Additional studies by Volkow and colleagues31 further suggest that the release of DA (as assessed by D2 receptor occupation) was a better predictor of high intensity than was DAT blockade. A general assumption guiding addiction research for much of the past two decades has therefore been that the addictive effects of the primary substances of abuse were contingent upon dopaminergic activation of the mesolimbic pathway.
THE TRANSITION FROM REWARD TO ADDICTION: A FOCUS ON DOPAMINE
The Dopamine Depletion Hypothesis of Addiction
The high or rush associated with the mesolimbic activation suggested that the neurotransmitter DA was itself responsible for the pleasurable feelings associated with rewards. The drive for continued drug administration (that is, addictive behaviors) was considered a consequence of the continued need to elicit heightened DA concentrations—and the resultant pleasure—in the mesolimbic pathway and associated brain regions. This hypothesis further predicted that the binge use of cocaine would result in a DA deficit state,3,32,33 resulting in a cocaine crash (i.e., anhedonia, depression) and a biologic demand (i.e., craving) for more cocaine to replenish the depleted DA stores. Drug-induced DA depletion has been variously referred to as the “dopamine depletion hypothesis”32 or the “general anhedonia model.”34 Evidence of increased striatal DAT receptors35 (suggesting upregulation in response to persistent DAT occupancy by cocaine) and decreased DA D2 receptors36,37 (suggesting downregulation in response to persistently elevated DA concentrations at the postsynaptic site) by PET imaging studies further supported dysregulation of the dopaminergic system. The attenuated D2 striatal concentrations were hypothesized to lessen the reinforcing salience of natural rewards and to heighten the need for substance-induced elevations in dopaminergic efflux. For example, an increase in D2 receptors decreases alcohol self-administration in rodents,38 whereas primates with lower D2 receptors demonstrate higher rates of cocaine self-administration.39 Cocaine-dependent subjects also demonstrated significantly reduced dopaminergic cell activity (as evidenced by reduced changes in striatal [11C]raclopride binding) following methylphenidate infusion relative to controls, as well as diminished reports of feeling “high.” Extracellular DA concentrations in the NAc were also reported to be inversely correlated with cocaine self-administration in rats, such that low DA levels produced moderate to high self-administration rates, and high DA levels produced moderate to low self-administration rates.40
To date, attempts to treat cocaine addiction by activating dopaminergic receptors—and thereby increasing dopaminergic tone—have not been successful. A number of dopaminergic agonists (e.g., pergolide,41 amantadine,42,43 bromocriptine,42,44,45 methylphenidate,46 and mazindol)47 did not attenuate relapse rates in cocaine-addicted subjects. Especially puzzling was a preclinical study exploring ventral striatal DA release and cocaine self-administration in DAT-deficient mice. Rocha and colleagues48 studied genetically altered mice in which the DAT receptor was not expressed (DAT −/− knockout mice). Since these mice did not have a DAT receptor to bind cocaine, cocaine administration did not induce an extracellular increase in ventral striatal DA. Unexpectedly, however, the DAT −/− mice self-administered similar amounts of cocaine as the wild-type mice (mice with intact DAT receptors). This finding revealed that neither the DAT nor an increase in synaptic DA was essential for the self-administration of cocaine. Additional studies by Rocha and colleagues48 (reported in the same article) suggested that the reinforcement properties of cocaine may have been mediated by cocaine’s occupancy of the serotonergic reuptake transporter. Other investigations, as reviewed by Spanagel and Weiss,49 also suggested that, except for the stimulants, mesolimbic DA neurotransmission does not play a crucial role in reinforcement maintained by drug of abuse.
Sensitization and Addiction
The sensitization hypothesis posited a contrasting view of dopamine’s relevance to the addictive process. This hypothesis predicted that the repeated administration of drugs would “sensitize” the DA system to the drug and the associated drug cues.50-52 This phenomenon was predicated upon observations that the intermittent, recurrent application of electrical stimuli to limbic brain regions induces a progressively excitable neuronal locus. The sensitized locus then exhibits a persistent, heightened sensitivity to the subsequent application of the original stimulus or its associated cues.53,54 Cocaine-induced sensitization was first observed by Grode in 1912,55 which was followed by pre-clinical studies showing both a progressive development in the likelihood of generalized seizures following daily cocaine administration51 and heightened sensitivity of dopaminergic receptors to psychomotor stimulants.56-58 Cocaine-induced limbic sensitization was suggested as a causal link between chronic cocaine use and cocaine-induced seizures,59 panic attacks,60,61 psychosis,62 and craving.50 The sensitization hypothesis of addiction (particularly with regard to cocaine) therefore predicted that relapse to drug use would decrease in response either to drugs used to suppress neuronal hyperexcitability (e.g., carbamazepine for temporal lobe epilepsy) or to dopaminergic antagonists that antagonize the hypersensitive dopaminergic response. Double-blind, placebo-controlled studies of carbamazepine demonstrated limited efficacy in the treatment of cocaine dependence,63-65 however, and dopaminergic antagonists (e.g., flupenthixol, risperidone, and ecopipam) were ineffective in decreasing relapse to cocaine use.66
Pharmacologic interventions guided by the DA-depletion or -sensitization hypotheses did not generate useful medications for the treatment of cocaine addiction. These clinical trials suggested that disruptions in dopaminergic transmission were subtler than simply an increase or decrease in extracellular release or dopaminergic sensitivity. A reevaluation of dopamine’s role in addictive disorders was therefore warranted.
INCENTIVE SALIENCE, LEARNING, AND NOVELTY: A RECONSIDERATION OF MESOLIMBIC DOPAMINE
In addition to the disappointing clinical response to dopaminergic agonists and antagonists, a paradigm shift in the role of DA in the addictive process occurred with the recognition that (1) DA does not, in and of itself, induce “pleasure,” (2) mesolimbic DA efflux increases not only in response to a reward, but also in anticipation of a potential reward and during aversive states, including foot shock, restraint stress, and the administration of anxiogenic drugs,17,24,34 (3) extracellular increases in accumbens DA are attenuated in rodents self-administering cocaine relative to “yoked” littermates (rodents that passively receive the same amount of cocaine that the other rodents self-administer),67 revealing that the self-administration of cocaine elicits less mesolimbic DA efflux compared to the passive administration of cocaine, and (4) DA plays critical, and overlapping, roles in the interpretation of stimuli and the acquisition of behaviors reinforced by natural rewards and drug stimuli.
Thus, the initial assumptions regarding the role of electrical brain stimulation in defining “reward” pathways were apparently overly simplistic. Berridge68 astutely noted that the early work of Heath7,8 (see above) reported that patients compulsively administered electrical stimuli. Rather than endorsing “pleasure” from the experience, however, these patients described a desire for more stimulation and other hedonic pursuits. It appeared that the “pleasure” pathway, identified primarily from studies of animals (who are notoriously reluctant to share their true mood states), may have been mislabeled. Instead, Berridge and colleagues23 and others69 have suggested that the mesolimbic pathway determines the incentive salience, or wanting, of a prospective reward—not the pleasurable experience of the reward itself. Stimulation of this pathway would therefore result in the motivational state of “wanting” (an expectation of pleasure) but would not mediate the reward’s hedonic, affective state, or “liking.”70 The distinction between “liking” and “wanting” was critical, as it segregated the cuphorigenic power of a substance from its addictive potential. In fact, previous studies have revealed that the drug self-administration could be maintained in the addicted subject in the absence of subjective pleasure—that is, drug liking was not a prerequisite for drug-seeking and drug-taking behavior.71 Di Chiara15 theorized a somewhat similar, though distinctive, role of the NAc, positing that mesolimbic dopaminergic activation affects motivational learning, not salience (see next section). Schulz and colleagues72 demonstrated that DA neurons fire only in response to novel rewards, regardless of the stimulus’s hedonic value; activation of the signal was dependent upon the predictability of the reward. Thus, an unexpected, novel, salient, and arousing stimulus elicits a strong dopaminergic signal, regardless of the motivational valence of the stimulus.73 With each repeated presentation of the stimulus, DA discharge lessens until the stimulus no longer produces a neuronal response. This role of DA is consistent with PET studies demonstrating that DA release in response to methylphenidate is attenuated in cocaine-addicted subjects relative to nonaddicted controls.74 Presumably, the cocaine stimulus is a more novel stimulus to nonaddicted subjects compared to cocaine-addicted volunteers. In an attempt to bridge these various roles of mesolimbic DA response, Salamone and colleagues34 have suggested that NAc DA is a “sensorimotor integrator” that “is involved in higher order motor and sensorimotor processes that are important for activational aspects of motivation, response allocation, and responsiveness to conditioned stimuli”—which, the authors acknowledge, “may not roll off the tongue as fluently as the word ‘reward.’”
The role of the mesolimbic pathway, and particularly the NAc, is therefore more complicated (and more contentious) than previously believed. Nevertheless, a common characteristic of the above constructs may be taken to suggest that the mesolimbic dopamine system mediates the interpretation or learning of prospective positive and negative reinforcers, and that DA signaling appears to promote goal-directed behaviors regardless of the reinforcer type. More specifically, the mesolimbic system assesses the salience, or value, of a potential reinforcer. To make this assessment, other cortical and limbic brain regions must work concurrently with the brain reward circuitry. An understanding of the role of the addictive process therefore also requires the inclusion of brain regions neuronally connected to the VTA and NAc. As noted earlier, the VTA provides dopaminergic innervation not only to the NAc, but also to the amygdala and BNST. In addition, the VTA projects DA from a third dopaminergic tract, the mesocortical pathway, which innervates prefrontal cortical regions that include the orbitofrontal cortex (OFC) and anterior cingulate.11 Coupled with glutaminergic and other reciprocal neurotransmitter connections, the NAc is integrated with the OFC, anterior cingulate, insular cortex, and hippocampus. Because of the importance of these regions in understanding the addictive processes discussed below, a brief description and diagram of these brain areas are provided in the text box and Figure 1.
FIGURE 1.
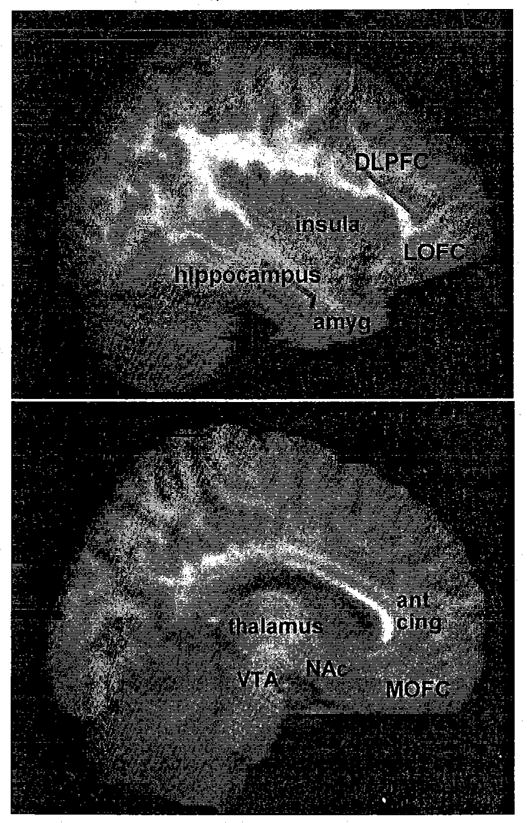
Brain regions relevant to the addictions (see text box for description of regions). Right panel represents an MRI of the sagittal brain (from SPM96) at Talairach coordinates x = 4–16; left panel, at x = 34–46. Each Talaraich coordinate represents a one mm MRI sagittal slice, and 13 slices were averaged for each displayed image. Amyg, amygdala; ant cing, anterior cingulate; DLPFC, dorsolateral prefrontal cortex; LOFC, lateral orbitofrontal cortex; MOFC, medial orbitofrontal cortex (ventromedial cortex); NAc, nucleus accumbens; VTA, ventral tegmental area. The MRI template was obtained from SPM96-MRI.
How, then, does this new understanding of the mesolimbic dopamine pathway, and the inclusion of other limbic and prefrontal cortical regions, guide us in our understanding of the addictive process? The progression from initial drug use to addiction may evolve as repeated activation of the mesolimbic pathway associated with persistent drug use heightens the incentive value, or salience, of the drug. As contextual cues become associated and strengthened with repetitive drug administration, a process initially under the purview of the mesolimbic DA system progressively incorporates the neurocircuitry involved in emotional memory, obsessive thoughts, stress response, decision making, and behavioral inhibition into the drug experience. These neuroadaptive responses remain engaged even in the absence of ongoing drug use and are thought to be a primary factor in drug relapse. The following two sections will discuss the brain mechanisms involved in the return to drug use, including the compulsive drug drive and the inhibitory dyscontrol observed in drug-addicted subjects, as well as the relationship of drug drive and inhibitory dyscontrol to dopaminergic dysregulation.
Brain Areas Involved in Addiction
Amygdala:Amygdalar activity is related to memory consolidation for emotionally arousing events. The amygdala is involved in assigning a reward value to stimuli and in the conditioning of fear to novel stimuli. For example, rodents favoring a specific cage that is identified with drug administration will lose this conditioned stimulus if the amygdala is ablated.
Anterior cingulate: Implicated in human disorders of emotion and attention, the anterior cingulate is involved in emotional self-control, focused problem-solving, error detection, performance monitoring, and adaptive response to changing conditions.75 It plays a role in the detection of processing conflicts, particularly when low-frequency responses are executed,76 but is influenced by both motivation and affective state.
Bed nucleus of the stria terminalis (BNST): Involved in autonomic and behavioral reactions to fearful stimuli, including the stress response, the BNST is considered part of the extended amygdala and shares with the nucleus accumbens a sensitivity to dopamine stimulation. In rats, the BNST is involved in the reinstatement of cocaine seeking after foot shock.77
Dorsolateral prefrontal cortex (DLPFC): Implicated in difficulties holding/maintaining several pieces of information “on line” or in short-term storage (i.e., “working memory”), the DLPFC is crucial for the control and regulation of cognitive activities, including the sequencing of events, planning, and the selection of goals.
Hippocampus: Critical for the acquisition of new factual information and the formation of new memories about personally experienced events (i.e., episodic memory), the hippocampus has been implicated in the loss of memory in Alzheimer’s disease. Damage to the hippocampus results in anterograde amnesia and, to a lesser degree, in retrograde amnesia.
Insular Cortex: Important for the processing of pain, the insular cortex receives visceral, olfactory, gustatory, and other somatosensory inputs. It probably plays an important role in relating interoceptive signals to information from other modalities, and often shows activation in neuroimaging studies producing acute anxiety.
Orbitofrontal cortex (OFC): In addition to being implicated in disorders of impulsivity and decision making, the OFC is involved in situations that are unpredictable or uncertain, and modulates the reinforcement value of stimuli in the context of recent experience. It assesses and decodes the likely value or behavioral relevance of available choices of action and is therefore activated when there is insufficient information available to determine an appropriate course of action. Recent evidence suggests that the medial OFC (ventromedial cortex), with connections to the hippocampus and cingulate, is involved in assessing the familiarity or “rightness” of a situation and in integrating outcome expectancies. The lateral OFC, with connections to the amygdala and insula, is associated with the suppression of previously rewarded responses and is required to change behavior (i.e., to provide “stop” signals).78
THE COMPULSIVE DRIVE TO RELAPSE
Compulsive drug use can be conceptualized (adapted from Koob & Moal)3 as arising from four overlapping brain regions or pathways, each delineating a distinctive pull toward substance use. The four regions/networks coincide with common inducements for relapse: (1) priming (i.e., a single drink that precipitates a binge),79 (2) drug cues, (3) cravings, and (4) stress. The sections on priming and drug cues also describe intracellular mechanisms involved in addictive processes.
Priming: The Nucleus Accumbens and Dopamine
Preclinical studies of priming confirm clinical observations—that a single drug administration is the most potent stimulus to renew drug use. Dopamine appears to play a critical role in priming, since the reinstatement of both opiate- and stimulant-seeking behaviors is elicited by the administration of directly acting dopaminergic agonists, and the priming effect of heroin, amphetamine, and cocaine is blocked by DA antagonists.58 (Reinstatement refers to the reinitiation of drug seeking in animal models after the extinction of previous drug administration. See the review by Shaham and colleagues).80 The effect of extracellular release of DA into the NAc upon drug-use behavior, however, is complicated by the heterogeneous responses of DA receptor subtypes. Dopaminergic receptors consist of two broad families (D1 and D2) and five subtypes (D1-like: D1, D5; D2-like: D2, D3, D4).81 Although both D1 and D2 agonists have reinforcing properties, the two receptors have distinct effects upon drug reinstatement. Stimulation of D2 receptors in the NAc induces drug-triggered relapse, whereas drugs that stimulate D1 receptors block drug-triggered relapse (see Self & Nestler58 for review). The differences between D1- and D2-like receptors can be best understood through an appreciation of the postreceptor perturbations induced by drug-induced neurotransmitter release upon second messenger pathways. D2 receptors inhibit intracellular adenylyl cyclase by coupling with inhibitory G proteins that reduce cAMP production, whereas D1 receptors stimulate cAMP formation by activating membrane G proteins stimulating adenylyl cyclase. The chronic exposure to cocaine, heroin, morphine, and ethanol all induce an upregulation of the NAc cAMP second messenger pathway, with resultant increases in adenylyl cyclase and, in turn, protein kinase. Thus, it appears that relatively persistent D2-induced reductions in intracellular cAMP following chronic drug administration may result in increased drug self-administration, whereas these effects can be opposed by stimulation of D1 receptors.
D2 receptor ligands have not proven useful in the treatment of stimulant dependence; D2 receptor agonists are reinforcing in animal models, and D2 receptor antagonists are not effective in human studies. However, since D2-agonists appear to be especially potent in the induction of priming, medications targeting D2-like D3 and D4 receptors have been explored. In addition, whereas D1 and D2 receptors are more diffusely concentrated throughout the brain, D3 receptors are preferentially expressed in the mesolimbic system, particularly in the NAc, and D4 receptors have their highest densities in the prefrontal cortex (PFC) and suprachiasmatic nucleus of hypothalamus.11 Preclinical studies reveal that D3 receptor antagonists block both cocaine’s reinforcing actions and cocaine-induced reinstatement of cocaine-seeking behaviors;82,83 a partially selective D3 receptor ligand is now undergoing assessment for human trials.84 In an exciting human study assessing the interaction between genetic factors and drug use, healthy subjects with different D4 variable-number-of-tandem-repeats (VNTR) polymorphisms were administered a priming dose of alcohol and then assessed for craving. Groups with different D4 VNTR polymorphisms demonstrated a differential response to the D4 antagonist,85 portending the developing importance of pharmacogenetics to inform the targeted use of medications based upon an individual’s genotype.
Drug Cues: The Nucleus Accumbens and Amygdala
The power of drug-related cues to induce a return to drug use is routinely observed in the clinical setting, prompting the admonition for addicted patients to avoid the “people, places, and things” that have been associated with their drug use. (Although such cues can induce craving, this phenomena is discussed in the following section). Di Chiara15 posits that the repeated use of drugs strengthens both stimulus-response and stimulus-reward associations, thus sensitizing the mesolimbic pathway and internally linking the association between the substance and its associated drug cues. The imbedding of the experience of substance use in tandem with the attendant conditioned environmental stimuli produces an “addiction memory”86 or “neural ghost” (Glenn Horwitz, personal communication). This neural ghost remains imbedded in the mesolimbic circuitry, particularly the amygdala87—often outside of conscious awareness. Upon stimulation of the mesolimbic pathway, either by conditioned drug cues88 or by drug priming, the circuit is activated, inducing a desire, or wanting, for additional drug.
The amygdala is implicated in the acquisition, storage, and expression of emotional memories. PET and fMRI neuroimaging studies of cocaine-89-92 and nicotine-addicted subjects93 show that cue exposure to drug-associated stimuli induces an activation of the amygdalar region. When animals are trained to associate a specific “place” with drug administration (i.e., conditioned place preference), they tend to return to the environment associated with receiving the drug. Following ablation of the amygdala, the animals “forget” this association.94-95 The reinforcing effect of the drug remains, however, as drug self-administration persists following amygdalar ablation. The formation of the associations between salient stimuli and internally rewarding (or aversive) events is facilitated by stimulation of dopaminergic neurons.49 However, glutamate also appears to be a primary mediator of cue-induced behavioral plasticity via glutamatergic connections that project from the amygdala to the NAc.96 The increase in glutamate release following repeated cocaine administration also mediates, at least in part, cocaine-induced sensitization.97,98
A relatively new area of investigation explores the intracellular mechanisms that accompany repeated drug use positing that the mnenomic connections that underlie cue-induced relapse are mediated by relatively long lasting cellular and molecular adaptations. The extracellular release of neurotransmitters can induce these alterations in intracellular processes by increasing or decreasing protein synthesis, including messenger, transcription, and scaffolding (or structural) proteins. Messenger (e.g., G) proteins were discussed in the previous section. Transcription factors regulate mRNA gene transcription by binding to the regulatory regions of specific genes. The two transcription factors most strongly associated with the chronic drug administration are ΔFosB and CREB (cAMP response element-binding protein) (see Nestler99 and Chao & Nestler100 for reviews). ΔFosB is a member of the Fos family of immediate early gene transcription factors. Most and possibly all members of this family are rapidly induced after the acute exposure to amphetamine cocaine, ethanol, nicotine, opiates, and PCP. ΔFosB is unique among these proteins in that it is very stable and persists intracellularly for several weeks or months.101 The repeated administration of drugs therefore results in an accumulation of ΔFosB, primarily in striatal GABAergic medium spiny neurons containing dynorphin and substance P.102,103 ΔFosB decreases the expression of dynorphin in these striatal projection neurons. The drug-induced accumulation of ΔFoxB enhances sensitivity to the rewarding effects of cocaine and morphine,103,104 possibly due to the feedback effect of striatal dynorphin upon kappa-opioid receptors on VTA dopaminergic neurons. Since the uniquely stable ΔFosB also accumulates in the amygdala and PFC, it has been suggested that ΔFosB may be the “molecular switch” that retains the connection between the experience of drug reward and the drug-associated cues long after drug use has ceased 105
The Obsessive Drive for Drugs: The Striato-Thalamo-Orbitofrontal Circuit
The pathway involved in the compulsive drive for substances is the striato-thalamo-orbitofrontal circuit. This circuit is tightly interconnected with other prefrontal and limbic regions, including the anterior cingulate, insula, dorsolateral prefrontal cortex (DLPFC), and amygdala. Innervation includes both the mesocortical dopaminergic pathway, which projects to PFC regions that include the OFC and anterior cingulate,11 and glutamate neurons that project reciprocally between the PFC and amygdala, as well as from the PFC to the NAc and VTA.96 This striato-thalamo-orbitofrontal circuit has been implicated in obsessive-compulsive disorder (OCD), a syndrome sharing common features with the addictive disorders; that is, the drive for drugs and alcohol includes a lack of control over intrusive thoughts and compulsive behaviors that are directed toward obtaining and administering substances.106,107
The obsessive-compulsive nature of substance use can be empirically assessed with the Obsessive Compulsive Drinking Scale,106 a predictive measure of treatment outcome.108 Single photon emission computerized tomography (SPECT) and PET imaging studies demonstrate increased activation of the OFC, anterior cingulate, and striatum in OCD, and this brain activity normalizes following successful pharmacologic or psychosocial treatment.109-111 Heightened activation of the OFC is similarly observed in cocaine-addicted subjects during cocaine craving112 and both procaine113 and methylphenidate114 administration, suggesting that the OFC is hypersensitive to a variety of psychological and pharmacologic challenges. In addition, other PET90-92,114 and fMRI112,115,116 studies during craving for cocaine, methylphenidate, and alcohol have demonstrated activation of the anterior cingulate, DLPFC, insula, and amygdala. Garavan and colleagues,117 however, compared regional brain activation with fMRI in cocaine-addicted and nonaddicted subjects while viewing films portraying individuals either smoking crack cocaine or engaged in sexual activity. The cocaine cues activated similar neuroanatomical substrates as the natural (sexual) stimuli in the cocaine-addicted subjects, suggesting that activation of these cortical and limbic regions may not be associated with a dedicated circuitry specific to drug cues. Of particular interest, however, was the finding that, whereas the cocaine-addicted subjects reacted more strongly to the cocaine cues than controls (as expected), the cocaine-addicted group showed an attenuated brain signal in response to the sexual stimuli relative to the non-addicted group. Consistent with clinical observations, this study implies that persistent drug use induces “wanting” only in response to drug-related cues—and not in response to natural cues. The chronic use of drugs therefore appears to co-opt higher-order neuronal circuitry, such that executive functioning responds primarily to drug-related stimuli and that planning, decision-making, and attentional processes all subserve drug acquisition and ingestion.
Stress-Induced Relapse: The Limbic-Hypothalamic-Pituitary-Adrenal Axis
Stress is a common precipitant of relapse in addicted patients,118 and intermittent stress is a powerful inducer for the reinstatement of substance use in animal models.58,119 Preclinical studies investigating this phenomena have revealed that stressors (including defeat stress, intermittent shock, maternal separation, prenatal stress, social isolation, tail pinch, unstable social environment, and food deprivation or restriction) are all important modulators of drug-seeking behavior, although the effect on any specific stressor is stressor-, procedure-, and drug-specific. Intermittent foot shock, however, tends to be the most consistent stress inducer of drug reinstatement (see Lu et al.119 for review). The stress circuit includes the hypothalamic-pituitary-adrenal (HPA) and extrahypothalamic corticotropin-releasing factor (CRF) stress systems, including the amygdala and BNST.3 External stressors stimulate a return to drug use through the BNST77 and amygdala,58 regions that are particularly sensitive to the anxiogenic effects of the neuropeptide CRF. Reinstatement to drug use following foot shock is blocked by the administration of CRF antagonists,120 revealing that CRF is a mediator in this relapse process. Norepinephrine (projecting from the locus coeruleus)121,122 and glutamate (projecting from the amygdala)123 are also involved in the stress-induced reinstatement of drug use. As described with drug-related cues and craving, reinstatement induced by foot shock likewise involves glutaminergic projections from the PFC and amygdala to the NAc.123
Stress interacts with mesolimbic DA through the peripheral release of glucocorticoids. Following stress-induced activation of the HPA axis, glucocorticoids cross the blood-brain barrier into the central nervous system and bind with VTA (and other) glucocorticoid receptors.124,125 Glucocorticoids have a permissive effect upon mesolimbic DA,126,127 and both stress and substances of abuse (e.g., amphetamine, cocaine, ethanol, morphine, and nicotine) cause a similar excitation of midbrain dopaminergic cells.128 Thus, the stress-induced release of both extrahypothalamic CRF and the glucocorticoids stimulates neuronal pathways involved in the compulsive drive for substance use. Conversely, abstinent alcohol-dependent. subjects demonstrate. an attenuated responsiveness of the HPA axis in response to pharmacologic and psychosocial stressors.129-131 This “inverse U” pattern, in which both the paucity and excess of glucocorticoids can be deleterious, is observed in a wide range of physiologic functions.132 Since preliminary studies suggest that an attenuation of HPA-axis functioning predicts relapse following treatment,133,134 medications that increase HPA tone may be useful in the treatment of alcohol-addicted subjects. For example, the opioid antagonists naltrexone and nalmefene block the inhibitory effect of the endogenous endorphins on paraventricular corticotropin-releasing hormone (CRH),135,136 thereby increasing corticotropin and cortisol. Opioid antagonists may subsequently decrease relapse by easing alcohol-related suppression of the HPA axis, resulting in normalization of the HPA-axis response to stress.
In summary, four overlapping networks of dopaminergic and glutaminergic projections integrate the brain regions mediating emotional memory, drug desire, and stress response with the primary repository of drug salience—that is, the VTA and NAc. Extracellular synaptic events induce intracellular changes that may underlie the persistence of the drug-associated cues long after drug use has ceased. The divergent precipitants of drug relapse and their associated extra- and intracellular perturbations suggest that pharmacologic interventions for drug relapse must intervene in multiple neuronal circuits, possibly targeting specific pathways for individual drive states.
INHIBITORY DYSCONTROL OF THE COMPULSIVE DRIVE STATE
The compulsive drug drive cannot fully account for recurrent relapse. Despite an enduring desire for alcohol or drugs, a sizable percentage of addicted patients maintain a lifetime of abstinence. The slip-induced relapse, the cue-induced cravings, the obsessive thoughts, and the return to drug use following traumatic events can be thwarted by a robust inhibitory control over the compulsive drive state. A deficit in inhibitory restraint (i.e., impulsivity), however, can provide a window through which the drive for drugs can express itself. Even in the absence of a compulsive drug drive, the relative absence of inhibitory control may lead to spontaneous drug use (see Figure 2). The relative absence of inhibitory restraint in addicted subjects is observed in standardized and experimental neurocognitive measures of impulsivity and decision making,137-142 revealing deficits in the ability of addicted subjects both to inhibit prepotent (or powerfully habituated) responses137 and to choose greater delayed rewards over lesser immediate ones.138,141,142
FIGURE 2.
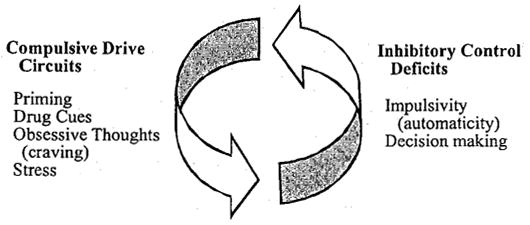
The compulsive drive toward drug use describes relapse in response to a priming dose of drug, drug cues, craving, or stress. These triggers for a return to drug use are mediated by overlapping brain regions/circuits: mesolimbic (priming), mesolimbic and amygdala (drug cues), striato-thalamo-orbitofrontal (obsessive thoughts), and extrahypothalamic CRF and the HPA axis (stress). A deficit in inhibitory control and poor decision making, mediated in part by the OFC cortex and anterior cingulate, may result in relapse even in the absence of a compulsive drug trigger. Adapted from Koob & Moal3 and Jenstch & Taylor.17
The OFC is critically involved in assessing the salience of potential rewards and punishments (i.e., high vs. low monetary rewards, immediate vs. delayed gain, similar or dissimilar objects) and is involved in both impulsivity and decision making. Patients with lesions of the OFC, for example, make irresponsible and impulsive decisions, yet their intellectual abilities—such as memory, learning, language, and attention—are often preserved. In general, there appears to be a perseveration of behavior, with continued responding to stimuli that are no longer rewarding; a reversal of reinforcement contingencies does not reverse behavioral responses.143 For example, subjects with lesions of the OFC perform poorly on the Gambling Task,144 which simulates real-life experiences involving uncertainty, reward, and punishment. Several investigators have demonstrated that drug- and alcohol-addicted subjects perform poorly on this task.142,145,146 Another key region involved in inhibitory restraint is the anterior cingulate, which monitors performance, detects conflicts, and assesses emotional self-control. Performance on the Gambling Task is highly correlated with resting rCBF of the anterior cingulate.147
Using PET imaging techniques, Volkow and colleagues have demonstrated a strong correlation between striatal D2 receptor number and energy utilization of the OFC and anterior cingulate in cocaine-36 and methamphetamine-addicted148 patients. The lower the number of D2 receptors, the lower the activity of the OFC and anterior cingulate. The decrease in basal OFC regional cerebral blood flow (rCBF) in cocaine-addicted subjects, relative to controls, has been confirmed in our laboratory using SPECT imaging techniques (see Figure 3).113 The correlation between D2 receptor number and OFC and anterior cingulate activity reported by Volkow and colleagues36,148 may suggest a neurobiologic connection between the compulsive drive state and the inhibitory deficit associated with drug addiction. Thus, a similar alteration in the mesocortical pathway may concurrently produce an impaired inhibitory control (due to decreased mesocortical input into the PFC) over a heightened desire for drug-induced stimulation (due to an attenuated response to natural reinforcers associated with the reduction in striatal D2 receptors).
FIGURE 3.
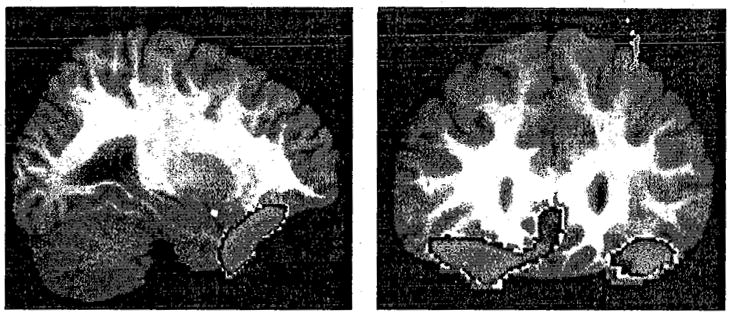
Decreased blood flow in the medial and lateral orbitofrontal cortex may contribute to the deficits in inhibitory control observed in addicted subjects. The figure demonstrates decreased rCBF (p < 0.01, in blue) in the orbitofrontal cortex of 37 two- to four-week abstinent cocaine-dependent men and women relative to 36 age-matched controls. Sagittal view (left panel) is at Talairach coordinate x = 28; coronal view (right panel), at y = 30 (right is to reader’s left). From Adinoff and colleagues, unpublished data.
Brain regions involved in the inhibitory processes, particularly the OFC and the anterior cingulate, have therefore been the focus of several neuroimaging studies of subjects with substance use disorders. As noted above, both PET and SPECT studies have shown that abstinent cocaine-,36,113 alcohol-,149 and methamphetamine-addicted150 subjects demonstrate decreased basal activity in the OFC. During the Stroop Interference Task, which assesses the ability to inhibit a prepotent response (i.e., response inhibition), the relationship between task performance and OFC activation is disrupted in cocaine- and alcohol-addicted subjects.161 OFC activation, as assessed by fMRI, is also muted in a decision-making task in methamphetamine-addicted subjects.152 The anterior cingulate shows decreased activation in addicted subjects during a task of response inhibition,153 procaine administration,113,154 and script-guided stress induction.155 Impaired functioning of the OFC156,157 and the anterior cingulate, particularly in response to cognitive tasks engaging either inhibitory processes or decision making, suggests that these brain areas may be involved in the addict’s inability to appropriately restrain the drive toward relapse.
The OFC and anterior cingulate also play a prominent role in the obsessive thoughts and craving described earlier. However, whereas these brain regions demonstrate increased regional brain activation (relative to controls) during craving induction, they generally show decreased activation (relative to controls) during other activating tasks (see above). These findings suggest that the mesocorticolimbic pathway does not appropriately engage non-drug-related cognitive or emotional stimuli, but is hyperresponsive to drug-related cues.
FUTURE DIRECTIONS: MEDICATION DEVELOPMENT
This review has described the progressive elucidation of neurobiologic mechanisms underlying uncontrolled drug use, focusing on the distinctive role that dopamine plays in reward and addiction. The ultimate aim of these efforts is to guide medication development for the addictive disorders. Several potential targets have been mentioned, with a focused intervention upon dopaminergic receptors. The review highlights the importance of the mesolimbic pathway in the development of an addiction—which thus may be an optimal site for intervention early in the addictive process. In high-risk individuals (i.e., those with strong genetic or environmental risk factors for the development of a substance use disorder), medications interacting with the mesolimbic pathway to decrease the incentive salience of drugs may even prove useful as a preventive measure. By contrast, in the case of repetitive drug use—which engages the prefrontal regions— the mesocortical pathway may be a more appropriate target for treatment. The review notes the relative importance of various types of triggers, and as mechanisms are developed to isolate the particular relapse style of an individual patient, targeted interventions (both psychosocial and pharmacologic) can be directed toward the trigger-relevant neurotransmitter system and neuronal circuit. For example, addicted patients who report an intense response to specific cues may best respond to disruptions in amygdalar-mesolimbic connections, whereas a patient identified as an impulsive relapser158 may require improved efficiency of orbitofrontal or anterior cingulate functioning—possibly by intensifying dopaminergic input. Isolating genotypes should lead to a pharmacogenetic approach to drug development, offering a specific drug treatment appropriate to an identified genetic polymorphism. Finally, drug-induced postsynaptic alterations in structural,159 messenger, and transcription proteins offer an exciting focus for future medication targets. In addition to the previously discussed prospective medications, dopaminergic agents presently being assessed include vanoxerine,160,161 a long-acting, noncompetitive inhibitor of the presynaptic DAT, and disulfiram, which inhibits dopamine-B-hydroxylase.162
Other receptor systems directly synapse upon dopaminergic neurons and offer excellent possibilities for pharmacologic intervention. As noted early in the review, most drugs of abuse have high affinity binding with G protein-mediated or ion channel receptors that activate dopaminergic release. Pharmacologic manipulations of these receptor systems have proven of some utility in drug treatment, and several new approaches are in development. Naltrexone, for example, appears to decrease alcohol craving163,164 by blocking the mu-opioid receptors that suppress GABAergic neurons. These neurons, in turn, tonically inhibit VTA dopaminergic neurons.165 Antagonism of mu-opioid receptors thus results in the disinhibition of the GABAergic neurons, causing a net decrease in VTA DA release. Opioid antagonists seem to have little effect, however, on the craving for other substances, including heroin. Baclofen, a GABAB receptor agonist that inhibits dopamine release, reduces stimulant self-administration in preclinical studies166 and has shown early promise in reducing cocaine use in human studies.167 Early suggestions of clinical efficacy in cocaine-dependent subjects have also been reported with vigabatrin, a selective, irreversible inhibitor of GABA transaminase.168 Enadoline, which binds to kappa-opioid receptors and inhibits DA release, attenuates the neurochemical and behavioral effects of cocaine in preclinical studies and is being assessed for human trials.169 Topiramate both facilitates GABA functioning through a non-benzodiazepine site on the GABAA receptor and antagonizes glutamate activity at the alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid (AMPA) and kianate receptors. This compound has demonstrated efficacy in both alcohol-170 and cocaine-dependent171 subjects. Glucocorticoid antagonists or agonists may prove useful by altering the permissive effects of mesolimbic DA release, particularly during periods of stress. Metyrapone, which blocks the synthesis of cortisol, is currently being assessed for cocaine addiction in phase I clinical trials, although suppression of HPA-axis activity by ketoconazole has not been effective in decreasing cocaine use.172 As discussed previously, however, medications that activate, not inhibit, HPA-axis functioning may prove more beneficial. 5HT3 receptors are abundant in central DA terminal areas, such as the NAc and striatum, and appear to mediate the excitatory effect of compounds acting upstream from DA neurons.173 Altering 5HT3 transmission with the 5HT3 antagonist ondansetron inhibits cocaine-induced sensitization,174 decreases alcohol use in early-onset alcohol-dependent subjects,175 and is being evaluated in phase II clinical trials for cocaine dependence.176 Cannabinoids activate mesolimbic DA through the CB1 receptor, and the CB1 receptor antagonist rimonabant decreases cocaine reinstatement.177 Whereas DA increases appetitive behavior, cholinergic agonists inhibit appetitive behavior and increase avoidance behavior.178 Behavioral reinforcement has therefore been envisioned as a balance between NAc DA and the cholinergic system179—mediated, at least in part, by cholinergic interneurons within the NAc. Preclinical studies have demonstrated an inhibitory effect of cholinergic agonists on cocaine self-administration.180,181 Ibogaine, a naturally occurring indole alkaloid, binds to kappa-opioid, NMDA glutamate, and nicotinic receptors, and blocks the expression of NAc DA in cocaine-sensitized animals.182 Ibogaine has been shown to decrease cocaine, ethanol, morphine, and nicotine self-administration in animal models,183 and extensive anecdotal evidence suggests this drug has treatment efficacy in cocaine and heroin addiction.184 Several of these new approaches are being targeted by the National Institute on Drug Abuse’s Clinical Research Efficacy Screening Trial.176
Experience with other chronic medical and psychiatric disorders, such as depression, epilepsy, hypertension, and schizophrenia, suggest that a cocktail of pharmacologic interventions will be required for the successful treatment of many addicted individuals. Medications targeting multiple receptor systems and designed for an individual’s specific relapse characteristics, genotype, and addiction severity may be the optimal intervention as medication development progresses.
Footnotes
Preparation of this manuscript was supported by National Institute on Drug Abuse grant no. DA11434 and National Institute on Alcohol Abuse and Alcoholism grant no. AA1570.
References
- 1.Rinaldi RC, Steindler EM, Wilford BB, Goodwin D. Clarification and standardization of substance abuse terminology. JAMA. 1988;259:555–7. [PubMed] [Google Scholar]
- 2.American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 4. Washington, DC: APA; 1994. [Google Scholar]
- 3.Koob GF, Moal M. Drug addiction, dysregulation of reward, and allostasis. Neuropsychopharmacology. 2001;24:97–129. doi: 10.1016/S0893-133X(00)00195-0. [DOI] [PubMed] [Google Scholar]
- 4.Adinoff B, O’Neil HK, Ballenger JC. Alcohol withdrawal and limbic kindling. Am J Addict. 1995;4:5–17. [Google Scholar]
- 5.Baker TB, Morse E, Sherman JE. The motivation to use drugs: a psychobiological analysis of urges. In: Dienstbier RA, Rivers PC, Lincoln NE, editors. Nebraska symposium on motivation, 1986: alcohol and addictive behavior. Lincoln: University of Nebraska Press; 1986. [PubMed] [Google Scholar]
- 6.Olds J, Milner P. Positive reinforcement produced by electrical stimulation of septal area and other regions of rat brain. J Comp Physiol Psychol. 1954;47:419–27. doi: 10.1037/h0058775. [DOI] [PubMed] [Google Scholar]
- 7.Heath RG. Electrical self-stimulation of the brain in man. Am J Psychiatry. 1963;120:571–7. doi: 10.1176/ajp.120.6.571. [DOI] [PubMed] [Google Scholar]
- 8.Heath RG. Pleasure and brain activity in man. Deep and surface electroencephalograms during orgasm. J Nerv Ment Dis. 1972;154:3–18. doi: 10.1097/00005053-197201000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Baumeister AA. The Tulane Electrical Brain Stimulation Program a historical case study in medical ethics. J Hist Neurosci. 2000;9:262–78. doi: 10.1076/jhin.9.3.262.1787. [DOI] [PubMed] [Google Scholar]
- 10.Gardner EL. Brain reward mechanisms. In: Lowinson JH, Ruiz P, Millman RB, Langrod JG, editors. Substance abuse: a comprehensive textbook. Baltimore, MD: Williams & Wilkins; 1997. [Google Scholar]
- 11.Gardner EL, Ashby CR., Jr Heterogeneity of the mesotelencephalic dopamine fibers: physiology and pharmacology. Neurosci Biobehav Rev. 2000;24:115–8. doi: 10.1016/s0149-7634(99)00048-2. [DOI] [PubMed] [Google Scholar]
- 12.Carlezon WA, Jr, Wise RA. Rewarding actions of phencyclidine and related drugs in nucleus accumbens shell and frontal cortex. J Neurosci. 1996;16:3112–22. doi: 10.1523/JNEUROSCI.16-09-03112.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fredholm BB, Svenningsson P. Adenosine-dopamine interactions: development of a concept and some comments on therapeutic possibilities. Neurology. 2003;61:S5–9. doi: 10.1212/01.wnl.0000095204.89871.ff. [DOI] [PubMed] [Google Scholar]
- 14.Wise RA. Neurobiology of addiction. Curr Opin Neurobiol. 1996;6:243–51. doi: 10.1016/s0959-4388(96)80079-1. [DOI] [PubMed] [Google Scholar]
- 15.Di Chiara G. A motivational learning hypothesis of the role of mesolimbic dopamine in compulsive drug use. J Psychopharmacol. 1998;12:54–67. doi: 10.1177/026988119801200108. [DOI] [PubMed] [Google Scholar]
- 16.Di Chiara G. Alcohol and dopamine. Alcohol Health Res World. 1997;21:108–14. [PMC free article] [PubMed] [Google Scholar]
- 17.Jentsch JD, Taylor JR. Impulsivity resulting from frontostriatal dysfunction in drug abuse: implications for the control of behavior by reward-related stimuli. Psychopharmacology (Berl) 1999;146:373–90. doi: 10.1007/pl00005483. [DOI] [PubMed] [Google Scholar]
- 18.Ritz MC, Lamb RJ, Goldberg SR, Kuhar MJ. Cocaine receptors on dopamine transporters are related to self-administration of cocaine. Science. 1987;237:1219–23. doi: 10.1126/science.2820058. [DOI] [PubMed] [Google Scholar]
- 19.Bunney BS, Aghajanian GK. d-Amphetamine-induced depression of central dopamine neurons: evidence for mediation by both autoreceptors and a striato-nigral feedback pathway. Naunyn Schmiedebergs Arch Pharmacol. 1978;304:255–61. doi: 10.1007/BF00507966. [DOI] [PubMed] [Google Scholar]
- 20.Johnson SW, North RA. Opioids excite dopamine neurons by hyperpolarization of local interneurons. J Neurosci. 1992;12:483–8. doi: 10.1523/JNEUROSCI.12-02-00483.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bozarth MA, Wise RA. Anatomically distinct opiate receptor fields mediate reward and physical dependence. Science. 1984;224:516–7. doi: 10.1126/science.6324347. [DOI] [PubMed] [Google Scholar]
- 22.Gardner EL. What we have learned about addiction from animal models of drug self-administration. Am J Addict. 2000;9:285–313. doi: 10.1080/105504900750047355. [DOI] [PubMed] [Google Scholar]
- 23.Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28:309–69. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- 24.Kelley AE, Berridge KC. The neuroscience of natural rewards: relevance to addictive drugs. J Neurosci. 2002;22:3306–11. doi: 10.1523/JNEUROSCI.22-09-03306.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pettit HO, Justice JB., Jr Effect of dose on cocaine self-administration behavior and dopamine levels in the nucleus accumbens. Brain Res. 1991;539:94–102. doi: 10.1016/0006-8993(91)90690-w. [DOI] [PubMed] [Google Scholar]
- 26.Bergman J, Kamien JB, Spealman RD. Antagonism of cocaine self-administration by selective dopamine D(l) and D(2) antagonists. Behav Pharmacol. 1990;1:355–60. doi: 10.1097/00008877-199000140-00009. [DOI] [PubMed] [Google Scholar]
- 27.Davis WM, Smith SG. Effect of haloperidol on (+)-amphetamine self-administration. J Pharm Pharmacol. 1975;27:540–2. doi: 10.1111/j.2042-7158.1975.tb09502.x. [DOI] [PubMed] [Google Scholar]
- 28.Caine SB, Koob GF. Effects of mesolimbic dopamine depletion on responding maintained by cocaine and food. J Exp Anal Behav. 1994;61:213–21. doi: 10.1901/jeab.1994.61-213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Breiter HC, Gollub RL, Weisskoff RM, et al. Acute effects of cocaine on human brain activity and emotion. Neuron. 1997;19:591–611. doi: 10.1016/s0896-6273(00)80374-8. [DOI] [PubMed] [Google Scholar]
- 30.Volkow ND, Wang GJ, Fischman MW, et al. Relationship between subjective effects of cocaine and dopamine transporter occupancy. Nature. 1997;386:827–30. doi: 10.1038/386827a0. [DOI] [PubMed] [Google Scholar]
- 31.Volkow ND, Wang GJ, Fowler JS, et al. Imaging endogenous dopamine competition with [11C]raclopride in the human brain. Synapse. 1994;16:255–62. doi: 10.1002/syn.890160402. [DOI] [PubMed] [Google Scholar]
- 32.Dackis CA, Gold MS. New concepts in cocaine addiction: the dopamine depletion hypothesis. Neurosci Biobehav Rev. 1985;9:469–77. doi: 10.1016/0149-7634(85)90022-3. [DOI] [PubMed] [Google Scholar]
- 33.Volkow ND, Fowler JS, Wang GJ, Goldstein RZ. Role of dopamine, the frontal cortex and memory circuits in drug addiction: insight from imaging studies. Neurobiol Learn Mem. 2002;78:610–24. doi: 10.1006/nlme.2002.4099. [DOI] [PubMed] [Google Scholar]
- 34.Salamone J, Cousins M, Snyder B. Behavioral functions of nucleus accumbens dopamine: Empirical and conceptual problems with the anhedonia hypothesis. Neurosci Biobehav Rev. 1997;21:341–59. doi: 10.1016/s0149-7634(96)00017-6. [DOI] [PubMed] [Google Scholar]
- 35.Malison RT, Best SB, van Dyck CH, et al. Elevated striatal dopamine transporters during acute cocaine abstinence as measured by [123I] beta-CIT SPECT. Am J Psychiatry. 1998;155:832–4. doi: 10.1176/ajp.155.6.832. [DOI] [PubMed] [Google Scholar]
- 36.Volkow ND, Fowler JS, Wang G-J, et al. Decreased dopamine D2 receptor availability is associated with reduced frontal metabolism in cocaine abusers. Synapse. 1993;14:169–77. doi: 10.1002/syn.890140210. [DOI] [PubMed] [Google Scholar]
- 37.Volkow ND, Chang L, Wang GJ, et al. Low level of brain dopamine d(2) receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. Am J Psychiatry. 2001;158:2015–21. doi: 10.1176/appi.ajp.158.12.2015. [DOI] [PubMed] [Google Scholar]
- 38.Thanos PK, Volkow ND, Freimuth P, et al. Overexpression of dopamine D2 receptors reduces alcohol self-administration. J Neurochem. 2001;78:1094–103. doi: 10.1046/j.1471-4159.2001.00492.x. [DOI] [PubMed] [Google Scholar]
- 39.Morgan D, Grant KA, Gage HD, et al. Social dominance in monkeys: dopamine D2 receptors and cocaine self-administration. Nat Neurosci. 2002;5:169–74. doi: 10.1038/nn798. [DOI] [PubMed] [Google Scholar]
- 40.Glick SD, Raucci J, Wang S, Keller RW, Jr, Carlson JN. Neurochemical predisposition to self-administer cocaine in rats: individual differences in dopamine and its metabolites. Brain Res. 1994;653:148–54. doi: 10.1016/0006-8993(94)90383-2. [DOI] [PubMed] [Google Scholar]
- 41.Malcolm R, Kajdasz DK, Herron J, Anton RF, Brady KT. A double-blind, placebo-controlled outpatient trial of pergolide for cocaine dependence. Drug Alcohol Depend. 2000;60:161–8. doi: 10.1016/s0376-8716(99)00151-9. [DOI] [PubMed] [Google Scholar]
- 42.Shoptaw S, Kintaudi PC, Charuvastra C, Ling W. A screening trial of amantadine as a medication for cocaine dependence. Drug Alcohol Depend. 2002;66:217–24. doi: 10.1016/s0376-8716(01)00205-8. [DOI] [PubMed] [Google Scholar]
- 43.Soares BG, Lima MS, Reisser AA, Farrell M. Dopamine agonists for cocaine dependence. Cochrane Database Syst Rev. 2001:CD003352. doi: 10.1002/14651858.CD003352. [DOI] [PubMed] [Google Scholar]
- 44.Eiler K, Schaefer MR, Salstrom D, Lowery R. Double blind comparison of bromocriptine and placebo in cocaine withdrawal. Am J Drug Alcohol Abuse. 1995;21:65–79. doi: 10.3109/00952999509095230. [DOI] [PubMed] [Google Scholar]
- 45.Handelsman L, Rosenblum A, Palij M, et al. Bromocriptine for cocaine dependence. A controlled clinical trial. Am J Addict. 1997;6:54–64. [PubMed] [Google Scholar]
- 46.Grabowski J, Roache JD, Schmitz JM, Rhoades H, Creson D, Korszun A. Replacement medication for cocaine dependence: methylphenidate. J Clin Psychopharmacol. 1997;17:485–8. doi: 10.1097/00004714-199712000-00008. [DOI] [PubMed] [Google Scholar]
- 47.Stine SM, Krystal JH, Kosten TR, Charney DS. Mazindol treatment for cocaine dependence. Drug Alcohol Depend. 1995;39:245–52. doi: 10.1016/0376-8716(95)01174-4. [DOI] [PubMed] [Google Scholar]
- 48.Rocha BA, Fumagalli F, Gainetdinov RR, et al. Cocaine self-administration in dopamine-transporter knockout mice. Nat Neurosci. 1998;1:132–7. doi: 10.1038/381. [DOI] [PubMed] [Google Scholar]
- 49.Spanagel R, Weiss F. The dopamine hypothesis of reward: past and current status. Trends Neurosci. 1999;22:521–7. doi: 10.1016/s0166-2236(99)01447-2. [DOI] [PubMed] [Google Scholar]
- 50.Halikas JA, Kuhn KL. A possible neurophysiological basis of cocaine craving. Ann Clin Psyhiatry. 1990;2:79–83. [Google Scholar]
- 51.Post RM, Weiss SR. Psychomotor stimulant vs. local anesthetic effects of cocaine: role of behavioral sensitization and kindling. NIDA Res Monogr. 1988;88:217–38. [PubMed] [Google Scholar]
- 52.Robinson TE, Berridge KC. The neural basis of drug craving: an incentive-sensitization theory of addiction. Brain Res Brain Res Rev. 1993;18:247–91. doi: 10.1016/0165-0173(93)90013-p. [DOI] [PubMed] [Google Scholar]
- 53.Goddard GV, McIntyre DC, Leech CK. A permanent change in brain function resulting from daily electrical stimulation. Exp Neurol. 1969;25:295–330. doi: 10.1016/0014-4886(69)90128-9. [DOI] [PubMed] [Google Scholar]
- 54.Janowsky JS, Laxer KD, Rushmer DS. Classical conditioning of kindled seizures. Epilepsia. 1980;21:393–8. doi: 10.1111/j.1528-1157.1980.tb04087.x. [DOI] [PubMed] [Google Scholar]
- 55.Grode J. Ueber die wirkung langerer cocainedarrechung bei tieren. Arch F Exp Path U Pharmakol. 1912;67:172–9. [Google Scholar]
- 56.Zahniser NR, Peris J, Dwoskin LP, et al. Sensitization to cocaine in the nigrostriatal dopamine system. NIDA Res Monogr. 1988;88:55–77. [PubMed] [Google Scholar]
- 57.Kalivas PW, Pierce RC, Cornish J, Sorg BA. A role for sensitization in craving and relapse in cocaine addiction. J Psychopharmacol (Oxf) 1998;12:49–53. doi: 10.1177/026988119801200107. [DOI] [PubMed] [Google Scholar]
- 58.Self DW, Nestler EJ. Relapse to drug-seeking: neural and molecular mechanisms. Drug Alcohol Depend. 1998;51:49–60. doi: 10.1016/s0376-8716(98)00065-9. [DOI] [PubMed] [Google Scholar]
- 59.Pascual-Leone A, Dhuna A, Anderson DC. Longterm neurological complications of chronic, habitual cocaine abuse. Neurotoxicology. 1991;12:393–400. [PubMed] [Google Scholar]
- 60.Washton AM, Gold MS. Chronic cocaine abuse: evidence for adverse effects on health and functioning. Psychiatr Ann. 1984;14:733–43. [Google Scholar]
- 61.Louie AK, Lannon RA, Ketter TA. Treatment of cocaine-induced panic disorder. Am J Psychiatry. 1989;146:40–4. doi: 10.1176/ajp.146.1.40. [DOI] [PubMed] [Google Scholar]
- 62.Satel SL, Edell WS. Cocaine-induced paranoia and psychosis proneness. Am J Psychiatry. 1991;148:1708–11. doi: 10.1176/ajp.148.12.1708. [DOI] [PubMed] [Google Scholar]
- 63.Halikas JA, Crosby RD, Pearson VL, Graves NM. A randomized double-blind study of carbamazepine in the treatment of cocaine abuse. Clin Pharmacol Ther. 1997;62:89–105. doi: 10.1016/S0009-9236(97)90155-7. [DOI] [PubMed] [Google Scholar]
- 64.Brady KT, Sonne SC, Malcolm RJ, et al. Carbamazepine in the treatment of cocaine dependence: subtyping by affective disorder. Exp Clin Psychopharmacol. 2002;10:276–85. doi: 10.1037//1064-1297.10.3.276. [DOI] [PubMed] [Google Scholar]
- 65.Lima AR, Lima MS, Soares BG, Farrell M. Carbamazepine for cocaine dependence. Cochrane Database Syst Rev. 2002:CD002023. doi: 10.1002/14651858.CD002023. [DOI] [PubMed] [Google Scholar]
- 66.Kleber HD. Pharmacologic treatments for heroin and cocaine dependence. Am J Addict. 2003;12(suppl 2):S5–18. [PubMed] [Google Scholar]
- 67.Hemby SE, Co C, Koves TR, Smith JE, Dworkin SI. Differences in extracellular dopamine concentrations in the nucleus accumbens during response-dependent and response-independent cocaine administration in the rat. Psychopharmacology (Berl) 1997;133:7–16. doi: 10.1007/s002130050365. [DOI] [PubMed] [Google Scholar]
- 68.Berridge KC. Pleasures of the brain. Brain Cogn. 2003;52:106–28. doi: 10.1016/s0278-2626(03)00014-9. [DOI] [PubMed] [Google Scholar]
- 69.Franken IH. Drug craving and addiction: integrating psychological and neuropsychopharmacological approaches. Prog Neuropsychopharmacol Biol Psychiatry. 2003;27:563–79. doi: 10.1016/S0278-5846(03)00081-2. [DOI] [PubMed] [Google Scholar]
- 70.Berridge KC, Robinson TE. Parsing reward. Trends Neurosci. 2003;26:507–13. doi: 10.1016/S0166-2236(03)00233-9. [DOI] [PubMed] [Google Scholar]
- 71.Lamb RJ, Preston KL, Schindler CW, et al. The reinforcing and subjective effects of morphine in post-addicts: a dose-response study. J Pharmacol Exp Ther. 1991;259:1165–73. [PubMed] [Google Scholar]
- 72.Schultz W, Tremblay L, Hollerman JR. Reward processing in primate orbitofrontal cortex and basal ganglia. Cereb Cortex. 2000;10:272–83. doi: 10.1093/cercor/10.3.272. [DOI] [PubMed] [Google Scholar]
- 73.Horvitz JC. Mesolimbocortical and nigrostriatal dopamine responses to salient non-reward events. Neuroscience. 2000;96:651–6. doi: 10.1016/s0306-4522(00)00019-1. [DOI] [PubMed] [Google Scholar]
- 74.Volkow ND, Wang GJ, Fowler JS, et al. Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature. 1997;386:830–3. doi: 10.1038/386830a0. [DOI] [PubMed] [Google Scholar]
- 75.Allman JM, Hakeem A, Erwin JM, Nimchinsky E, Hof P. The anterior cingulate cortex. The evolution of an interface between emotion and cognition. Ann N Y Acad Sci. 2001;935:107–17. [PubMed] [Google Scholar]
- 76.Braver TS, Barch DM, Gray JR, Molfese DL, Snyder A. Anterior cingulate cortex and response conflict: effects of frequency, inhibition and errors. Cereb Cortex. 2001;11:825–36. doi: 10.1093/cercor/11.9.825. [DOI] [PubMed] [Google Scholar]
- 77.Erb S, Stewart J. A role for the bed nucleus of the stria terminalis, but not the amygdala, in the effects of corticotropin-releasing factor on stress-induced reinstatement of cocaine seeking. J Neurosci. 1999;19:RC35. doi: 10.1523/JNEUROSCI.19-20-j0006.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Elliott R, Dolan RJ, Frith CD. Dissociable functions in the medial and lateral orbitofrontal cortex: evidence from human neuroimaging studies. Cereb Cortex. 2000;10:308–17. doi: 10.1093/cercor/10.3.308. [DOI] [PubMed] [Google Scholar]
- 79.Ludwig AM, Wikler A, Stark LH. The first drink: psychobiological aspects of craving. Arch Gen Psychiatry. 1974;30:539–47. doi: 10.1001/archpsyc.1974.01760100093015. [DOI] [PubMed] [Google Scholar]
- 80.Shaham Y, Shalev U, Lu L, De Wit H, Stewart J. The reinstatement model of drug relapse: history, methodology and major findings. Psychopharmacology (Berl) 2003;168:3–20. doi: 10.1007/s00213-002-1224-x. [DOI] [PubMed] [Google Scholar]
- 81.Strange PG. The molecular biology of dopamine receptors. In: Stone TW, editor. CNS neurotransmitters and neuromodulators: dopamine. Boca Rouge, FL: CRC; 1996. pp. 65–87. [Google Scholar]
- 82.Xi ZX, Gilbert J, Campos AC, et al. Blockade of mesolimbic dopamine D(3) receptors inhibits stress-induced reinstatement of cocaine-seeking in rats. Psychopharmacology (Berl) 2004;176:57–65. doi: 10.1007/s00213-004-1858-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vorel SR, Ashby CR, Jr, Paul M, et al. Dopamine D3 receptor antagonism inhibits cocaine-seeking and cocaine-enhanced brain reward in rats. J Neurosci. 2002;22:9595–603. doi: 10.1523/JNEUROSCI.22-21-09595.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Garcia-Ladona FJ, Cox BF. BP 897, a selective dopamine D3 receptor ligand with therapeutic potential for the treatment of cocaine-addiction. CNS Drug Rev. 2003;9:141–58. doi: 10.1111/j.1527-3458.2003.tb00246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hutchison KE, Wooden A, Swift RM, et al. Olanzapine reduces craving for alcohol: a DRD4 VNTR polymorphism by pharmacotherapy interaction. Neuropsychopharmacology. 2003;28:1882–8. doi: 10.1038/sj.npp.1300264. [DOI] [PubMed] [Google Scholar]
- 86.Boening JA. Neurobiology of an addiction memory. J Neural Transm. 2001;108:755–65. doi: 10.1007/s007020170050. [DOI] [PubMed] [Google Scholar]
- 87.See RE, Fuchs RA, Ledford CC, McLaughlin J. Drug addiction, relapse, and the amygdala. Ann N Y Acad Sci. 2003;985:294–307. doi: 10.1111/j.1749-6632.2003.tb07089.x. [DOI] [PubMed] [Google Scholar]
- 88.O’Brien C, Childress A, Ehrman R, Robbins S. Conditioning factors in drug abuse: can they explain compulsion? J Psychopharmacol. 1998;12:15–22. doi: 10.1177/026988119801200103. [DOI] [PubMed] [Google Scholar]
- 89.Breiter HC, Aharon I, Kahneman D, Dale A, Shizgal P. Functional imaging of neural responses to expectancy and experience of monetary gains and losses. Neuron. 2001;30:619–39. doi: 10.1016/s0896-6273(01)00303-8. [DOI] [PubMed] [Google Scholar]
- 90.Childress AR, Mozley PD, McElgin W, Fitzgerald J, Reivich M, O’Brien CP. Limbic activation during cue-induced cocaine craving. Am J Psychiatry. 1999;156:11–8. doi: 10.1176/ajp.156.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Kilts CD, Schweitzer JB, Quinn CK, et al. Neural activity related to drug craving in cocaine addiction. Arch Gen Psychiatry. 2001;58:334–41. doi: 10.1001/archpsyc.58.4.334. [DOI] [PubMed] [Google Scholar]
- 92.Grant S, London ED, Newlin DB, et al. Activation of memory circuits during cue-elicited cocaine craving. Proc Natl Acad Sci US A. 1996;93:12040–5. doi: 10.1073/pnas.93.21.12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Due DL, Huettel SA, Hall WG, Rubin DC. Activation in mesolimbic and visuospatial neural circuits elicited by smoking cues: evidence from functional magnetic resonance imaging. Am J Psychiatry. 2002;159:954–60. doi: 10.1176/appi.ajp.159.6.954. [DOI] [PubMed] [Google Scholar]
- 94.Hiroi N, White NM. The lateral nucleus of the amygdala mediates expression of the amphetamine-produced conditioned place preference. J Neurosci. 1991;11:2107–16. doi: 10.1523/JNEUROSCI.11-07-02107.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meil WM, See RE. Lesions of the basolateral amygdala abolish the ability of drug associated cues to reinstate responding during withdrawal from self-administered cocaine. Behav Brain Res. 1997;87:139–48. doi: 10.1016/s0166-4328(96)02270-x. [DOI] [PubMed] [Google Scholar]
- 96.Kalivas PW. Glutamate systems in cocaine addiction. Curr Opin Pharmacol. 2004;4:23–9. doi: 10.1016/j.coph.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 97.Kalivas PW, Duffy P. Repeated cocaine administration alters extracellular glutamate in the ventral tegmental area. J Neurochem. 1998;70:1497–502. doi: 10.1046/j.1471-4159.1998.70041497.x. [DOI] [PubMed] [Google Scholar]
- 98.Carlezon WA, Jr, Nestler EJ. Elevated levels of GluR1 in the midbrain: a trigger for sensitization to drugs of abuse? Trends Neurosci. 2002;25:610–5. doi: 10.1016/s0166-2236(02)02289-0. [DOI] [PubMed] [Google Scholar]
- 99.Nestler EJ. Common molecular and cellular substrates of addiction and memory. Neurobiol Learn Mem. 2002;78:637–7. doi: 10.1006/nlme.2002.4084. [DOI] [PubMed] [Google Scholar]
- 100.Chao J, Nestler EJ. Molecular neurobiology of drug addiction. Annu Rev Med. 2004;55:113–32. doi: 10.1146/annurev.med.55.091902.103730. [DOI] [PubMed] [Google Scholar]
- 101.Chen J, Kelz MB, Hope BT, Nakabeppu Y, Nestler EJ. Chronic Fos-related antigens: stable variants of deltaFosB induced in brain by chronic treatments. J Neurosci. 1997;17:4933–41. doi: 10.1523/JNEUROSCI.17-13-04933.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moratalla R, Elibol B, Vallejo M, Graybiel AM. Network-level changes in expression of inducible Fos-Jun proteins in the striatum during chronic cocaine treatment and withdrawal. Neuron. 1996;17:147–56. doi: 10.1016/s0896-6273(00)80288-3. [DOI] [PubMed] [Google Scholar]
- 103.Kelz MB, Nestler EJ. deltaFosB: a molecular switch underlying long-term neural plasticity. Curr Opin Neurol. 2000;13:715–20. doi: 10.1097/00019052-200012000-00017. [DOI] [PubMed] [Google Scholar]
- 104.Kelz MB, Chen J, Carlezon WA, Jr, et al. Expression of the transcription factor deltaFosB in the brain controls sensitivity to cocaine. Nature. 1999;401:272–6. doi: 10.1038/45790. [DOI] [PubMed] [Google Scholar]
- 105.Nestler EJ. Molecular basis of long-term plasticity underlying addiction. Nat Rev Neurosci. 2001;2:119–28. doi: 10.1038/35053570. [DOI] [PubMed] [Google Scholar]
- 106.Anton RF, Moak DH, Latham P. The Obsessive Compulsive Drinking Scale: a self-rated instrument for the quantification of thoughts about alcohol and drinking behavior. Alcohol Clin Exp Res. 1995;19:92–9. doi: 10.1111/j.1530-0277.1995.tb01475.x. [DOI] [PubMed] [Google Scholar]
- 107.Verheul R, van den Brink W, Geerlings P. A three-pathway psychobiological model of craving for alcohol. Alcohol Alcohol. 1999;34:197–222. doi: 10.1093/alcalc/34.2.197. [DOI] [PubMed] [Google Scholar]
- 108.Roberts JS, Anton RF, Latham PK, Moak DH. Factor structure and predictive validity of the Obsessive Compulsive Drinking Scale. Alcohol Clin Exp Res. 1999;23:1484–91. [PubMed] [Google Scholar]
- 109.Schwartz JM. Neuroanatomical aspects of cognitive-behavioural therapy response in obsessive-compulsive disorder. An evolving perspective on brain and behaviour. Br J Psychiatry Suppl. 1998:38–44. [PubMed] [Google Scholar]
- 110.Perani D, Colombo C, Bressi S, et al. [18F]FDG PET study in obsessive-compulsive disorder. A clinical/metabolic correlation study after treatment. Br J Psychiatry. 1995;166:244–50. doi: 10.1192/bjp.166.2.244. [DOI] [PubMed] [Google Scholar]
- 111.Rubin RT, Ananth J, Villanueva-Meyer J, Trajmar PG, Mena I. Regional 133xenon cerebral blood flow and cerebral 99mTc-HMPAO uptake in patients with obsessive-compulsive disorder before and during treatment. Biol Psychiatry. 1995;38:429–37. doi: 10.1016/0006-3223(94)00305-m. [DOI] [PubMed] [Google Scholar]
- 112.Wang GJ, Volkow ND, Fowler JS, et al. Regional brain metabolic activation during craving elicited by recall of previous drug experiences. Life Sci. 1999;64:775–84. doi: 10.1016/s0024-3205(98)00619-5. [DOI] [PubMed] [Google Scholar]
- 113.Adinoff B, Devous MDS, Best SM, George MS, Alexander D, Payne K. Limbic responsiveness to procaine in cocaine-addicted subjects. Am J Psychiatry. 2001;158:390–8. doi: 10.1176/appi.ajp.158.3.390. [DOI] [PubMed] [Google Scholar]
- 114.Volkow ND, Wang GJ, Fowler JS, et al. Association of methylphenidate-induced craving with changes in right striato-orbitofrontal metabolism in cocaine abusers: implications in addiction. Am J Psychiatry. 1999;156:19–26. doi: 10.1176/ajp.156.1.19. [DOI] [PubMed] [Google Scholar]
- 115.Maas LC, Lukas SE, Kaufman MJ, et al. Functional magnetic resonance imaging of human brain activation during cue-induced cocaine craving. Am J Psychiatry. 1998;155:124–6. doi: 10.1176/ajp.155.1.124. [DOI] [PubMed] [Google Scholar]
- 116.Schneider F, Habel U, Wagner M, et al. Subcortical correlates of craving in recently abstinent alcoholic patients. Am J Psychiatry. 2001;158:1075–83. doi: 10.1176/appi.ajp.158.7.1075. [DOI] [PubMed] [Google Scholar]
- 117.Garavan H, Pankiewicz J, Bloom A, et al. Cue-induced cocaine craving: neuroanatomical specificity for drug users and drug stimuli. Am J Psychiatry. 2000;157:1789–98. doi: 10.1176/appi.ajp.157.11.1789. [DOI] [PubMed] [Google Scholar]
- 118.Sinha R. How does stress increase risk of drug abuse and relapse? Psychopharmacology (Berl) 2001;158:343–59. doi: 10.1007/s002130100917. [DOI] [PubMed] [Google Scholar]
- 119.Lu L, Shepard JD, Scott Hall F, Shaham Y. Effect of environmental stressors on opiate and psychostimulant reinforcement, reinstatement and discrimination in rats: a review. Neurosci Biobehav Rev. 2003;27:457–91. doi: 10.1016/s0149-7634(03)00073-3. [DOI] [PubMed] [Google Scholar]
- 120.Shaham Y, Erb S, Leung S, Buczek Y, Stewart J. CP-154,526, a selective, non-peptide antagonist of the corticotropin-releasing factor1 receptor attenuates stress-induced relapse to drug seeking in cocaine- and heroin-trained rats. Psychopharmacology (Berl) 1998;137:184–90. doi: 10.1007/s002130050608. [DOI] [PubMed] [Google Scholar]
- 121.Erb S, Hitchcott PK, Rajabi H, Mueller D, Shaham Y, Stewart J. Alpha-2 adrenergic receptor agonists block stress-induced reinstatement of cocaine seeking. Neuropsychopharmacology. 2000;23:138–50. doi: 10.1016/S0893-133X(99)00158-X. [DOI] [PubMed] [Google Scholar]
- 122.Shaham Y, Highfield D, Delfs J, Leung S, Stewart J. Clonidine blocks stress-induced reinstatement of heroin seeking in rats: an effect independent of locus coeruleus noradrenergic neurons. Eur J Neurosci. 2000;12:292–302. doi: 10.1046/j.1460-9568.2000.00899.x. [DOI] [PubMed] [Google Scholar]
- 123.McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. J Neurosci. 2004;24:1551–60. doi: 10.1523/JNEUROSCI.4177-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Harfstrand A, Fuxe K, Agnati LF, Benfenati F, Goldstein M. Receptor autoradiographical evidence for high densities of 125I-neuropeptide Y binding sites in the nucleus tractus solitarius of the normal male rat. Acta Physiol Scand. 1986;128:195–200. doi: 10.1111/j.1748-1716.1986.tb07966.x. [DOI] [PubMed] [Google Scholar]
- 125.Deutch AY, Bean AJ. Colocalization in dopamine neurons. In: Bloom FE, Kupfer DJ, editors. Psychopharmacology: the fourth generation of progress. New York: Raven; 1995. [Google Scholar]
- 126.Barrot M, Marinelli M, Abrous DN, et al. The dopaminergic hyper-responsiveness of the shell of the nucleus accumbens is hormone-dependent. Eur J Neurosci. 2000;12:973–9. doi: 10.1046/j.1460-9568.2000.00996.x. [DOI] [PubMed] [Google Scholar]
- 127.Marinelli M, Aouizerate B, Barrot M, Le Moal M, Piazza PV. Dopamine-dependent responses to morphine depend on gluco-corticoid receptors. Proc Natl Acad Sci U S A. 1998;95:7742–7. doi: 10.1073/pnas.95.13.7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003;37:577–82. doi: 10.1016/s0896-6273(03)00021-7. [DOI] [PubMed] [Google Scholar]
- 129.Adinoff B, Iranmanesh A, Veldhuis JD, Fisher L. Disturbances of the stress response: the role of the HPA axis during alcohol withdrawal and abstinence. Alcohol Health Res World. 1998;22:67–72. [PMC free article] [PubMed] [Google Scholar]
- 130.Lovallo WR, Dickensheets SL, Myers DA, Thomas TL, Nixon SJ. Blunted stress cortisol response in abstinent alcoholic and polysubstance-abusing men. Alcohol Clin Exp Res. 2000;24:651–8. [PubMed] [Google Scholar]
- 131.Coiro V, Vescovi PP. Effect of cigarette smoking on ACTH/cortisol secretion in alcoholic after short- and medium-term abstinence. Alcohol Clin Exp Res. 1999;23:1515–8. [PubMed] [Google Scholar]
- 132.Sapolsky RM. McEwen-induced modulation of endocrine history: a partial review. Stress. 1997;2:1–12. doi: 10.3109/10253899709014733. [DOI] [PubMed] [Google Scholar]
- 133.Kiefer F, Jahn H, Schick M, Wiedemann K. Alcohol self-administration, craving and HPA-axis activity: an intriguing relationship. Psychopharmacology (Berl) 2002;164:239–40. doi: 10.1007/s00213-002-1255-3. [DOI] [PubMed] [Google Scholar]
- 134.Junghanns K, Backhaus J, Tietz U, et al. Impaired serum cortisol stress response is a predictor of early relapse. Alcohol Alcohol. 2003;38:189–93. doi: 10.1093/alcalc/agg052. [DOI] [PubMed] [Google Scholar]
- 135.O’Malley SS, Krishnan-Sarin S, Farren C, Sinha R, Kreek J. Naltrexone decreases craving and alcohol self-administration in alcohol-dependent subjects and activates the hypothalamo-pituitary-adrenocortical axis. Psychopharmacology (Berl) 2002;160:19–29. doi: 10.1007/s002130100919. [DOI] [PubMed] [Google Scholar]
- 136.Schluger JH, Ho A, Borg L, Porter M, et al. Nalmefene causes greater hypothalamic-pituitary-adrenal axis activation than naloxone in normal volunteers: implications for the treatment of alcoholism. Alcohol Clin Exp Res. 1998;22:1430–6. doi: 10.1111/j.1530-0277.1998.tb03931.x. [DOI] [PubMed] [Google Scholar]
- 137.Fillmore MT, Rush CR. Impaired inhibitory control of behavior in chronic cocaine users. Drug Alcohol Depend. 2002;66:265–73. doi: 10.1016/s0376-8716(01)00206-x. [DOI] [PubMed] [Google Scholar]
- 138.Petry NM, Bickel WK, Arnett M. Shortened time horizons and insensitivity to future consequences in heroin addicts. Addiction. 1998;93:729–38. doi: 10.1046/j.1360-0443.1998.9357298.x. [DOI] [PubMed] [Google Scholar]
- 139.Heyman GM, Dunn B. Decision biases and persistent illicit drug use: an experimental study of distributed choice and addiction. Drug Alcohol Depend. 2002;67:193–203. doi: 10.1016/s0376-8716(02)00071-6. [DOI] [PubMed] [Google Scholar]
- 140.Moeller FG, Dougherty DM, Barratt ES, et al. Increased impulsivity in cocaine dependent subjects independent of antisocial personality disorder and aggression. Drug Alcohol Depend. 2002;68:105–11. doi: 10.1016/s0376-8716(02)00106-0. [DOI] [PubMed] [Google Scholar]
- 141.Rogers RD, Everitt BJ, Baldacchino A, et al. Dissociable deficits in the decision-making cognition of chronic amphetamine abusers, opiate abusers, patients with focal damage to prefrontal cortex, and tryptophan-depleted normal volunteers: evidence for monoaminergic mechanisms. Neuropsy-chopharmacology. 1999;20:322–39. doi: 10.1016/S0893-133X(98)00091-8. [DOI] [PubMed] [Google Scholar]
- 142.Grant S, Contoreggi C, London ED. Drug abusers show impaired performance in a laboratory test of decision making. Neuropsychologia. 2000;38:1180–7. doi: 10.1016/s0028-3932(99)00158-x. [DOI] [PubMed] [Google Scholar]
- 143.Meunier M, Bachevalier J, Mishkin M. Effects of orbital frontal and anterior cingulate lesions on object and spatial memory in rhesus monkeys. Neuropsychologia. 1997;35:999–1015. doi: 10.1016/s0028-3932(97)00027-4. [DOI] [PubMed] [Google Scholar]
- 144.Bechara A, Damasio H, Tranel D, Anderson SW. Dissociation of working memory from decision making within the human prefrontal cortex. J Neuroscience. 1998;18:428–37. doi: 10.1523/JNEUROSCI.18-01-00428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Bechara A, Dolan S, Denburg N, Hindes A, Anderson SW, Nathan PE. Decision-making deficits, linked to a dysfunctional ventromedial prefrontal cortex, revealed in alcohol and stimulant abusers. Neuropsychologia. 2001;39:376–89. doi: 10.1016/s0028-3932(00)00136-6. [DOI] [PubMed] [Google Scholar]
- 146.Ernst M, Grant SJ, London ED, Contoreggi CS, Kimes AS, Spurgeon L. Decision making in adolescents with behavior disorders and adults with substance abuse. Am J Psychiatry. 2003;160:33–40. doi: 10.1176/appi.ajp.160.1.33. [DOI] [PubMed] [Google Scholar]
- 147.Adinoff B, Devous MD, Sr, Cooper DB, et al. Resting regional cerebral blood flow and gambling task performance in cocaine-dependent subjects and healthy comparison subjects. Am J Psychiatry. 2003;160:1892–4. doi: 10.1176/appi.ajp.160.10.1892. [DOI] [PubMed] [Google Scholar]
- 148.Volkow ND, Wang GJ, Fowler JS, et al. Effects of methylphenidate on regional brain glucose metabolism in humans: relationship to dopamine D2 receptors. Am J Psychiatry. 1997;154:50–5. doi: 10.1176/ajp.154.1.50. [DOI] [PubMed] [Google Scholar]
- 149.Volkow ND, Hitzemann R, Wang GJ, et al. Decreased brain metabolism in neurologically intact healthy alcoholics. Am J Psychiatry. 1992;149:1016–22. doi: 10.1176/ajp.149.8.1016. [DOI] [PubMed] [Google Scholar]
- 150.Volkow ND, Wang G-J, Fowler JS, et al. Effects of methylphenidate on regional brain glucose metabolism in humans: Relationship to dopamine D2 receptors. Am J Psychiatry. 1997;154:50–5. doi: 10.1176/ajp.154.1.50. [DOI] [PubMed] [Google Scholar]
- 151.Goldstein RZ, Volkow ND, Wang GJ, Fowler JS, Rajaram S. Addiction changes orbitofrontal gyrus function: involvement in response inhibition. Neuroreport. 2001;12:2595–9. doi: 10.1097/00001756-200108080-00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Paulus MP, Hozack NE, Zauscher BE, et al. Behavioral and functional neuroimaging evidence for prefrontal dysfunction in methamphetamine-dependent subjects. Neuropsychopharmacology. 2002;26:53–63. doi: 10.1016/S0893-133X(01)00334-7. [DOI] [PubMed] [Google Scholar]
- 153.Kaufman JN, Ross TJ, Stein EA, Caravan H. Cingulate hypoactivity in cocaine users during a GO-NOGO task as revealed by event-related functional magnetic resonance imaging. J Neurosci. 2003;23:7839–43. doi: 10.1523/JNEUROSCI.23-21-07839.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Adinoff B, Devous MD, Best SE, et al. Regional cerebral blood flow in female cocaine-addicted subjects following limbic activation. Drug Alcohol Depend. 2003;71:255–68. doi: 10.1016/s0376-8716(03)00138-8. [DOI] [PubMed] [Google Scholar]
- 155.Sinha R. Probing stress-reward circuitry to understand cocaine craving and relapse. Paper presented at the 2003 annual meeting of the American College of Neuropsychopharmacology; San Juan, Puerto Rico. December 2003; [Abstract no. THAM13] [Google Scholar]
- 156.London ED, Ernst M, Grant S, Bonson K, Weinstein A. Orbitofrontal cortex and human drug abuse: functional imaging. Cereb Cortex. 2000;10:334–42. doi: 10.1093/cercor/10.3.334. [DOI] [PubMed] [Google Scholar]
- 157.Volkow ND, Fowler JS. Addiction, a disease of compulsion and drive: involvement of the orbitofrontal cortex. Cereb Cortex. 2001;10:318–25. doi: 10.1093/cercor/10.3.318. [DOI] [PubMed] [Google Scholar]
- 158.Krebaum SR, Jackley PK, Adinoff B. The Impulsive Relapse Questionnaire: development and validation. Drug Alcohol Depend. 2002;66(suppl):S96. [Google Scholar]
- 159.Robinson TE, Gorny G, Mitton E, Kolb B. Cocaine self-administration alters the morphology of dendrites and dendritic spines in the nucleus accumbens and neocortex. Synapse. 2001;39:257–66. doi: 10.1002/1098-2396(20010301)39:3<257::AID-SYN1007>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 160.Rothman RB, Glowa JR. A review of the effects of dopaminergic agents on humans, animals, and drug-seeking behavior, and its implications for medication development. Focus on GBR 12909. Mol Neurobiol. 1995;11:1–19. doi: 10.1007/BF02740680. [DOI] [PubMed] [Google Scholar]
- 161.Preti A. Vanoxerine National Institute on Drug Abuse. Curr Opin Investig Drugs. 2000;1:241–51. [PubMed] [Google Scholar]
- 162.Carroll KM, Fenton LR, Ball SA, et al. Efficacy of disulfiram and cognitive behavior therapy in cocaine-dependent outpatients: a randomized placebo-controlled trial. Arch Gen Psychiatry. 2004;61:264–72. doi: 10.1001/archpsyc.61.3.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Volpicelli JR, Alterman AI, Hayashida M, O’Brien CP. Naltrexone in the treatment of alcohol dependence. Arch Gen Psychiatry. 1992;49:876–80. doi: 10.1001/archpsyc.1992.01820110040006. [DOI] [PubMed] [Google Scholar]
- 164.O’Malley SS, Jaffe AJ, Chang G, Schottenfeld RS, Meyer RE, Rounsaville B. Naltrexone and coping skills therapy for alcohol dependence. Arch Gen Psychiatry. 1992;49:881–7. doi: 10.1001/archpsyc.1992.01820110045007. [DOI] [PubMed] [Google Scholar]
- 165.Benjamin D, Grant ER, Pohorecky LA. Naltrexone reverses ethanol-induced dopamine release in the nucleus accumbens in awake, freely moving rats. Brain Res. 1993;621:137–40. doi: 10.1016/0006-8993(93)90309-b. [DOI] [PubMed] [Google Scholar]
- 166.Brebner K, Childress AR, Roberts DC. A potential role for GABA(B) agonists in the treatment of psychostimulant addiction. Alcohol Alcohol. 2002;37:478–84. doi: 10.1093/alcalc/37.5.478. [DOI] [PubMed] [Google Scholar]
- 167.Shoptaw S, Yang X, Rotheram-Fuller EJ, et al. Randomized placebo-controlled trial of baclofen for cocaine dependence: preliminary effects for individuals with chronic patterns of cocaine use. J Clin Psychiatry. 2003;64:1440–8. doi: 10.4088/jcp.v64n1207. [DOI] [PubMed] [Google Scholar]
- 168.Brodie JD, Figueroa E, Dewey SL. Treating cocaine addiction: from preclinical to clinical trial experience with gamma-vinyl GABA. Synapse. 2003;50:261–5. doi: 10.1002/syn.10278. [DOI] [PubMed] [Google Scholar]
- 169.Walsh SL, Geter-Douglas B, Strain EC, Bigelow GE. Enado-line and butorphanol: evaluation of kappa-agonists on cocaine pharmacodynamics and cocaine self-administration in humans. J Pharmacol Exp Ther. 2001;299:147–58. [PubMed] [Google Scholar]
- 170.Johnson BA, Ait-Daoud N, Bowden CL, et al. Oral topiramate for treatment of alcohol dependence: a randomised controlled trial. Lancet. 2003;361:1677–85. doi: 10.1016/S0140-6736(03)13370-3. [DOI] [PubMed] [Google Scholar]
- 171.Kampman KM, Pettinati H, Lynch KG, et al. A pilot trial of topiramate for the treatment of cocaine dependence. Drug Alcohol Depend. 2004;75:233–40. doi: 10.1016/j.drugalcdep.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 172.Kosten TR, Oliveto A, Sevarino KA, Gonsai K, Feingold A. Ketoconazole increases cocaine and opioid use in methadone maintained patients. Drug Alcohol Depend. 2002;66:173–80. doi: 10.1016/s0376-8716(01)00198-3. [DOI] [PubMed] [Google Scholar]
- 173.Montgomery AM, Rose IC, Herberg LJ. The effect of a 5-HT3 receptor antagonist, ondansetron, on brain stimulation reward, and its interaction with direct and indirect stimulants of central dopaminergic transmiss. J Neural Transm Gen Sect. 2001;91:1–11. doi: 10.1007/BF01244914. [DOI] [PubMed] [Google Scholar]
- 174.King GR, Xiong Z, Douglass S, Ellinwood EH. Long-term blockade of the expression of cocaine sensitization by ondansetron, a 5-HT(3) receptor antagonist. Eur J Pharmacology. 2000;394:97–101. doi: 10.1016/s0014-2999(99)00926-7. [DOI] [PubMed] [Google Scholar]
- 175.Johnson BA, Roache JD, Javors MA, et al. Ondansetron for reduction of drinking among biologically predisposed alcoholic patients. JAMA. 2000;284:963–71. doi: 10.1001/jama.284.8.963. [DOI] [PubMed] [Google Scholar]
- 176.Gorelick DA, Gardner EL, Xi ZX. Agents in development for the management of cocaine abuse. Drugs. 2004;64:1547–73. doi: 10.2165/00003495-200464140-00004. [DOI] [PubMed] [Google Scholar]
- 177.De Vries TJ, Shaham Y, Homberg JR, et al. A cannabinoid mechanism in relapse to cocaine seeking. Nat Med. 2001;7:1151–4. doi: 10.1038/nm1001-1151. [DOI] [PubMed] [Google Scholar]
- 178.Rada PV, Hoebel BG. Aversive hypothalamic stimulation releases acetylcholine in the nucleus accumbens, and stimulation-escape decreases it. Brain Res. 2001;888:60–5. doi: 10.1016/s0006-8993(00)02865-1. [DOI] [PubMed] [Google Scholar]
- 179.Hajnal A, Mark GP, Rada PV, Lenard L, Hoebel BG. Nore-pinephrine microinjections in the hypothalamic paraventricular nucleus increase extracellular dopamine and decrease acetylcholine in the nucleus accumbens: relevance to feeding reinforcement. J Neurochem. 1997;68:667–74. doi: 10.1046/j.1471-4159.1997.68020667.x. [DOI] [PubMed] [Google Scholar]
- 180.Smith JE, Co C, Yin X, et al. Involvement of cholinergic neuronal systems in intravenous cocaine self-administration. Neurosci Biobehav Rev. 2004;27:841–50. doi: 10.1016/j.neubiorev.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 181.Rasmussen T, Sauerberg P, Nielsen EB, et al. Muscarinic receptor agonists decrease cocaine self-administration rates in drug-naive mice. Eur J Pharmacol. 2000;402:241–6. doi: 10.1016/s0014-2999(00)00442-8. [DOI] [PubMed] [Google Scholar]
- 182.Szumlinski KK, Maisonneuve IM, Glick SD. Iboga interactions with psychomotor stimulants: panacea in the paradox? Toxicon. 2001;39:75–86. doi: 10.1016/s0041-0101(00)00158-6. [DOI] [PubMed] [Google Scholar]
- 183.Glick SD, Maisonneuve IS. Mechanisms of antiaddictive actions of ibogaine. Ann N YAcad Sci. 1998;844:214–26. [PubMed] [Google Scholar]
- 184.Mash DC, Kovera CA, Buck BE, Norenberg MD, Shapshak P, Hearn WL, Sanchez-Ramos J. Medication development of ibogaine as a pharmacotherapy for drug dependence. Ann N Y Acad Sci. 1998;844:274–92. [PubMed] [Google Scholar]