

Abstract
Herpes simplex virus type 1 (HSV-1) entry into permissive cells involves attachment to cell-surface glycosaminoglycans (GAGs) and fusion of the virus envelope with the cell membrane triggered by the binding of glycoprotein D (gD) to cognate receptors. In this study, we characterized the observation that soluble forms of the gD ectodomain (sgD) can mediate entry of gD-deficient HSV-1. We examined the efficiency and receptor specificity of this activity and used sequential incubation protocols to determine the order and stability of the initial interactions required for entry. Surprisingly, virus binding to GAGs did not increase the efficiency of sgD-mediated entry and gD-deficient virus was capable of attaching to GAG-deficient cells in the absence of sgD. These observations suggested a novel binding interaction that may play a role in normal HSV infection.
Keywords: HSV-1, glycoprotein D (gD), glycosaminoglycans, nectin-1, HVEM, soluble glycoprotein, virus entry
Introduction
Infection of permissive cells by herpes simplex virus type 1 (HSV-1) generally requires virus attachment to the cell surface and fusion of the viral envelope with the cytoplasmic or endosomal membrane. HSV attachment involves the coordinated binding of multiple viral glycoproteins to their cognate cellular receptors. Glycoproteins B (gB) and C (gC) bind to herparan sulfate (HS) glycosaminoglycan (GAG) side chains of cell-surface proteoglycans, while glycoprotein D (gD) engages one of three HSV-1 entry receptors, HVEM (HveA), nectin-1 (Hve-C), or 3-O-sulfated HS (Geraghty et al., 1998; Montgomery et al., 1996; Shukla et al., 1999). Receptor binding by gD is believed to set in motion the fusion process, which requires gB and the gH:gL heterodimer for completion reviewed in (Campadelli-Fiume et al., 2000; Spear, 2004).
Glycoprotein B and C binding to HS represents approximately 80% of the HSV-1 binding capacity to fully permissive cells (Laquerre et al., 1998). Accordingly, mutant viruses defective for these activities bind to and infect cells with reduced efficiency (Banfield et al., 1995; Laquerre et al., 1998; Shieh and Spear, 1994). Likewise, HSV binding and entry are reduced approximately 10-fold on cells that lack surface HS (Shieh et al., 1992). It is generally believed that the interaction of gD with its receptor(s) is responsible for the residual cell-binding activity of HSV in the absence of cell-surface HS, but this has not been directly demonstrated. A recent report suggesting gB binding to a novel receptor (Bender et al., 2005) illustrates the possibility that other interactions are also involved.
Mutational analyses and mapping of antibody binding sites have revealed distinct, but overlapping regions of gD that contribute to the binding of the different cognate receptors. An array of gD mutants have been described that are competent for binding and entry via nectin-1, but not HVEM (Carfi et al., 2001; Connolly et al., 2003; Krummenacher et al., 1998; Lopez et al., 2000; Milne et al., 2003; Montgomery et al., 1996; Warner et al., 1998; Whitbeck et al., 1997). Rare mutants with the reciprocal profile have now been identified as well (Connolly et al., 2005; Manoj et al., 2004) (Q. Bai, W. Ali Shah, J.B. Cohen, R.J. Eisenberg, G.H. Cohen and J.C. Glorioso, 26th International Herpesvirus Workshop, abstr. 2.10, 2001). In addition, individual antibodies have been described that impair HSV-1 infection through one, but not the other receptor (Nicola et al., 1998). Elucidation of the crystal structures of a C-terminally truncated version of the gD ectodomain in free form and complexed with HVEM has identified an N-terminal hairpin domain as a major component of the HVEM binding site (Carfi et al., 2002; Carfi et al., 2001), consistent with the location of HVEM-specific loss-of-function mutations. Although the nectin-1 binding site remains poorly defined, current evidence supports the view that it is formed by several discontinuous sequences, some shared with the HVEM binding site, others unique.
Recent studies have identified a region in gD that is required for virus entry, but not binding of gD to HVEM or nectin-1 (Cocchi et al., 2004; Zago, Jogger, and Spear, 2004). This region, roughly spanning amino acids 260–310 in the C-terminal portion of the ectodomain, was divided into two sub-regions that were found to bind independently to sequences in the upstream portion of the gD ectodomain. Importantly, it was shown that these interactions were disrupted by binding of gD to nectin-1 or HVEM (Fusco, Forghieri, and Campadelli-Fiume, 2005). These observations suggested that receptor binding liberates the C-terminal portion of the gD ectodomain for interaction with components of the membrane fusion machinery, thereby setting in motion the virus entry process. Compelling support for this model has since been provided by the elucidation of the structure of the complete ectodomain of gD, demonstrating multiple contacts between the C-terminal region and important components of the HVEM and nectin-1 binding sites in unliganded gD (Krummenacher et al., 2005).
Cocchi and coworkers reported that the gD ectodomain in soluble form (gD285) can mediate entry of gD-deficient HSV-1 into susceptible cells, suggesting that the ectodomain can interact in trans with the virus (Cocchi et al., 2004). Along with the demonstration that this activity was abolished by truncation of the ectodomain at position 260, this result represents compelling evidence that the proximal target of gD within the fusion machinery is another viral protein, such as gB or gH. However, this study did not report whether receptor-deficient cells were resistant to gD285-mediated entry, leaving open the possibility that the soluble molecule did not require activation by receptor binding to interact with cell-attached virus and initiate the entry process. In addition, the efficiency of entry was not addressed, thus prohibiting any conclusions as to whether the proposed interaction between gD residues 260–285 and a separate viral component may in fact be the principal signal to initiate membrane fusion.
We independently observed infection of HSV-1-susceptible cells by a gD-deficient virus in the presence of soluble gD ectodomain, in this instance a molecule truncated after residue 287 (sgD287, hereafter also referred to as sgD). To characterize this activity, we showed that infection was dependent on the presence of a cell-surface receptor for gD and specific recognition of this receptor by soluble gD. We acquired data indicating that sgD287-mediated infection was approximately 10-fold less efficient than infection by gD+ virus, consistent with the suggestion that the process is comparable to that initiated by virion gD. Variation of the infection conditions showed that exposure of cell-bound sgD to 37°C impaired entry of subsequently added virus. We found that this was due to gradual sgD dissociation from the cells at this temperature in the absence of virus, indicating that entry requires virus interaction with receptor-bound sgD at the cell surface. However, since entry was not abruptly abolished by pre-exposure of cell-bound sgD to 37°C, we conclude that the entry-mediating conformation of sgD was stable as long as the soluble molecule remained attached to its receptor. Remarkably, we observed that sgD-mediated gD− virus entry was not dependent on either of the known mechanisms for HSV attachment to cells, namely gB and gC binding to cellular GAGs and virion gD binding to cognate cellular receptors. A novel binding function was suggested by experiments showing adsorption of gD-deficient virus to GAG-deficient cells in the absence of sgD. Together, these results illustrate the utility of soluble gD to explore the early events in the HSV infectious process.
Results
Soluble gD mediates entry of a gD-deficient virus into CHO-K1 cells expressing entry receptors
Soluble gD ectodomain, comprised of the first 287 amino acids of mature gD followed by a histidine tag (sgD287), was prepared by transfection of 293T cells with a suitable mammalian expression construct and recovery of the histidine-tagged products by passage of the growth media over a Ni2+ affinity column, as previously described (Nakano et al., 2005). Analysis by SDS-PAGE and Coomassie staining revealed a single band at a molecular size of approximately 38 kD for wild-type (wt) sgD287 and two mutants (data not shown). These products were recognized on Western blots by both a His tag-specific monoclonal antibody (Fig. 1A) and a gD-specific polyclonal antibody, R7 (Krummenacher et al., 2000) (data not shown).
Fig. 1.
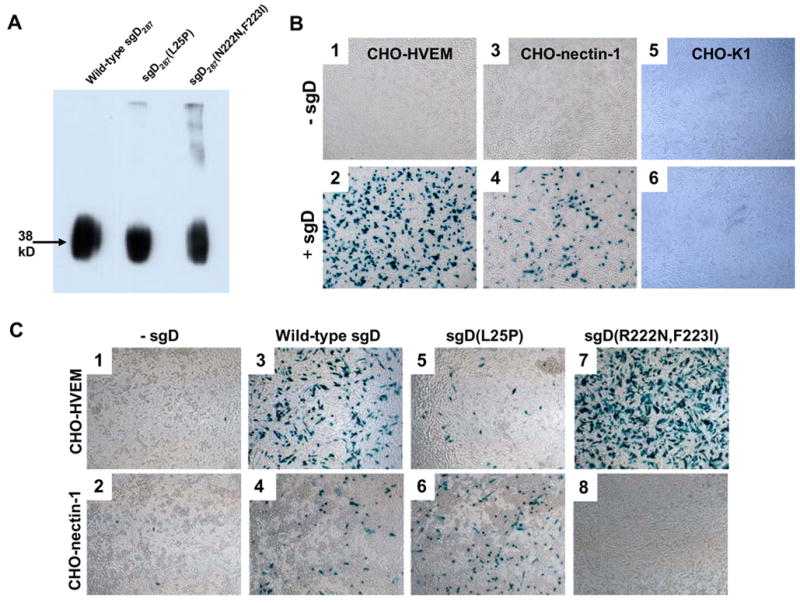
Soluble gD proteins mediate entry of gD-deficient KΔUs3-8Z virus into receptor-positive cells. (A) sgD287, sgD287(L25P), and sgD287(R222N,F223I) were detected on a Western blot using an anti-histidine tag antibody, as described in Materials and Methods. (B) Sub-confluent monolayers of CHO-HVEM (1 and 2), CHO-nectin-1 (3 and 4), and CHO-K1 cells (5 and 6) in 96-well plates were incubated with gD-deficient KΔUs3-8Z in the absence (1, 3, and 5) or presence of 500 ng sgD287 (2, 4, and 6) for 3 h at 37°C. Infected cells were identified by X-gal staining 16 h after infection. (C) gD-deficient KΔUs3-8Z was incubated with CHO-HVEM or CHO-nectin-1 cells in the absence (1 and 2) or presence of wild-type sgD287 (3 and 4), sgD287(L25P) (5 and 6), or sgD287(R222N,F223I) (7 and 8) for 3 h at 37°C. Infected cells were identified by X-gal staining at 16 h after infection.
The gD-deficient HSV-1 recombinant KΔUs3-8Z, containing a lacZ reporter cassette in place of the Us3-Us8 region of HSV-1 KOS, was described previously (Anderson et al., 2000). The deletion in this virus extends at both ends beyond the limits of the complementing region in the genome of VD60 cells and the virus can therefore be propagated on these cells without detectable rescue by homologous recombination. KΔUs3-8Z virions lacking gD were produced by infection of Vero cells with gD-complemented KΔUs3-8Z, removal of input extracellular virus, and collection of cell lysates 3–5 days later.
HSV-resistant CHO-K1 cells and susceptible derivatives expressing HVEM or nectin-1 were incubated for 3 h with gD-deficient KΔUs3-8Z in the presence of sgD287. Entry was visualized 16 h later by staining the cells for β-galactosidase activity expressed by the virus. Fig. 1B demonstrates that wt sgD enabled entry of the gD-deficient virus into CHO-HVEM and CHO-nectin-1 cells, but not CHO-K1 cells. The degree of infection increased linearly with both the virus dose and the amount of sgD (data not shown).
To determine the relative efficiency of infection mediated by sgD compared to virion gD, we used quantitative TaqMan PCR to estimate the number of gD-deficient virus particles used in our experiments. The results showed a reduction of approximately 10-fold in transducing events per viral particle for gD-deficient KΔUs3-8Z in the presence of sgD compared to a KΔUs3-8Z preparation harvested from gD-complementing VD60 cells (see Materials and Methods). Thus we estimate that the efficiency of sgD-mediated infection is approximately one order of magnitude lower than infection mediated by virion-incorporated gD.
Soluble gD-mediated entry of gD-deficient HSV requires specific interaction with a membrane-bound gD receptor
Productive HSV-1 entry into cells depends on the interaction of virion gD with a cognate receptor. To confirm that the observed entry of gD-deficient virus required a similar interaction and was not mediated by contaminants in our sgD preparations, we tested receptor-specific versions of the soluble protein. The L25P mutation in gD impairs virus entry through HVEM without diminishing entry via nectin-1 (Yoon et al., 2003). In addition, we previously isolated a rare gD mutant with the reciprocal properties [gD(R222N,F223I); Q. Bai, W.A. Shah, J.B. Cohen, R.J. Eisenberg, G.H. Cohen and J.C. Glorioso. Abstr. 26th International Herpesvirus Workshop, abstr. 2.10, 2001]; the characteristics of this mutant have been confirmed and described in more detail by others (Manoj et al., 2004). As shown in Fig. 1C, sgD287(L25P) displayed a substantially reduced ability to mediate entry into CHO-HVEM cells (compare panels 3 and 5) while its ability to mediate entry into CHO-nectin-1 cells was unaltered compared to wild-type sgD287 (panels 4 and 6). Conversely, sgD287(R222N,F223I) mediated entry into CHO-HVEM, but not CHO-nectin-1 cells (panels 7 and 8). These results showed that sgD-mediated entry of gD-deficient virus required specific receptor recognition in a manner similar to infection by complete virions; they argued strongly against the possibility that entry was mediated by an unrelated contaminant in our sgD preparations.
Establishment of parameters for sgD-mediated entry
By analogy to current models for gD+ virus entry, we hypothesized that sgD binding to its receptor enabled a subsequent interaction between the soluble molecule and the viral envelope that initiated membrane fusion to deliver de-enveloped particles to the cytoplasm. To test this prediction, CHO-HVEM cells were incubated with sgD for 1 h at 37°C, washed several times to remove unbound sgD, and exposed to gD-deficient KΔUs3-8Z at 37°C. The results, summarized in Table 1, showed that infection was reduced to background levels under these conditions (Table 1 protocol 3 and Fig. 2C), compared to readily detectable infection when sgD and the virus were added to the cells either simultaneously (Table 1 protocol 1 and Fig. 2B) or in the reverse order (Table 1 protocol 2 and Fig. 2D). Negligible infection was also observed when sgD was bound to the cells at 4°C prior to the washes and the virus was added at 37°C (Table 1 protocol 4). However, when sgD binding at 4°C was followed by virus binding at 4°C and a second washing step before the temperature was raised, infection occurred (Table 1 protocol 5). These results suggested that the virus must interact with receptor-bound sgD prior to entry and that the enabling activity of sgD is unstable.
Table 1.
Incubation protocols for gD-deficient KΔUs3-8Z entry into CHO-HVEM cells
| Protocol # | Incubation 1 | Wash | Incubation 2 | Wash | Infection | Entry* |
|---|---|---|---|---|---|---|
| 1. | Virus+sgD/37°C | +++ | ||||
| 2. | Virus/37°C | yes | sgD/37°C | +++ | ||
| 3. | sgD/37°C | yes | Virus/37°C | - | ||
| 4. | sgD/4°C | yes | Virus/37°C | - | ||
| 5. | sgD/4°C | yes | Virus/4°C | yes | 37°C | ++ |
+++, > 50% infected cells; ++, 20–50% infected cells; -, < 5% infected cells
Fig. 2.
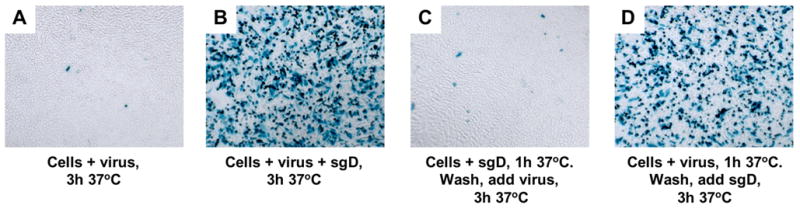
Pre-attachment of sgD to CHO-HVEM cells eliminates efficient infection by gD− KΔUs3-8Z. Cells were incubated with (A) gD− KΔUs3-8Z at 37°C for 3 h, (B) gD− KΔUs3-8Z plus sgD for 3 h at 37°C, (C) sgD at 37°C for 1 h, washed, and then incubated with gD− KΔUS3-8Z for 3 h, or (D) gD− KΔUs3-8Z at 37°C for 1 h, washed, and then incubated with sgD for 3 h. Media were replaced with fresh medium and infected cells identified by X-gal staining 16 h later.
Additional experiments showed that the loss of sgD activity under the conditions of protocols 3 and 4 (Table 1) could be attributed to sgD dissociation from the cells at 37°C. First, the levels of cell-associated sgD before and after incubation at 37°C were compared by immunofluorescence. The cells were incubated with sgD for 2 h at 4°C, washed, and fixed either immediately or after incubation at 37°C for 30 min in fresh medium. Staining with a pool of gD-specific monoclonal antibodies (Highlander et al., 1987) showed abundant cell-associated sgD before, but not after the 37°C incubation period for both CHO-HVEM (Fig. 3A, B) and CHO-nectin-1 cells (Fig. 3C, D). Second, the amount of CHO-HVEM-associated sgD before and after incubation for 10 or 30 min at 37ºC was determined by immunoblotting. Samples were prepared by incubation of the cells with sgD for 2 h at 4ºC, followed by several washes, incubation at 37ºC, and collection of cell lysates. This analysis, as well, showed a loss of cell-associated sgD over time (Fig. 4A, lanes 2–4). In a separate experiment, we collected both the cells and the medium after the 30-min incubation period at 37ºC and compared the total amount of sgD in both samples with the amount of cell-associated sgD at the start of the 37ºC incubation. The results showed that the entire amount of sgD associated with the cells at time zero could be recovered from the medium at the end of the 37ºC incubation period (Fig. 4B). Consistent with this result, we found that all cell-associated sgD at the different time points was sensitive to treatment of the cells with trypsin prior to cell lysis (Fig. 4A, lanes 5–7), supporting the conclusion that the loss of sgD from the cell surface at 37ºC was due to dissociation rather than internalization by the cells.
Fig. 3.
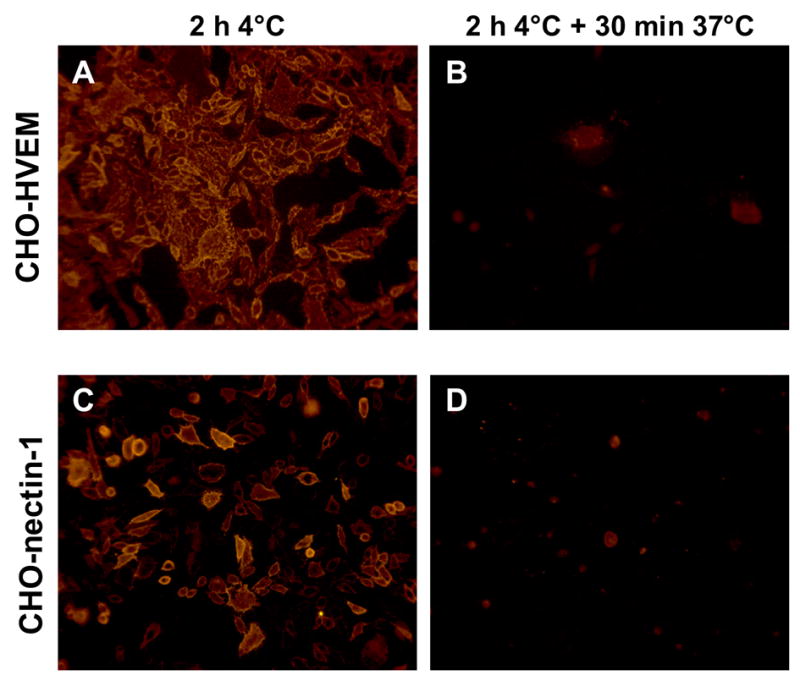
Detection of cell-associated sgD. sgD was bound to CHO-HVEM (A and B) or CHO-nectin-1 (C and D) cells at 4°C for 2 h. Cells were washed and either fixed immediately (A and C) or the temperature was shifted to 37°C for 30 min prior to fixation (B and D). Cell-associated sgD was detected with a pool of monoclonal anti-gD antibodies and a goat anti-mouse secondary antibody linked to Cy3.
Fig. 4.
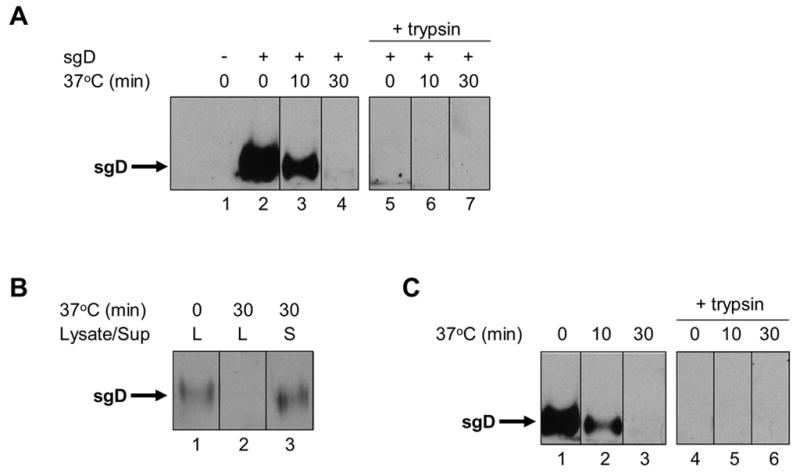
sgD localization by Western blot analysis with R7 anti-gD antibody. (A) sgD or no protein was bound to CHO-HVEM cells for 2 h at 4°C. Cells were washed with cold PBS and either lysed immediately (lanes 1 and 2) or incubated with fresh, pre-warmed media at 37°C for 10 min (lane 3) or 30 min (lane 4). To identify internalized sgD, duplicate cultures were treated with trypsin prior to cell lysis (lanes 5–7). (B) sgD was bound to CHO-HVEM cells for 2 h at 4°C. The cells were washed and lysed either immediately (lane 1) or after incubation at 37°C for 30 min with fresh, pre-warmed media (lane 2). The media was collected at the end of the 37°C incubation and the protein contents precipitated with TCA (lane 3). (C) sgD and gD− KΔUs3-8Z were bound to CHO-HVEM cells for 2 h at 4°C. Cells were washed with cold PBS and either lysed immediately (lane 1) or incubated with fresh, pre-warmed media at 37°C for 10 min (lane 2) or 30 min (lane 3). For identification of internalized sgD, duplicate cultures were treated with trypsin prior to cell lysis (lanes 4–6).
Additional Western blot analyses demonstrated that sgD was not stabilized at the cell surface by prior virus attachment and that all cell-associated sgD detected under these conditions before or after incubation at 37ºC remained fully sensitive to trypsin (Fig. 4C). These observations argued that cell-associated virus and sgD did not engage in a stable interaction at 37°C and that sgD was not endocytosed in the presence of virus. Instead, they suggested that sgD interacts transiently with the virus prior to virus entry.
Nicola and colleagues have reported that productive HSV infection of susceptible CHO cells occurs by pH-dependent endocytosis and requires intra-endosomal interaction of the virus with a gD receptor (Nicola, McEvoy, and Straus, 2003; Nicola and Straus, 2004). Our evidence that sgD interacts transiently with gD-deficient virus at the cell surface to mediate virus entry, but is not itself internalized, argued against sgD mediating virus release from endosomes. Accordingly, we asked whether productive sgD-mediated infection of CHO cells requires the preceding step in endocytosis, endosome acidification. CHO-HVEM and CHO-nectin-1 cells were treated with bafilomycin A1, a specific inhibitor of vacuolar H+-ATPase, for 30 min and infected in the continued presence of the drug for 5.5 h. For comparison, we used a replication-defective vesicular stomatitis virus (VSV) recombinant that expresses green fluorescent protein (GFP) (Bergman et al., 2003) and recorded GFP expression at 4 h post-infection, prior to widespread VSV-mediated cell death. As illustrated in Fig. 5 (second column), treatment of the cells with bafilomycin did not reduce productive sgD-mediated infection. Surprisingly, the drug also failed to inhibit infection by gD-complemented KΔUs3-8Z (Fig. 5, third column) or an ICP4-deleted KOS derivative (SHZ.1, fourth column) (Mester, Pitha, and Glorioso, 1995) whereas infection by VSV-GFP was completely abolished (fifth column). Although we used a higher concentration of bafilomycin (400 nM) than is common in the literature (≤ 200 nM; (Akula et al., 2003; Bergman et al., 2003; Gianni, Campadelli-Fiume, and Menotti, 2004; Milne et al., 2005; Nicola, McEvoy, and Straus, 2003; Nicola and Straus, 2004), we observed no clear changes in cell survival or morphology in mock-infected cultures treated with the drug (e.g. Fig. 5, first column). These results indicated that productive infection by gD-deficient HSV through either complementation or sgD did not require exposure to a low-pH environment. Although we have no direct evidence demonstrating the specific virus entry pathway, our combined results favor the suggestion that sgD-mediated productive infection occurs by fusion at the cell surface rather than by endocytosis.
Fig. 5.
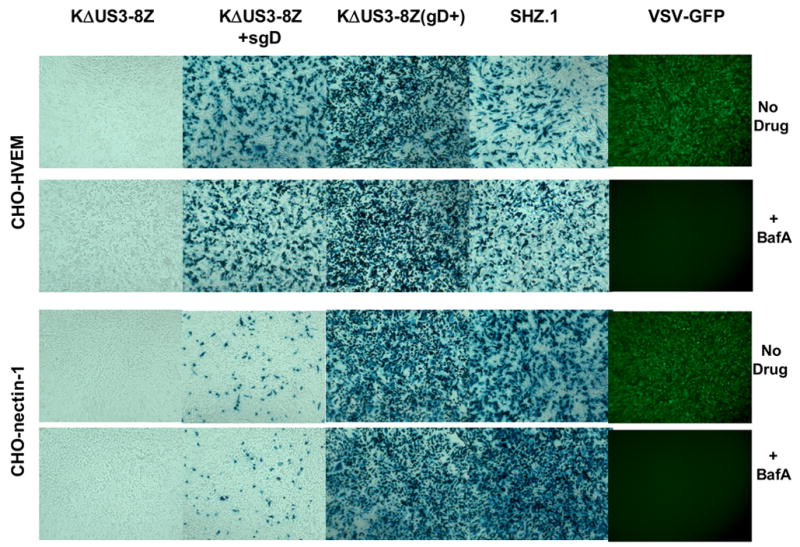
Sensitivity of viral reporter gene expression to bafilomycin A1 (BFLA) treatment of the cells. CHO-HVEM and CHO-nectin-1 cells were incubated for 30 min at 37°C in medium with or without 400 nM BFLA. The cells were infected with gD-deficient KΔUs3-8Z alone or in the presence of 500 ng sgD. Control infections were performed with gD-complemented KΔUs3-8Z, replication-deficient SHZ.1, or VSV-GFP. After 4 h in the continued presence or absence of BFLA, VSV-infected cells were photographed under a fluorescent microscope. HSV-exposed cultures were incubated for 5.5 h in the continued presence or absence of the drug and stained with X-gal 24 h later.
Conformational stability of receptor-bound sgD
In our Western blot analysis of the fate of cell-bound sgD at 37°C (Fig. 4A), a trypsin-sensitive signal was still detectable at the 10-min time point. We asked whether this material retained the ability to mediate gD-deficient virus entry. As outlined in Fig. 6A, we bound sgD to the cells at 4°C, washed the cells, raised the temperature to 37°C for 0–30 min, and then chilled the cells, added gD-deficient KΔUs3-8Z for a 2-h incubation at 4°C, washed the cells again, and brought the temperature back to 37°C for a 3-h infection period. The results (Fig. 6B) showed a gradual decline in infection with progressively longer incubation times at 37°C prior to virus binding. This gradual decrease appeared comparable to the rate of sgD dissociation from the cells observed under the same conditions (Fig. 4A), suggesting that the entry-mediating conformation of sgD is stable as long as the soluble molecule remains bound to the cells.
Fig. 6.
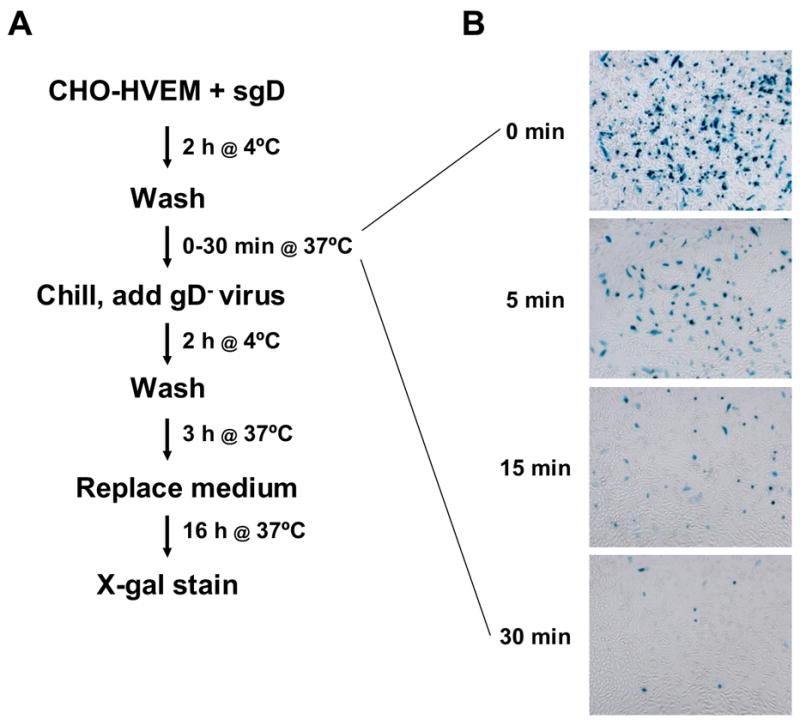
sgD retains its ability to mediate entry of gD-deficient KΔUs3-8Z while cell-associated. (A) Sequential incubation protocol. (B) Infected cells identified by X-gal staining.
sgD-mediated entry into glycosaminoglycan (GAG)-deficient cells
Since gD-deficient HSV lacks the ability to interact with gD receptors, we expected that sgD-mediated entry would be absolutely dependent on virus attachment to cell-surface GAGs. To test this prediction, we generated a GAG-deficient, nectin-1-expressing cell line (pgsA745-nectin-1) by stable transfection of a selectable nectin-1 expression plasmid (Takahashi et al., 1999) into CHO mutant cells (pgsA745) that are defective in GAG synthesis (Esko, Stewart, and Taylor, 1985). Characterization of the new line by flow-cytometry for nectin-1 expression and by X-gal staining for susceptibility to gD+ virus infection is presented in Fig. 7. These analyses showed that the fraction of cells expressing cell-surface nectin-1 was much greater for the pgsA745-nectin-1 line (Fig. 7B) than for the GAG-expressing CHO-nectin-1 line used in our earlier experiments (Fig. 7A). Furthermore, pgsA745-nectin-1 cells expressed more surface nectin-1 per cell than their CHO-nectin-1 counterparts. Nevertheless, the GAG+ CHO-nectin-1 line was more susceptible to HSV-1 infection, as illustrated using KHZ.1, a recombinant KOS virus that expresses lacZ (Mester, Pitha, and Glorioso, 1995; Rasty, Goins, and Glorioso, 1995) (Fig. 7C and 7D). These characteristics may suggest that GAGs lower the threshold level of nectin-1 required for cellular susceptibility to HSV-1, consistent with previous observations that GAGs increase the efficiency of infection (Banfield et al., 1995; Gruenheid et al., 1993; Herold et al., 1994; Herold et al., 1991; Laquerre et al., 1998; Shieh and Spear, 1994). In addition, high levels of nectin-1 may not translate into greater receptor availability for viral gD as an increased abundance of nectin-1 may be expected to favor the formation of cell junctions. Sequestration of nectin-1 in cell junctions is known to reduce virus infection (Yoon and Spear, 2002).
Fig. 7.
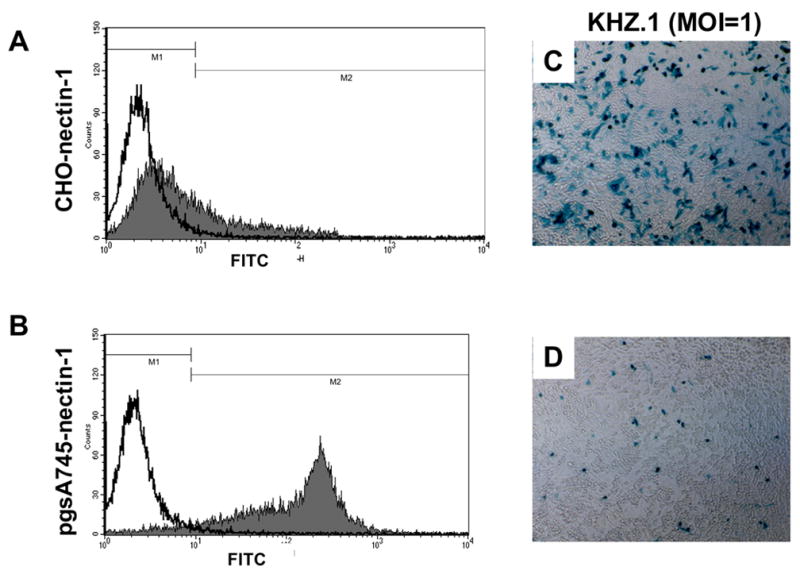
Nectin-1 expression on CHO-nectin-1 and pgsA745-nectin-1 cells, and susceptibility of both lines to lacZ-expressing HSV-1 KOS recombinant KHZ.1. (A) CHO-nectin-1 or (B) pgsA745-nectin-1 cells were incubated with CK41 monoclonal antibody for 1 h at 4°C, followed by incubation with anti-mouse FITC-conjugated antibody for 30 min. Cells were fixed and receptor levels on each cell line were quantitated by flow cytometry. (C) CHO-nectin-1 or (D) pgsA745-nectin-1 cells were infected with KHZ.1 (MOI=1) for 2 h at 37°C and X-gal stained at 16 h post-infection.
Remarkably, we observed that the pgsA745-nectin-1 line was permissive for sgD-mediated infection by gD-deficient KΔUs3-8Z despite the absence in this system of established mechanisms for virus attachment (Fig. 8C and D). Moreover, infection was typically more widespread on pgsA745-nectin-1 cells than on the GAG-expressing CHO-nectin-1 line (compare Figs. 8D and F). One possible explanation for these observations is the broader expression of nectin-1 on pgsA745-nectin-1 cells, suggesting that the rate-limiting step in sgD-mediated entry is not virus attachment, as in gD+ virus entry, but a nectin-1-dependent event, such as the establishment of an interaction between nectin-1-bound sgD and the virus. Alternatively, if the availability of nectin-1 on pgsA-nectin-1 cells is limited due to widespread formation of cell junctions, sgD may invade and disrupt these junctions more readily than complete gD+ virus particles, thus explaining the observation that nectin-1 levels correlate with productive entry in sgD-mediated, but not gD+ virus infection. Indeed, Krummenacher and colleagues have reported that a soluble gD similar to ours [gD(285t)] efficiently disrupts nectin-1-dependent aggregation of B78H1-C10 cells, whereas wild-type virus (HSV-1 KOS) or a soluble gD that has lower affinity for nectin-1 [gD(306t)] does not (Krummenacher et al., 2002).
Fig. 8.
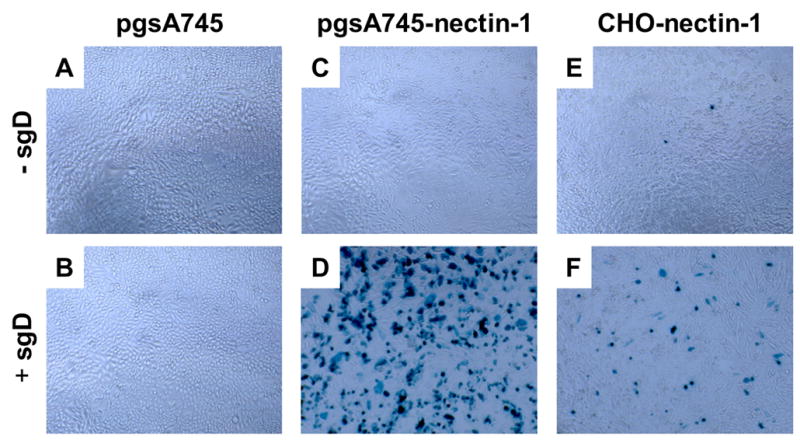
sgD mediated infection of receptor-bearing CHO-K1 cells by gD-deficient HSV-1 is not dependent on the presence of cell-surface GAGs. pgsA745, pgsA745-nectin-1 (GAG−), and CHO-nectin-1 (GAG+) cells were infected with gD-deficient KΔUs3-8Z in the presence (B, D, F) or absence (A, C, E) of sgD287. Virus entry was visualized 16 h later by X-gal staining.
Binding of gD-deficient virus to GAG-deficient cells
The unexpected finding of virus entry in the absence of GAGs and virion gD, along with the suggestion that virus attachment is not rate-limiting in sgD-mediated infection, indicated that HSV-1 may have a GAG- and gD-independent means of binding to cells. To examine this possibility, we incubated pgsA745-nectin-1 cells with gD-deficient KΔUs3-8Z for 2 h at 4ºC, washed the cells to remove unbound virus, and lysed the cells for detection of cell-associated viral protein. Western blot analysis showed that the lysate contained the major capsid protein VP5 (Fig. 9A, lane 3), supporting the existence of a previously unknown binding interaction between the virus and cells. The VP5 signal was not increased by the presence of sgD during the binding step (Fig. 9A, lane 2), indicating that sgD interaction with its receptor did not significantly contribute to virus binding at 4°C.
Fig. 9.
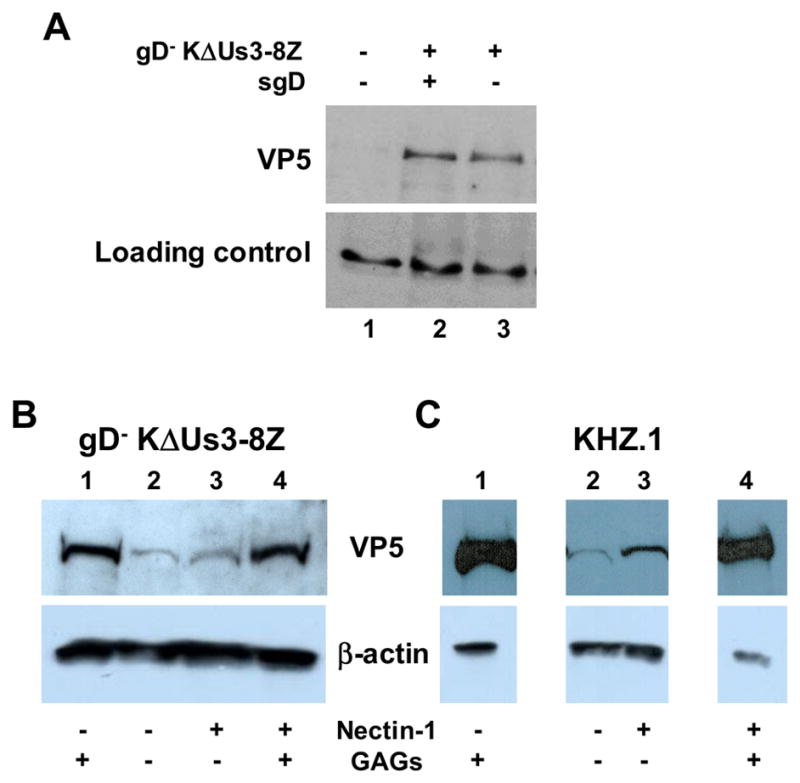
gD− KΔUs3-8Z attachment to GAG-deficient cells. (A) pgsA745-nectin-1 cells were mock treated (lane 1) or incubated with gD− KΔUs3-8Z and sgD (lane 2) or KΔUs3-8Z alone (lane 3) for 2 h at 4°C. Cells were washed, lysed, and analyzed by Western blot analysis using anti-HSV-1 VP5 antibody. A prominent, non-specific cellular band is shown as a loading control. (B, C) CHO-K1 (lanes 1), pgsA745 (lanes 2), pgsA745-nectin-1 (lanes 3), and CHO-nectin-1 cells (lanes 4) were incubated with gD− KΔUs3-8Z (B) or KHZ.1 (C) for 2 h at 4°C. Cells were washed to remove unbound virus, lysed, and the total cell lysate analyzed by Western blot analysis using anti-VP5 antibody. β-actin was visualized as a loading control.
To further investigate the distinct effects of GAGs on infection by gD+ virus (KHZ.1) and sgD-mediated infection by gD− virus, we compared the binding of both viruses to GAG+ and GAG− cells. As shown in Fig. 9B, GAGs enhanced the binding of gD− KΔUs3-8Z to both nectin-1-bearing cells (lanes 3, 4) and nectin-1-deficient cells (lanes 1, 2). Since sgD-mediated infection was not increased by the presence of GAGs (Fig. 8), these results supported the earlier suggestion that virus attachment is not a limiting step in sgD-mediated entry. As expected, nectin-1 expression had no detectable effect on the binding of gD− KΔUs3-8Z (Fig. 9B, lanes 2, 3), demonstrating that the interaction of this virus with pgsA745-nectin-1 cells did not involve nectin-1 and thus could not be attributed to residual envelope gD in the virus preparation or to previously unknown interactions of nectin-1 with other envelope components. The results for KHZ.1 showed GAG- and gD receptor-independent binding as well (Fig. 9C, lane 2), indicating that both gD− and gD+ virus possess a cell attachment mechanism that requires neither cell surface GAGs nor a gD-receptor interaction. As expected, KHZ.1 binding to both nectin-1-bearing and nectin-1-deficient cells was greatly augmented by GAGs (Fig. 9C, lanes 4 and 1). We used this enhancement to confirm the GAG- and gD-independent interaction. Thus, we found that the binding of KHZ.1 to CHO-K1 cells was reduced to the level observed for KHZ.1 binding to pgsA745 cells by pre-treatment of the cells with a mixture of heparinases I and III, whereas the binding of KHZ.1 to pgsA745 cells was resistant to the same pre-treatment (data not shown). This control experiment showed that KHZ.1 binding to pgsA745 cells was not due to residual GAGs on the GAG-deficient cells. KHZ.1 attachment to pgsA745 cells was detectably enhanced by nectin-1 (Fig. 9C, lane 3), providing direct evidence that nectin-1 can function as an attachment receptor for gD+ virus, at least in the absence of GAGs. Together, these results supported the suggestion that virus attachment is rate-limiting for gD+ virus infection, but not for sgD-mediated infection, and that virus attachment in sgD-mediated infection occurs by a novel binding interaction.
Discussion
HSV infection involves several stages consisting of attachment, initiation of fusion of the virus envelope with the cell-surface or endosomal membrane, and completion of the fusion reaction with release of de-enveloped particles into the cytoplasm. Virus attachment is mediated by the binding of viral envelope components gB and gC to glycosaminoglycan moieties (GAGs) at the cell surface with subsequent or simultaneous binding of viral envelope gD to one of several possible cell-surface co-receptors, including HVEM, nectin-1, or 3-OST-3 modified heparan sulphate (Banfield et al., 1995; Geraghty et al., 1998; Laquerre et al., 1998; Montgomery et al., 1996; Shieh et al., 1992; Shukla et al., 1999). Engagement of a gD receptor is thought to provide a signal to the fusion apparatus launching the fusion cascade. In this model, gD does not play a direct role in the fusion mechanism. Rather, the fusion apparatus most likely consists of the envelope components gB and gH since deletion of either glycoprotein blocks fusion, but not virus attachment (Cai, Gu, and Person, 1988; Cai et al., 1987; Forrester et al., 1992; Roop, Hutchinson, and Johnson, 1993). Indeed, recent evidence suggests that gH contains an active fusion domain (Gianni et al., 2005; Gianni, Menotti, and Campadelli-Fiume, 2005; Gianni et al., 2006). We sought to develop an experimental approach whereby gD engagement of its receptor provides an activating signal to the fusion machinery in trans. This approach was intended to clarify the order of events leading to fusion and allow examination of the role of GAGs in the absence of virus attachment to the cells through gD-receptor interactions.
We previously demonstrated that a soluble, gD-binding domain of nectin-1 was capable of efficient induction of entry provided that the virus was bound to the cells via GAGs. These experiments supported the conclusion that engagement of a gD receptor was sufficient to initiate envelope fusion, arguing against the need for a receptor-transduced signal through the target membrane (Kwon et al., 2006). In the current study, we asked whether soluble gD interacting with HVEM or nectin-1 expressed on the cell surface could promote entry of virions that lack gD. While this proved to be the case, others reported a similar observation (Cocchi et al., 2004), leading us to address aspects of the process that were not examined in the published study. These included the efficiency, receptor dependence, and receptor specificity of sgD-mediated infection. In addition, we sought to gain a better understanding of the steps involved in this mode of entry. Our results showed that sgD-mediated entry depends on sgD binding to HSV entry receptors with a specificity similar to that of virion gD binding. Although sgD-mediated entry was less efficient than virus entry mediated by virion gD, this reduction was most likely caused by the slower assembly of fusion-initiating complexes from three separate components (cells, gD− virus and sgD) rather than two (cells and gD+ virus). These observations supported the conclusion that sgD engagement of its receptor resulted in signaling to the fusion apparatus in a manner similar to that occurring in natural infection. However, two surprising observations were made during the course of this study. First, GAGs on the cell surface did not increase the efficiency of sgD-mediated infection of gD-deficient virus, indicating that entry occurred independently of virus binding to GAGs. Second, gD-deficient virus was capable of attaching to GAG-deficient cells, suggesting that virus binding occurred via an unknown receptor in a manner that involved neither gD nor GAGs.
In experiments to define the temporal order of the initial interactions required for sgD-mediated virus entry, we sought to determine whether sgD binding to HVEM or nectin-1 created a stable receptor complex for gD-deficient virus entry. We observed that sequential incubation of the cells with sgD and virus, separated by a washing step to remove free sgD, resulted in entry only when both steps were completed before the temperature was raised. Subsequent studies showed that sgD dissociated from the cells at 37ºC in the absence of abundant sgD in the medium, suggesting that virus added to sgD-coated cells at 37ºC did not diffuse rapidly enough to the cell surface to engage sgD prior to dissociation of the soluble molecule from its receptor. This interpretation implied that sgD-mediated infection requires the simultaneous presence of virus and sgD at the cell surface, as also proposed by Cocchi et al. (Cocchi et al., 2004), suggesting that the intact sgD-receptor complex serves as an entry receptor for gD-deficient HSV.
We considered the possibility that the dependence of sgD-mediated entry on the simultaneous presence of virus and sgD at the cell surface could additionally reflect instability of the entry-mediating conformation of receptor-bound sgD at 37°C, causing inactivation of the intact sgD-receptor complex; such instability could prevent productive interaction with newly added virus even before sgD dissociation occurred. Experiments to examine this possibility showed that incubation of cell-bound sgD for 15 min at 37°C in sgD- and virus-free medium did not abolish entry of subsequently added gD− virus. Although entry was reduced due to sgD dissociation, the remaining activity indicated that the active conformation of sgD is stable as long as the soluble molecule remains bound to its receptor. Thus we propose that receptor binding converts sgD to an active conformation at 37°C that persists until the complex dissociates or sgD interacts with virus to initiate the entry cascade.
The use of CHO cells in our experiments clearly demonstrated that cells other than those examined by Cocchi and colleagues are susceptible to sgD-mediated infection. Several reports have demonstrated that infection of CHO cells occurs by an endocytic pathway (Milne et al., 2005; Nicola, McEvoy, and Straus, 2003; Nicola and Straus, 2004), whereas the different lines used in the study of Cocchi et al. undergo infection by fusion at the cell surface (Cocchi et al., 2004). In our study, sgD was not stably bound to its cognate receptor and the addition of virus did not result in retention of sgD at the cell surface at 37°C. While the virus was able to utilize receptor-bound sgD for entry, infection did not appear to be accompanied by sgD internalization. These results indicated that receptor-bound sgD interacted transiently with the virus and did not function as a receptor for stable virus attachment to the cells. Furthermore, they suggested that sgD did not act intracellularly to mediate receptor-dependent virus release from endosomes. We observed no inhibition of productive infection by bafilomycin A1, suggesting that capsid transport to the nucleus did not require virus exposure to an acidic pH. In combination, these results are most consistent with the possibility that sgD-mediated productive infection does not occur by endocytosis, but instead takes place by envelope fusion with the cytoplasmic membrane. We have initiated additional studies to strengthen this conclusion and better understand apparent inconsistencies with previous reports. First, it is possible that our trypsin-protection assay was not sensitive enough to detect small amounts of internalized sgD that could be sufficient to trigger capsid exit from endosomes. Second, Nicola and colleagues showed that productive infecton of receptor-bearing CHO cells by gD+ HSV-1 is sensitive to various lysosomotropic agents (Nicola, McEvoy, and Straus, 2003). Our finding that CHO-cell infection by gD-complemented KΔUs3-8Z is resistant to a relatively high concentration of bafilomycin raised the question whether this mutant virus behaves in an atypical manner. However, preliminary results in our laboratory indicate that other HSV-1 recombinants may also be resistant to the drug (e.g. SHZ.1, Fig. 5). While Nicola and colleagues did not report the effect of bafilomycin on viral reporter gene expression or other indicators of productive infection, they did provide visual and biological evidence that this drug, considered one of the most specific lysosomotropic agents available, blocked the delivery of capsids to the nuclei of permissive CHO cells (Nicola, McEvoy, and Straus, 2003: Nicola, 2004 #2733). It is conceivable, although not predictable, that the outcome of their microscopy experiments was influenced by the cycloheximide treatment included during infection (Nicola, McEvoy, and Straus, 2003). It is also noteworthy that HSV infection of B78-C10 cells reportedly involves a bafilomycin-resistant endocytic pathway (Milne et al., 2005).
Analogous to normal HSV infection, it was anticipated that sgD-mediated infection would be enhanced by virus binding to cell-surface GAGs. In support of this prediction, we previously reported that GAGs are required for efficient HSV infection of gD receptor-deficient CHO cells mediated by a soluble form of nectin-1 or a soluble, bispecific adapter (Kwon et al., 2006; Nakano et al., 2005). Surprisingly, we observed that GAG-deficient CHO-K1 cells expressing nectin-1 were at least as susceptible to sgD-mediated infection as GAG-bearing CHO-nectin-1 cells. By comparing the amounts of gD-deficient virus bound to pgsA745-nectin-1 cells in the absence and presence of sgD, we obtained evidence for GAG- and sgD-independent virus attachment, suggesting the existence of a previously unknown attachment mechanism. Since the gD-deficient virus used in these experiments also lacked gG, gJ, gI, and gE, we could rule out a role for these glycoproteins in the proposed novel attachment process. We also found that a gD+ virus could bind to receptor-deficient pgsA745 cells and that this binding was resistant to pre-treatment of the cells with heparinases.
In a recent report, Bender and co-workers described heparin-resistant binding of a soluble gB ectodomain to cells, along with blocking of this interaction by virus-neutralizing anti-gB monoclonal antibodies and reduced infection of heparan sulfate-deficient cells in the presence of soluble gB (Bender et al., 2005). These results suggested a novel binding interaction between gB and target cells that plays a role in entry. It seems likely that this is the same interaction we report. While we show that both gD+ and gD− virus can bind to GAG-deficient cells that lack entry receptors (Fig. 9), we noticed a difference between the two viruses when their binding to GAG-deficient cells (Fig. 9B and 9C, lanes 2) was normalized to their binding to GAG+ cells (Fig. 9B and 9C, lanes 1). This observation raises the possibility that virion gD controls the novel binding interaction, blocking it until gD changes conformation by binding to a receptor. In this manner, gD might ensure orderly interaction of gB with GAGs and the predicted novel gB receptor, keeping the attachment and entry processes separate. Since gD-deficient virus is not infectious for GAG-deficient cells, it is nevertheless clear that the novel interaction is not sufficient for entry and that gD carries out functions besides controlling this interaction.
In conclusion, the combination of gD-deficient virus, GAG-deficient cells, and entry triggered by the addition of sgD should facilitate further characterization of the complex roles of gD and other envelope glycoproteins in the different pathways utilized by HSV to gain entry into cells. While it remains to be established whether sgD faithfully mimics the activities of envelope-embedded gD in these different situations, the availability of a trans-acting system that is operational across multiple types of host cells (Cocchi et al., 2004) (and this work) adds a new dimension to the study of the initial events leading to HSV infection.
Materials and Methods
Cells and viruses
Vero and 293T cells were obtained from the ATCC. VD60, a gD-complementing cell line (Ligas and Johnson, 1988), was a gift from Dr. David C. Johnson (Oregon Health Sciences University). Vero, VD60, and 293T cells were grown in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen, Carlsbad, CA) supplemented with 5% fetal bovine serum (FBS). Chinese hamster ovary cells (CHO-K1), and CHO-K1 cells stably expressing HVEM or nectin-1 were kindly provided by Dr. Patricia Spear (Northwestern University), and grown in F-12K medium (Invitrogen) supplemented with 10% FBS. PgsA745 cells (Esko, Stewart, and Taylor, 1985) were obtained from ATCC (CRL-2242). PgsA745-nectin-1 cells were derived from pgsA745 cells by transfection with a full-length nectin-1 cDNA expression plasmid (Takahashi et al., 1999) and cloning of hygromycin B-resistant cells.
KΔUs3-8Z is derived from HSV-1 KOS. This recombinant virus contains a lacZ reporter gene under transcriptional control of a human cytomegalovirus (HCMV) promoter in place of the unique short (US) region 3 through 8 genes of the HSV-1 genome, including the glycoprotein D (Us6) gene (Anderson et al., 2000). KΔUs3-8Z was propagated and titered on VD60 cells and passaged through Vero cells to obtain gD-deficient virions. KHZ.1 is a KOS-derived virus containing an HCMV-lacZ expression cassette in the thymidine kinase locus (Mester, Pitha, and Glorioso, 1995; Rasty, Goins, and Glorioso, 1995). SHZ.1, which is isogenic with KHZ.1 except for the deletion of both copies of the ICP4 gene (Mester, Pitha, and Glorioso, 1995), was propagated on ICP4-complementing E5 cells (DeLuca, McCarthy, and Schaffer, 1985a). VSV-EGFP-δG coated with VSV-G protein (Bergman et al., 2003) was kindly provided by Dr. Patricia Whitaker-Dowling (University of Pittsburgh).
Construction of sgD plasmids
The HSV-1 SacI fragment containing the gD promoter and coding sequence was cloned into pSP72 (Promega, Madison, WI) and the resulting plasmid designated pgDSac. The transmembrane region of gD was removed after amino acid 287 by digesting pgDSac with NarI and EcoRI and replacement of the excised fragment with annealed complementary oligonucleotides specifying 6 histidine residues followed by a stop codon (primer 1: 5′ CG CAC CAT CAC CAT CAC CAT TAG TTT AAA CGG GGG 3′; primer 2: 5′ A ATT CCC CCG TTT AAA CTA ATG GTG ATG GTG ATG GTG 3′). The truncated gD-His6 sequence was isolated from a selected recombinant by digestion with HindIII and EcoRI and placed under transcriptional control of the HCMV promoter in expression vector pcDNA3.1+ (Invitrogen). The resulting plasmid, psgD287, was confirmed by DNA sequencing.
Mutant sgD constructs were obtained by transfer of appropriate regions from pgDSac mutant constructs. The L25P mutation was introduced into pgDSac using the Gene Editor in vitro site-directed mutagenesis kit (Promega). The R222N,F223I double mutation was obtained by cloning a degenerate oligonucleotide between codons 221 and 230 of pgDSac, screening of recombinant plasmids for complementation of gD-deficient KΔUs3-8Z entry into CHO-HVEM, but not CHO-nectin-1 cells, and sequencing of selected constructs (W.A.S., J.B.C. and J.C.G., unpublished results). In order to transfer the L25P and R222N,F223I mutations from pgDSac to psgD287, an acceptor plasmid (psgD287) was first derived from psgD287 by deletion of an internal 216-bp BstZ17I-NaeI fragment. The desired mutations were then transferred from the pgDSac-based plasmids as HindIII-PpuMI fragments, restoring the deleted region in the acceptor plasmid. Recombinants were identified by digestion with FspI, which cuts in the deleted region and distinguishes between pgDSac and psgD287.
Purification of sgD proteins
293T cells were transfected with psgD287, psgD287(L25P) or psgD287(R222N,F223I) plasmids using LipofectAMINE-PLUS (Invitrogen). Twenty-four hours post-transfection, medium was replaced with serum-free medium. Two days later, supernatants were collected and loaded onto ProBond (Invitrogen) nickel-chelating columns for purification of the His-tagged proteins according to the manufacturer’s protocol. Proteins were concentrated using a Centricon YM-10 centrifugal filter device (Amicon, Bedford, MA) and dialyzed against PBS at 4°C overnight. The purity and molecular weights of each protein were estimated by Western blot analysis using polyclonal anti-gD antibody R7 (kindly provided by Drs. G.H. Cohen and R.J. Eisenberg, University of Pennsylvania) and an anti-rabbit HRP-conjugated secondary antibody (Santa Cruz Biotechnology, Santa Cruz, CA), or using an anti-His HRP-conjugated antibody (Invitrogen). Protein concentrations were determined by Bradford assay (Bio-Rad, Hercules, CA).
sgD287 mediated infection of CHO-K1-derived cells by KΔUs3-8Z
CHO-K1, CHO-HVEM, CHO-nectin-1, pgsA745 and pgsA745-nectin-1 cells were plated in 96-well plates at 5x104 cells/well. Sub-confluent monolayers were incubated with gD-deficient KΔUs3-8Z in the presence of 500 ng sgD287, sgD287(L25P), or sgD287(R222N,F223I) for various times at 4°C or 37°C. The monolayers were then washed, overlaid with F-12K/10% FBS medium, and incubated at 37°C. After 16 h, β-galactosidase activity was visualized by X-gal staining (Research Products International Corp., Mt. Prospect, IL). Over the course of this work, different preparations of gD-deficient KΔUs3-8Z virus were used. The activities of these preparations were standardized as transducing units/ml, measured as the number of blue cells per unit volume induced on CHO-HVEM cells in the presence of sgD.
Bafilomycin inhibition
2 × 104 CHO-HVEM or CHO-nectin-1 cells were seeded in a 96-well plate. The following day, the cells were washed once with medium and incubated for 30 min at 37°C with fresh medium alone or medium containing 400 nM bafilomycin A1 (Sigma, St. Louis, MO). The cells were then infected in the continued presence or absence of the drug with (i) gD-deficient KΔUs3-8Z with or without 500 ng sgD; (ii) gD-complemented (gD+) KΔUs3-8Z; (iii) SHZ.1; or (iv) VSV-GFP, the last three at an MOI of 5 determined on Vero cells. GFP expression in VSV-infected cultures was visualized at 4 h post-infection using a fluorescent microscope. HSV-infected cultures were washed at 5.5 h post-infection, incubated for an additional 24 h in fresh medium, and stained with X-gal.
Detection of cell-bound sgD by immunofluorescence
Subconfluent monolayers of CHO-HVEM or CHO-nectin-1 cells in a 48-well plate were incubated with 1 μg of sgD for 2 h at 4°C. The cells were then washed and either fixed immediately with 2% paraformaldehyde, or the temperature was raised to 37°C for 30 min before fixation. The fixed cells were washed and incubated with a pool of monoclonal anti-gD antibodies (Highlander et al., 1987) at a 1:5000 dilution for 1 h at room temperature (RT). The cells were then washed again, incubated for 1 h with a 1:500 diluted, Cy3-conjugated anti-mouse secondary antibody (Sigma), and visualized at 200x magnification under a Nikon Diaphot TMS fluorescent microscope (Nikon, Melville, NY). Photographs were taken with a Leica Microsystems AG DFC 300F digital camera (Leica, Deerfield, IL)
Flow cytometry
Mouse monoclonal antibody CK41 (Krummenacher et al., 2000) at a 1:250 dilution was used to identify surface nectin-1 in adherent cell cultures. Antibody binding and washes were performed at 4ºC in PBS containing 1% horse serum (Invitrogen). Cell bound CK41 was detected using FITC-conjugated anti-mouse antibody (Sigma) and analyzed on a FACSCalibur (Becton-Dickinson, San Diego, CA).
Detection of cell-bound virus by Western blot analysis
Virus (KHZ.1 at an MOI of 100 or gD-deficient KΔUs3-8Z at approximately 0.7 transducing units/cell) was adsorbed to confluent monolayers of CHO-HVEM, CHO-nectin-1, pgsA745, pgsA745-nectin-1, or CHO-K1 cells in a 24-well plate for 2 h at 4°C, either in the presence or absence of 1 μg sgD. The cells were then washed with cold buffer and lysed with 50 μl PARP buffer (6 M urea, 2% SDS, 10% glycerol, 62.5 mM Tris-HCl, pH 6.8, and 5% β-mercaptoethanol) (Pink et al., 2000). Each sample was sonicated for 20 sec with a Fisher Scientific 60 Sonic Dismembrator (Fisher Scientific, Hampton, NH). Unless indicated otherwise, half of each lysate was electrophoresed on a 10% SDS-polyacrylamide gel and the proteins were transferred to an Immobilon PVDF membrane (Millipore, Billerica, MA). The membrane was blocked for 1 h in 5% non-fat dry milk (NFDM) in PBS-Tween (PBST) at RT and reacted with NC1 polyclonal rabbit anti-VP5 antiserum (kindly provided by Dr. Fred Homa, University of Pittsburgh) at a 1:5000 dilution in 5% NFDM/PBST at RT for 1.5 h. The membrane was washed, incubated with HRP-conjugated goat anti-rabbit secondary antibody (Santa Cruz Biotechnology) at a 1:2000 dilution, and developed using an Amersham ECL kit (Amersham, Buckinghamshire, England). As a control for the amount of protein loaded in each lane, the membrane was re-probed with a mouse anti-actin monoclonal antibody (Chemicon International, Temecula, CA) at a 1:5000 dilution.
Trypsin sensitivity assay
Confluent monolayers of CHO-HVEM cells in a 24-well plate were incubated with 1 μg sgD/well for 2 h at 4°C in the presence or absence of gD-deficient KΔUs3-8Z (~0.7 tranducing units/cell). Cells were washed with ice cold PBS and lysed in PARP buffer either immediately or after 10–30 min incubation at 37°C in fresh medium; supernatants were collected at the 10 and 30 min time points prior to lysis and the protein contents precipitated at 4°C with 10% TCA. Duplicate samples were treated with 100 μg/ml TPCK-trypsin (Sigma) for 2.5 min prior to lysis. Trypsin was quenched with 200 μg/ml soybean trypsin inhibitor (Sigma) and the cells lysed in PARP buffer. Equal fractions of lysates and TCA-precipitated supernatants were electrophoresed on a 10% SDS-PAGE gel and sgD was detected by Western blot with R7 antibody, as described above.
Real-time quantitative PCR
Viral preparations were quantified for the immediate early gene ICP47. All assays were conducted in 50 μl PCR volumes containing viral samples (2μl), 200nM of each primer, 200nM probe, and 25 μl TaqMan Fast Universal PCR Master Mix (2x) (PE Applied Biosystems, Foster City, CA). Primer sequences for ICP47 (forward: 5′ CAC GAC ATG CTT TTC CCG A 3′; reverse: 5′ TTC CCG CAG GAG GAA CG 3′) were designed using the Primer Express program (PE Applied Biosystems). The TaqMan probe for detection of ICP47 (5′ CGC CGG TCG CCT CGA CGA 3′) was labeled with fluorescent reporter dye 6-carboxyfluorescein (6-FAM) at the 5′ end and the quencher dye carboxytetramethylrhodamine (TAMRA) at the 3′ end (PE Applied Biosystems). All PCR reactions were set up in a MicroAmp Optical 96 well Reaction Plate (PE Applied Biosystems). Amplification conditions were 2 min at 50°C and 10 min at 95°C for the first cycle, followed by 40 cycles of 95°C for 15 sec and 60°C for 1 min. The TaqMan probes were cleaved during the amplification of target sequence, generating fluorescent emission specific for FAM-labeled probes. All samples and standards were run at least in duplicate and each run contained both negative (reaction mix with no sample and a sample known to contain no HSV DNA) and positive controls (samples known to contain HSV sequence, i.e. an ICP47 plasmid and an HSV vector). Standard curves were generated using 10-fold serial dilutions of the ICP47 plasmid. Efficiency of ICP47 primer-probe set was confirmed previously by side-by-side TaqMan PCR runs with other HSV genes such as ICP27 present in other KOS-derived viruses that have also been quantified by negative staining electron microscope analysis (A. Ozuer, unpublished data). The emission data was collected in real-time from an ABIPRISM 7000 Sequence Detector System and analyzed using Sequence Detector Software (PE Applied Biosystems).
Calculation of relative entry efficiencies
Using the TaqMan standard curves described above, the particle concentrations of our virus stocks were determined as 8.39 ± 1.26 × 109/ml for gD− KΔUs3-8Z and 9.03 ± 1.91 × 1010/ml for gD-complemented KΔUs3-8Z. By counting transducing events (blue cells) induced by these preparations on CHO-HVEM cells, we derived the following transduction efficiencies (particles/blue cells): approximately 1.1 × 103 for the gD− stock in the presence of saturating amounts of sgD and 1.1 × 102 for the gD-complemented stock. Thus, the difference in the transduction efficiencies of the two stocks was approximately 10-fold.
Acknowledgments
We thank Drs. Claude Krummenacher, Roselyn Eisenberg, and Gary Cohen (University of Pennsylvania) for CK6 anti-nectin-1α and R-7 anti-HSV gD antibodies, Dr. Patricia Spear (Northwestern University) for CHO-nectin-1 and CHO-HVEM cells, Dr. David Johnson (Oregon Health Sciences University) for the VD60 gD-complementing cell line, Dr. Fred Homa (University of Pittsburgh) for the VP5 antibody, and Dr. Patricia Whitaker-Dowling (University of Pittsburgh) for VSV-GFP. We are also grateful to Drs. Fred Homa, Hiroaki Uchida, and Ora Weisz for helpful discussions. This work was funded by grants to J.C.G. from the NIH/NHLBI (“Program in Excellence in Gene Therapy”, grant HL66949), the NIH/NIGMS (GM34534), and the NIH/NIDDK (DK44935-08).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akula SM, Naranatt PP, Walia NS, Wang FZ, Fegley B, Chandran B. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J Virol. 2003;77(14):7978–90. doi: 10.1128/JVI.77.14.7978-7990.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson DB, Laquerre S, Ghosh K, Ghosh HP, Goins WF, Cohen JB, Glorioso JC. Pseudotyping of glycoprotein D-deficient herpes simplex virus type 1 with vesicular stomatitis virus glycoprotein G enables mutant virus attachment and entry. J Virol. 2000;74(5):2481–7. doi: 10.1128/jvi.74.5.2481-2487.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banfield BW, Leduc Y, Esford L, Schubert K, Tufaro F. Sequential isolation of proteoglycan synthesis mutants by using herpes simplex virus as a selective agent: evidence for a proteoglycan-independent virus entry pathway. J Virol. 1995;69(6):3290–3298. doi: 10.1128/jvi.69.6.3290-3298.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender FC, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J Virol. 2005;79(18):11588–97. doi: 10.1128/JVI.79.18.11588-11597.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergman I, Whitaker-Dowling P, Gao Y, Griffin JA, Watkins SC. Vesicular stomatitis virus expressing a chimeric Sindbis glycoprotein containing an Fc antibody binding domain targets to Her2/neu overexpressing breast cancer cells. Virology. 2003;316(2):337–47. doi: 10.1016/j.virol.2003.07.010. [DOI] [PubMed] [Google Scholar]
- Cai W, Gu B, Person S. Role of glycoprotein B of herpes simplex virus type 1 in viral entry and cell fusion. J Virol. 1988;62:2596–2604. doi: 10.1128/jvi.62.8.2596-2604.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai W, Person S, Warner S, Zhou J, Glorioso J. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J Virol. 1987;61:714–721. doi: 10.1128/jvi.61.3.714-721.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campadelli-Fiume G, Cocchi F, Menotti L, Lopez M. The novel receptors that mediate the entry of herpes simplex viruses and animal alphaherpesviruses into cells. Rev Med Virol. 2000;10(5):305–19. doi: 10.1002/1099-1654(200009/10)10:5<305::aid-rmv286>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- Carfi A, Gong H, Lou H, Willis SH, Cohen GH, Eisenberg RJ, Wiley DC. Crystallization and preliminary diffraction studies of the ectodomain of the envelope glycoprotein D from herpes simplex virus 1 alone and in complex with the ectodomain of the human receptor HveA. Acta Crystallogr D Biol Crystallogr. 2002;58(Pt 5):836–8. doi: 10.1107/s0907444902001270. [DOI] [PubMed] [Google Scholar]
- Carfi A, Willis SH, Whitbeck JC, Krummenacher C, Cohen GH, Eisenberg RJ, Wiley DC. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol Cell. 2001;8(1):169–79. doi: 10.1016/s1097-2765(01)00298-2. [DOI] [PubMed] [Google Scholar]
- Cocchi F, Fusco D, Menotti L, Gianni T, Eisenberg RJ, Cohen GH, Campadelli-Fiume G. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc Natl Acad Sci U S A. 2004;101(19):7445–50. doi: 10.1073/pnas.0401883101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Whitbeck JC, Zuo Y, Wiley DC, Cohen GH, Eisenberg RJ. Potential nectin-1 binding site on herpes simplex virus glycoprotein d. J Virol. 2005;79(2):1282–95. doi: 10.1128/JVI.79.2.1282-1295.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connolly SA, Landsburg DJ, Carfi A, Wiley DC, Cohen GH, Eisenberg RJ. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J Virol. 2003;77(14):8127–40. doi: 10.1128/JVI.77.14.8127-8140.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLuca NA, McCarthy AM, Schaffer PA. Isolation and characterization of deletion mutants of herpes simplex virus type 1 in the gene encoding immediate-early regulatory protein ICP4. J Virol. 1985;56:558–570. doi: 10.1128/jvi.56.2.558-570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esko JD, Stewart TE, Taylor WH. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc Natl Acad Sci U S A. 1985;82(10):3197–201. doi: 10.1073/pnas.82.10.3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester A, Farrell H, Wilkinson G, Kaye J, Davis-Poynter N, Minson T. Construction and properties of a mutant of herpes simplex virus type 1 with glycoprotein H coding sequences deleted. J Virol. 1992;66:341–348. doi: 10.1128/jvi.66.1.341-348.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusco D, Forghieri C, Campadelli-Fiume G. The pro-fusion domain of herpes simplex virus glycoprotein D (gD) interacts with the gD N terminus and is displaced by soluble forms of viral receptors. Proc Natl Acad Sci U S A. 2005;102(26):9323–8. doi: 10.1073/pnas.0503907102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science. 1998;280(5369):1618–20. doi: 10.1126/science.280.5369.1618. [DOI] [PubMed] [Google Scholar]
- Gianni T, Campadelli-Fiume G, Menotti L. Entry of herpes simplex virus mediated by chimeric forms of nectin1 retargeted to endosomes or to lipid rafts occurs through acidic endosomes. J Virol. 2004;78(22):12268–76. doi: 10.1128/JVI.78.22.12268-12276.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Martelli PL, Casadio R, Campadelli-Fiume G. The ectodomain of herpes simplex virus glycoprotein H contains a membrane alpha-helix with attributes of an internal fusion peptide, positionally conserved in the herpesviridae family. J Virol. 2005;79(5):2931–40. doi: 10.1128/JVI.79.5.2931-2940.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Menotti L, Campadelli-Fiume G. A heptad repeat in herpes simplex virus 1 gH, located downstream of the alpha-helix with attributes of a fusion peptide, is critical for virus entry and fusion. J Virol. 2005;79(11):7042–9. doi: 10.1128/JVI.79.11.7042-7049.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gianni T, Piccoli A, Bertucci C, Campadelli-Fiume G. Heptad repeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J Virol. 2006;80(5):2216–24. doi: 10.1128/JVI.80.5.2216-2224.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenheid S, Gatzke L, Meadows H, Tufaro F. Herpes simplex virus infection and propagation in a mouse L cell mutant lacking heparan sulfate proteoglycans. J Virol. 1993;67(1):93–100. doi: 10.1128/jvi.67.1.93-100.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold B, Visalli R, Susmarski N, Brandt C, Spear P. Glycoprotein C-independent binding of herpes simplex virus to cells requires cell surface heparan sulfate and glycoprotein B. J Gen Virol. 1994;75:1211–1222. doi: 10.1099/0022-1317-75-6-1211. [DOI] [PubMed] [Google Scholar]
- Herold BC, WuDunn D, Soltys N, Spear PG. Glycoprotein C of herpes simplex virus type 1 plays a principal role in the adsorption of virus to cells and in infectivity. J Virol. 1991;65:1090–1098. doi: 10.1128/jvi.65.3.1090-1098.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Highlander SL, Sutherland SL, Gage PJ, Johnson DC, Levine M, Glorioso JC. Neutralizing monoclonal antibodies specific for herpes simplex virus glycoprotein D inhibit virus penetration. J Virol. 1987;61:3356–3364. doi: 10.1128/jvi.61.11.3356-3364.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Ponce de Leon M, Whitbeck JC, Lou H, Cohen GH, Eisenberg RJ. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J Virol. 2000;74(23):10863–72. doi: 10.1128/jvi.74.23.10863-10872.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Baribaud I, Sanzo JF, Cohen GH, Eisenberg RJ. Effects of herpes simplex virus on structure and function of nectin-1/HveC. J Virol. 2002;76(5):2424–33. doi: 10.1128/jvi.76.5.2424-2433.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Nicola AV, Whitbeck JC, Lou H, Hou W, Lambris JD, Geraghty RJ, Spear PG, Cohen GH, Eisenberg RJ. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J Virol. 1998;72(9):7064–74. doi: 10.1128/jvi.72.9.7064-7074.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krummenacher C, Supekar VM, Whitbeck JC, Lazear E, Connolly SA, Eisenberg RJ, Cohen GH, Wiley DC, Carfi A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. Embo J. 2005;24(23):4144–53. doi: 10.1038/sj.emboj.7600875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H, Bai Q, Baek HJ, Felmet K, Burton EA, Goins WF, Cohen JB, Glorioso JC. Soluble V Domain of Nectin-1/HveC Enables Entry of Herpes Simplex Virus Type 1 (HSV-1) into HSV-Resistant Cells by Binding to Viral Glycoprotein D. J Virol. 2006;80(1):138–48. doi: 10.1128/JVI.80.1.138-148.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laquerre S, Argnani R, Anderson DB, Zucchini S, Manservigi R, Glorioso JC. Heparan sulfate proteoglycan binding by herpes simplex virus type 1 glycoproteins B and C, which differ in their contributions to virus attachment, penetration, and cell-to-cell spread. J Virol. 1998;72(7):6119–30. doi: 10.1128/jvi.72.7.6119-6130.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ligas M, Johnson D. A herpes simplex virus mutant in which glycoprotein D sequences are replaced by β-galactosidase sequences binds to but is unable to penetrate into cells. J Virol. 1988;62:1486–1494. doi: 10.1128/jvi.62.5.1486-1494.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez M, Cocchi F, Menotti L, Avitabile E, Dubreuil P, Campadelli-Fiume G. Nectin2alpha (PRR2alpha or HveB) and nectin2delta are low-efficiency mediators for entry of herpes simplex virus mutants carrying the Leu25Pro substitution in glycoprotein D. J Virol. 2000;74(3):1267–74. doi: 10.1128/jvi.74.3.1267-1274.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoj S, Jogger CR, Myscofski D, Yoon M, Spear PG. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc Natl Acad Sci U S A. 2004;101(34):12414–21. doi: 10.1073/pnas.0404211101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mester JC, Pitha PM, Glorioso JC. Antiviral activity of herpes simplex virus vectors expressing murine alpha 1-interferon. Gene Ther. 1995;2(3):187–96. [PubMed] [Google Scholar]
- Milne RS, Hanna SL, Rux AH, Willis SH, Cohen GH, Eisenberg RJ. Function of herpes simplex virus type 1 gD mutants with different receptor-binding affinities in virus entry and fusion. J Virol. 2003;77(16):8962–72. doi: 10.1128/JVI.77.16.8962-8972.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne RS, Nicola AV, Whitbeck JC, Eisenberg RJ, Cohen GH. Glycoprotein D receptor-dependent, low-pH-independent endocytic entry of herpes simplex virus type 1. J Virol. 2005;79(11):6655–63. doi: 10.1128/JVI.79.11.6655-6663.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery RI, Warner MS, Lum BJ, Spear PG. Herpes simplex virus 1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell. 1996;87(3):427–436. doi: 10.1016/s0092-8674(00)81363-x. [DOI] [PubMed] [Google Scholar]
- Nakano K, Asano R, Tsumoto K, Kwon H, Goins WF, Kumagai I, Cohen JB, Glorioso JC. Herpes simplex virus targeting to the EGF receptor by a gD-specific soluble bridging molecule. Mol Ther. 2005;11(4):617–26. doi: 10.1016/j.ymthe.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Nicola AV, McEvoy AM, Straus SE. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J Virol. 2003;77(9):5324–32. doi: 10.1128/JVI.77.9.5324-5332.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, Ponce de Leon M, Xu R, Hou W, Whitbeck JC, Krummenacher C, Montgomery RI, Spear PG, Eisenberg RJ, Cohen GH. Monoclonal antibodies to distinct sites on herpes simplex virus (HSV) glycoprotein D block HSV binding to HVEM. J Virol. 1998;72(5):3595–601. doi: 10.1128/jvi.72.5.3595-3601.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola AV, Straus SE. Cellular and viral requirements for rapid endocytic entry of herpes simplex virus. J Virol. 2004;78(14):7508–17. doi: 10.1128/JVI.78.14.7508-7517.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pink JJ, Planchon SM, Tagliarino C, Varnes ME, Siegel D, Boothman DA. NAD(P)H:Quinone oxidoreductase activity is the principal determinant of beta-lapachone cytotoxicity. J Biol Chem. 2000;275(8):5416–24. doi: 10.1074/jbc.275.8.5416. [DOI] [PubMed] [Google Scholar]
- Rasty S, Goins WF, Glorioso JC. Site-specific integration of multigenic shuttle plasmids into the herpes simplex virus type 1 (HSV-1) genome using a cell-free Cre-lox recombination system. In: Adolph K, editor. Methods in Molecular Genetics. Academic Press; 1995. [Google Scholar]
- Roop C, Hutchinson L, Johnson D. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J Virol. 1993;67(4):2285–2297. doi: 10.1128/jvi.67.4.2285-2297.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh M, Wudunn D, Montgomery R, Esko J, Spear P. Cell surface receptors for herpes simplex virus are heparan sulfate proteoglycans. J Cell Biol. 1992;116:1273–1281. doi: 10.1083/jcb.116.5.1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh M-T, Spear P. Herpes virus-induced cell fusion that is dependent on cell surface heparan sulfate on soluble heparin. J Virol. 1994;68:1224–1228. doi: 10.1128/jvi.68.2.1224-1228.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla D, Liu J, Blaiklock P, Shworak NW, Bai X, Esko JD, Cohen GH, Eisenberg RJ, Rosenberg RD, Spear PG. A novel role for 3-O-sulfated heparan sulfate in herpes simplex virus 1 entry. Cell. 1999;99(1):13–22. doi: 10.1016/s0092-8674(00)80058-6. [DOI] [PubMed] [Google Scholar]
- Spear PG. Herpes simplex virus: receptors and ligands for cell entry. Cell Microbiol. 2004;6(5):401–10. doi: 10.1111/j.1462-5822.2004.00389.x. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Nakanishi H, Miyahara M, Mandai K, Satoh K, Satoh A, Nishioka H, Aoki J, Nomoto A, Mizoguchi A, Takai Y. Nectin/PRR: an immunoglobulin-like cell adhesion molecule recruited to cadherin-based adherens junctions through interaction with Afadin, a PDZ domain-containing protein. J Cell Biol. 1999;145(3):539–49. doi: 10.1083/jcb.145.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner MS, Geraghty RJ, Martinez WM, Montgomery RI, Whitbeck JC, Xu R, Eisenberg RJ, Cohen GH, Spear PG. A cell surface protein with herpesvirus entry activity (HveB) confers susceptibility to infection by mutants of herpes simplex virus type 1, herpes simplex virus type 2, and pseudorabies virus. Virology. 1998;246(1):179–89. doi: 10.1006/viro.1998.9218. [DOI] [PubMed] [Google Scholar]
- Whitbeck JC, Peng C, Lou H, Xu R, Willis SH, Ponce de Leon M, Peng T, Nicola AV, Montgomery rI, Warner MS, Soulika AM, Spruce LA, Moore WT, Lambris JD, Spear PG, Cohen GH, Eisenberg RJ. Glycoprotein D of herpes simplex virus (HSV) binds directly to HVEM, a mediator of HSV entry. J Virol. 1997;71 doi: 10.1128/jvi.71.8.6083-6093.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Spear PG. Disruption of adherens junctions liberates nectin-1 to serve as receptor for herpes simplex virus and pseudorabies virus entry. J Virol. 2002;76(14):7203–8. doi: 10.1128/JVI.76.14.7203-7208.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon M, Zago AY, Shukla D, Spear PG. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J Virol. 2003;77(17):9221–31. doi: 10.1128/JVI.77.17.9221-9231.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zago A, Jogger CR, Spear PG. Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc Natl Acad Sci U S A. 2004;101(50):17498–503. doi: 10.1073/pnas.0408186101. [DOI] [PMC free article] [PubMed] [Google Scholar]