

Abstract
Susceptibility to most human diseases is polygenic, with complex interactions between functional polymorphisms of single genes governing disease incidence, phenotype, or both. In this context, the contribution of any discrete gene is generally modest for a single individual, but may confer substantial attributable risk on a population level. Environmental exposure can modify the effects of a polymorphism, either by providing a necessary substrate for development of human disease or because the effects of a given exposure modulate the effects of the gene. In several diseases, genetic polymorphisms have been shown to be context-dependent, i.e. the effects of a genetic variant are realized only in the setting of a relevant exposure. Since sarcoidosis susceptibility is dependent on both genetic and environmental modifiers, the study of gene-environment interactions may yield important pathogenetic information and will likely be crucial for uncovering the range of genetic susceptibility loci. However, the complexity of these relationships implies that investigations of gene-environment interactions will require the study of large cohorts with carefully-defined exposures and similar clinical phenotypes. A general principle is that the study of gene-environment interactions requires a sample size at least several-fold greater than for either factor alone. To date, the presence of environmental modifiers has been demonstrated for one sarcoidosis susceptibility locus, HLA-DQB1, in African-American families. This article reviews general considerations obtaining for the study of gene-environment interactions in sarcoidosis. It also describes the limited current understanding of the role of environmental influences on sarcoidosis susceptibility genes.
Introduction
Sarcoidosis is a systemic inflammatory syndrome of unknown etiology characterized by accumulation of immune effector cells in affected organs [1]. Noncaseating granulomas are the pathologic hallmark of the disease, and the clinical course is extremely heterogeneous. There are accumulating parallel data suggesting important roles for both genetic susceptibility (reviewed by DuBois) and specific transmissible environmental agents (reviewed by Crouser) (Table 1). A variety of investigators have reported important susceptibility and protective roles for genes mediating immune responses, especially for the human leukocyte antigen (HLA) genes located on Chromosome 6p [2-4]. The HLA genes govern the expression of the Type II major histocompatibility complex (MHC) on antigen presenting cells, which mediates antigen-specific responses to exogenous agents by presenting the relevant antigen to a cognate T-cell antigen receptor that is expressed on the surface of T-cells. These observations fit well with the current concepts of disease pathogenesis, reserving a central role for activation of antigen-specific oligoclonal CD4+ T-cells by MHC Class II-restricted antigen presenting cells, which then amplify immune mechanisms that lead to granuloma formation. At the same time, there are epidemiologic and experimental observations suggesting a causative role for several environmental exposures, likely in the form of respirable particles. These two avenues of investigation, genetic and environmental, support the general hypothesis that development of sarcoidosis depends on an appropriate exposure in a genetically susceptible host (Figure 1). Bridging the two approaches is a challenge for sarcoidosis research.
Table 1.
Summary of evidence for environmental and genetic etiologies
| Environmental |
| Case clustering |
| Transmission via solid organ or stem cell transplant |
| Case-control association studies (e.g. ACCESS) |
| Analogy to chronic beryllium disease |
| Molecular analysis (micro-organism nucleic acids or proteins) |
| Genetic |
| Familial aggregation |
| Ethnic susceptibility |
| Case-control association studies |
| Non-parametric studies (e.g. genome scans) |
Figure 1.
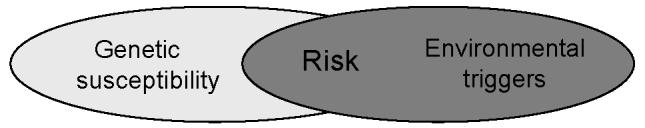
Proposed model for development of sarcoidosis. Individuals inherit a spectrum of genetic polymorphisms that may influence susceptibility to disease, but that are only realized in the context of a relevant exposure. Disease-associated susceptibility genes may influence position on this matrix differentially, depending on the function of the individual gene product, and on the influence of gene-gene interactions. The relevance of environmental exposures may be influenced by their duration, intensity, timing or context. A similar model might also describe disease phenotype.
It is clear that there is not one set of genetic susceptibility markers that is sufficient to explain all sarcoidosis cases. In this regard, numerous attempts to define disease susceptibility genes, usually by candidate gene approaches, although more recently using haplotype analysis, have yielded results that are poorly reproducible [5]. More recently, two whole genome scans have yielded differing results, possibly because two different populations were studied [6, 7]. Recent successful genetic analyses have relied on defining very clear phenotypes, such as Lofgren’s syndrome, to demonstrate reproducibility between populations [8, 9]. These findings illuminate the need for assiduous clinical phenotyping, careful study design and circumspection when extrapolating results between groups. Likewise, the influence of variable environmental exposures on the results of most genetic studies has not been tested.
Several exposures have been suggested to mediate disease risk, but there has also been poor reproducibility of the findings, leading to the hypothesis that a range of agents may mediate sarcoidosis, and that epidemiologic risk factors may vary between disparate populations. It is possible that the correct term is “sarcoidoses”, connoting a set of stereotyped immune responses to various agents that may display phenotypic differences and for which various pathogenetic mechanisms might obtain. Therefore, dissecting the interactions between genetic polymorphisms and environmental exposures will likely be highly relevant to advancing understanding of pathogenesis. However, several considerations present obstacles to these lines of investigation.
Gene-environment interactions
General considerations
Progress in genetic epidemiology and genotyping technology has yielded tremendous advances in recent years. Useful information on disease pathogenesis, genetic susceptibility profiling, and pharmacogenomics have been derived from these endeavors. In contrast, although it is widely recognized that many human diseases are influenced by both genes and environment, the study of gene-environment interactions is in its infancy. Many susceptibility genes do not have a substantial primary etiologic role but rather modulate an individual’s response to environmental cues—i.e. they function as response modifiers in an appropriate environmental context [10]. The variants in complex genetic diseases such as sarcoidosis are usually relatively common polymorphisms that might not result in overt recognizable phenotypes until the appropriate conditions are available.
Defined broadly, as either additive or multiplicative interactions, gene-environment interactions influence many familiar human diseases. Examples include breast cancer, asthma, venous thrombosis, and dyslipidemia [11-15]. That these conditions are influenced by both genes and environment implies a statistical, but not necessarily a biologic, interaction between the gene and the environmental factor. A statistical interaction implies that both the gene and the environmental factor independently contribute main effects to the risk of developing disease.
Statistical gene-environment relationships might be categorized by defining them as requisite vs. modifier and additive vs. multiplicative [16, 17]. All these relationships are likely to be present in sarcoidosis, a complex human disease with multiple genetic and environmental modifiers. A requisite relationship implies that the exposure (or gene) is a necessary causative agent of disease or phenotype among individuals with the susceptibility gene (or exposure); in a modifier relationship, the exposure or gene modifies the likelihood of disease susceptibility or phenotype but is not the triggering etiology (Fig 3). The additive versus multiplicative term refers to the mathematical effect of the relationship; additive modifiers are generally used to describe the effects of continuous variables, while multiplicative terms more often are used for stratified analyses [18]. Malignant melanoma is an example of a “requisite” gene-environment relationship: incidence is influenced by the degree of ultraviolet exposure in carriers of a mutation in a cell-cycle regulatory protein, CDKN2A [19]. Tobacco exposure and the risk of granulomatous lung diseases represent a susceptibility modifier. For example, the development of hypersensitivity pneumonitis due to avian antigens is more likely in pigeon breeders with specific HLA-DR and DQ haplotypes [20]. However, individuals who smoke tobacco are relatively protected from sensitization and this disease [21]. The phenotype of disease may also be altered by environmental or genetic modifiers. For example, tobacco smoking increases the risk of pulmonary metastases among breast cancer patients from 1.06-3.73 fold depending on the cumulative dose [22]. Statistical gene-environment relationships can generally be examined by conventional regression analysis and are not the focus of this review.
Figure 3.
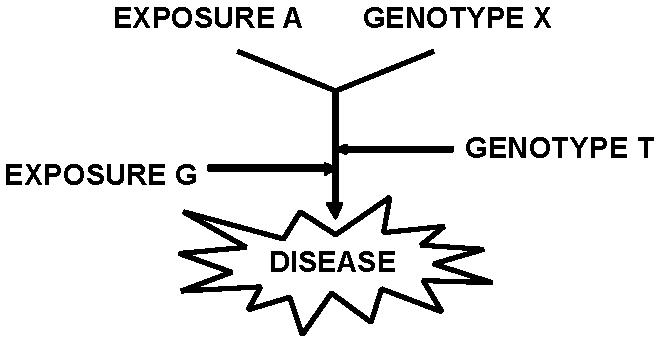
Possible gene-environment relationships. Exposure “A” and Genotype “X” represent a gene or environmental exposure requisite for development of the disease. Modifier genes (T) or exposures (G) also influence susceptibility or phenotypic determination.
In a more narrow sense, a gene-environment interaction implies a relationship that influences a disease phenotype more than would be expected from the additive or multiplicative combination of the genetic and environmental components (Figure 2d). In other words, a true interaction implies that there is a biologic relationship between the environmental exposure and the gene. A hypothetical example is provided in Table 2. To date, relatively few gene-environment interactions of this type have been described. An example of these types of gene-environment interactions is illustrated by the effect of endothelin-1 on systolic blood pressure. Tiret et al. studied the effect of a functional polymorphism that causes an amino acid change (Lys? Asn) at codon 198 [10]. The presence of any Asn allele was more strongly associated with systolic blood pressure at high body mass index, but the effect was absent or possibly reversed in thin individuals (similar to Figure 2d). Similarly, it has been suggested that the CD14 (LPS binding protein receptor) C159T promoter polymorphism may influence asthma risk, but in opposite directions depending on the level of environmental endotoxin exposure [13]. Uncovering these biologic gene-environment interactions for sarcoidosis will likely require larger sample sizes and non-traditional statistical approaches to succeed broadly. Fortunately, the general requirement for larger sample-sizes when evaluating interactions may be mitigated in special circumstances, such as when the gene or the exposure is very rare.
Figure 2.
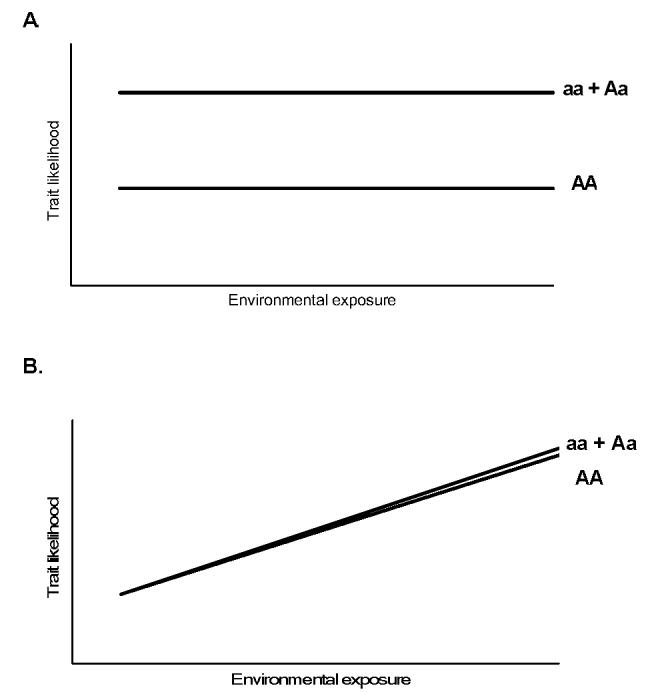
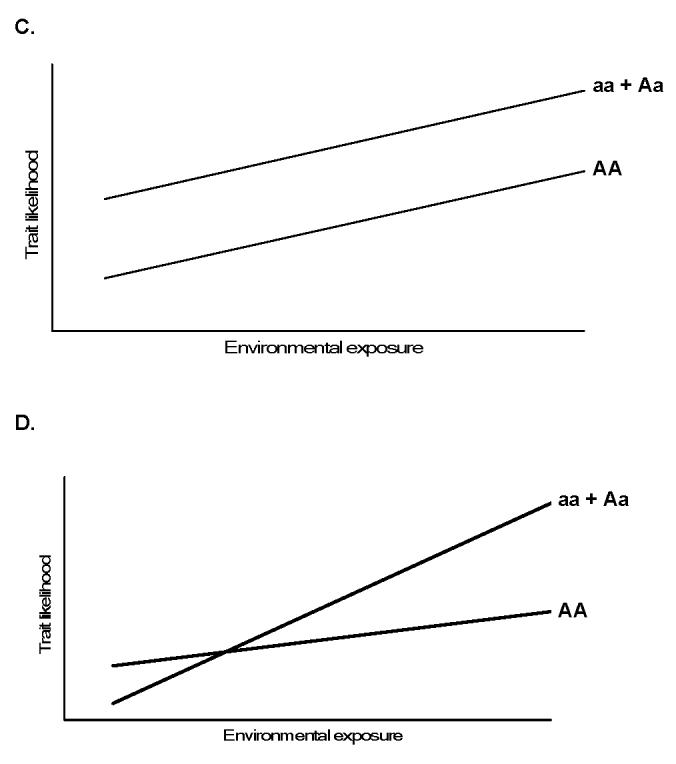
Theoretical gene-environment relationships. Panels A and B depict diseases influenced only by the gene or environmental variable, while Panel C demonstrates an additive relationship for a dominant mutation “a” with environmental exposure modeled as a continuous variable. Panel D, a true (biologic) gene-environment interaction, where the effect of the genotype is variably influenced by the exposure. Modified from [18].
Table 2.
Example of hypothetical calculation for gene-environment interaction for a dichotomous variable (e.g sarcoidosis risk). The example demonstrates a multiplicative relationship (no interaction) for the gene of interest with exposure to yttrium, but a suggestion of biologic gene-environment interaction for bohrium.
| Exposure | Genotype | OR for sarcoidosis | |
|---|---|---|---|
| Yttrium: | N | aa | 1.0 |
| Y | aa | 1.5 | |
| N | ab or bb | 4.0 | |
| Y | ab or bb | 6.0 | |
| Multiplicative relationship | |||
| Bohrium: | N | aa | 1.0 |
| Y | aa | 1.5 | |
| N | ab or bb | 4.0 | |
| Y | ab or bb | 8.0 | |
| Deviates from multiplicative relationship |
Strategies to study gene-environment interaction
Study designs
As a general principle, exposure can be assessed using cohort, case-control, or family-based designs. The strengths and weaknesses of these approaches for sarcoidosis are summarized in Table 3. Population cohort studies have the advantages of minimizing selection bias, ascertainment bias, and avoiding recall error when assessing exposures. However, in the case of uncommon diseases with variable age of onset and unclear disease latency, cohort studies are problematic [23]. To be effective in sarcoidosis research, very large groups would need to be recruited and periodically screened for disease. In addition, epidemiologic risk factors would need to be prospectively identified and measured at the inception of the study.
Table 3.
Consideration for design choices in studies of gene-environment interactions in sarcoidosis
| Design |
|||
|---|---|---|---|
| concern | family-based | case-control | cohort |
| Population stratification bias | nil | Potential problem; minimized by careful design or genomic control | Generally less than for case-control studies; minimized by careful design |
| Recall bias | Moderate to high | Moderate to high | Nil if exposure assessed at inception |
| Survival bias | Low | Low | Nil if DNA obtained at baseline |
| Latency bias | Possible | Low risk with good design | Low risk with good design |
| External validity | Least generalizable type | Confirmation required in several populations | Confirmation required in several populations |
| Sample size | Achievable | Achievable | Difficult except for common exposures or polymorphisms; would require pooling of multiple studies |
Case-control studies are employed more commonly since they are less complex. When using case-control designs, however, a major challenge is minimizing classification error for environmental exposures [24]. Unlike genetic studies, where accuracy for common genotyping methods typically exceeds 99%, accurate assessment of exposure in epidemiologic studies may be less than 80% [24]. A number of factors influence the ability to define exposure—including appreciation of threshold effects (e.g. the minimum dose to induce a gene), the pattern of dose-response curve (e.g. logarithmic vs linear), and the latency from exposure to a gene-driven response. Misclassification of individuals’ exposure, classification error, can be expressed as a correlation coefficient between an observer and a reference standard (r=0.8 implies a 20% error rate). The effects of even small classification errors are not trivial, as illustrated by the example in Figure 4. For a theoretical exposure conferring a relative risk of 2.0, an assessment tool that misclassified exposures in 20% of cases would estimate the risk as 1.7.
Figure 4.
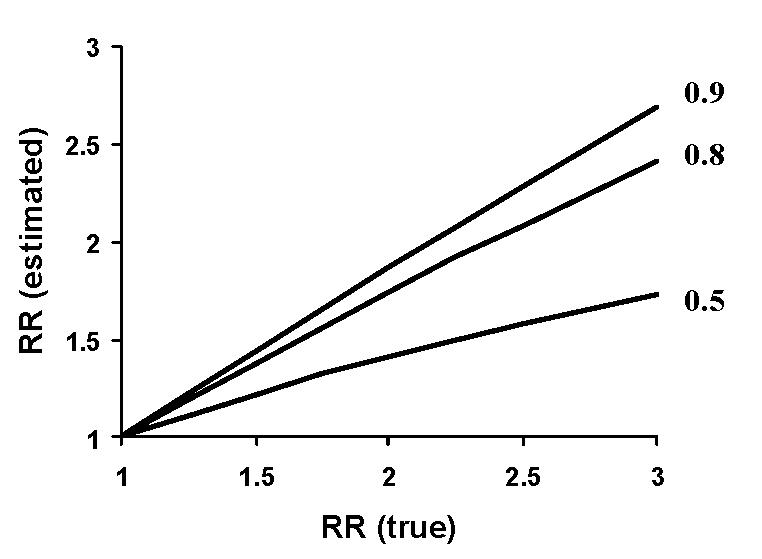
Influence of random misclassification error on relative risk estimates. The estimated relative risk for the outcome depends on the degree actual risk (RR true) and the accuracy of assessing the exposure of interest [24]. For a hypothetical observer correctly classifying exposure 80% of the time, a true relative risk of 2.5 would be estimated to be 2.0.
The implications of exposure misclassification errors include underestimates of actual risk, failure to find true associations due to inadequate sample size, publication and funding bias favoring gene-only studies (since genetic misclassification rates are substantially lower), and poor reproducibility of genetic studies in instances where environmental factors truly do interact with a polymorphism. For example, low ozone concentrations appear to be necessary to develop the effects of the -308 TNF promoter polymorphism on asthma prevalence, perhaps accounting for inconsistent results among prior studies of this gene [12]. Exposure misclassification errors are magnified when intercalated into calculations of interactions with genetic polymorphisms, since testing for interaction necessarily requires several fold-increases in sample size [23].
Family-based designs are an attractive option for studying gene-environment interactions. A major weakness of case-control studies, population stratification, can be avoided entirely by using family-based designs. Population stratification refers to segregation of a candidate susceptibility polymorphism differently in cases versus controls due to differences in ancestral origin. For example, consider a situation where African-American case subjects from Cleveland (predominantly West African origin) are compared with control subjects from Columbus, Ohio, which has a substantial Sudanese population. If the population frequency of genotype A in blacks of West African descent is 0.2, but it is 0.4 in individuals originating from Sudan, the putative sarcoidosis “susceptibility gene” may actually be a marker of the divergent genetic ancestry of the two populations. Population stratification may skew results for interactions more than for independent effects of genes or environment, especially when the interaction is strong [25].
In family-based designs, assuming consanguity, population stratification issues can be avoided entirely. When the carriage of an allele is associated more strongly with risk of a trait than predicted for Mendelian transmission among family members, then that locus may be potentially associated with the trait or in linkage disequilibrium with a gene that confers risk for the trait. Affected sibling pair methods are a subtype of family based study that rely on similar assumptions, and can be accomplished without obtaining parental DNA. Family-based designs may require a higher sample size than case-control studies for testing genetic main effects due to overmatching, but when looking for gene-environment interactions, overmatching is actually helpful, and reduces sample size requirements [26]. Family-based designs are also attractive in sarcoidosis because of the presence of familial sarcoidosis, with odds ratios for disease ranging from 3.8 to 5.8 among affected family members [27]. Weaknesses of family-based designs for studying gene-environment interactions include difficulties obtaining sibling or parental control DNA and exposure histories, questions of consanguity, and statistical power for exposures that are not frequent. Also, these studies will share the same recall biases that occur in case-control studies.
Modeling interactions
A general issue that complicates studies of gene-environment interaction is the need for much larger sample sizes, especially when multiple or less common genes or exposures are tested. One potential strategy may be to use parametric models incorporating pre-existing gene data to support or refute specific hypotheses [23]. Candidate pathways or exposures can then be investigated in detail, either as discrete variables or functional groups. For example, it may be useful to examine environmental factors in relation to genes for which marginal effects on disease have previously been estimated, or to use “case-only” designs [28]. This type of “two-step” analysis risks underestimating gene-environment interactions in cases where the interaction is strongly multiplicative, but may often be sufficient for additive relationships. More complex modeling may be required to study the effects of multiple allelic variables, such as haplotypes of linked single nucleotide polymorphisms (SNPs), or multiple environmental exposures. In these circumstances, stratified models may fail to provide enough observations for all but the most common gene-environment combinations [23].
A similar strategy, Mendelian randomization, is to use traditional genetic association studies to define polymorphisms that inform “internal phenotypes” lying on the causal pathway between the environmental exposure and the disease (Figure 5) [29, 30]. For example, Davey Smith and Ebrahim described the use of Mendelian randomization to assess the risk of developing neural tube defects in relation to maternal folate ingestion. This association might be difficult to prove in epidemiologic studies since multiple dietary and socioeconomic confounders would complicate the analysis. However, the importance of the environmental exposure (folate level) can be inferred by examining the trait risks associated with maternal methylene tetrahydrofolate reductase polymorphisms and the functional effects of the polymorphisms on folate levels [29]. This approach has the advantage of avoiding all the problems of accurate diet assessment and socioeconomic confounders, since the presence of the genetic polymorphisms is unlikely to be associated with these confounding variables. It may also be useful for choosing among competing environmental factors when there are multiple exposures; if a relevant gene can be linked to an intermediate phenotype, then the importance of a specific exposure can be inferred. However, caution will still need to be used, since population stratification, linkage disequilibrium, and inadequate sample size can all lead to incorrect results.
Figure 5.
Concept of Mendelian randomization. The relationship of the exposure to development of disease can be estimated by measuring the association of genotype with disease and on development of an internal phenotype (IP) that is directly related to the disease, thereby allowing an estimate of the relation between IP and the disease. If the gene can mediate other internal phenotypes, then no causality can be assigned to the exposure. Modified from [30].
A more recent example is for psoriasis, where this approach was used to suggest a role for non-metabolized tobacco products as etiologic risk factors. Epidemiologic studies of psoriasis had suggested a potential role for several environmental exposures, including tobacco consumption, alcohol, infections and medications, however the results were anecdotal and inconclusive. Richter-Hintz et al. conducted a case-control association study for a panel of polymorphisms among xenobiotic metabolizing enzymes [31]. The results suggested that a common (hypoactive) CYP1A1 variant conferred disease susceptibility. CYP1A1 is important for metabolism of tobacco compounds, and in subsequent analysis, the at-risk genotype was found to interact strongly with tobacco use, where it conferred risk more strongly in smokers (O.R. 3.6, p=0.01) than ex-smokers (O.R. 2.88, p=0.04) or never-smokers (O.R. 1.6, p=0.18) [32].
This approach can also be used to study the mechanism of gene-environment interactions. For example, Padyukov et al. investigated smoking status and several HLA-DR alleles, called shared epitope (SE) genes, in a population-based case-control study of rheumatoid arthritis [33]. A strong gene-environment interaction was found for seropositive disease, with 40% of incident seropositive cases attributable to the gene-environment interaction alone. The pathophysiology of the interaction was then elucidated by exploring the development of an internal phenotype for rheumatoid arthritis—the presence of antibodies to citrullinated proteins. Since citrullination of proteins increases the likelihood of binding with SE-containing HLA-DR residues, this process may render self proteins more immunogenic. In this situation, the shared epitope HLA genes can be associated with the development of antibodies to citrullinated proteins, an internal phenotype that is driven by tobacco exposure in a dose dependent manner [34]. The relationship between the susceptibility genes, development of anticitrulline antibodies, and the presence of smoking is powerful enough to explain all the findings of the initial association study [34].
For sarcoidosis, polymorphisms in several genes (NRAMP, TLR4, HLA, Vitamin D receptor) involved in the immune response to exogenous pathogens have been investigated [1, 35-37], suggesting that exposure to environmental factors capable of eliciting these pathways may be important in development of sarcoidosis. Polymorphisms in the vitamin D receptor are intriguing, and have been used in conjunction with an internal phenotype (Vitamin D deficiency) to suggest the presence of a gene-environment interaction between diet and VDR for risk of pulmonary tuberculosis among Gujaratis [38]. Vitamin D receptor activity has multiple potential roles in granulomatous disease, including activation of macrophages, inhibition of intracellular organisms, and regulation of interferon gamma activity. A strong negative gene-environment relationship has been demonstrated between one polymorphism and the presence of 25-Vitamin D deficiency on the risk of tuberculosis [38]. In this example, the odds ratios for the polymorphism, ff, was 3.6 (p=NS), and for 25-Vitamin D deficiency was 5.7. However, the interaction of both risk factors unexpectedly reduced the odds ratio to 3.1. The mechanism for his effect is unclear, but it illustrates at least two important aspects of studying gene-environment interactions. One is the possibility of dismissing the role of a gene (or biologic pathway) when environmental modifiers are not considered. Second, the relationships between genes and environment may be complex and unpredictable.
A limitation of using Mendelian randomization approaches for studying gene-environment interactions in sarcoidosis is the need to better define intermediate phenotypes that relate in a reasonably specific way to exposures of interest. Humoral or cell-mediated responses to environmental antigens, such as mycobacterial peptides [39], could be used as an internal phenotype to search for genetic polymorphisms that interact with exposure to those agents. However, if the effects of a given polymorphism result in a second (unmeasured) intermediate phenotype that can also influence the disease but is unrelated to environmental exposure, the results are difficult to interpret.
Gene-environment interactions in sarcoidosis
Evidence for main effects
Observational data suggesting familial clustering and ethnic susceptibility for sarcoidosis led early investigators to hypothesize that there might be a genetic predisposition to sarcoidosis. The inheritance pattern in familial sarcoidosis is complex, suggesting polygenic susceptibility [40]. More recently, genetic studies of sarcoidosis have demonstrated that specific gene polymorphisms are involved in both susceptibility to and phenotypic determination of the disease [41]. Until recently, however, all the genetic analyses in sarcoidosis were restricted to candidate gene approaches, most of which yielded either negative or weakly significant results that could not be replicated in other populations.
A model for gene-environment relationships in sarcoidosis may be inferred from another granulomatous lung disorder, chronic beryllium disease. Chronic beryllium disease is also CD4+ T-cell mediated, occurs in 2-8% of exposed workers, and is histologically identical to sarcoidosis [42]. In a seminal observation, Richeldi et al. suggested that carriage of a glutamic acid residue at position 69 of the HLA-DPB1 gene conferred susceptibility in exposed workers [43]. The substitution of glutamate for lysine in this position alters the putative antigen-binding pocket, favoring beryllium binding [44]. It appears that the major effect of the polymorphism is to mediate sensitization rather than disease, with other genetic loci likely necessary for progression to overt disease [45].
Similar to chronic beryllium disease, development of sarcoidosis requires both a susceptible host and a relevant exposure. Much genetic research to date has focused on the roles of genes governing the molecules necessary for antigen-specific T-cell stimulation. In this regard, numerous investigators have described disease associations with HLA polymorphisms, especially those coding for Class II molecules [3, 4]. Predictably, analysis of T-cell receptor variants has also revealed patterns associated with disease susceptibility or phenotype [46]. However, poor reproducibility has characterized these studies, with substantial inter-population variability due to population stratification, sampling error, or unsuspected differences in modifier genes/exposures between populations [2, 5]. Epidemiologic observations have suggested multiple potential etiologic triggers for sarcoidosis, with the possibility that susceptibility to any putative agent may be a function of the individuals’ genetic predisposition.
The most suggestive description of gene-environment interactions comes from the recent A Case Control Etiologic Study of Sarcoidosis (ACCESS) study. The ACCESS study evaluated epidemiologic risk factors among 706 newly diagnosed patients and a cohort of age-, race-, and sex-matched control subjects. Several occupational and environmental risk factors were identified, although their relationship to genetic factors is unknown [47]. The strongest negative interaction, for smoking (O.R. 0.65), was consistent with prior experience in chronic beryllium disease and hypersensitivity pneumonitis, where a gene and environment-driven immune response is negatively influenced by tobacco, a modifier exposure. The data also suggested the presence of gene-environment interactions by demonstrating that environmental factors influence the risk of familial sarcoidosis [27]. For siblings of affected Caucasians, the adjusted familial risk was 20.5 (C.I. 1.8-231.2). Shared environmental exposure, assessed with the surrogate parameter of time living together prior to the diagnosis of sarcoidosis, conferred independent risk in this population (O.R. 1.1, C.I. 1.0-1.3) [27]. However, these types of analyses do not allow consideration of whether the interaction is purely statistical (e.g. multiplicative) or operates on a biologic level.
Evidence for interaction
The first study to systematically examine gene-environment interactions employed family-based association analysis to identify HLA-DQB1 susceptibility alleles in 704 individuals from 225 African-American families [3]. Unclear consanguity, suspected by finding non-Mendelian segregation for a panel of unlinked genomic markers, resulted in exclusion of 8% of the families. The authors employed a strategy of low-resolution genotyping of DQB1 alleles. Two alleles, DQB1*02 (protective) and DQB1*06 (susceptibility) deviated significantly from expected Mendelian frequencies. Fine mapping confirmed the presence of one protective allele, DQB1*0201, with approximately 50% expected transmission to affected offspring, and one susceptibility allele, DQB1*0602, transmitted about 20% more often than expected to affected probands. Sixty-one environmental exposures were assessed with univariable testing in sib pairs, controlled for age and sex, and modeled for gene-environment interactions if univariate analysis suggested significance at the p<0.15 level. Testing for gene interaction with ten exposures revealed the presence of two risk factors, exposure to high humidity in the workplace for > 1 year and exposure to water damage, that modified the effect of the DQB1*0201 allele; none of the exposures interacted with DQB1*0602. For DQB1*0201- individuals, exposure to high humidity increased the chance of developing sarcoidosis (O.R. 1.62, 95% CI 1.03-2.52); in contradistinction, the exposure was actually protective in DQB1*0201+ individuals (O.R. 0.47, 95% CI 0.16-1.40). On the other hand, DQB1*0201, a protective allele when examined for the group, interacted with exposure to water damage in the opposite way. Carriers of the allele had higher chances of developing sarcoidosis with this exposure than did non-carriers (O.R. 4.44 vs 1.53). These data represent the strongest published observations to date documenting a potential true (biologic) gene-environment interaction for sarcoidosis.
Barriers to study of gene-environment interactions in sarcoidosis
A major limitation to study of gene-environment interactions in sarcoidosis is sample size. As a guideline, the sample size needed to evaluate the effect of a single interaction will be about four times the number needed for evaluation of either the gene or the exposure [48]. Inadequate sample size is already an important reason for poor reproducibility of genetic association studies [49]. In complex diseases like sarcoidosis, modest effect sizes for any given gene will also necessitate larger study cohorts, unless functional groups of genes can be linked together for analysis to create functional “haplotypes”. Sample sizes in the hundreds are likely to miss most effects; the majority of significant findings in such studies are likely to be false positive [50]. Factors that introduce measurement errors will tend to increase the sample size requirements, leading to the conclusion that well-done studies in this area will require very careful planning [51]. Restriction of such studies to strictly-defined clinical phenotypes and homogenous populations may improve the odds of finding relationships.
Racial admixture may be an additional important factor [52], either tending to mitigate the chances of finding significant relationships between genes and environment or leading to false positive findings through unsuspected population stratification. In African-Americans, admixture may be as high as 22% [53]. This issue is especially relevant in complex polygenic diseases where the effect size of any given gene is likely to be small. This is not a trivial issue, since variants of key genes such as HLA and TNF may be influenced in African-Americans depending on ancestral origin or variable admixture [54]. One potential strategy to deal with population admixture is to include a panel of putatively non-interacting genes (ancestry informative markers) that are known to exhibit different allelic frequencies in ancestral populations [55]. For sarcoidosis, family-based approaches may be the optimal approach to avoid these concerns.
For assessment of environmental exposures, study design issues will include consideration of the effects of the intensity, timing and duration of the putative environmental exposure. Most environmental exposures will require characterization as dichotomous variables, although this may not be satisfactory from a biologic viewpoint. In addition, recall bias is a substantial limitation for case-control or family-based studies. Use of a population-based cohort design reduces this risk substantially, but is not practical for sarcoidosis because of the low prevalence of the disease and ascertainment biases. Recall bias is especially an issue if development of the disease alters the individuals’ ascertainment or reporting of an exposure. For example, individuals with newly diagnosed pulmonary sarcoidosis commonly arrive at the clinic with a list of occupational agents and a history of noticing “mold” in the basement. Very careful instrument construction may help alleviate this issue. Another potential solution for some exposures is the use of serum biomarkers, such as cotinine for tobacco exposure or antibodies to mycobacterial catalase for tuberculosis [39].
A final issue related to environmental exposure is the rarity of some potentially important exposures. For example in ACCESS, sample size requirements dictated enrollment of 720 cases and matched controls to attain 80% power to discover a 2.0 fold relative risk for exposures present in at least 5% of the population [47]. However, all but one of the environmental factors associated with disease risk was present in at least 10% of the controls, and approximately half of the a priori hypotheses could not be tested at the 90% confidence level due to sample size limitations. Several factors that had previously been associated with development of sarcoidosis (naval service, fire-fighting) could not be investigated. The chance of detecting a true association between environment and disease is thereby lessened, compared to associations due to widespread environmental exposures.
Conclusion
The study of gene-environment interaction in sarcoidosis is nascent but holds the promise of dramatically improving our understanding of sarcoidosis. A number of benefits can be envisioned. Specific pathophysiologic mechanisms will be illuminated, including some that are not obvious until one considers the interaction of genes and environment. Specific combinations of genetic susceptibility markers may be linked to individual antigens, allowing better disease characterization and risk management of populations. It may become evident that sarcoidosis phenotype is predictable, depending on all three components of the “trimolecular complex”—the HLA molecule, the T-cell receptor, and the specific antigen [56]. In the next several decades, when DNA typing is widespread, individuals with genetic susceptibility may receive counseling about avoidance of specific environmental risks [57]. These advances will require the combined efforts of geneticists, biologists, experts in exposure characterization, and epidemiologists and will be facilitated by improvements in statistical modeling of complex interactions. Studies of genetics and epidemiology have yielded important advances in sarcoidosis; the study of gene-environment interactions promises to be even more challenging, yet ultimately it should lead to insights that can barely be imagined now.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Hunninghake GW, et al. ATS/ERS/WASOG statement on sarcoidosis. American Thoracic Society/European Respiratory Society/World Association of Sarcoidosis and other Granulomatous Disorders. Sarcoidosis Vasc Diffuse Lung Dis. 1999;16(2):149–73. [PubMed] [Google Scholar]
- 2.Martinetti M, et al. “The sarcoidosis map”: a joint survey of clinical and immunogenetic findings in two European countries. Am J Respir Crit Care Med. 1995;152(2):557–64. doi: 10.1164/ajrccm.152.2.7633707. [DOI] [PubMed] [Google Scholar]
- 3.Iannuzzi MC, et al. Sarcoidosis Susceptibility and Resistance HLA-DQB1 Alleles in African Americans. Am J Respir Crit Care Med. 2003;167(9):1225–31. doi: 10.1164/rccm.200209-1097OC. [DOI] [PubMed] [Google Scholar]
- 4.Rossman MD, et al. HLA-DRB1*1101: a significant risk factor for sarcoidosis in blacks and whites. Am J Hum Genet. 2003;73(4):720–35. doi: 10.1086/378097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rybicki BA, et al. Sarcoidosis and granuloma genes: a family-based study in African-Americans. Eur Respir J. 2004;24(2):251–7. doi: 10.1183/09031936.04.00005904. [DOI] [PubMed] [Google Scholar]
- 6.Iannuzzi MC, et al. Genome-wide search for sarcoidosis susceptibility genes in African Americans. Genes Immun. 2005;6(6):509–18. doi: 10.1038/sj.gene.6364235. [DOI] [PubMed] [Google Scholar]
- 7.Schurmann M, et al. Results from a genome-wide search for predisposing genes in sarcoidosis. Am J Respir Crit Care Med. 2001;164(5):840–6. doi: 10.1164/ajrccm.164.5.2007056. [DOI] [PubMed] [Google Scholar]
- 8.Spagnolo P, et al. C-C chemokine receptor 5 gene variants in relation to lung disease in sarcoidosis. Am J Respir Crit Care Med. 2005;172(6):721–8. doi: 10.1164/rccm.200412-1707OC. [DOI] [PubMed] [Google Scholar]
- 9.Berlin M, et al. HLA-DR predicts the prognosis in Scandinavian patients with pulmonary sarcoidosis. Am J Respir Crit Care Med. 1997;156(5):1601–5. doi: 10.1164/ajrccm.156.5.9704069. [DOI] [PubMed] [Google Scholar]
- 10.Tiret L. Gene-environment interaction: a central concept in multifactorial diseases. Proc Nutr Soc. 2002;61(4):457–63. doi: 10.1079/pns2002178. [DOI] [PubMed] [Google Scholar]
- 11.Brandt B, et al. Modification of breast cancer risk in young women by a polymorphic sequence in the egfr gene. Cancer Res. 2004;64(1):7–12. doi: 10.1158/0008-5472.can-03-2623. [DOI] [PubMed] [Google Scholar]
- 12.Li YF, et al. Associations of Tumor Necrosis Factor G-308A with Childhood Asthma and Wheezing. Am J Respir Crit Care Med. 2006;173(9):970–6. doi: 10.1164/rccm.200508-1256OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Martinez FD. Gene-environment interactions in asthma and allergies: a new paradigm to understand disease causation. Immunol Allergy Clin North Am. 2005;25(4):709–21. doi: 10.1016/j.iac.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 14.van Boven HH, et al. Gene-gene and gene-environment interactions determine risk of thrombosis in families with inherited antithrombin deficiency. Blood. 1999;94(8):2590–4. [PubMed] [Google Scholar]
- 15.Fumeron F, et al. Alcohol intake modulates the effect of a polymorphism of the cholesteryl ester transfer protein gene on plasma high density lipoprotein and the risk of myocardial infarction. J Clin Invest. 1995;96(3):1664–71. doi: 10.1172/JCI118207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mucci LA, et al. The role of gene-environment interaction in the aetiology of human cancer: examples from cancers of the large bowel, lung and breast. J Intern Med. 2001;249(6):477–93. doi: 10.1046/j.1365-2796.2001.00839.x. [DOI] [PubMed] [Google Scholar]
- 17.Grigorenko EL.The inherent complexities of gene-environment interactions J Gerontol B Psychol Sci Soc Sci 200560 Spec No 1:53–64. [DOI] [PubMed] [Google Scholar]
- 18.Talmud PJ. How to identify gene-environment interactions in a multifactorial disease: CHD as an example. Proc Nutr Soc. 2004;63(1):5–10. doi: 10.1079/PNS2003311. [DOI] [PubMed] [Google Scholar]
- 19.Hayward NK. Genetics of melanoma predisposition. Oncogene. 2003;22(20):3053–62. doi: 10.1038/sj.onc.1206445. [DOI] [PubMed] [Google Scholar]
- 20.Camarena A, et al. Major histocompatibility complex and tumor necrosis factor-alpha polymorphisms in pigeon breeder′s disease. Am J Respir Crit Care Med. 2001;163(7):1528–33. doi: 10.1164/ajrccm.163.7.2004023. [DOI] [PubMed] [Google Scholar]
- 21.McSharry C, Banham SW, Boyd G. Effect of cigarette smoking on the antibody response to inhaled antigens and the prevalence of extrinsic allergic alveolitis among pigeon breeders. Clin Allergy. 1985;15(5):487–94. doi: 10.1111/j.1365-2222.1985.tb02299.x. [DOI] [PubMed] [Google Scholar]
- 22.Scanlon EF, et al. Influence of smoking on the development of lung metastases from breast cancer. Cancer. 1995;75(11):2693–9. doi: 10.1002/1097-0142(19950601)75:11<2693::aid-cncr2820751109>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 23.Kraft P, Hunter D. Integrating epidemiology and genetic association: the challenge of gene-environment interaction. Philos Trans R Soc Lond B Biol Sci. 2005;360(1460):1609–16. doi: 10.1098/rstb.2005.1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vineis P. A self-fulfilling prophecy: are we underestimating the role of the environment in gene-environment interaction research? Int J Epidemiol. 2004;33(5):945–6. doi: 10.1093/ije/dyh277. [DOI] [PubMed] [Google Scholar]
- 25.Wang Y, Localio R, Rebbeck TR. Evaluating bias due to population stratification in epidemiologic studies of gene-gene or gene-environment interactions. Cancer Epidemiol Biomarkers Prev. 2006;15(1):124–32. doi: 10.1158/1055-9965.EPI-05-0304. [DOI] [PubMed] [Google Scholar]
- 26.Kauffmann F. Post-genome respiratory epidemiology: a multidisciplinary challenge. Eur Respir J. 2004;24(3):471–80. doi: 10.1183/09031936.04.00076803. [DOI] [PubMed] [Google Scholar]
- 27.Rybicki BA, et al. Familial aggregation of sarcoidosis. A case-control etiologic study of sarcoidosis (ACCESS) Am J Respir Crit Care Med. 2001;164(11):2085–91. doi: 10.1164/ajrccm.164.11.2106001. [DOI] [PubMed] [Google Scholar]
- 28.Khoury MJ, Flanders WD. Nontraditional epidemiologic approaches in the analysis of gene-environment interaction: case-control studies with no controls! Am J Epidemiol. 1996;144(3):207–13. doi: 10.1093/oxfordjournals.aje.a008915. [DOI] [PubMed] [Google Scholar]
- 29.Davey Smith G, Ebrahim S. ′Mendelian randomization′: can genetic epidemiology contribute to understanding environmental determinants of disease? Int J Epidemiol. 2003;32(1):1–22. doi: 10.1093/ije/dyg070. [DOI] [PubMed] [Google Scholar]
- 30.Thomas DC, Conti DV. Commentary: the concept of ′Mendelian Randomization′. Int J Epidemiol. 2004;33(1):21–5. doi: 10.1093/ije/dyh048. [DOI] [PubMed] [Google Scholar]
- 31.Richter-Hintz D, et al. Allelic variants of drug metabolizing enzymes as risk factors in psoriasis. J Invest Dermatol. 2003;120(5):765–70. doi: 10.1046/j.1523-1747.2003.12124.x. [DOI] [PubMed] [Google Scholar]
- 32.Kramer U, Esser C. Cigarette smoking, metabolic gene polymorphism, and psoriasis. J Invest Dermatol. 2006;126(3):693–4. doi: 10.1038/sj.jid.5700161. author reply 695.
- 33.Padyukov L, et al. A gene-environment interaction between smoking and shared epitope genes in HLA-DR provides a high risk of seropositive rheumatoid arthritis. Arthritis Rheum. 2004;50(10):3085–92. doi: 10.1002/art.20553. [DOI] [PubMed] [Google Scholar]
- 34.Klareskog L, et al. A new model for an etiology of rheumatoid arthritis: smoking may trigger HLA-DR (shared epitope)-restricted immune reactions to autoantigens modified by citrullination. Arthritis Rheum. 2006;54(1):38–46. doi: 10.1002/art.21575. [DOI] [PubMed] [Google Scholar]
- 35.Dubaniewicz A, et al. Association between SLC11A1 (formerly NRAMP1) and the risk of sarcoidosis in Poland. Eur J Hum Genet. 2005;13(7):829–34. doi: 10.1038/sj.ejhg.5201370. [DOI] [PubMed] [Google Scholar]
- 36.Pabst S, et al. Toll-like receptor (TLR) 4 polymorphisms are associated with a chronic course of sarcoidosis. Clin Exp Immunol. 2006;143(3):420–6. doi: 10.1111/j.1365-2249.2006.03008.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niimi T, et al. Vitamin D receptor gene polymorphism in patients with sarcoidosis. Am J Respir Crit Care Med. 1999;160(4):1107–9. doi: 10.1164/ajrccm.160.4.9811096. [DOI] [PubMed] [Google Scholar]
- 38.Wilkinson RJ, et al. Influence of vitamin D deficiency and vitamin D receptor polymorphisms on tuberculosis among Gujarati Asians in west London: a case-control study. Lancet. 2000;355(9204):618–21. doi: 10.1016/S0140-6736(99)02301-6. [DOI] [PubMed] [Google Scholar]
- 39.Song Z, et al. Mycobacterial catalase-peroxidase is a tissue antigen and target of the adaptive immune response in systemic sarcoidosis. J Exp Med. 2005;201(5):755–67. doi: 10.1084/jem.20040429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rybicki BA, et al. Heterogeneity of familial risk in sarcoidosis. Genet Epidemiol. 1996;13(1):23–33. doi: 10.1002/(SICI)1098-2272(1996)13:1<23::AID-GEPI3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 41.Rybicki BA, et al. Epidemiology, demographics, and genetics of sarcoidosis. Semin Respir Infect. 1998;13(3):166–73. [PubMed] [Google Scholar]
- 42.Culver DA, Dweik RA. Chronic beryllium disease. Clin Pulm Med. 2003;10:72–79. [Google Scholar]
- 43.Richeldi L, Sorrentino R, Saltini C. HLA-DPB1 glutamate 69: a genetic marker of beryllium disease. Science. 1993;262(5131):242–4. doi: 10.1126/science.8105536. [DOI] [PubMed] [Google Scholar]
- 44.Amicosante M, et al. Beryllium binding to HLA-DP molecule carrying the marker of susceptibility to berylliosis glutamate beta 69. Hum Immunol. 2001;62(7):686–93. doi: 10.1016/s0198-8859(01)00261-0. [DOI] [PubMed] [Google Scholar]
- 45.Maier LA, et al. Influence of MHC class II in susceptibility to beryllium sensitization and chronic beryllium disease. J Immunol. 2003;171(12):6910–8. doi: 10.4049/jimmunol.171.12.6910. [DOI] [PubMed] [Google Scholar]
- 46.Grunewald J, et al. Lung restricted T cell receptor AV2S3+ CD4+ T cell expansions in sarcoidosis patients with a shared HLA-DRbeta chain conformation. Thorax. 2002;57(4):348–52. doi: 10.1136/thorax.57.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newman LS, et al. A case control etiologic study of sarcoidosis: environmental and occupational risk factors. Am J Respir Crit Care Med. 2004;170(12):1324–30. doi: 10.1164/rccm.200402-249OC. [DOI] [PubMed] [Google Scholar]
- 48.Smith PG, Day NE. The design of case-control studies: the influence of confounding and interaction effects. Int J Epidemiol. 1984;13(3):356–65. doi: 10.1093/ije/13.3.356. [DOI] [PubMed] [Google Scholar]
- 49.Hirschhorn JN, et al. A comprehensive review of genetic association studies. Genet Med. 2002;4(2):45–61. doi: 10.1097/00125817-200203000-00002. [DOI] [PubMed] [Google Scholar]
- 50.Garcia-Closas M, Lubin JH. Power and sample size calculations in case-control studies of gene-environment interactions: comments on different approaches. Am J Epidemiol. 1999;149(8):689–92. doi: 10.1093/oxfordjournals.aje.a009876. [DOI] [PubMed] [Google Scholar]
- 51.Garcia-Closas M, Rothman N, Lubin J. Misclassification in case-control studies of gene-environment interactions: assessment of bias and sample size. Cancer Epidemiol Biomarkers Prev. 1999;8(12):1043–50. [PubMed] [Google Scholar]
- 52.Barnes KC. Genetic epidemiology of health disparities in allergy and clinical immunology. J Allergy Clin Immunol. 2006;117(2):243–54. doi: 10.1016/j.jaci.2005.11.030. quiz 255-6.
- 53.Nickel RG, et al. Determination of Duffy genotypes in three populations of African descent using PCR and sequence-specific oligonucleotides. Hum Immunol. 1999;60(8):738–42. doi: 10.1016/s0198-8859(99)00039-7. [DOI] [PubMed] [Google Scholar]
- 54.Kuffner T, et al. HLA class II and TNF genes in African Americans from the Southeastern United States: regional differences in allele frequencies. Hum Immunol. 2003;64(6):639–47. doi: 10.1016/s0198-8859(03)00056-9. [DOI] [PubMed] [Google Scholar]
- 55.Bonilla C, et al. Admixture in the Hispanics of the San Luis Valley, Colorado, and its implications for complex trait gene mapping. Ann Hum Genet. 2004;68(Pt 2):139–53. doi: 10.1046/j.1529-8817.2003.00084.x. [DOI] [PubMed] [Google Scholar]
- 56.Moller DR, Chen ES. Genetic basis of remitting sarcoidosis: triumph of the trimolecular complex? Am J Respir Cell Mol Biol. 2002;27(4):391–5. doi: 10.1165/rcmb.2002-0164PS. [DOI] [PubMed] [Google Scholar]
- 57.Rybicki BA. Genetic epidemiological approaches to the study of lung disease. Semin Respir Crit Care Med. 2003;24(2):137–50. doi: 10.1055/s-2003-39029. [DOI] [PubMed] [Google Scholar]