

Abstract
Mutations in the parkin gene cause autosomal recessive familial Parkinson’s disease (PD). Parkin-deficient mouse models fail to recapitulate nigrostriatal dopaminergic neurodegeneration as seen in PD, but produce deficits in dopaminergic neurotransmission and noradrenergic-dependent behavior. Since sporadic PD is thought to be caused by a combination of genetic susceptibilities and environmental factors, we hypothesized that neurotoxic insults from catecholaminergic toxins would render parkin knockout mice more vulnerable to neurodegeneration. Accordingly, we investigated the susceptibility of catecholaminergic neurons in parkin knockout mice to the potent dopaminergic and noradrenergic neurotoxins 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) respectively. We report that nigrostriatal dopaminergic neurons in parkin knockout mice do not show increased susceptibility to the parkinsonian neurotoxin, MPTP, in acute, subacute and chronic dose regimens of the neurotoxin. Additionally, parkin knockout mice do not show increased vulnerability to the noradrenergic neurotoxin, DSP-4, regarding levels of norepinephrine in cortex, brain stem and spinal cord. These findings suggest that absence of parkin in mice does not increase susceptibility to the loss of catecholaminergic neurons upon exposure to both dopaminergic and noradrenergic neurotoxins.
Keywords: Parkinson’s disease, parkin, alpha-synuclein, MPTP, DSP-4, substantia nigra, locus coeruleus, dopamine, norepinephrine
Introduction
Parkinson’s disease (PD) is a debilitating disorder marked by a prominent and progressive loss of dopaminergic neurons in the substantia nigra pars compacta (SNpc) with the presence of intraneuronal protein inclusions called Lewy bodies (Forno, 1987, Lang and Lozano, 1998). Clinical manifestations of PD include motor impairments involving resting tremor, bradykinesia, postural instability and rigidity. In addition to SNpc, other catecholaminergic neurons, including the locus coeruleus, as well as olfactory nuclei, sympathetic ganglia and also a substantial number of neurons in the pedunculopontine nucleus and median raphe degenerate in PD (Halliday et al., 1990, Braak et al., 2003, Micieli et al., 2003, Zarow et al., 2003). The molecular mechanisms leading to the degeneration of nigral dopaminergic neurons in PD are unclear; however a combination of genetic susceptibilities and environmental factors appears to play a critical role (Greenamyre and Hastings, 2004). Although the majority of PD cases are sporadic, identification of genes linked to rare forms of familial PD has yielded significant insights into pathogenesis of PD.
Inherited mutations in PARK2, the gene encoding parkin, an E3 ubiquitin protein ligase, cause selective degeneration of catecholaminergic neurons in the substantia nigra and locus coeruleus of the brainstem, resulting in early-onset parkinsonism (Kitada et al., 1998, Zhang et al., 2000). A number of putative substrates for parkin have been identified, and the accumulation of one or several of these substrates may account for the neurodegeneration in patients with loss of function-parkin mutations by impairment of ubiquitin-proteasomal degradation (von Coelln et al., 2004a, Ko et al., 2005). Parkin has been suggested to function as a multipurpose neuroprotective agent against a variety of toxic insults and is thought to be critical for survival of dopaminergic neurons (Feany and Pallanck, 2003). These include alpha-synuclein toxicity (Petrucelli et al., 2002, Lo Bianco et al., 2004), proteasomal dysfunction (Tsai et al., 2003), Pael-R (Yang et al., 2003), accumulation of the authentic parkin substrate p38/JTV-1 (Ko et al., 2005), kainate-induced excitoxicity (Staropoli et al., 2003), dopamine toxicity (Jiang et al., 2004), and toxins that impair mitochondrial function (Darios et al., 2003). Additionally, impairment of the catalytic activity of parkin either due to familial PD-linked mutations or due to nitrosative, oxidative or dopaminergic stress contributes to parkin dysfunction (Chung et al., 2004, Kalia et al., 2004, LaVoie et al., 2005, Sriram et al., 2005, Wang et al., 2005a, Wang et al., 2005b). Moreover, parkin may be a critical factor in the normal functioning of key cellular processes that involve maintenance of mitochondrial function (Greene et al., 2003, Palacino et al., 2004), expression of monoamine oxidases (Jiang et al., 2006), stability of microtubules (Yang et al., 2005), and signaling of cell survival pathways (Cha et al., 2005). The dysfunction of these processes could potentially impair dopamine neuron survival. Understanding how lack of parkin activity or its dysfunction is linked to the degeneration of catecholaminergic brainstem neurons in individuals with parkin-associated familial PD should provide insights into the pathogenic mechanisms causing the much more common sporadic form of PD.
Epidemiological studies indicate that exposure to pesticides, rural living, farming, and drinking well water are associated with an increased risk of developing PD (Gorell et al., 1998). There is increasing evidence, which suggests that these environmental factors cause oxidative stress by affecting mitochondrial functions and key cell survival pathways that play important roles in the pathogenesis of PD. These environmental agents include toxins that interfere with normal functioning of mitochondria, increase oxidative stress, and impair the ubiquitin-proteasome system in dopaminergic neurons, leading to their degeneration (Dawson and Dawson, 2003, Warner and Schapira, 2003). The lack of cardinal pathological features relevant to PD in mouse models of parkin deficiency prompted us to investigate the existence of a potential link between environmental factors and how mutations in parkin cause parkinsonism, by using the previously described mouse model with targeted disruption of the mouse parkin gene (Von Coelln et al., 2004b). Our parkin knockout mice show partial loss of catecholaminergic locus coeruleus neurons associated with reduced levels of norepinephrine in discrete brain regions and a marked reduction of the norepinephrine-dependent acoustic startle response; however, the nigrostriatal dopaminergic system appears to be intact (Von Coelln et al., 2004b).
In the present study we investigated how neurotoxins selective to central catecholaminergic neurons affect survival of these PD-associated neuronal populations in a mouse model of parkin deficiency. The parkin knockout mice were treated either with the potent parkinsonian neurotoxin, MPTP (Dauer and Przedborski, 2003), that causes selective degeneration of nigral dopaminergic neurons, or with the central noradrenergic neurotoxin, DSP-4, that induces selective degeneration of noradrenergic axon terminals originating in the locus coeruleus (Jonsson et al., 1981, Fritschy and Grzanna, 1989). We show that lack of parkin fails to increase the vulnerability of SNpc dopaminergic neurons to MPTP toxicity in acute, subacute and chronic dose regimens both in young and old animals. Furthermore, compared to age-matched wild-type littermates, parkin-deficient mice are not more susceptible to the DSP-4-induced degeneration of noradrenergic axon terminals, as determined by measuring norepinephrine levels in discrete brain regions. Taken together, we provide evidence that absence of parkin does not render murine central catecholaminergic neurons more susceptible to neurotoxins commonly used for animal models of neurodegenerative disease. These results therefore argue against a critical role of parkin in the neuronal defense mechanism against exogenous, and possibly also environmental, toxic factors.
Materials and Methods
Animals
All mice were housed and treated in strict accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals. Mice were housed in a pathogen-free facility, 4–5 animals per cage in a temperature-controlled room with a 12-hour light/dark cycle and access to food and water ad libitum. All procedures were approved by and conformed to the guidelines of the Institutional Animal Care Committee of Johns Hopkins University. We generated parkin knockout mice and age-matched littermate controls by crossing parkin heterozygous mice as described previously (Von Coelln et al., 2004b). The wild type and parkin knockout mice used in the present study were backcrossed to C57Bl6 mice for (N6) six generations. Age and sex matched littermate cohorts were used for all the experiments to account for potential strain differences.
PCR and Western blot analysis
Genotyping was performed by PCR analysis of genomic DNA, using the following primers: exon 7 sense, AATGGATGAGTTCAAGGTTGCACAG; exon 7 antisense, AACTCCAGAGCTAGGATAGGGCATA. The amplification products were separated on a 1% agarose gel for visualization (Von Coelln et al., 2004b). To assess parkin expression, wild-type, heterozygous and parkin null mice were decapitated, and the brains were dissected. Lysis was performed in buffer A (10 mM Tris·HCl, pH 7.4/150 mM NaCl/5 mM EDTA/0.5% Nonidet P-40/10 mM Na-β-glycerophosphate/Phosphatase Inhibitor Mixture 1 and 2 (Sigma, St. Louis, MO)/Complete Protease Inhibitor Mixture (Roche, Indianapolis, IN)), by using a Diax 900 homogenizer (Heidolph, Cinnaminson, NJ). After homogenization, samples were rotated at 4°C for 30 min for complete lysis, and then centrifuged (100,000 × g, 4°C, 20 min). The pellet was discarded. Proteins were separated by SDS/PAGE and electroblotted onto a poly (vinylidene difluoride) membrane (Bio-Rad, Hercules, CA). Immunolabeling was carried out by using an anti-parkin primary antibody [PRK8, mouse monoclonal] (Pawlyk et al., 2003, Von Coelln et al., 2004b), , a horseradish peroxidase-conjugated secondary antibody (Amersham Pharmacia Biosciences, Piscataway, NJ), and ECL solutions (Amersham Pharmacia). Chemiluminescence was visualized by using Hyperfilm (Amersham Pharmacia).
MPTP and DSP-4 treatment in mice
Male mice from different age groups were used for neurotoxic challenges induced by the dopaminergic neurotoxin MPTP, and the noradrenergic neurotoxin DSP-4. Four different paradigms of MPTP treatment were used in the present study. In the first paradigm, eight-week-old mice (n=5 per group) were administered an acute dose of 20 mg/kg free base MPTP every two hours four times a day, and toxicity analyses were done at day 7. In the second paradigm, three-month-old mice were treated with an acute dose of 15 mg/kg free base MPTP (n=6 per group) every two hours four times a day, and twelve-month-old mice were treated with an acute dose of 12.5 mg/kg free base MPTP every two hours four times a day (n=5 per group), and toxicity analyses performed at two weeks. A third paradigm consisted of a subacute treatment in which 3-month-old mice were treated with 25 mg/kg free base MPTP once daily for a total of 5 days (n=6 per group) and toxicity analyses performed two weeks after the last MPTP injection. In the fourth paradigm, MPTP was administered in 3-month-old mice (n=6–9 per group) in a chronic fashion at a dose of 20 mg/kg free base MPTP every day for 28 days using Alzet mini osmotic pumps (Model 2004, Durect Corporation, CA), and toxicity analyses were performed 7 days later (Fornai et al., 2005).
To assess neurotoxicity to noradrenergic neurons of the locus coeruleus, eighteen-month-old mice were treated with DSP-4 with a single injection of 10 mg/kg, (n=5 per group) and the toxicity analyses were performed one week after DSP-4 injection (Jonsson et al., 1981).
Control cohorts of mice received equivalent volumes of saline at the same frequency as the respective paradigms of MPTP or DSP-4 used in the study (n=5 per group). All systemic injections were administered through the intraperitoneal route and the neurotoxins were dissolved in normal saline. All procedures involving MPTP injections in mice were performed according to standard procedures (Przedborski et al., 2001).
Tyrosine hydroxylase and alpha-synuclein immunostaining
Immunohistochemical analyses were performed by anesthetizing mice using a lethal dose of pentobarbital (100 mg/kg) and by perfusing them intra-aortically with 50 ml of ice-cold phosphate-buffered saline (PBS, pH 7.4) followed by 75 ml of 4% paraformaldehyde in PBS (pH 7.4). Brains were removed, postfixed over night in the same fixative, and cryopreserved in 30% sucrose for 36 to 48 hours at 4°C. They were then frozen in dry ice and stored in −80°C until further analysis. Thirty micrometer-thick free-floating coronal sections were cut on a sliding microtome HM440E (Microm, Waldorf, Germany). For tyrosine hydroxylase (TH) and alpha-synuclein immunostaining sections were permeabilized and blocked in PBS containing 0.3% Triton X-100 and 10% normal goat serum at room temperature for 1 h. Sections were then incubated in 1:1000 rabbit anti-tyrosine hydroxylase antibody (Novus Biologicals, Littleton, CO) or alpha synuclein monoclonal antibody (Syn-1 mAb, BD Biosciences Pharmingen, San Jose, CA, 1:1000), in PBS containing 2% normal goat serum and 0.2% Triton X-100 and 0.03% sodium azide overnight at 4°C and washed with PBS successively. Next, sections were incubated for 2 h in secondary antibody (1:1000 biotin-labeled F(ab′)2 fragment goat-anti rabbit and goat-anti mouse antibody (Jackson Immunoresearch, West Grove, PA) in PBS, 0.2% Triton X-100, and 1.5% goat serum, washed, and visualized by incubation in biotin-streptavidin-HRP (Vector Laboratories, Burlingame, CA), followed by incubation with SigmaFast™ DAB Peroxidase Substrate (Sigma, St. Louis, MO) as per the manufacturer’s instructions. Sections were mounted on glass slides, dried and dehydrated in graded ethanol, cleared in xylene, and mounted with permount (Von Coelln et al., 2004b).
Stereological Cell counts
Stereological methods were employed to determine an unbiased estimate of total neurons and TH immunopositive neurons within SNpc. Briefly, mice treated with different paradigms of saline and MPTP were processed for TH immunohistochemistry and Nissl counter stain on every fourth midbrain section throughout the entire extent of SNpc as described previously (Mandir et al., 1999). Neurons were counted using the optical fractionator, an unbiased method for cell counting that is not affected by either the volume of reference (SNpc) or the size of the counted elements (neurons) (West, 1993). This method was carried out using a computer-assisted image analysis system, consisting of an Axiophot photomicroscope (Carl Zeiss Vision, Hallbergmoos, Germany) comprising of a Zeiss planapochromat 100 X oil objective equipped with a computer-controlled motorized stage, a video camera, and the Stereo Investigator software (MicroBrightField, Williston, VT). Cell counts were performed by counting the number of neurons on the left SNpc of every fourth section throughout the entire extent of the SNpc using a standard mouse atlas (Franklin and Paxinos, 1997) as anatomical reference. The total number of TH-positive neurons was calculated using the formula previously described for this method (West et al., 1991). In order to count TH-positive neurons of the locus coeruleus (LC), TH and Nissl counter staining was done on every hindbrain section from the rostral part of the pons to the caudal part of the medulla. All LC-containing sections were selected and counted using the optical fractionator, following the same method as described for the SNpc. The LC was identified referring to a commonly used mouse brain atlas (Franklin and Paxinos, 1997).
Measurement of biogenic amines by HPLC
To determine the concentration of biogenic amines in discrete brain regions by HPLC with electrochemical detection, mice were sacrificed by decapitation and the brain was quickly removed and discrete brain regions dissected on ice and immediately frozen and stored at −80°C. The tissue was weighed and sonicated in 0.2 ml of 0.1 M perchloric acid containing 0.01% EDTA and 25 μg/ml 3,4-dihydroxybenzylamine (DHBA) (Sigma, St. Louis, MO) as an internal standard. After centrifugation (15,000 × g, 10 min, 4 °C), 20 μl of the supernatant were injected onto a C-18 reverse phase Spheri 5, RP-18, 4.6mm × 25 cm catecholamine column (BASi, West Lafayette, IN). The mobile phase consisted of 0.15 M chloroacetic acid, 0.2 mM EDTA, 0.86 mM sodium octyl sulphate, 4% acetonitrile and 2.5% tetrahydrofuran (pH 3). The flow rate was kept at 1.5 ml/min. Biogenic amines and their metabolites were detected by an electrochemical detector, Prostar ECD Model 370 (Varian, Palo Alto, CA), with the working electrode kept at 0.6 V. Data were collected and processed on a Star Chromatography Workstation 5.52 (Varian) (Von Coelln et al., 2004b).
Measurement of Striatal MPP+ levels
To assess if MPTP metabolism is altered due to lack of parkin, striatal levels of MPP+ were measured in three-month-old male wild-type and parkin knock out mice (n=5 per group) after single intraperitoneal injections of MPTP-HCl (20 mg/kg free base). Ninety minutes after the injection, striata were weighed and processed by sonication followed by centrifugation in chilled 0.1 M perchloric acid and centrifuged (15,000 × g, 10 min, 4°C). Ten microlitres of the supernatant was analyzed by HPLC using 4.6 mm × 250 mm Waters Xterra MSC18 5 μm column, Waters 515 pump, and a Waters 474 Scanning Fluorometer set at 295 nm excitation and 375 nm emission. The mobile phase was a 20 mM borate buffer (pH 8.0) containing 3 mM tetrabutylammonium hydrogen sulfate and 0.25 mM 1-heptanesulfonic acid with 10% isopropanol (Naoi et al., 1987). Concentrations of MPP+ are expressed as nanograms per milligram of wet tissue.
Statistical analysis
Throughout the experiments the investigators were blinded to genotype and treatment of the mice. Data represent mean ± SEM from groups of animals. Statistical analysis for MPP+ levels were assessed by Students t test (two tailed probability) and for all other experiments were assessed by Student-Newman-Keuls multiple comparison test. Differences were considered significant when p < 0.05. All statistical analyses were performed using GraphPad InStat-version 3 and Prism software (San Diego, CA).
Results
Absence of parkin does not increase vulnerability of dopaminergic neurons to MPTP-intoxication
Identification of Parkin Knockout Mice
We first determined the genotypes of the mice from parkin heterozygote matings by PCR using genomic DNA (Figure 1A); these results were then confirmed by Western blot analysis for parkin expression after the mice had been sacrificed (Figure 1B).
Figure 1. PCR and western blot analysis for parkin expression in wild-type, heterozygotes and parkin knockout mice.
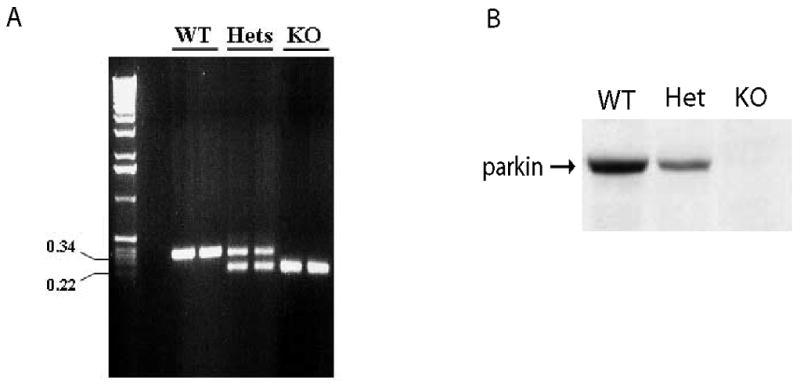
(A) PCR analysis of genomic DNA showing various genotypes of mice, i.e. wild-type, heterozygotes and parkin knockout mice. (B) To assess expression of parkin in wild-type (WT), heterozygous (hets), and knockout (KO) mice, total protein isolated from 6-month-old whole brains was subjected to immunoblot analysis for parkin with a monoclonal parkin antibody.
Acute MPTP paradigm
We determined if lack of parkin in mice affects the metabolism and trafficking of MPTP in the nigrostriatal system. MPP+ levels were measured in age-matched wild-type and parkin knockout mice 90 min after a single injection of MPTP (20mg/kg free base). MPP+ levels in parkin knockout mice (9.90 ± 0.74 ng/mg tissue) are not significantly different compared to those in wild-type mice (9.77 ± 0.69 ng/mg tissue), suggesting that lack of parkin does not impair the conversion of MPTP to MPP+.
To assess vulnerability of nigral dopaminergic neurons to the potent parkinsonian neurotoxin MPTP, eight-week-old wild-type or parkin knockout mice received four injections of MPTP (20 mg MPTP/kg every two hours) or saline. One week later, TH staining shows dramatic loss of SNpc dopaminergic neurons and striatal dopaminergic nerve terminals in both wild-type and parkin knockout mice (Figure 2A). No significant differences are observed between wild-type and parkin knockout mice. Unbiased stereologic counts of total (i.e. Nissl-positive) and TH-immunopositive SNpc neurons show a statistically significant loss of neurons compared to saline control (Wild type= 50.2% and parkin knockout= 50.4%) (Figure 2B). However, no differences in susceptibility to MPTP are observed between wild-type and parkin knockout mice (Figure 2B). Consistent with the loss of striatal TH-positive fibers (Figure 2A), HPLC analysis reveals a profound reduction in striatal dopamine and its metabolites (DOPAC and HVA) in this acute paradigm of MPTP treatment, in both wild-type (Wild type; DA= 84.5%, DOPAC= 60.4%, HVA= 84% compared to saline controls) and parkin knockout mice (parkin knockout; DA= 85.2%, DOPAC= 65.3%, HVA= 84%), compared to saline controls (Figure 2C). However, no statistically significant differences in the striatal concentrations of dopamine or its metabolites are seen in wild-type compared to parkin knockout mice following MPTP administration (Figure 2C). MPTP treatment results in an increased striatal dopamine turnover (DOPAC+HVA/DA) in wild-type and parkin knockout mice compared to saline controls; however, no statistically significant differences between wild-type and parkin knockout mice are observed following MPTP treatment (data not shown).
Figure 2. Nigrostriatal dopaminergic system in parkin knockout mice does not show increased sensitivity to acute MPTP intoxication.
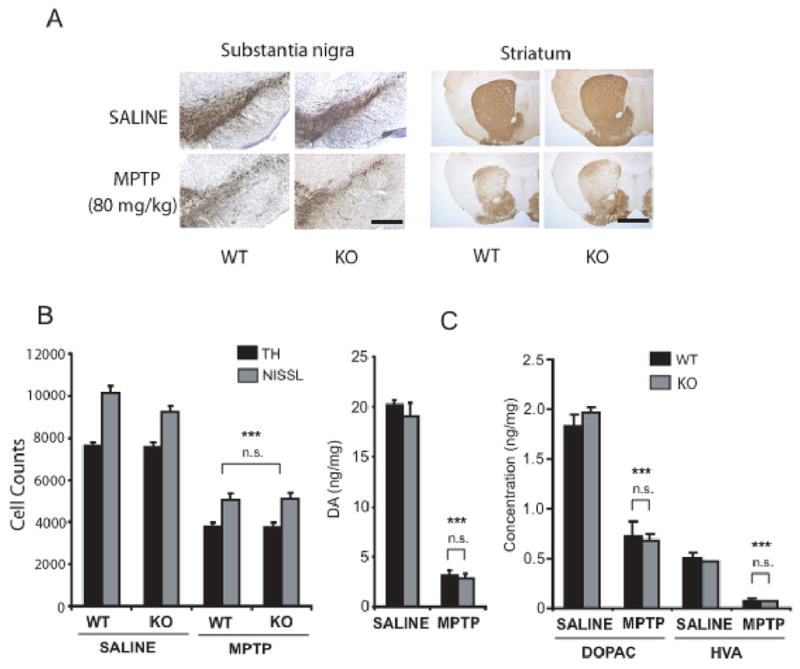
Eight-week-old male wild-type and parkin knockout mice were subjected to the acute paradigm of either saline or MPTP injections (20mg MPTP/kg four times, once every 2h). (A) TH immunostaining of SN and striatum shows significant loss of TH staining after MPTP. No significant differences are observed between wild-type and parkin knockout mice receiving the same treatment (saline or MPTP) (Scale bar=200μm). (B) Stereological neuronal counts of TH-immunopositive and total (i.e. Nissl-positive) neurons in wild-type and parkin knockout mice were analyzed 7 days after the last MPTP injection. While MPTP treatment causes a significant reduction in TH-immunopositive and total neurons, no significant differences in neuronal counts are observed between wild-type and parkin knockout mice receiving the same treatment (saline or MPTP). (C) Striatal levels of dopamine and its metabolites DOPAC and HVA in wild-type and parkin knockout mice were analyzed 7 days after last MPTP injection. No differences are seen among wild-type and parkin knockout mice receiving the same treatments (saline or MPTP). Data represent mean ± SEM. *p< 0.05, statistical significance versus saline controls using ANOVA followed by Student-Newman-Keuls test, n=5–6 per group, n.s not significant.
Subacute MPTP paradigm
The paradigm of MPTP delivery in mice may have an effect upon the mechanisms of neuronal death (Przedborski and Jackson-Lewis, 1998, Dauer and Przedborski, 2003). The acute intoxication paradigm of MPTP is known to cause cell death due to necrosis, whereas the subacute MPTP paradigms may cause apoptotic mechanisms of neuronal death (Jackson-Lewis et al., 1995, Tatton and Kish, 1997). To investigate if a different MPTP paradigm would result in differential effects between wild-type and parkin knockout mice, we treated 3-month-old wild-type and parkin knockout mice with a 5-day subacute MPTP paradigm. Stereologic neuronal counts were performed 2 weeks following the final MPTP dose (Figure 3). Although a significant loss of total and TH-immunopositive neurons (wild type= 35% and parkin knockout= 34.5%) is observed following MPTP administration, there is no significant difference between wild-type and parkin knockout mice.
Figure 3. Nigral dopaminergic neurons in parkin knockout mice do not show increased sensitivity to subacute MPTP intoxication.
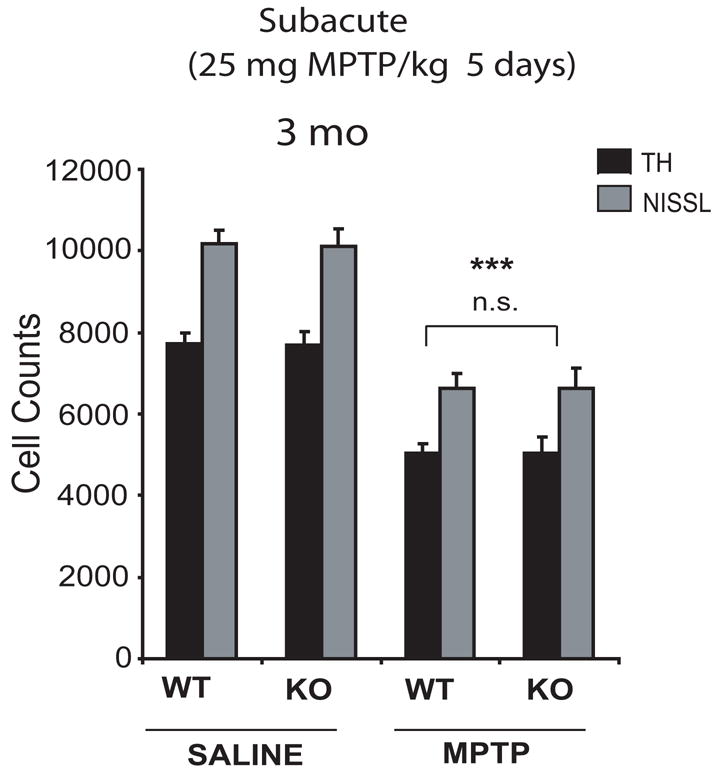
Stereological neuronal counts of TH-immunopositive and total (i.e. Nissl-positive) neurons in wild-type and parkin knockout mice were analyzed 2 weeks following a subacute paradigm of either saline or MPTP injections (25mg MPTP/kg for 5 days) in 3-month-old mice. While MPTP treatment causes a significant reduction in TH-immunopositive and total neurons, no significant differences in neuronal counts are observed between wild-type and parkin knockout mice receiving same amount of MPTP. Data represent mean ± SEM. *p< 0.05, statistical significance versus saline controls using ANOVA followed by Student-Newman-Keuls test, n=5–6 per group, n.s not significant.
Effect of age on susceptibility of dopaminergic neurons to MPTP
Increase in age is associated with increased vulnerability to MPTP-intoxication (Ali et al., 1993). Accordingly, we examined if age might have a synergistic effect with lack of parkin on susceptibility of nigral dopaminergic neurons to MPTP treatment in mice. For these studies we had to use a different dose of MPTP (15 mg/kg q2h × 4) for young (3 month old) mice versus 12 month old mice (12.5 mg/kg q2h ×4) as 100% of the 12 month old littermate wild type and parkin knockout mice died following 15 mg MPTP/kg q2h × 4 (data not shown). Following an acute paradigm (four injections, every 2 hours), MPTP was administered to wild-type and parkin knockout mice at 3 months of age (15 mg MPTP/kg) and 12 months of age (12.5 mg MPTP/kg). Mice were sacrificed and analyzed two weeks later (Figure 4A, B). Although MPTP-treated wild-type and parkin knockout mice demonstrate a significant loss of total and TH-immunopositive neurons of SNpc compared to saline treated mice both in 3 months (Wild type= 32% and parkin knockout= 31.8%) and 12 months old animals (Wild type= 25% and parkin knockout= 25%), no increase in susceptibility to MPTP toxicity is observed in parkin knockout compared to wild-type animals (Figure 4A, B).
Figure 4. Nigral dopaminergic neurons in parkin knockout mice do not show abnormally increased sensitivity to acute MPTP intoxication with an increase in age.
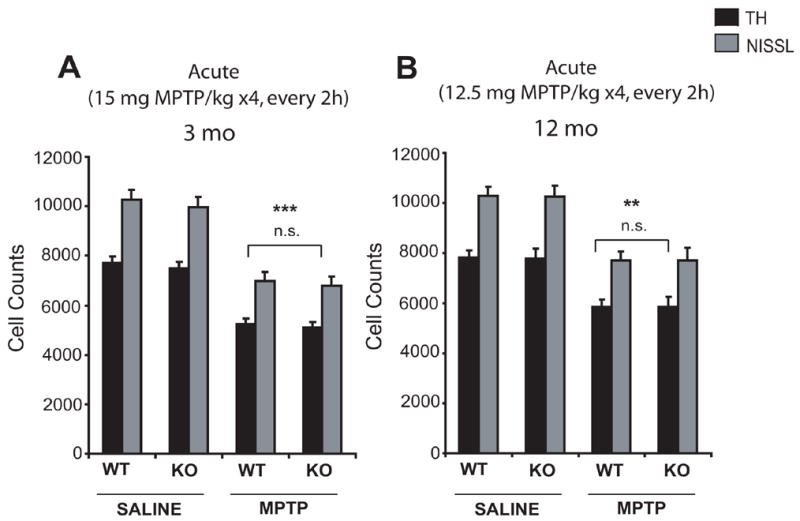
Stereological neuronal counts of TH-immunopositive and total (i.e. Nissl-positive) neurons in wild-type and parkin knockout mice, 2 weeks following an acute paradigm of either saline or MPTP injections in (A) 3-month-old mice (15mg MPTP/kg four times, every 2h), and (B) 12–month-old mice (12.5mg MPTP/kg four times, every 2h). While MPTP treatment causes a significant reduction in TH-immunopositive and total neurons, no significant differences in neuronal counts are observed between wild-type and parkin knockout mice receiving the same amount of MPTP. Data represent mean ± SEM. *p< 0.05, statistical significance versus saline controls using ANOVA followed by Student-Newman-Keuls test, n=5–6 per group, n.s not significant.
Continuous MPTP infusion
Chronic continuous infusion of MPTP employing a subcutaneous implantation of Alzet mini osmotic pumps in mice causes severe nigrostriatal dopaminergic toxicity, noradrenergic neuronal loss of locus coeruleus, accompanied by motor deficits recapitulating cardinal features seen in PD (Fornai et al., 2005). Accordingly, mini osmotic pumps that delivered either saline or 20 mg MPTP/kg/day for 28 days were subcutaneously implanted in 3-month-old wild-type and parkin knockout mice. One week after termination of chronic continuous MPTP treatment, TH staining of mesencephalic brain sections shows significant loss of SNpc dopaminergic neurons, compared to saline-treated control animals (Figure 5A). However, no significant differences in TH staining of SNpc are observed between wild-type and parkin knockout mice following MPTP (Figure 5A). Consistent with the impression from TH immunostaining, the stereologic cell counts show a significant loss of total and TH-immunopositive SNpc neurons in both wild-type (56%) and parkin knockout mice (59.8%) following MPTP administration compared to saline controls, with no significant differences between wild-type and parkin knockout mice (Figure 5B). In the current paradigm of chronic MPTP infusion we also fail to observe loss of TH-immunopositive neurons of the locus coeruleus in wild type and parkin knockout mice compared to saline controls (Table 1). However, as previously reported parkin knockout mice show a significant loss (24%) of TH-immunopositive neurons compared to wild type mice both in saline and MPTP groups.
Figure 5. Nigral dopaminergic neurons in parkin knockout mice do not show increased sensitivity to chronic continuous infusion of MPTP.
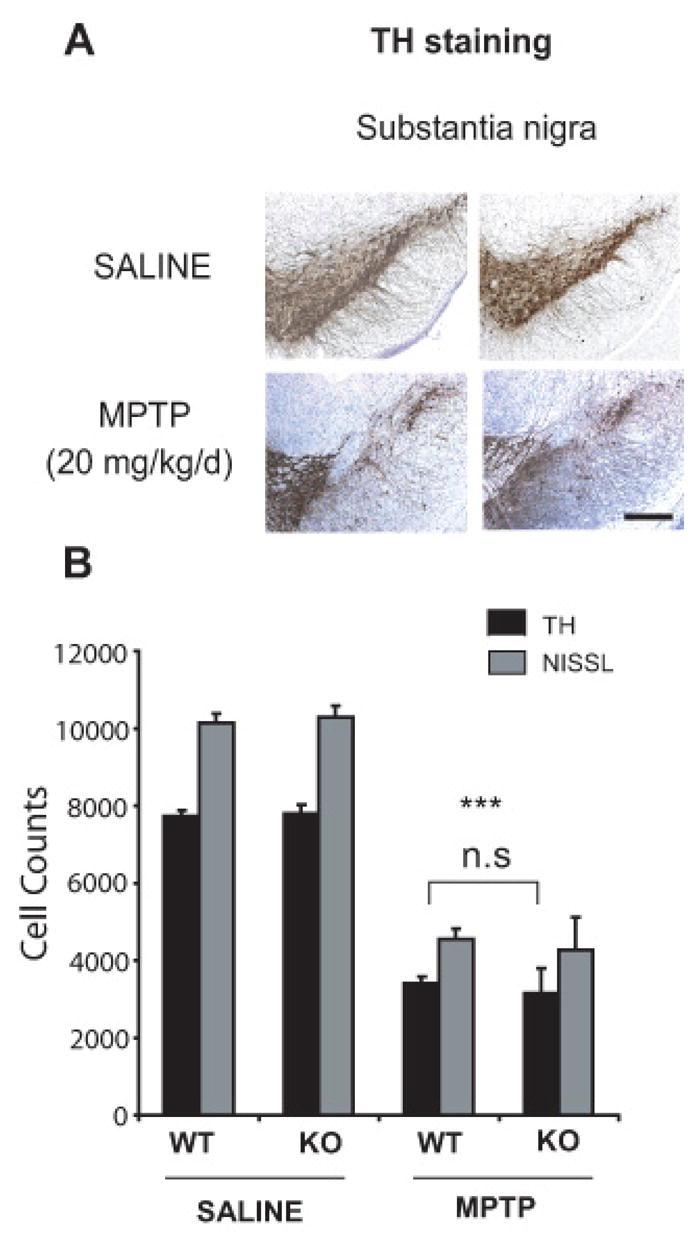
Three-month-old male wild-type and parkin knockout mice were subjected to a chronic continuous exposure of either saline or MPTP (20mg MPTP/kg/day for 28 days). Analysis was performed 7 days after the end of the 28-day-period of MPTP infusion. (A) TH immunostainings in wild-type and parkin knockout mice show significant loss in TH staining after MPTP. No significant differences are observed between wild-type and parkin knockout mice receiving the same treatment (saline or MPTP) (Scale bar= 200μm). (B) Stereological neuronal counts of TH-immunopositive and total (i.e. Nissl-positive) neurons in wild-type and parkin knockout mice. While MPTP treatment causes a significant loss of TH-immunopositive and total neurons, no significant differences in neuronal counts are observed between wild-type and parkin knockout mice receiving the same treatment (saline or MPTP). Data represent mean ± SEM. *p< 0.05, statistical significance versus saline controls using ANOVA followed by Student-Newman-Keuls test, n=5–6 per group, n.s not significant.
Table 1.
Stereological analysis of TH-positive neurons in Locus coeruleus (LC) of wild-type and parkin knockout mice treated with either saline and/or chronic continuous MPTP 20 mg/kg/day for 28 days and analyzed one week after chronic continuous administration of MPTP. Data are expressed as mean (n=5–9) ± SEM, and as ratio of the respective mean to wild type saline controls mean in percent. Statistical analysis was performed by ANOVA, revealing no significant differences among group means. Statistically significant differences in parkin knockout saline controls (*p<0.01) and in parkin knockout MPTP (*p<0.01) were observed compared to wild type saline controls.
| Genotypes/Treatment | TH+ neurons of LC | |
|---|---|---|
| Cell counts | [%] | |
| WT Saline | 1250 ± 55.8 | 100.0 |
| KO Saline | 949.1 ± 51.4** | 75.9 |
| WT MPTP (20mg/kg/d) | 1230 ± 61.8 | 98.4 |
| KO MPTP (20mg/kg/d) | 950.4 ± 55.37** | 76.0 |
The chronic continuous infusion of MPTP using mini osmotic pumps in mice causes accumulation of alpha-synuclein in nigral dopaminergic neurons (Fornai et al., 2005). Although we failed to observe Lewy-body-like alpha-synuclein-immunoreactive inclusions in nigral dopaminergic neurons following the chronic infusion of MPTP, as previously reported by Fornai et al. (2005), we did observe accumulation of alpha-synuclein in dopaminergic neurons following MPTP in both wild-type and parkin knockout mice (Figure 6). Lack of parkin did not impact on the qualitative accumulation of alpha-synuclein in nigral neurons compared to wild-type mice (Figure 6), suggesting that accumulation of alpha-synuclein in a chronic MPTP pump infusion model is independent of the presence or absence of parkin. Moreover, the accumulated alpha synuclein in nigral dopaminergic neurons following chronic MPTP did not stain for ubiquitin using a variety of antigen retrieval methods in both wild-type and parkin knockout mice (data not shown).
Figure 6. Alpha-synuclein accumulates in both wild-type and parkin knockout mice after chronic infusion of MPTP.
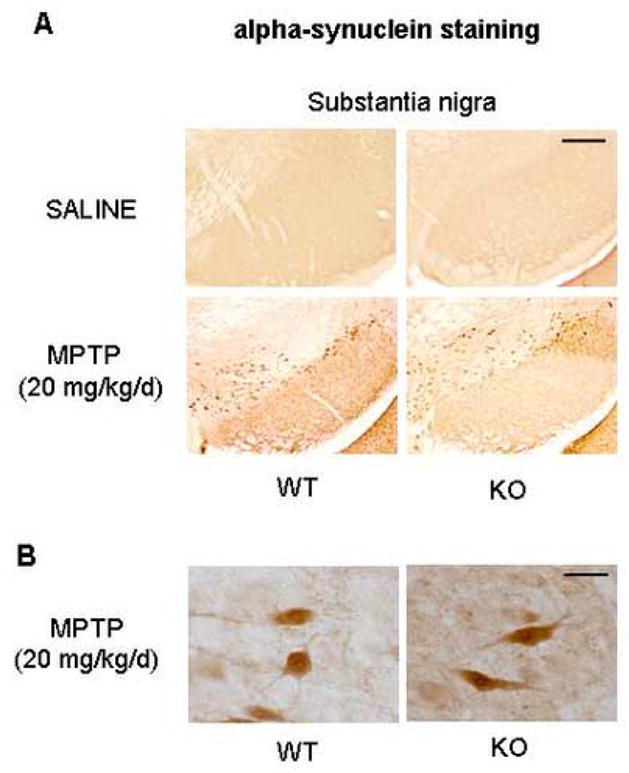
Alpha-synuclein-immunostaining was performed 7 days after the end of the 28-day-period of chronic continuous MPTP infusion. (A) SNpc of wild-type as well as parkin knockout mice shows significant accumulation of alpha synuclein induced by MPTP treatment. No significant differences are observed between wild-type and parkin knockout mice receiving same treatment (saline or MPTP) (Scale bar= 200μm). (B) High magnification image shows no difference between wildtype and parkin knockout mice following MPTP (Scale bar= 10 μm).
Noradrenergic neurons in parkin knockout mice are not abnormally vulnerable to DSP-4
DSP-4 is a selective neurotoxin to central noradrenergic terminals which depletes noradrenaline levels in target regions innervated by the noradrenergic neurons of locus coeruleus (Jonsson et al., 1981). Lesions of the locus coeruleus either by DSP-4 or by chemical lesions affects the acoustic startle response in rodents (Adams and Geyer, 1981, Kehne and Davis, 1985). We reported previously that exon 7-deleted parkin knockout mice show a loss of locus coeruleus neurons associated with reduction in noradrenaline levels in discrete regions of the central nervous system including spinal cord and olfactory bulb (Von Coelln et al., 2004b). To assess if locus coeruleus neurons in parkin knockout mice are more vulnerable to the noradrenergic neurotoxin DSP-4, eighteen-month-old wild-type and parkin knockout mice were administered a single intraperitoneal injection of DSP-4 (10 mg/kg). Norepinephrine levels in locus coeruleus-innervated target regions of the central nervous system were measured by HPLC electrochemistry after 7 days. As previously reported, saline-treated parkin knockout mice show a statistically significant decrease in norepinephrine levels in olfactory bulb (24%) and spinal cord (25%), but not cortex, compared to wild-type mice (Fig. 7A–C). Treatment with DSP-4 results in a significant loss of norepinephrine levels in olfactory bulb (Wild type= 36% and parkin knockout= 40%), spinal cord (Wild type= 37.5% and parkin knockout= 40.6%) and cortex (Wild type= 36.2% and parkin knockout= 37.8%) compared to saline treated mice; however, susceptibility of noradrenaline levels to DSP-4-toxicity is not significantly increased in parkin knockout mice, compared to wild-type control animals (Figure 7). We failed to observe any loss of dopamine and DOPAC levels or changes in the DA/DOPAC ratio in our intoxication paradigm of DSP-4 in discrete brain regions in both wild type and parkin knockout mice (data not shown). These results suggest that lack of parkin does not confer increased susceptibility of noradrenergic neurons to toxic stimuli.
Figure 7. Noradrenergic axon terminals in parkin knockout mice do not show increased sensitivity to DSP-4 intoxication.
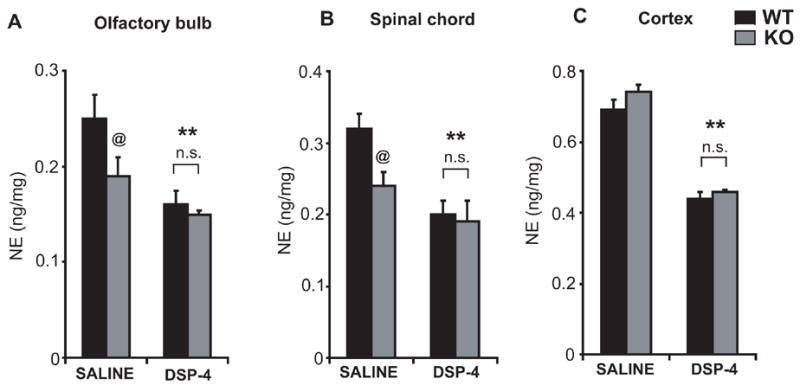
Eighteen-month-old wild-type and parkin knockout mice received a single injection of DSP-4 (10 mg/kg), and noradrenaline levels were measured after 7 days in (A) olfactory bulb, (B) spinal cord, and (C) cortex. While saline-treated parkin knockout mice, compared to wild-type control animals, show a significant reduction in noradrenaline levels in olfactory bulb and spinal cord, DSP-4 treatment causes a significant reduction in noradrenaline levels in all brain regions of both wild-type and parkin knockout mice. No significant differences in noradrenaline levels are observed between wild-type and parkin knockout mice receiving the same amount of DSP-4. Data represent mean ± SEM.*p< 0.05, statistical significance versus saline controls and @p< 0.05, statistical significance compared to wild-type using ANOVA followed by Student-Newman-Keuls test, n=5–6 per group, n.s not significant.
Discussion
In the present study, we examined the susceptibility of catecholaminergic neurons of SNpc and locus coeruleus in parkin-deficient mice to MPTP and DSP-4, respectively. MPTP is a potent parkinsonian neurotoxin which causes selective degeneration of nigral dopaminergic neurons, while DSP-4 possesses selective toxicity to central noradrenergic neurons of the locus coeruleus, affecting noradrenaline levels in diverse target regions of this brainstem nucleus. Our results indicate that parkin knockout mice do not show increased vulnerability of their dopaminergic nigrostriatal system to acute, subacute and chronic administration of MPTP. Furthermore, our data suggest that, although parkin knockout mice have a significant loss of noradrenaline in discrete regions of the central nervous system, lack of parkin fails to increase the susceptibility of the noradrenergic system to DSP-4 in mice.
There is considerable evidence to suggest that a combination of environmental factors and genetic susceptibilities cause PD (Steece-Collier et al., 2002, Dawson and Dawson, 2003). The present study, however, suggests that parkin deficiency fails to have an impact in mouse models administered with neurotoxins directed against central catecholaminergic neurons that are vulnerable to degeneration in PD. In recent years, it has become clear that parkin plays a pivotal role in survival of dopaminergic neurons. We and others have previously demonstrated that impairment of the ubiquitin E3 ligase activity of parkin either due to nitrosative or oxidative damage or due to familial PD linked mutations significantly blocks its ability to protect against a variety of toxic insults (Chung et al., 2001, Chung et al., 2004, Pesah et al., 2004, Wang et al., 2005b). Studies from parkin-deficient mouse models indicate small, but significant abnormalities specific to catecholaminergic neuronal systems. These include mitochondrial dysfunction and oxidative damage (Goldberg et al., 2003, Itier et al., 2003, Palacino et al., 2004, Casarejos et al., 2005), impairment in dopaminergic neurotransmission (Goldberg et al., 2003), increases in striatal dopamine turnover due to an increase in DOPAC/DA and DOPAC/3-MT levels, significant decreases in levels of striatal dopamine transporter (DAT) and vesicular monoamine transporter-2 (VMAT-2 (Itier et al., 2003), and loss of locus coeruleus neurons and deficits in noradrenergic-dependent acoustic startle response (Von Coelln et al., 2004b). Based on these findings we expected that challenging with the appropriate environmental trigger that specifically targets pathways already impaired in parkin-deficient mice would reveal and recapitulate robust behavioral and pathological features cardinal to PD.
The potent parkinsonian neurotoxin MPTP causes degeneration of nigral dopaminergic neurons by blocking oxidative phosphorylation through inhibition of the multienzyme complex I of the mitochondrial electron transport chain (Nicklas et al., 1985). Since parkin-deficient mice show defects in mitochondrial function (Greene et al., 2003, Palacino et al., 2004) we expected that this genetic defect would render their nigral dopaminergic neurons more sensitive to the intoxication with MPTP. Administration of MPTP according to the acute intoxication paradigm in younger animals failed to reveal an increased vulnerability to loss of striatal dopamine in parkin-deficient mice. Moreover, the lack of parkin failed to increase the sensitivity to loss of TH-positive neurons in SNpc. Older age heightens vulnerability of dopaminergic neurons to MPTP-intoxication (Ali et al., 1993). Absence of parkin also failed to exacerbate susceptibility to MPTP-induced loss of nigral dopaminergic neurons in older (i.e. 12-month-old) animals. Furthermore, it has been suggested that the choice of paradigm of MPTP delivery may have an effect on mechanisms of neuronal death, with the acute toxicity causing neuronal necrosis, whereas the subacute intoxication is thought to kill dopamine neurons primarily by apoptosis (Jackson-Lewis et al., 1995, Tatton and Kish, 1997). Both the subacute and chronic modes of MPTP administration failed to show an increased susceptibility of nigral dopaminergic neurons in parkin knockout mice. Furthermore, it has been shown that deficits in the central noradrenergic system exacerbate MPTP toxicity (Marien et al., 1993). We previously reported that our parkin-deficient mice show loss of locus coeruleus neurons as early as 2 months of age (Von Coelln et al., 2004b). Curiously, this basal impairment in the noradrenergic system in our parkin knockout mice failed to enhance MPTP-induced neurotoxicity. One possible explanation would be that loss of noradrenergic neurons in parkin-deficient mice is not sufficient to result in an increased sensitivity to MPTP; also, there might be compensatory mechanisms by surviving neurons of the locus coeruleus. Additionally, in the chronic MPTP toxicity model, the accumulation of alpha-synuclein in nigral dopaminergic neurons was unaltered irrespective of the presence or absence or parkin. This is consistent with our recent finding, that, in a double-mutant mouse model, neurodegeneration in a mouse model of alpha-synucleinopathy is independent of parkin-mediated ubiquitin E3 ligase activity (von Coelln et al., 2006). Interestingly, our findings are also consistent with a recent study in which parkin-deficient mice failed to show heightened vulnerability to 6-hydroxydopamine and methamphetamine, two commonly used dopaminergic neurotoxins (Perez et al., 2005).
Since parkin knockout mice show morphological and biochemical deficits of the central noradrenergic system (Von Coelln et al., 2004b) we reasoned that these neurons might be more vulnerable to DSP-4, which selectively damages axonal terminals of noradrenergic neurons located in the locus coeruleus (Fritschy and Grzanna, 1989). However, our study fails to show increased susceptibility to DSP-4 in parkin knockout mice.
Why MPTP and DSP-4 fail to reveal an increased vulnerability of nigrostriatal dopaminergic neurons and noradrenergic neurons of the locus coeruleus in parkin-deficient mice is not clear. One possible explanation from our findings and the study by Perez F et al. (2005) is that there might be compensatory mechanisms in mice that complement for lack of parkin in parkin knockout mice, thereby preventing a heightened sensitivity to neurotoxins. The second possibility is that parkin function in mice could be fundamentally different from that in humans and hence parkin knockout mice do not develop robust signs of parkinsonism. The third possibility is that parkin deficiency may render vulnerability to other forms of toxic insults not yet studied. Therefore, further studies will be required to uncover the compensatory mechanisms in parkin knockout mice and/or characterize the appropriate toxic stimuli that cause catecholaminergic brainstem neurons to degenerate. Such studies would not only provide crucial insight into the mechanisms leading to the parkin-associated autosomal-recessive form of PD, but would also help clarifying if there is a link between environmental factors and parkin deficiency / dysfunction in the pathogenesis of sporadic PD.
Acknowledgments
This work was supported by NIH grants NS38377 and NS48206. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. Thanks to Beverly Lorenzo for help with striatal MPP+ measurements.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams LM, Geyer MA. Effects of 6-hydroxydopamine lesions of locus coeruleus on startle in rats. Psychopharmacology (Berl) 1981;73:394–398. doi: 10.1007/BF00426474. [DOI] [PubMed] [Google Scholar]
- Ali SF, David SN, Newport GD. Age-related susceptibility to MPTP-induced neurotoxicity in mice. Neurotoxicology. 1993;14:29–34. [PubMed] [Google Scholar]
- Braak H, Del Tredici K, Rub U, de Vos RA, Jansen Steur EN, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging. 2003;24:197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- Casarejos MJ, Solano RM, Menendez J, Rodriguez-Navarro JA, Correa C, Garcia de Yebenes J, Mena MA. Differential effects of l-DOPA on monoamine metabolism, cell survival and glutathione production in midbrain neuronal-enriched cultures from parkin knockout and wild-type mice. J Neurochem. 2005;94:1005–1014. doi: 10.1111/j.1471-4159.2005.03249.x. [DOI] [PubMed] [Google Scholar]
- Cha GH, Kim S, Park J, Lee E, Kim M, Lee SB, Kim JM, Chung J, Cho KS. Parkin negatively regulates JNK pathway in the dopaminergic neurons of Drosophila. Proc Natl Acad Sci U S A. 2005;102:10345–10350. doi: 10.1073/pnas.0500346102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung KK, Thomas B, Li X, Pletnikova O, Troncoso JC, Marsh L, Dawson VL, Dawson TM. S-nitrosylation of parkin regulates ubiquitination and compromises parkin’s protective function. Science. 2004;304:1328–1331. doi: 10.1126/science.1093891. [DOI] [PubMed] [Google Scholar]
- Chung KK, Zhang Y, Lim KL, Tanaka Y, Huang H, Gao J, Ross CA, Dawson VL, Dawson TM. Parkin ubiquitinates the alpha-synuclein-interacting protein, synphilin-1: implications for Lewy-body formation in Parkinson disease. Nat Med. 2001;7:1144–1150. doi: 10.1038/nm1001-1144. [DOI] [PubMed] [Google Scholar]
- Darios F, Corti O, Lucking CB, Hampe C, Muriel MP, Abbas N, Gu WJ, Hirsch EC, Rooney T, Ruberg M, Brice A. Parkin prevents mitochondrial swelling and cytochrome c release in mitochondria-dependent cell death. Hum Mol Genet. 2003;12:517–526. doi: 10.1093/hmg/ddg044. [DOI] [PubMed] [Google Scholar]
- Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron. 2003;39:889–909. doi: 10.1016/s0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL. Molecular pathways of neurodegeneration in Parkinson’s disease. Science. 2003;302:819–822. doi: 10.1126/science.1087753. [DOI] [PubMed] [Google Scholar]
- Feany MB, Pallanck LJ. Parkin: a multipurpose neuroprotective agent? Neuron. 2003;38:13–16. doi: 10.1016/s0896-6273(03)00201-0. [DOI] [PubMed] [Google Scholar]
- Fornai F, Schluter OM, Lenzi P, Gesi M, Ruffoli R, Ferrucci M, Lazzeri G, Busceti CL, Pontarelli F, Battaglia G, Pellegrini A, Nicoletti F, Ruggieri S, Paparelli A, Sudhof TC. Parkinson-like syndrome induced by continuous MPTP infusion: convergent roles of the ubiquitin-proteasome system and alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:3413–3418. doi: 10.1073/pnas.0409713102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forno LS. The Lewy body in Parkinson’s disease. Adv Neurol. 1987;45:35–43. [PubMed] [Google Scholar]
- Franklin KBJ, Paxinos G. The mouse brain in stereotaxic coordinates. Academic Press; San Diego: 1997. [Google Scholar]
- Fritschy JM, Grzanna R. Immunohistochemical analysis of the neurotoxic effects of DSP-4 identifies two populations of noradrenergic axon terminals. Neuroscience. 1989;30:181–197. doi: 10.1016/0306-4522(89)90364-3. [DOI] [PubMed] [Google Scholar]
- Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem. 2003;278:43628–43635. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- Gorell JM, Johnson CC, Rybicki BA, Peterson EL, Richardson RJ. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology. 1998;50:1346–1350. doi: 10.1212/wnl.50.5.1346. [DOI] [PubMed] [Google Scholar]
- Greenamyre JT, Hastings TG. Biomedicine. Parkinson’s--divergent causes, convergent mechanisms. Science. 2004;304:1120–1122. doi: 10.1126/science.1098966. [DOI] [PubMed] [Google Scholar]
- Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci U S A. 2003;100:4078–4083. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halliday GM, Li YW, Blumbergs PC, Joh TH, Cotton RG, Howe PR, Blessing WW, Geffen LB. Neuropathology of immunohistochemically identified brainstem neurons in Parkinson’s disease. Ann Neurol. 1990;27:373–385. doi: 10.1002/ana.410270405. [DOI] [PubMed] [Google Scholar]
- Itier JM, Ibanez P, Mena MA, Abbas N, Cohen-Salmon C, Bohme GA, Laville M, Pratt J, Corti O, Pradier L, Ret G, Joubert C, Periquet M, Araujo F, Negroni J, Casarejos MJ, Canals S, Solano R, Serrano A, Gallego E, Sanchez M, Denefle P, Benavides J, Tremp G, Rooney TA, Brice A, Garcia de Yebenes J. Parkin gene inactivation alters behaviour and dopamine neurotransmission in the mouse. Hum Mol Genet. 2003;12:2277–2291. doi: 10.1093/hmg/ddg239. [DOI] [PubMed] [Google Scholar]
- Jackson-Lewis V, Jakowec M, Burke RE, Przedborski S. Time course and morphology of dopaminergic neuronal death caused by the neurotoxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Neurodegeneration. 1995;4:257–269. doi: 10.1016/1055-8330(95)90015-2. [DOI] [PubMed] [Google Scholar]
- Jiang H, Jiang Q, Liu W, Feng J. Parkin suppresses the expression of monoamine oxidases. J Biol Chem. 2006 doi: 10.1074/jbc.M510926200. [DOI] [PubMed] [Google Scholar]
- Jiang H, Ren Y, Zhao J, Feng J. Parkin protects human dopaminergic neuroblastoma cells against dopamine-induced apoptosis. Hum Mol Genet. 2004;13:1745–1754. doi: 10.1093/hmg/ddh180. [DOI] [PubMed] [Google Scholar]
- Jonsson G, Hallman H, Ponzio F, Ross S. DSP4 (N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine)--a useful denervation tool for central and peripheral noradrenaline neurons. Eur J Pharmacol. 1981;72:173–188. doi: 10.1016/0014-2999(81)90272-7. [DOI] [PubMed] [Google Scholar]
- Kalia SK, Lee S, Smith PD, Liu L, Crocker SJ, Thorarinsdottir TE, Glover JR, Fon EA, Park DS, Lozano AM. BAG5 inhibits parkin and enhances dopaminergic neuron degeneration. Neuron. 2004;44:931–945. doi: 10.1016/j.neuron.2004.11.026. [DOI] [PubMed] [Google Scholar]
- Kehne JH, Davis M. Central noradrenergic involvement in yohimbine excitation of acoustic startle: effects of DSP4 and 6-OHDA. Brain Res. 1985;330:31–41. doi: 10.1016/0006-8993(85)90005-8. [DOI] [PubMed] [Google Scholar]
- Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- Ko HS, von Coelln R, Sriram SR, Kim SW, Chung KK, Pletnikova O, Troncoso J, Johnson B, Saffary R, Goh EL, Song H, Park BJ, Kim MJ, Kim S, Dawson VL, Dawson TM. Accumulation of the authentic parkin substrate aminoacyl-tRNA synthetase cofactor, p38/JTV-1, leads to catecholaminergic cell death. J Neurosci. 2005;25:7968–7978. doi: 10.1523/JNEUROSCI.2172-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang AE, Lozano AM. Parkinson’s disease. First of two parts. N Engl J Med. 1998;339:1044–1053. doi: 10.1056/NEJM199810083391506. [DOI] [PubMed] [Google Scholar]
- LaVoie MJ, Ostaszewski BL, Weihofen A, Schlossmacher MG, Selkoe DJ. Dopamine covalently modifies and functionally inactivates parkin. Nat Med. 2005;11:1214–1221. doi: 10.1038/nm1314. [DOI] [PubMed] [Google Scholar]
- Lo Bianco C, Schneider BL, Bauer M, Sajadi A, Brice A, Iwatsubo T, Aebischer P. Lentiviral vector delivery of parkin prevents dopaminergic degeneration in an alpha-synuclein rat model of Parkinson’s disease. Proc Natl Acad Sci U S A. 2004;101:17510–17515. doi: 10.1073/pnas.0405313101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, Smulson ME, Hoffman BE, Guastella DB, Dawson VL, Dawson TM. Poly(ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1, 2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci U S A. 1999;96:5774–5779. doi: 10.1073/pnas.96.10.5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marien M, Briley M, Colpaert F. Noradrenaline depletion exacerbates MPTP-induced striatal dopamine loss in mice. Eur J Pharmacol. 1993;236:487–489. doi: 10.1016/0014-2999(93)90489-5. [DOI] [PubMed] [Google Scholar]
- Micieli G, Tosi P, Marcheselli S, Cavallini A. Autonomic dysfunction in Parkinson’s disease. Neurol Sci. 2003;24(Suppl 1):S32–34. doi: 10.1007/s100720300035. [DOI] [PubMed] [Google Scholar]
- Naoi M, Takahashi T, Nagatsu T. A fluorometric determination of N-methyl-4-phenylpyridinium ion, using high-performance liquid chromatography. Anal Biochem. 1987;162:540–545. doi: 10.1016/0003-2697(87)90431-3. [DOI] [PubMed] [Google Scholar]
- Nicklas WJ, Vyas I, Heikkila RE. Inhibition of NADH-linked oxidation in brain mitochondria by 1-methyl-4-phenyl-pyridine, a metabolite of the neurotoxin, 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. Life Sci. 1985;36:2503–2508. doi: 10.1016/0024-3205(85)90146-8. [DOI] [PubMed] [Google Scholar]
- Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem. 2004;279:18614–18622. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- Pawlyk AC, Giasson BI, Sampathu DM, Perez FA, Lim KL, Dawson VL, Dawson TM, Palmiter RD, Trojanowski JQ, Lee VM. Novel monoclonal antibodies demonstrate biochemical variation of brain parkin with age. J Biol Chem. 2003;278:48120–48128. doi: 10.1074/jbc.M306889200. [DOI] [PubMed] [Google Scholar]
- Perez FA, Curtis WR, Palmiter RD. Parkin-deficient mice are not more sensitive to 6-hydroxydopamine or methamphetamine neurotoxicity. BMC Neurosci. 2005;6:71. doi: 10.1186/1471-2202-6-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pesah Y, Pham T, Burgess H, Middlebrooks B, Verstreken P, Zhou Y, Harding M, Bellen H, Mardon G. Drosophila parkin mutants have decreased mass and cell size and increased sensitivity to oxygen radical stress. Development. 2004;131:2183–2194. doi: 10.1242/dev.01095. [DOI] [PubMed] [Google Scholar]
- Petrucelli L, O’Farrell C, Lockhart PJ, Baptista M, Kehoe K, Vink L, Choi P, Wolozin B, Farrer M, Hardy J, Cookson MR. Parkin protects against the toxicity associated with mutant alpha-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36:1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V. Mechanisms of MPTP toxicity. Mov Disord. 1998;13(Suppl 1):35–38. [PubMed] [Google Scholar]
- Przedborski S, Jackson-Lewis V, Naini AB, Jakowec M, Petzinger G, Miller R, Akram M. The parkinsonian toxin 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP): a technical review of its utility and safety. J Neurochem. 2001;76:1265–1274. doi: 10.1046/j.1471-4159.2001.00183.x. [DOI] [PubMed] [Google Scholar]
- Sriram SR, Li X, Ko HS, Chung KK, Wong E, Lim KL, Dawson VL, Dawson TM. Familial-associated mutations differentially disrupt the solubility, localization, binding and ubiquitination properties of parkin. Hum Mol Genet. 2005;14:2571–2586. doi: 10.1093/hmg/ddi292. [DOI] [PubMed] [Google Scholar]
- Staropoli JF, McDermott C, Martinat C, Schulman B, Demireva E, Abeliovich A. Parkin is a component of an SCF-like ubiquitin ligase complex and protects postmitotic neurons from kainate excitotoxicity. Neuron. 2003;37:735–749. doi: 10.1016/s0896-6273(03)00084-9. [DOI] [PubMed] [Google Scholar]
- Steece-Collier K, Maries E, Kordower JH. Etiology of Parkinson’s disease: Genetics and environment revisited. Proc Natl Acad Sci U S A. 2002;99:13972–13974. doi: 10.1073/pnas.242594999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatton NA, Kish SJ. In situ detection of apoptotic nuclei in the substantia nigra compacta of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice using terminal deoxynucleotidyl transferase labelling and acridine orange staining. Neuroscience. 1997;77:1037–1048. doi: 10.1016/s0306-4522(96)00545-3. [DOI] [PubMed] [Google Scholar]
- Tsai YC, Fishman PS, Thakor NV, Oyler GA. Parkin facilitates the elimination of expanded polyglutamine proteins and leads to preservation of proteasome function. J Biol Chem. 2003;278:22044–22055. doi: 10.1074/jbc.M212235200. [DOI] [PubMed] [Google Scholar]
- von Coelln R, Dawson VL, Dawson TM. Parkin-associated Parkinson’s disease. Cell Tissue Res. 2004a;318:175–184. doi: 10.1007/s00441-004-0924-4. [DOI] [PubMed] [Google Scholar]
- von Coelln R, Thomas B, Andrabi SA, Lim KL, Savitt JM, Saffary R, Stirling W, Bruno K, Hess EJ, Lee MK, Dawson VL, Dawson TM. Inclusion body formation and neurodegeneration are parkin independent in a mouse model of alpha-synucleinopathy. J Neurosci. 2006;26:3685–3696. doi: 10.1523/JNEUROSCI.0414-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Von Coelln R, Thomas B, Savitt JM, Lim KL, Sasaki M, Hess EJ, Dawson VL, Dawson TM. Loss of locus coeruleus neurons and reduced startle in parkin null mice. Proc Natl Acad Sci U S A. 2004b;101:10744–10749. doi: 10.1073/pnas.0401297101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Ko HS, Thomas B, Tsang F, Chew KC, Tay SP, Ho MW, Lim TM, Soong TW, Pletnikova O, Troncoso J, Dawson VL, Dawson TM, Lim KL. Stress-induced alterations in parkin solubility promote parkin aggregation and compromise parkin’s protective function. Hum Mol Genet. 2005a;14:3885–3897. doi: 10.1093/hmg/ddi413. [DOI] [PubMed] [Google Scholar]
- Wang C, Tan JM, Ho MW, Zaiden N, Wong SH, Chew CL, Eng PW, Lim TM, Dawson TM, Lim KL. Alterations in the solubility and intracellular localization of parkin by several familial Parkinson’s disease-linked point mutations. J Neurochem. 2005b;93:422–431. doi: 10.1111/j.1471-4159.2005.03023.x. [DOI] [PubMed] [Google Scholar]
- Warner TT, Schapira AH. Genetic and environmental factors in the cause of Parkinson’s disease. Ann Neurol. 2003;53(Suppl 3):S16–23. doi: 10.1002/ana.10487. discussion S23–15. [DOI] [PubMed] [Google Scholar]
- West MJ. New stereological methods for counting neurons. Neurobiol Aging. 1993;14:275–285. doi: 10.1016/0197-4580(93)90112-o. [DOI] [PubMed] [Google Scholar]
- West MJ, Slomianka L, Gundersen HJ. Unbiased stereological estimation of the total number of neurons in thesubdivisions of the rat hippocampus using the optical fractionator. Anat Rec. 1991;231:482–497. doi: 10.1002/ar.1092310411. [DOI] [PubMed] [Google Scholar]
- Yang F, Jiang Q, Zhao J, Ren Y, Sutton MD, Feng J. Parkin stabilizes microtubules through strong binding mediated by three independent domains. J Biol Chem. 2005;280:17154–17162. doi: 10.1074/jbc.M500843200. [DOI] [PubMed] [Google Scholar]
- Yang Y, Nishimura I, Imai Y, Takahashi R, Lu B. Parkin suppresses dopaminergic neuron-selective neurotoxicity induced by Pael-R in Drosophila. Neuron. 2003;37:911–924. doi: 10.1016/s0896-6273(03)00143-0. [DOI] [PubMed] [Google Scholar]
- Zarow C, Lyness SA, Mortimer JA, Chui HC. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch Neurol. 2003;60:337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gao J, Chung KK, Huang H, Dawson VL, Dawson TM. Parkin functions as an E2-dependent ubiquitin- protein ligase and promotes the degradation of the synaptic vesicle-associated protein, CDCrel-1. Proc Natl Acad Sci U S A. 2000;97:13354–13359. doi: 10.1073/pnas.240347797. [DOI] [PMC free article] [PubMed] [Google Scholar]