

Abstract
While the structures of skeletal and smooth muscle myosins are homologous, they differ functionally from each other in several respects, i.e., motor activities and regulation. To investigate the molecular basis for these differences, we have produced a skeletal/smooth chimeric myosin molecule and analyzed the motor activities and regulation of this myosin. The produced chimeric myosin is composed of the globular motor domain of skeletal muscle myosin (Met1–Gly773) and the C-terminal long α-helix domain of myosin subfragment 1 as well as myosin subfragment 2 (Gly773–Ser1104) and light chains of smooth muscle myosin. Both the actin-activated ATPase activity and the actin-translocating activity of the chimeric myosin were completely regulated by light chain phosphorylation. On the other hand, the maximum actin-activated ATPase activity of the chimeric myosin was the same as skeletal myosin and thus much higher than smooth myosin. These results show that the C-terminal light chain-associated domain of myosin head solely confers regulation by light chain phosphorylation, whereas the motor domain determines the rate of ATP hydrolysis. This is the first report, to our knowledge, that directly determines the function of the two structurally separated domains in myosin head.
Myosin, an actin-dependent motor protein, is distributed in diverse cell types and plays a crucial role in cell contractility and motility (1). While the structure of myosins is highly conserved, their functions are quite diversified, and each myosin molecule is characterized by its unique enzymatic activity, actin-translocating activity, and mode of regulation. However, the molecular basis of the functional divergence has not been understood yet.
Myosin found in striated muscle has high ATPase activity and fast actin sliding velocity, and requires thin filament regulatory proteins, troponin/tropomyosin, for its regulation (2). That is, striated muscle myosin shows its motor activity without phosphorylation, and the troponin/tropomyosin system inhibits myosin motor activity when Ca2+ is dissociated from troponin C molecule, a subunit of troponin complex. On the other hand, motor activity of vertebrate smooth muscle and nonmuscle myosins is regulated by phosphorylation of its regulatory subunit (light chain) and generally has a much lower ATPase activity and actin-translocating activity (3, 4). The phosphorylation takes place at Ser19 of the 20-kDa light chain subunit catalyzed by a Ca2+/calmodulin-dependent protein kinase, myosin light chain kinase, and the phosphorylation is prerequisite for smooth muscle/nonmuscle myosin motor activity. Furthermore, smooth muscle/nonmuscle myosins are characterized by their change in the conformation, known as 10S–6S conformational transition (5–13). 10S myosin has a folded conformation that is not found in striated muscle myosin, and the formation of this conformation is characterized by the inhibition of Mg2+- or Ca2+-ATPase activity (7, 10) and thick filament formation (13). Of interest is that phosphorylation destabilizes the formation of 10S conformation (7–9).
According to the known three-dimensional structure of chicken adult skeletal muscle myosin subfragment 1 (S1) (14), the myosin head domain is composed of an N-terminal globular motor domain from Met1 to Gly770 and a long α-helix domain that extends from Leu771 to Val826 of the C-terminal domain of the S1 heavy chain. The long α-helix domain contains the two classes of light chain-binding regions and is clearly separated from the dense globular motor domain, suggesting that the protein folding at the globular motor head domain is not influenced from the long α-helix domain. This view is supported by the finding that the globular motor head domain by itself exhibits motility activity (15, 16). In this study, we produced a chimeric myosin heavy chain (MHC) containing a globular motor domain (Met1–Gly773) of skeletal myosin and a light chain-binding domain as well as myosin subfragment 2 (S2) (Gly773–Ser1104) of smooth myosin to clarify the molecular basis of dictating the differences in the function between skeletal and smooth myosins. The results show that the C-terminal light chain-associated domain of myosin head solely confers regulation by light chain phosphorylation, whereas the motor domain determines the rate of ATP hydrolysis.
MATERIALS AND METHODS
Protein Preparation.
Actin was prepared from rabbit skeletal muscle acetone powder (17). Smooth and skeletal myosin light chain kinases were prepared as described (18). Calmodulin was prepared from bull testes (19). Smooth muscle myosin was prepared from turkey gizzards (20), and heavy meromyosin (HMM) as well as S1 was prepared by Staphylococcus aureus protease digestion (21). Skeletal muscle myosin was prepared from chicken pectoralis, and HMM was prepared by α-chymotrypsin digestion (22).
Expression and Purification of Chimeric Myosin.
An ApaI site was created 2319 bp from the initiation codon in chicken skeletal MHC by using a transformer site-directed mutagenesis kit (CLONTECH). Another ApaI site was introduced 2337 bp in a truncated chicken gizzard MHC cDNA containing an introduced stop codon at base pair 3331 (23, 24). These unique ApaI sites were used to generate a cDNA encoding a 127-kDa skeletal/smooth chimeric MHC (Met1–Gly773 are skeletal and Gly773–Ser1104 are smooth) (Fig. 1). One amino acid residue change consequent to generation of restriction sites, i.e., Val774 → Pro was corrected by the second mutagenesis after the chimeric MHC cDNA was subcloned into pBluescript II SK(−) (Stratagene). This cDNA was digested with SpeI and subcloned into a baculovirus transfer vector pBlueBacM (Invitrogen) using an unique NheI site.
Figure 1.

Construction of skeletal/smooth chimeric MHC. Schematic drawing of skeletal/smooth chimeric MHC construct. A chimeric MHC cDNA was produced by ligating skeletal MHC cDNA containing the 5′ 2319 bp corresponding to the globular motor domain and a smooth MHC cDNA containing 993 bp corresponding to the C-terminal long α-helix domain of S1 and S2. ELC, essential light chain; RLC, regulatory light chain.
Two cDNA clones encoding 20-kDa RLC and 17-kDa ELC were obtained from a chicken gizzard cDNA library and subcloned into pT7-7 Escherichia coli expression vector (25). The cDNAs were excised with XbaI digestion and subcloned into the pBlueBacM using an NheI site.
Recombinant baculovirus was obtained for each cDNA construct by the protocols recommended by the manufacturer (Invitrogen). Sf9 cells were coinfected with the recombinant viruses expressing chimeric MHC, smooth RLC, and smooth ELC (23, 24, 26, 27). The expressed chimeric myosin was purified as described (24).
Gel Electrophoresis, ATPase Assay, and in Vitro Motility Assay.
SDS/PAGE was carried out on a 7.5–20% polyacrylamide gradient slab gel. Immunoblot and nondenaturing gel electrophoresis were performed as described (24). ATPase activity was measured as described by Ikebe and Hartshorne (20).
The in vitro motility assay was performed as described (24). A monoclonal antibody, MM9, which recognizes the S2 portion (Ala873–Ser944) of chicken smooth myosin (28) was used to bind the chimeric myosin as well as smooth HMM to the nitrocellulose surface.
Analytical Ultracentrifugation.
Sedimentation velocity analysis was performed at 20°C on a Beckman model-E analytical ultracentrifuge. Sedimentation patterns were acquired with an on-line Rayleigh system (29) and converted into concentration versus radius every 20 s. Sapphire cell windows were used, and the camera was focused at the 2/3 plane of the cell. The apparent sedimentation coefficient distribution functions were computed as described by Stafford (30, 31).
Electron Microscopy.
Myosin sample (≈3 μg/ml) in a solution containing 2 mM MgCl2, 0.3 mM DTT, 1 mM ATP, 20 mM Tris·HCl (pH 7.5), 30% glycerol, and 0.01 or 0.4 M KCl was adsorbed onto a freshly cleaved mica surface for 30 s. Unbound protein was rinsed away, then the specimen was stabilized by brief exposure to uranyl acetate as described (32). The specimens were visualized by the rotary shadowing technique according to Mabuchi (33) with an electron microscope (Phillips Electronic Instruments, Mahwah, NJ; model EM 300) at 60 kV.
Statistical Analysis.
Student’s t test was used for statistical comparison of mean values. A value of P < 0.01 was considered to be significant.
RESULTS
Expression and Purification of the Chimeric Myosin.
The skeletal/smooth chimeric MHC contains 1104 amino acid residues with calculated molecular mass of 127 kDa, thus containing entire S1 and S2. We chose to produce the heavy chain construct containing both S1 and S2 portions as it has been shown that the two-headed structure of myosin is critical for the phosphorylation-mediated regulation of smooth myosin function (23, 24, 34), and the S2 portion is necessary for a stable two-headed structure (23, 24, 27).
The chimeric MHC was coexpressed with smooth RLC and ELC. The purified chimeric myosin contained an MHC with an apparent molecular mass of 127 kDa, a 20-kDa RLC, and a 17-kDa ELC, respectively (Fig. 2A). Stoichiometry of the three polypeptides was 1:1.11:1.02, based upon gel densitometry, indicating that the recombinant myosin consisted of one heavy chain and one of each class of light chains, as in naturally isolated myosin. To confirm the authenticity of the chimeric MHC, immunoblot analysis was performed using antibodies recognizing the 50-kDa motor head domain of skeletal MHC and the S2 portion of smooth MHC, respectively. The former antibody recognized the chimeric MHC and the naturally isolated skeletal MHC but not the smooth MHC (Fig. 2B). The antibody recognizing the 25-kDa N-terminal domain of skeletal MHC also reacted with the chimeric MHC (data not shown). On the other hand, the anti-smooth muscle S2 antibody reacted with both chimeric and smooth myosins but not with skeletal myosin (Fig. 2C). These results clearly show that the purified chimeric myosin contains the skeletal myosin globular motor domain and the smooth myosin S2 domain, as expected from the cDNA construct.
Figure 2.

Isolation of the chimeric myosin.(A) SDS/PAGE (7.5–12% gel gradient) of myosin fragments. Lanes: 1, skeletal muscle myosin; 2, skeletal muscle HMM; 3, purified skeletal/smooth chimeric myosin; 4, smooth muscle myosin; 5, smooth muscle HMM; 6, molecular weight standard. (B) Immunoblot of the myosin fragments using a monoclonal antibody, 50KD, which recognizes a 50-kDa fragment of chicken skeletal muscle myosin S1. Lanes: 1, skeletal muscle myosin; 2, chimeric myosin; 3, smooth muscle myosin. (C) Immunoblot of the myosin fragments using a monoclonal antibody, MM9, which recognizes the S2 portion of chicken smooth muscle myosin. Lanes: 1, skeletal muscle myosin; 2, chimeric myosin; 3, smooth muscle myosin. (D) Nondenaturing gel electrophoresis of myosin fragments. Lanes: 1, smooth muscle HMM; 2, chimeric myosin; 3, smooth muscle myosin S1.
It has been shown that the two-headed structure is critical for the phosphorylation-dependent regulation of smooth muscle myosin motor activity (23, 24, 34). Nondenaturing gel electrophoresis was employed to determine whether the expressed chimeric myosin forms a double-headed structure or a single-headed structure. As shown in Fig. 2D, the entire fraction of the chimeric myosin migrated at the same position as naturally isolated smooth HMM but much slower than naturally isolated smooth S1, suggesting that the chimeric myosin forms a stable double-headed structure.
Vmax of the Actin-Activated ATPase Activity of the Chimeric Myosin.
The actin-activated ATPase activity of the chimeric myosin was significantly enhanced by RLC phosphorylation, as was the naturally isolated smooth HMM but not the skeletal HMM (Table 1), whereas the apparent dissociation constant for actin, Ka, of the chimeric myosin was similar to skeletal HMM (data not shown). On the other hand, the rate of ATP hydrolysis of the phosphorylated form of the chimeric myosin was much higher than that of the smooth HMM and did not significantly differ from that of the skeletal HMM. These results clearly demonstrate that the C-terminal domain of S1 composed of a long α-helix of the MHC with two classes of light chains is responsible for regulation by phosphorylation of the RLC regardless of the nature of the globular motor domain. The results also suggest that the rate of ATP hydrolysis is solely determined by the nature of the globular motor domain, and the C-terminal domain of S1 including the two classes of light chains does not apparently influence the ATPase cycle rate.
Table 1.
Actin-activated ATPase activity of the myosin fragments
| Myosin fragments |
Vmax, S−1
|
|
|---|---|---|
| Phosphorylated | Dephosphorylated | |
| Skeletal HMM | 4.67 ± 0.40 | 4.50 ± 0.41 |
| Smooth HMM | 0.86 ± 0.09 | 0.06 ± 0.01 |
| Chimeric myosin | 4.32 ± 0.03 | 0.48 ± 0.01 |
Actin-activated ATPase activity was measured at 25°C in 0.01 mg/ml myosin fragment, 0.03 mM ATP, 60 mM KCl, 30 mM Tris·HCl, pH 7.5, 8 mM MgCl2 with various concentrations of F-actin. To measure the activity of phosphorylated myosin, 0.2 mM CaCl2, 15 μg/ml myosin light chain kinase, and 10 μg/ml calmodulin were added, whereas 1 mM EGTA was added for the dephosphorylated one. Results are means ± SD of three independent preparations. A computed nonlinear least-squares curve-fitting program was used to estimate the maximum actin-activated ATPase activity (Vmax) and the apparent dissociation constant for actin (Ka) based on the equation V = Vmax/(1 + Ka/[actin]).
Actin-Translocating Activity of the Chimeric Myosin.
Table 2 shows the actin-translocating velocity of the recombinant myosin fragments as well as the naturally isolated HMMs. Consistent with the actin-activated ATPase activity, the in vitro motility activity was completely regulated for the naturally isolated smooth HMM but not for the skeletal HMM. The recombinant smooth 128-kDa MHC fragment containing both light chains showed the same phosphorylation-dependent actin-translocating activity as the naturally isolated smooth HMM, indicating that the expressed myosin is properly folded in Sf9 cells and retains native conformation. Consistent with the actin-activated ATPase activity, actin-translocating activity of the chimeric myosin was completely dependent on the RLC phosphorylation. On the other hand, the actin-translocating velocity of the chimeric myosin was much slower than that of the skeletal HMM in contrast to the ATPase activity, indicating that the actin sliding velocity is not solely determined by the globular motor domain but is influenced by the C-terminal regulatory domain of myosin head.
Table 2.
Sliding velocity of actin filament on the myosin fragments
| Myosin fragment | Sliding velocity,* μm/s
|
|
|---|---|---|
| Phosphorylated | Dephosphorylated | |
| Skeletal HMM | 4.78 ± 0.78 | 4.67 ± 0.58 |
| Smooth HMM | 0.58 ± 0.06 | 0 |
| Expressed smooth 128-kDa fragment† | 0.59 ± 0.11 | 0 |
| Chimeric myosin | 0.12 ± 0.03 | 0 |
Actin movement was observed in 30 mM KCl, 5 mM MgCl2, 25 mM imidazole, 1 mM EGTA, 1% 2-mercaptoethanol, 0.5% metylcellulose, 4.5 mg/ml glucose, 216 μg/ml glucose oxidase, 36 μg/ml catalase, 2 mM ATP, pH 7.5 at 25°C. All values are mean velocities ± SD for three independent preparations. For each preparation, 30–40 actin filaments were measured to obtain an average velocity for each condition.
Effect of Ionic Strength on the ATPase Activity of the Chimeric Myosin.
It is known that smooth myosin or HMM forms a flexed conformation in low ionic strength (5–9) which is characterized by low Ca2+ and Mg2+ ATPase activity of myosin (7, 10, 12, 35). As KCl concentration increased, the EDTA-ATPase activity of the chimeric myosin increased as is found in both skeletal and smooth HMMs (data not shown). On the other hand, the Ca2+-ATPase activity of the chimeric myosin was decreased at low ionic strength similar to the smooth HMM (Fig. 3). In contrast, the Ca2+-ATPase activity of the skeletal HMM increased at low ionic strength. Since depression of Ca2+-ATPase is known to be correlated to the formation of the flexed conformation (7), the result suggests that the chimeric myosin forms a flexed conformation as smooth HMM does.
Figure 3.

KCl dependence of Ca2+-ATPase activity of myosin fragments. Ca2+-ATPase activity was measured in 0.01 mg/ml myosin fragment, 5 mM CaCl2, 3 mM EDTA, 50 mM Tris·HCl (pH 7.5), and various concentrations of KCl at 25°C. The reaction was started by adding ATP, and the liberated inorganic phosphate was measured as described (20).
Effect of Ionic Strength on the Sedimentation Velocity of the Chimeric Myosin.
To confirm the conformational transition of the chimeric myosin, sedimentation patterns for smooth HMM and chimeric myosin were measured in 0.01 and 0.4 M KCl (Fig. 4). The sedimentation pattern of the smooth HMM revealed a single symmetric peak under both conditions, whose sedimentation coefficient (s20,w value) increased from 7.73 to 8.29 with decreasing KCl concentration. This is consistent with the observation by Suzuki et al. (35) demonstrating the conformational transition of smooth HMM. Similarly, the s20,w value of the chimeric myosin increased from 7.74 to 8.37 as the KCl concentration decreased, indicating that the chimeric myosin also adopts a flexed conformation at low ionic strength.
Figure 4.

Apparent sedimentation coefficient distributions for myosin fragments. Sedimentation velocity was determined in solutions containing 2 mM MgCl2, 0.3 mM DTT, 1 mM ATP, 20 mM Tris·HCl (pH 7.5), 0.1 mg/ml myosin fragment, and either 0.01 M or 0.4 M KCl. The velocity runs were carried out at 56,000 rpm at 20°C for smooth HMM (A) and chimeric myosin (B). The x axis, 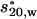 is the apparent sedimentation coefficient computed as described by Stafford (30, 31).
is the apparent sedimentation coefficient computed as described by Stafford (30, 31).
Electron Micrographs of the Chimeric Myosin.
Smooth HMM and chimeric myosin were also subjected to electron microscopic study to further confirm the conformational transition. Consistent with the result of nondenaturing gel electrophoresis, the chimeric myosin was predominantly double-headed (Fig. 5 A and B). The structure of the chimeric myosin molecules could be categorized into two types, i.e., an extended form and a flexed form. In the extended form, the heads extended away from the tail, whereas the heads oriented back toward the tail in the flexed form. To statistically analyze the distribution of these conformations, the height of the globule/α-helix junction from the C-terminal end of the tail (h) was measured for 300 molecules under each condition. The average h value for the chimeric myosin in 0.01 M KCl (46.1 ± 4.1 nm) was significantly smaller than that in 0.4 M KCl (52.7 ± 2.8 nm), indicating that the chimeric myosin tends to adopt a flexed structure at low ionic strength, whereas a high ionic strength favors the formation of a extended form (Fig. 5 C and D), as was found for naturally isolated smooth HMM (data not shown). Based upon these findings, it is concluded that the formation of a folded structure of smooth myosin is determined by the regulatory domain and is not influenced by the nature of the globular domain.
Figure 5.

Electron micrographs of the chimeric myosin molecules. The rotary-shadowed images of the chimeric myosin in 0.4 M KCl (A) and 0.01 M KCl (B). (A and B, ×100,000; bars = 0.1 μm.) The distribution of the height of the globule/α-helix junction (h) in 0.4 M KCl and 0.01 M KCl are summarized in C and D, respectively. The height was measured according to the schematic drawings for the extended form (E) and the flexed form (F).
DISCUSSION
The present results clearly demonstrate that the light chain-associated regulatory region solely confers the phosphorylation mediated regulation of the smooth muscle myosin motor activity. Recently, it was found that the two-headed structure of myosin is critical for the phosphorylation-mediated regulation (23, 24, 34). Therefore, the present results imply that the interaction between the two heads at the C-terminal portion of S1 (the regulatory domain) but not at the N-terminal globular motor domain is operating and critical for the regulation of myosin motor activity by phosphorylation.
The production of chimeric smooth HMM, in which the actin-binding loop is replaced with that of striated muscle myosin, was recently reported (36). Their chimeric HMM was unregulated constitutively, and it was claimed that the actin-binding loop could confer isoform-specific properties onto myosin. However, these substitutions of actin-binding loop significantly decreased both actin-activated ATPase activity and actin-translocating activity of the phosphorylated forms. Hence, it is plausible that the artificial substitution in the closely packed globular motor domain of myosin affected the proper folding of this region. Therefore, it is possible that the chimeric myosins with foreign actin-binding loop are unregulated due to the disruption of myosin conformation, which is crucial for physiological regulation of smooth myosin. In contrast, the phosphorylated form of the chimeric myosin produced in this study has high actin-activated ATPase activity similar to that of skeletal HMM, indicating that the substitution of the C-terminal long α-helix domain of S1 and S2 does not affect the proper folding of the globular motor domain.
It is known that a folded structure of smooth myosin is destabilized by light chain phosphorylation (8, 13), whereas stabilization of the 6S conformation is not directly coupled with activation of smooth myosin in vitro (37–39). Because folded myosin cannot form thick filament, it has been hypothesized that phosphorylation could dynamically control filament assembly in certain cell types (7, 13), whereas assembly/disassembly is unlikely to play a major role in regulating the contraction/relaxation cycle in smooth muscle cells (40, 41). Several studies have revealed the molecular basis of the conformational transition which is unique to smooth muscle and nonmuscle myosins. Quite recently, it was reported that two-headed structure is also critical for the conformational transition (24, 42). It was shown that Arg16 and Arg13 of the RLC are critical for the formation of a folded structure of smooth myosin (38, 39), and it was proposed that these basic residues interact with the acidic residues in myosin tail to stabilize a folded conformation and that the negative charges of the phosphate moiety on Ser19 interfere with this charge interaction to destabilize the folded structure (38). Consistent with the previous finding, the present results demonstrate that the light chain-associated region of myosin is critical and sufficient to dictate 10S–6S conformational transition.
Another important finding is that ATPase cycle rate, thus ATP hydrolysis rate or phosphate release rate, is uniquely determined by the globular head domain of myosin. It should be noted that the actin-translocating activity of the phosphorylated chimeric myosin was significantly slower than that of the smooth HMM. Uncoupling of the ATPase and actin-translocating activity has been observed in Dictyostelium myosin mutants (43), truncated S1s (15, 16), and single-headed smooth myosins (24, 34). It is generally accepted that the actomyosin ATPase cycle is divided into two states, i.e., the weak binding state and the strong binding state (44). Upon ATP binding, myosin forms the weak binding state, which is then followed by ATP hydrolysis. The conversion from the weak binding state to the strong binding state is achieved by phosphate release from myosin. The rate-limiting step of ATPase cycle is thought to be in the weak binding state (45). On the other hand, the actin sliding velocity is related to the strong bound state but is not related much to the weakly bound state. Therefore, it may be reasonable that actin translocation and ATPase activity are determined by distinct structural elements. The present finding is consistent with this notion and supports the idea that the actin-translocating velocity is not solely determined by the globular motor head domain, in contrast to the enzymatic activity of myosin. While no artificial amino acid residues are introduced into the chimeric HMM, we cannot exclude the possibility that the two segregated domains interact with each other and that the actin sliding activity is somewhat inhibited due to the artificial junction created in this chimera. Further study will be required to clarify the molecular basis of these uncouplings between ATPase and motility activities.
Acknowledgments
We thank Dr. Jeffrey Robbins (Children’s Hospital Medical Center) for providing a chicken skeletal MHC cDNA. We also thank Dr. Morris Burke (Case Western Reserve University) for providing anti-skeletal myosin antibodies. We are grateful to Dr. Motoi Matsuura for his valuable comments on this study. This work was supported by Grants AR 41653, HL37117, and HL47530 from the National Institutes of Health. M.S. is supported by research fellowships from the American Heart Association, Massachusetts Affiliate, Inc., the Fukuda Memorial Foundation, and the Ueda Memorial Trust Fund for Research of Heart Diseases.
Footnotes
Abbreviations: HMM, heavy meromyosin; S1, myosin subfragment 1; S2, myosin subfragment 2; MHC, myosin heavy chain; ELC, essential light chain; RLC, regulatory light chain.
References
- 1.Mooseker M S, Cheney R E. Annu Rev Cell Dev Biol. 1995;11:633–675. doi: 10.1146/annurev.cb.11.110195.003221. [DOI] [PubMed] [Google Scholar]
- 2.Zot A S, Potter J D. Annu Rev Biophys Biophys Chem. 1987;16:535–559. doi: 10.1146/annurev.bb.16.060187.002535. [DOI] [PubMed] [Google Scholar]
- 3.Hartshorne D J. In: Physiology of the Gastrointestinal Tract. 2nd Ed. Johnson L R, editor. Vol. 1. New York: Raven; 1987. pp. 423–482. [Google Scholar]
- 4.Sellers J R, Adelstein R A. In: The Enzymes. Boyer P, Krebs E G, editors. Vol. 18. San Diego: Academic; 1987. pp. 381–418. [Google Scholar]
- 5.Trybus K M, Huiatt T M, Lowey S. Proc Natl Acad Sci USA. 1982;79:6151–6155. doi: 10.1073/pnas.79.20.6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Onishi H, Wakabayashi T. J Biochem (Tokyo) 1982;92:871–879. doi: 10.1093/oxfordjournals.jbchem.a134001. [DOI] [PubMed] [Google Scholar]
- 7.Ikebe M, Hinkins S, Hartshorne D J. Biochemistry. 1983;22:4580–4587. doi: 10.1021/bi00288a036. [DOI] [PubMed] [Google Scholar]
- 8.Craig R, Smith R, Kendrick-Jones J. Nature (London) 1983;302:436–439. doi: 10.1038/302436a0. [DOI] [PubMed] [Google Scholar]
- 9.Trybus K M, Lowey S. J Biol Chem. 1984;259:8564–8571. [PubMed] [Google Scholar]
- 10.Cross R A, Cross K E, Sobieszek A. EMBO J. 1986;5:2637–2641. doi: 10.1002/j.1460-2075.1986.tb04545.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Trybus K M, Lowey S. J Biol Chem. 1988;263:16485–16492. [PubMed] [Google Scholar]
- 12.Trybus K M, Chatman T A. J Biol Chem. 1993;268:4412–4419. [PubMed] [Google Scholar]
- 13.Suzuki H, Onishi H, Takahashi K, Watanabe S. J Biochem (Tokyo) 1978;84:1529–1542. doi: 10.1093/oxfordjournals.jbchem.a132278. [DOI] [PubMed] [Google Scholar]
- 14.Rayment I, Rypniewski W R, Schmidt-Base K, Smith R, Tomchick D R, Benning M M, Winkelman D A, Wesenberg G, Holden M M. Science. 1993;261:50–58. doi: 10.1126/science.8316857. [DOI] [PubMed] [Google Scholar]
- 15.Itakura S, Yamakawa H, Yano-Toyoshima Y, Ishijima A, Kojima T, Harada Y, Yanagida T, Wakabayashi T, Sutoh K. Biochem Biophys Res Commun. 1993;196:1504–1510. doi: 10.1006/bbrc.1993.2422. [DOI] [PubMed] [Google Scholar]
- 16.Waller G S, Ouyang G, Swafford J, Vibert P, Lowey S. J Biol Chem. 1995;270:15348–15352. doi: 10.1074/jbc.270.25.15348. [DOI] [PubMed] [Google Scholar]
- 17.Spudich J A, Watt S. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 18.Ikebe M, Stepinska M, Kemp B E, Means A R, Hartshorne D J. J Biol Chem. 1987;262:13828–13834. [PubMed] [Google Scholar]
- 19.Walsh M P, Hinkins S, Dabrowska R, Hartshorne D J. Methods Enzymol. 1983;99:279–288. doi: 10.1016/0076-6879(83)99063-8. [DOI] [PubMed] [Google Scholar]
- 20.Ikebe M, Hartshorne D J. J Biol Chem. 1985;260:13146–13153. [PubMed] [Google Scholar]
- 21.Ikebe M, Hartshorne D J. Biochemistry. 1985;24:2380–2387. doi: 10.1021/bi00330a038. [DOI] [PubMed] [Google Scholar]
- 22.Margossian S S, Lowey S. Methods Enzymol. 1982;85:55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 23.Matsu-ura M, Ikebe M. FEBS Lett. 1995;363:246–250. doi: 10.1016/0014-5793(95)00326-5. [DOI] [PubMed] [Google Scholar]
- 24.Sata M, Matsuura M, Ikebe M. Biochemistry. 1996;35:11113–11118. doi: 10.1021/bi960435s. [DOI] [PubMed] [Google Scholar]
- 25.Kamisoyama H, Araki Y, Ikebe M. Biochemistry. 1994;33:840–847. doi: 10.1021/bi00169a027. [DOI] [PubMed] [Google Scholar]
- 26.Sweeney H L, Straceski A J, Leinwand L A, Tikunov B A, Faust L. J Biol Chem. 1993;269:1603–1605. [PubMed] [Google Scholar]
- 27.Trybus K M. J Biol Chem. 1994;269:20819–20822. [PubMed] [Google Scholar]
- 28.Higashihara M, Ikebe M. FEBS Lett. 1990;263:241–244. doi: 10.1016/0014-5793(90)81383-y. [DOI] [PubMed] [Google Scholar]
- 29.Yphantis D A, Lary J W, Stafford W F, Liu S, Olsen P H, Hayes D B, Moody T P, Ridgeway T M,, Lyons D A, Laue T M. In: Acquisition and Interpretation of Data for Biological and Synthetic Polymer Systems. Schuster T M, Laue T M, editors. Boston: Birkhaeuser; 1994. pp. 209–226. [Google Scholar]
- 30.Stafford W F. Anal Biochem. 1992;203:295–301. doi: 10.1016/0003-2697(92)90316-y. [DOI] [PubMed] [Google Scholar]
- 31.Stafford W F. Methods Enzymol. 1994;240:478–501. doi: 10.1016/s0076-6879(94)40061-x. [DOI] [PubMed] [Google Scholar]
- 32.Mabuchi K. J Struct Biol. 1991;107:22–28. doi: 10.1016/1047-8477(91)90027-t. [DOI] [PubMed] [Google Scholar]
- 33.Mabuchi K. J Struct Biol. 1990;103:249–256. doi: 10.1016/1047-8477(90)90043-c. [DOI] [PubMed] [Google Scholar]
- 34.Cremo C R, Sellers J R, Facemyer K C. J Biol Chem. 1995;270:2171–2175. doi: 10.1074/jbc.270.5.2171. [DOI] [PubMed] [Google Scholar]
- 35.Suzuki H, Stafford W F, III, Slayter H S, Seidel J C. J Biol Chem. 1985;260:14810–14817. [PubMed] [Google Scholar]
- 36.Rovner A S, Freyzon Y, Trybus K M. J Biol Chem. 1995;270:30260–30263. doi: 10.1074/jbc.270.51.30260. [DOI] [PubMed] [Google Scholar]
- 37.Trybus K M. J Cell Biol. 1989;109:2887–2894. doi: 10.1083/jcb.109.6.2887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ikebe M, Ikebe R, Kamisoyama H, Reardon S, Schwonek J P, Sanders C R, II, Matsuura M. J Biol Chem. 1994;269:28173–28180. [PubMed] [Google Scholar]
- 39.Sweeney H L, Yang Z, Zhi G, Stull J T, Trybus K M. Proc Natl Acad Sci USA. 1994;91:1490–1494. doi: 10.1073/pnas.91.4.1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horowitz A, Trybus K M, Bowman D S, Fay F S. J Cell Biol. 1994;126:1195–1200. doi: 10.1083/jcb.126.5.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Somlyo A V, Butler T M, Bond M, Somlyo A P. Nature (London) 1981;294:567–569. doi: 10.1038/294567a0. [DOI] [PubMed] [Google Scholar]
- 42.Olney J J, Sellers J R, Cremo C R. J Biol Chem. 1996;271:20375–20384. doi: 10.1074/jbc.271.34.20375. [DOI] [PubMed] [Google Scholar]
- 43.Uyeda T Q P, Ruppel K M, Spudich J A. Nature (London) 1994;368:567–569. doi: 10.1038/368567a0. [DOI] [PubMed] [Google Scholar]
- 44.Eisenberg E, Hill T L. Science. 1985;227:999–1006. doi: 10.1126/science.3156404. [DOI] [PubMed] [Google Scholar]
- 45.Rosenfeld S S, Taylor E W. J Biol Chem. 1984;259:11920–11929. [PubMed] [Google Scholar]