

Abstract
E-selectin is a major adhesion molecule expressed by endothelial cells (ECs), which are exposed to shear stress and neighboring smooth muscle cells (SMCs). We investigated the mechanisms underlying the modulation of EC E-selectin expression by SMCs and shear stress. SMC coculture induced rapid and sustained increases in expression of E-selectin and phosphorylation of interleukin-1 (IL-1) receptor-associated kinase glycoprotein-130, as well as the downstream mitogen-activated protein kinases (MAPKs) and Akt. By using specific inhibitors, dominant-negative mutants, and small interfering RNA, we demonstrated that activations of c-Jun-NH2-terminal kinase (JNK) and p38 of the MAPK pathways are critical for the coculture-induced E-selectin expression. Gel shifting and chromatin immunoprecipitation assays showed that SMC coculture increased the nuclear factor-κB (NF-κB)–promoter binding activity in ECs; inhibition of NF-κB activation by p65-antisense, lactacystin, and N-acetyl-cysteine blocked the coculture-induced E-selectin promoter activity. Protein arrays and blocking assays using neutralizing antibodies demonstrated that IL-1β and IL-6 produced by EC/SMC cocultures are major contributors to the coculture induction of EC signaling and E-selectin expression. Preshearing of ECs at 12 dynes/cm2 inhibited the coculture-induced EC signaling and E-selectin expression. Our findings have elucidated the molecular mechanisms underlying the SMC induction of EC E-selectin expression and the shear stress protection against this SMC induction.
Introduction
During the development of atherosclerotic lesions, vascular smooth muscle cells (SMCs) change from their physiologic contractile phenotype to the pathophysiologic synthetic phenotype and migrate into the intima, where they release proinflammatory cytokines and interact with vascular endothelial cells (ECs) to regulate their gene expression and function, including the modulation of leukocyte recruitment.1–3 ECs are constantly subjected to blood flow–induced shear stress, which can modulate leukocyte-EC interaction and the subsequent leukocyte extravasation into inflamed tissue, mainly by modulating EC surface expression of adhesion molecules.3,4
E-selectin is a major EC adhesion molecule that regulates binding and extravasation of leukocytes from the bloodstream to sites of inflammation. The effects of cytokines and shear stress on EC E-selectin expression have been extensively studied. The E-selectin gene is rapidly expressed by ECs in response to proinflammatory cytokines, and it is more responsive to disturbed and oscillatory flows5,6 than to laminar shear stress.7 Most studies on effects of shear stress on EC gene expression have been performed on EC monolayers, which may not reflect the in vivo environment of ECs, which exist in close proximity to SMCs. By using our newly developed EC/SMC coculture flow system8 in which ECs and SMCs are grown on opposite sides of a porous membrane, we demonstrated that coculture with SMCs induced E-selectin expression in ECs under static condition and that this coculture-induced E-selectin expression was inhibited by application of shear stress (12 dynes/cm2) to ECs.8 These results suggest a protective role of shear stress in vascular homeostasis by inhibiting the proinflammatory gene expression in ECs located in close proximity to SMCs. The aim of this investigation was to elucidate the mediator(s) and signaling pathway(s) that regulate the SMC-induced E-selectin expression in ECs and the mechanism of its inhibition by shear stress.
To gain insights into the mechanisms by which SMCs and shear stress regulate EC E-selectin expression, we used a cytokine protein array that contains antibodies against 120 cytokines and other proteins to analyze the proinflammatory factors produced by EC/SMC coculture. We found that the cytokines interleukin-1β (IL-1β) and IL-6 produced by EC/SMC can exert paracrine effects on ECs to elevate their E-selectin expression. The E-selectin expression induced by IL-1β and IL-6 produced by EC/SMC is mediated through the receptor-interacting molecules IL-1 receptor-associated kinase (IRAK) and glycoprotein-130 (gp130), the intracellular signaling cascades c-Jun-NH2-terminal kinase (JNK) and p38 mitogen-activated protein kinase (MAPK), and the transcription factor nuclear factor-κB (NF-κB). Before coculture with SMCs, pre-exposure of ECs to a high shear stress at 12 dynes/cm2, but not a low shear stress at 0.5 dynes/cm2, inhibits the coculture-induced signaling and E-selectin expression. Our findings provide a molecular basis for the mechanisms by which (1) SMCs induce E-selectin expression in ECs in close proximity and (2) high shear stress inhibits this SMC-induced E-selectin expression to effect its protective role in vascular homeostasis.
Materials and methods
Approval for these studies was obtained from the Institutional Review Board of National Health Research Institutes of Taiwan.
Materials
Mouse monoclonal antibodies against extracellular signal-regulated kinase 2 (ERK2; sc-1647), JNK1 (sc-7345), IκBα (sc-1643), p50 (sc-8414), p65 (sc-8008), and p-Tyr (sc-508), mouse monoclonal phospho-ERK (sc-7383), and phospho-JNK (sc-6254) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibodies against p38 and Akt, mouse monoclonal phospho-p38 antibody, and rabbit polyclonal phospho-Akt antibody were purchased from Cell Signaling Technology (Beverly, MA). The monoclonal E-selectin antibody and neutralizing antibodies against IL-1β, IL-6, basic fibroblast growth factor (bFGF), monocyte chemotactic protein-1 (MCP-1), growth-related oncogene (GRO), regulated-on-activation normal T-cell–expressed and secreted chemokine (RANTES), stromal cell–derived factor (SDF-1), interferon-inducible T-cell-α chemoattractant (I-TAC), and IL-4 were obtained from R & D Systems (Minneapolis, MN). The rabbit polyclonal antibodies against gp130 and IRAK were obtained from Upstate (Lake Placid, NY). The E-selectin promoter construct was a gift from Dr P.E. DiCorleto (Case Western Reserve University School of Medicine). The catalytically inactive mutant of ERK2 (mERK), RasN17, Raf310, JNK(K-R), and RacN17 were previously described.9 The ERK-, JNK-, p38-, Akt-, IRAK-, and gp130-specific small interfering RNA (siRNA) and control siRNA were purchased from Invitrogen (Carlsbad, CA). All other chemicals of reagent grade were obtained from Sigma (St Louis, MO).
Cell culture
ECs were isolated from fresh human umbilical cords by collagenase perfusion10 and grown in Petri dishes in medium 199 (M199; Gibco, Grand Island, NY) supplemented with 20% fetal bovine serum (FBS; Gibco) for 3 days. Secondary cultures were used in all experiments. Third-passage human umbilical artery SMCs were obtained commercially (Clonetics, Palo Alto, CA) and maintained in F12K medium (Gibco) supplemented with 10% FBS. Cells between passages 4 to 6 were used.
Shear stress experiment
ECs were seeded onto the outer side of a 10-μm-thick membrane containing 0.4-μm pores (5 × 105 cells/cm2; Falcon cell culture inserts; Becton Dickinson, Lincoln Park, NJ) (precoated with fibronectin at 30 μg/cm2).8 After incubation in M199 containing 2% FBS for 24 hours, the membrane with ECs was incorporated into a parallel-plate flow chamber containing a polycarbonate insert holder8 and connected to a perfusion loop system for the application of shear stress at a high (HSS; 12 dynes/cm2) or low level (LSS; 0.5 dynes/cm2) for 4 or 24 hours.
Coculture of ECs and SMCs
After the completion of EC shearing, the opposite (inner) side of the membrane was seeded with SMCs (2 × 105 cells/cm2) under static condition, thus forming an EC/SMC coculture system (the adjacent-bilayer model; Figure 1A).8 Before seeding onto the membrane, the SMCs collected after trypsinization were incubated in an Ultra Low Attachment Microplate (Costar 3471; Corning Inc, Corning, NY) for 2 hours to eliminate the effect of trypsinization. Controls had ECs, but no SMCs on the opposite side of the membrane (EC/∅). To study the effect of separation of ECs and SMCs, the ECs seeded on the outer side of the membrane were separated from the SMCs plated on the bottom surface of the outer chamber by 1 mm (EC/M/SMC; the media-separation model; Figure 1B). ECs and SMCs were maintained in the shared culture medium containing 2% FBS.
Figure 1.

Preshearing of ECs at HSS, but not LSS, inhibits SMC-induced EC E-selectin expression. ECs were kept as controls (EC/∅) or cocultured with SMCs in an adjacent-bilayer model (EC/SMC) (A,C,E-G) or a media-separation model (EC/M/SMC) (B,D) for 4 hours (E,F), 24 hours (G), or the times indicated (C,D). The E-selectin mRNA (C-F) and surface protein (G) expressions of these ECs were determined by using Northern blot and flow cytometric analyses, respectively. In some experiments, ECs were presheared at HSS (12 dynes/cm2) for 4 hours (HS4) or 24 hours (HS24) or LSS (0.5 dynes/cm2) for 24 hours (LS24) before SMC coculture (E-G). Control ECs were cocultured with SMCs without preshearing (CL) (E-F). Data are presented as percentage changes in band density from control EC/℘ normalized to GAPDH RNA level (C-F) and are shown as mean ± standard of the mean (SEM) from 3 independent experiments. *P < .05 versus control EC/∅. #P < .05 versus control EC/SMC. Results of flow cytometric analysis (G) are representative of triplicate experiments with similar results. ECs incubated with FITC-conjugated control IgG or FITC-conjugated antibody alone were used as IgG or negative controls (ie, Blanks: B). Numbers are mean ± SEM of mean fluorescent intensity for all experiments determined by comparison with corresponding negative controls.
The procedures of (1) RNA isolation; (2) Northern blots; (3) reverse-transcription–polymerase chain reaction (RT-PCR); (4) real-time PCR; (5) Western blots; (6) flow cytometric analysis; (7) electrophoretic mobility shift assay (EMSA); (8) immunoprecipitation; (9) chromatin immunoprecipitation (ChIP) assay; (10) reporter gene construct, DNA plasmids, siRNA, transfection, and luciferase assays; (11) protein array assay for detecting cytokines in conditioned media; (12) antisense oligonucleotides; and (13) statistic analysis are provided in Document S1 (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Results
Pre-exposure of ECs to HSS, but not LSS, for 24 hours inhibits SMC-induced E-selectin expression in ECs
EC/SMC coculture in the adjacent-bilayer model (Figure 1A) induced an increase in E-selectin mRNA expression in ECs (detectable within 1 hour; Figure 1C). When ECs were separated from the cocultured SMCs by 1 mm filled with media (media-separation model; Figure 1B), the increase in the EC E-selectin mRNA expression was much slower (detected at 4 hours; Figure 1D). The increases in EC E-selectin mRNA expression in both coculture models were sustained for 24 hours. Pre-exposure of ECs to HSS at 12 dynes/cm2 for 24 hours, but not 4 hours, significantly inhibited the coculture-induced E-selectin mRNA expression (Figure 1E). However, LSS at 0.5 dynes/cm2 did not have such an inhibitory effect (Figure 1F). As controls, HSS and LSS per se did not alter the E-selectin mRNA expression in monocultured ECs in comparison to static cells (Figure S1A). Flow cytometric analysis showed that adjacent coculture for 24 hours resulted in an increase in expression of E-selectin protein on EC surface, with a mean fluorescence intensity of 99.8 compared with 11.3 in EC monocultures (Figure 1G). Twenty-four–hour preshearing of ECs at HSS reduced the coculture-induced E-selectin expression to a mean fluorescence intensity of 18.7.
SMC-induced EC expression of E-selectin and its inhibition by shear stress are mediated by the JNK and p38 pathways
The MAPK superfamily (ie, ERK, JNK, and p38) and phosphatidylinositol 3-kinase (PI3K)/Akt are known to regulate gene expression and cellular functions.11,12 The phosphorylation of ERK, JNK, p38, and Akt in ECs increased rapidly (within 5 minutes) after coculture with SMCs, reaching maximal levels at 10 minutes for Akt, 30 minutes for ERK and JNK, and 1 hour for p38 (Figure 2). After transient increases, the levels of phosphorylation decreased to nearly basal levels (ERK and p38 at 6 hours) or even below (JNK at 4 hours and Akt at 2 hours). To determine whether the SMC-induced E-selectin expression is mediated through the MAPK- or PI3K/Akt-dependent pathway, ECs were incubated with the specific inhibitor for ERK (PD98059; 30 μM), JNK (SP600125; 20 μM), p38 (SB203580; 10 μM), or PI3K/Akt (LY294002; 30 μM) for 1 hour before and during coculture with SMCs. The coculture-induced mRNA (Figure 3A) and surface protein (Figure 3B) expressions of EC E-selectin were significantly inhibited by SP600125 and SB203580 but not by PD98059 and LY294002. Treatment of ECs with SP600125 and SB203580 simultaneously did not result in additive inhibition in coculture-induced E-selectin mRNA (Figure 3A) and surface protein (data not shown) expressions. Pre-exposure of ECs to HSS, but not LSS, for 24 hours significantly inhibited the coculture-induced JNK and p38 phosphorylation (Figure 3C), suggesting that the inhibitory effect of HSS on coculture-induced E-selectin expression was attributable, at least in part, to its inhibition of the coculture-induced JNK and p38 activation. As controls, HSS and LSS per se had only minor effects on the activations of ERK, JNK, p38, and Akt in ECs in comparison to static controls (Figure S1B-E).
Figure 2.

Coculture with SMCs induces ECs to increase their phosphorylation. Coculture with SMCs induces ECs to increase their phosphoryalation of ERK (A), JNK (B), p38 (C), and Akt (D). ECs were kept as controls (EC/∅) or cocultured with SMCs in the adjacent-bilayer model (EC/SMC) for the times indicated, and the phosphorylations of their ERK, JNK, p38, and Akt were determined by using Western blot analysis. The amounts of phosphorylated ERK, JNK, p38, and Akt proteins in EC/SMC are presented as band densities (normalized to the total protein levels) relative to those in control EC/∅. The results are mean ± SEM from 3 independent experiments. *P < .05 versus control EC/∅.
Figure 3.

Induction of EC E-selectin expression by SMC-coculture and its inhibition by shear stress are mediated by JNK and p38 pathways. ECs were kept as controls (EC/∅) or cocultured with SMCs in the adjacent-bilayer model (EC/SMC) for 30 minutes (C,E), 4 hours (A,D), or 24 hours (B,F). Before kept as controls or coculture with SMCs, ECs were (1) pretreated with PD98059 (PD; 30 μM), SP600125 (SP; 20 μM), SB203580 (SB; 10 μM), or LY294002 (LY; 30 μM) individually or SP600125 and SB203580 simultaneously (CB) for 1 hour (A-B); (2) presheared at HSS (HS) or LSS (LS) for 24 hours (C); or (3) transfected with control siRNA or a specific siRNA of ERK, JNK, p38, or Akt (100 μmol/mL for each), or empty vector control PSRα (1 μg/mL), RasN17 (1 μg/mL), RacN17 (0.5 μg/mL), JNK(K-R) (1 μg/mL), Raf301 (1 μg/mL), or mERK (0.25 μg/mL) for 48 hours (D-F). Control ECs (CL) were cocultured with SMCs without any pretreatment (A) or preshearing (C). In some experiments, ECs were transfected with different siRNA and chimera at various concentrations for 48 hours (E). (F) ECs were cotransfected with chimera (2 μg) containing 540 bp (base pair) of E-selectin promoter region and the reporter gene luciferase. The mRNA (A,D) or surface protein (B) expression or promoter activity (F) of E-selectin or the expression or phosphorylation (C,E) of different MAPKs or Akt in these ECs was determined by using Northern blot (A), flow cytometric (B), Western blot (C,E), real-time PCR (D), or luciferase assay analysis (F), respectively, as described in “Materials and methods.” The results are shown as mean ± SEM from 3 or 4 separate experiments (A,C,D,F) or are representative of triplicate experiments with similar results (B,E). Data are presented as percentage changes in band density from control EC/∅ (A,C); normalized to 18S RNA (A) or JNK or p38 protein level (C). (B) ECs incubated with FITC-conjugated control IgG or FITC-conjugated antibody alone were used as IgG controls or negative controls (ie, Blanks: B). Numbers are mean ± SEM of mean fluorescent intensity for all experiments determined by comparison with corresponding negative controls. *P < .05 versus control EC/∅. #P < .05 versus control EC/SMC. $P < .05 versus cells transfected with control siRNA or empty vector control PSRα.
To further confirm the involvement of JNK and p38, but not ERK and PI3K/Akt, pathways in the modulation of E-selectin expression in ECs by SMC coculture and shear stress, we examined the effects of the dominant-negative mutants or siRNA of these signaling pathways on E-selectin expression in ECs cocultured with SMCs in the presence or absence of preshearing. The coculture-induced E-selectin mRNA expression was inhibited by transfections of ECs with JNK- or p38-specific siRNA (100 μmol/mL for each), the dominant-negative mutant of Ras (RasN17; 1 μg/mL) or Rac (RacN17; 0.5 μg/mL), or a catalytically inactive mutant of JNK [JNK(K-R); 1 μg/mL[, but not by transfecting with ERK- or Akt-specific siRNA (100 μmol/mL for each), the dominant-negative mutant of Raf-1 (Raf301; 1 μg/mL), or a catalytically inactive mutant of ERK2 (mERK; 0.25 μg/mL) (Figure 3D). The effectiveness of these treatments was validated: ERK-, JNK-, p38-, and Akt-specific siRNA (compared with control siRNA) caused a 75% reduction in ERK, JNK, p38, and Akt protein expressions, respectively (Figure 3E). The RasN17, RacN17, and JNK(K-R) inhibited coculture-induced JNK phosphorylation (compared with empty vector control PSRα), and the Raf301 and mERK caused inhibitions in coculture-induced ERK phosphorylation (compared with control PSRα) (Figure 3E). ECs transfected with the E-selectin–Luc showed an increase in promoter activity by the SMC coculture to 3.3-fold compared with the EC monocultures (Figure 3F). Preshearing of ECs at HSS, but not LSS, abolished this SMC-increased E-selectin promoter activity. Cotransfection of ECs with the empty vector control PSRα or control siRNA had no effect on coculture-induced E-selectin promoter activity. Cotransfection of ECs with p38-specific siRNA, RasN17, RacN17, or JNK(K-R) resulted in a significant inhibition in the coculture-induced E-selectin promoter activity. However, cotransfection with the Akt-specific siRNA, Raf301, or mERK had no effect on the SMC coculture inducibility. These results provide additional evidence that the JNK and p38 pathways play significant roles in the regulatory effects of SMCs and shear stress on E-selectin expression in ECs.
SMC-induced EC expression of E-selectin and its inhibition by shear stress are dependent on NF-κB
NF-κB is an important mediator for cellular responses to inflammatory stimuli.13 We examined the effects of interference with NF-κB expression on the SMC induction of E-selectin expression in ECs by using the antisense oligonucleotides to NF-κB subunit p65 (p65-antisense), the transcriptional inhibitor lactacystin, and the antioxidant N-acetyl-cysteine (NAC). The addition of both NAC and lactacystin to ECs transfected with the E-selectin–Luc completely abolished the coculture-induced E-selectin promoter activity (Figure 4A). Moreover, cotransfection of the cells with p65 antisense, but not p65 sense, oligonucleotides significantly inhibited the coculture-induced promoter activity. The results of EMSA showed that coculture with SMCs caused the NF-κB–DNA binding activity in the EC nucleus to increase within 10 minutes (Figure 4B) and remain elevated for at least 2 hours. This increase in NF-κB–DNA binding activity was accompanied by a concomitant decrease in IκBα, an inhibitory protein that prevents translocation of NF-κB dimers into the nucleus (Figure 4C). Preshearing of ECs at HSS (but not LSS) significantly inhibited the increase in NF-κB–DNA binding activity (Figure 4D) and the decrease in IκBα protein expression induced by SMC-coculture (Figure 4F). Preshearing at HSS for 24 hours had no effects on NF-κB–DNA binding activity in ECs in comparison to static controls; in contrast, preshearing at LSS resulted in an increase in NF-κB–DNA binding activity in ECs (Figure S1F). SP600125 and SB203580 did not have significant effects on the coculture-mediated changes in NF-κB–DNA binding activity (Figure 4E) and IκBα protein expression (Figure 4F). The formation of the NF-κB–DNA complex required the presence of a wild-type NF-κB–binding site, as demonstrated by the lack of direct binding to or competition by the mutant oligonucleotide, whereas excess unlabeled wild-type oligonucleotide was able to effectively compete with 32P-labeled oligonucleotide for NF-κB binding (Figure 4G). The specificity of this binding for NF-κB was further substantiated by the supershifting in gel mobility of the NF-κB–DNA complex after preincubation of nuclear proteins with an antibody to p65.
Figure 4.

NF-κB is involved in SMC and shear stress modulations of E-selectin expression in ECs. ECs were kept as controls (EC/∅) or cocultured with SMCs in the adjacent-bilayer model (EC/SMC) for 30 minutes (D-G,I), 24 hours (A), or indicated times (B,C,H). Before kept as controls or coculture with SMCs, ECs were (1) transfected with E-selectin–Luc for 48 hours and/or pretreated with antisense (p65-a; 1 μg/mL) or sense (p65-s; 1 μg/mL) oligonucleotides to the NF-κB subunit p65 for 24 hours or with lactacystin (Lac; 20 μM) or N-acetyl-cysteine (NAC; 20 mM) for 1 hour (A), (2) presheared at HSS (HS) or LSS (LS) for 24 hours (D,F,I), or (3) pretreated with SP600125 (SP; 20 μM) or SB203580 (SB; 10 μM) individually or in combination (CB) for 1 hour (E,F,I). Control ECs (CL) were cocultured with SMCs without any preshearing (D,F,I) or pretreatment (D,F,I). The E-selectin promoter activity (A), NF-κB–DNA binding activity (B,D,E,G), IκBα protein expression (C,F), and in vivo NF-κB–promoter binding (H-I) in these ECs were determined by using luciferase assay (A), EMSA (B,D,E,G), Western blot analysis (C,F), and ChIP assay (H,I), respectively, as described in “Materials and methods.” In some experiments (G), EMSA was performed using total nuclear extracts and 32P-labeled oligonucleotides containing wild-type (CL) or mutant (Mut) human E-selectin NF-κB binding sites. The specificity of the retarded complexes (NF-κB) was assessed by preincubating the nuclear extracts either with 20-fold excess unlabeled oligonucleotides (wild-type or mutant) as a competitor or with p50 and/or p65 antibodies (1 μg). Nuclear extracts preincubated with the p65 antibody show a super shift band (SH) (G). (B-G) The results are representative of 2 or 3 independent experiments with similar results. (A,H-I) Data are represented as mean ± SEM from 3 to 5 independent experiments. *P < .05 versus control EC/∅. #P < .05 versus control EC/SMC.
To further assess the in vivo regulation of NF-κB binding to the promoter regions of the E-selectin gene in ECs cocultured with SMCs in the presence or absence of preshearing, we performed ChIP assays in these ECs by using an antibody against p65 and the promoter-specific primers. ECs cocultured with SMCs increased the in vivo NF-κB binding to their E-selectin promoter as early as 10 minutes after the coculture, reaching maximal levels (∼ 4.8-fold in comparison to monocultured ECs) within 30 minutes (Figure 4H). The levels of NF-κB promoter binding declined after 2 hours of coculture with SMCs but still remained higher than monocultured ECs. This coculture-induced in vivo NF-κB binding to the E-selectin promoter was abolished by preshearing of ECs at HSS (but not LSS), and it was not blocked by pretreatment of ECs with SP600125, SB203580, or both (Figure 4I).
IL-1β and IL-6 produced by EC/SMC are the major factors contributing to the SMC-induced signaling and E-selectin expression in ECs
The increases in E-selectin expression in ECs by SMC coculture with the media-separation model suggest that the coculture caused the release of certain mediator(s) to exert paracrine effects on ECs to induce their E-selectin expression. To address this possible paracrine effect, we examined the expression levels of cytokines in the conditioned media of EC/SMC and EC/EC by using a human cytokine array system containing antibodies against 120 cytokines and other proteins (Figure S2; Table S1). Using this array, we identified the proteins whose expression was significantly different between the media bathing the EC/SMC versus EC/EC (ie, for P ≤ .05 and the mean coculture/monoculture ratio ≥ 2 or ≤ .5). By applying these criteria to analyze the results on 120 cytokines present on the array, we identified IL-1β and IL-6 as proteins that were released at significantly higher levels from EC/SMC than EC/EC (Figure 5A; Table S1), with the protein ratios of 4.16 ± 0.12 and 4.13 ± 0.19, respectively. Incubation of ECs with a neutralizing antibody against IL-6 or IL-1β (or in combination) significantly inhibited the coculture-induced E-selectin mRNA expression (Figure 5B), as well as the coculture-induced increases in JNK and p38 phosphorylation (Figure 5C) and NF-κB-DNA binding activity (Figure 5D), and decreases in IκBα protein levels (Figure 5E). In concert with these results, ChIP assays revealed that incubation of ECs with antibodies against IL-1β, IL-6, or both blocked the coculture-induced in vivo binding of NF-κB to the E-selectin promoter in ECs (Figure 5F). As controls, incubation of ECs with a neutralizing antibody against bFGF, MCP-1, GRO, RANTES, or SDF-1 (whose expression was elevated in the EC/SMC versus EC/EC), I-TAC (whose expression was not changed), or IL-4 (whose expression was reduced) did not inhibit the coculture-induced E-selectin mRNA expression (Figure S3).
Figure 5.

IL-1β and IL-6 produced by EC/SMC are the major factors contributing to the SMC-induced signaling and E-selectin expression in ECs. (A) Detection of protein levels of cytokines in conditioned media of EC/EC or EC/SMC coculture. The membranes spotted with antibodies against 120 different cytokines and other proteins (Figure S2) were incubated with 2-fold–diluted conditioned media of EC/EC or EC/SMC coculture and then incubated with a mixture of biotin-labeled antibodies, as described in “Materials and methods.” Signal detection by enhanced chemiluminescence (ECL) shows that the expression levels of IL-1β (spots in solid boxes) and IL-6 (spots in dash boxes) produced by EC/SMC were significantly higher than that of EC/EC. Results are representative of 4 independent experiments with similar results. (B-F) ECs were kept as controls (EC/∅) or cocultured with SMCs in the adjacent-bilayer model (EC/SMC) for 30 minutes (C-F) or 4 hours (B). In parallel experiments, ECs were preincubated with a neutralizing antibody against IL-1β or IL-6 (5 μg/mL for each) or their combination for 1 hour and then cocultured with SMCs in the presence of the antibodies. Control ECs were cocultured with SMCs in the presence of control IgG (CL). The E-selectin mRNA expression (B), JNK and p38 phosphorylations (C), NF-κB-DNA binding activity (D), IκBα protein expression (E), and in vivo NF-κB–promoter binding (F) were determined by using Northern blot analysis, Western blot analysis, EMSA, and ChIP assay, respectively, as described in “Materials and methods.” (B-C,E) Data are presented as percentage changes in band densities from control EC/∅ normalized to GAPDH RNA (B), JNK or p38 protein (C), or actin protein (E). The results shown are mean ± SEM from 3 to 4 independent experiments. *P < .05 versus control EC/∅. #P < .05 versus control EC/SMC.
IRAK and gp130 are involved in regulatory effects of SMC coculture and shear stress on EC E-selectin expression
Given our findings that IL-6 and IL-1β produced by EC/SMC cocultures are the major factors contributing to the SMC-induced EC signaling and E-selectin expression and that preshearing (at HSS) of ECs inhibited these SMC-induced changes, we postulated that the modulation of EC signaling and gene expression by SMC coculture and shear stress may be mediated via the IL-6 and IL-1β receptors in ECs. To test this possibility, we used the specific siRNA against gp130, which is an IL-6 receptor, and IRAK, which forms a complex with the receptor of IL-1β on its stimulation, to suppress the expressions of gp130 and IRAK and examined their effect on the SMC-induced E-selectin expression. Transfection of ECs with gp130- and IRAK-specific siRNA at concentrations of 100 μmol/mL reduced the expressions of gp130 and IRAK mRNAs by 80% compared with the cells transfected with control siRNA (Figure 6A). These reductions of gp130 (Figure 6B) and IRAK (Figure 6C) were accompanied by a decrease of the SMC-induced E-selectin expression in ECs. To further examine the effect of SMC coculture and shear stress on the activation of gp130 and IRAK in ECs, extracts of ECs cocultured with SMCs in the presence or absence of preshearing were immunoprecipitated with an antibody against gp130 or IRAK, followed by Western blot analysis with an antibody against p-Tyr or IRAK. Coculturing ECs with SMCs induced phosphorylation of gp130 and IRAK over the 30-minute period tested (Figure 6D), and such increases in gp130 and IRAK phosphorylation were inhibited by preshearing ECs at HSS (but not LSS) for 24 hours (Figure 6E). These results suggest that the effects of SMC coculture and shear stress on EC signaling and gene expression were mediated, at least in part, through the modulations of IL-6 and IL-1β receptor activation in ECs.
Figure 6.

IRAK and gp130 contribute to the modulation of EC signaling and E-selectin expression by SMC-coculture and shear stress. (A) ECs were kept as controls or transfected with control siRNA (siCL) or a specific siRNA of gp130 (sigp130) or IRAK (siIRAK) at indicated concentrations for 48 hours, and their gp130 or IRAK mRNA expression was determined by RT-PCR analysis, as described in “Materials and methods.” (B-E) ECs were kept as controls (EC/∅) or cocultured with SMCs in the adjacent-bilayer model (EC/SMC) for 10 minutes (E), 4 hours (B-C), or indicated times (D). Before coculture with SMCs, ECs were transfected with control siRNA or a specific siRNA of gp130 (B) or IRAK (C) (100 μmol/mL for each) for 48 hours or were presheared at HSS (HS) or LSS (LS) for 24 hours (E). Control ECs (CL) were cocultured with SMCs without transfection (B-C) or preshearing (E). The mRNA expressions of E-selectin and gp130 (B) or IRAK (C), or the phosphorylations of gp130 and IRAK (D-E) in these ECs were determined by using RT-PCR analysis or immunoprecipitation assay and Western blot analysis, respectively. Results in (A-E) are representative of triplicate experiments with similar results.
Discussion
E-selectin, which plays significant roles in atherosclerosis, is a major adhesion molecule expressed by vascular ECs, which exist in close proximity to vascular SMCs and are constantly subjected to blood flow–induced shear stress. Our present study aims at elucidating the molecular mechanisms underlying the roles of SMCs and shear stress in modulating E-selectin expression in ECs. Using our newly developed EC/SMC coculture flow system, we demonstrated that coculture of ECs with SMCs under static condition induced rapid and sustained increases of EC E-selectin expression. This increase in E-selectin expression was attributable, at least in part, to the paracrine effects of cytokines IL-1β and IL-6 produced by the EC/SMC coculture, acting on the ECs to activate their receptor-interacting molecules IRAK and gp130, as well as the downstream JNK/p38 and NF-κB pathways. Pre-exposure of ECs to a high level, but not a low level, of shear stress significantly inhibited such coculture-induced signaling and E-selectin expression. Our findings provide a molecular basis for the mechanisms underlying the SMC-induction of EC E-selectin expression and the protective function of shear stress against this SMC induction.
The paracrine effect exerted by SMCs on ECs to induce E-selectin expression was substantiated by the increased expression of E-selectin in ECs cocultured with SMCs in a media-separation model (Figure 1B,D), in which the 2 types of cells were separated by 1 mm of media. The time needed to induce E-selection expression, as expected, was longer (4 hours) than that in the adjacent-bilayer model (1 hour; Figure 1A,C). This slower induction of E-selectin in the media-separation model may be attributed to the longer distance between ECs and SMCs and the lower concentration of paracrine substances reaching the ECs, in comparison to the adjacent-bilayer model. By using protein arrays to perform a systematic analysis of the expression levels of cytokines produced in the cocultures, we identified IL-1β and IL-6 as proteins whose expression in the conditioned media of EC/SMC was significantly higher than that of EC/EC. The higher levels of IL-1β and IL-6 in the media of EC/SMC may not be due to increased productions of IL-1β and IL-6 by cocultured ECs, because the expressions of these 2 cytokines in ECs were not altered by their coculture with SMCs (data not shown). IL-1β and IL-6 have been shown to be highly expressed in human and experimental atherosclerotic lesions and are recognized as biomarkers of activated SMCs.14 Thus, the SMCs used in the present study exhibit characteristics resembling neointimal SMCs, which display a synthetic phenotype characterized by increased cytokine expression.2,3 Our recent data showed that SMCs under the condition of the present experiments had lower levels of expression of contractile marker proteins (eg, smooth muscle α-actin, myosin heavy chain, h-caldesmon, and calponin) than those cultured in the medium containing only 0.5% FBS.4 We have found by using protein arrays that the SMCs used in the present study had higher levels of IL-1β and IL-6 expression than the ECs (data not shown). Treatment of monocultured ECs with IL-1β or IL-6, or with the supernatant of SMCs, mimicked the effect of SMC coculture in inducing EC E-selectin expression (data not shown). These results suggest that the SMCs in our present study were in a synthetic phenotype, which may affect the adjacent ECs to induce their proinflammatory gene expression and function through the paracrine release of IL-1β and IL-6.
By using neutralizing antibodies against IL-1β and IL-6, we have demonstrated that these 2 cytokines produced by EC/SMC are the major factors contributing to the coculture-induced E-selectin expression in ECs. This coculture-induced E-selectin expression was mediated by the IRAK/gp130 and downstream JNK/p38 and NF-κB pathways. Several lines of evidence support this finding. First, ECs cocultured with SMCs induced a rapid phosphorylation of IRAK/gp130, which has been shown to activate several intracellular signaling pathways, including MAPKs, PI3K/Akt, and NF-κB.15 Second, coculture with SMCs induced rapid phosphorylations of ERK, JNK, p38, and Akt in ECs; however, only specific inhibitors to JNK and p38 (ie, SP600125 and SB203580) inhibited the coculture-induced E-selectin expression, suggesting that the activation of JNK/p38 is critical for the coculture-induced E-selectin expression. The involvement of the JNK/p38 pathway in coculture-induced E-selectin expression was confirmed by the inhibition of coculture-induced E-selectin mRNA expression and promoter activity in ECs by transfecting with a dominant-negative mutant of JNK, Rac, or Ras, or a specific siRNA of JNK or p38. Third, the results of EMSA and ChIP assays showed that the SMC coculture increased the binding activity and in vivo promoter binding of NF-κB in EC nuclei. This increase in NF-κB binding activity was accompanied by a reduction of IκBα. NF-κB inhibitors, including p65-antisense, lactacystin, and NAC, inhibited the coculture-induced E-selectin promoter activity, suggesting the involvement of NF-κB in the coculture-induced E-selectin expression. Finally, the inhibitory effects of neutralizing antibodies against IL-1β and IL-6 on the coculture-induced activations of JNK/p38 and NF-κB and E-selectin expression in ECs indicate that the effects of SMC coculture are mediated by the binding of IL-1β and IL-6 to their corresponding receptors in ECs. Moreover, the involvement of IRAK and gp130 in coculture-induced E-selectin expression was confirmed by the reduction of this E-selectin expression in ECs by transfecting with a specific siRNA of IRAK or gp130.
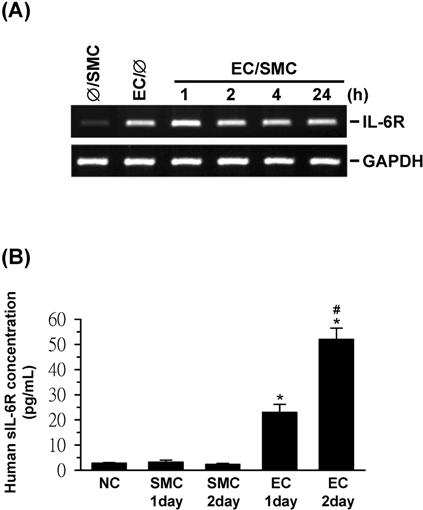
It has been demonstrated that IL-6 exerts its biologic activity through binding to its specific receptor (IL-6R) and gp130, which serves as a signal-transducing unit.16 However, some studies have suggested that ECs do not express IL-6R and require exogenous soluble IL-6R (sIL-6R) to trigger gp130 signaling,17 whereas others have reported that ECs do express IL-6R and that IL-6 can directly activate EC signaling and gene expression.18,19 These differences in results could be due to the functional and molecular heterogeneity that exists among different types of ECs.20 Because direct addition of IL-6 (2.5 ng/mL; Sigma) to EC monocultures without exogenous addition of sIL-6R can mimic the effects of SMC coculture in inducing EC activation of gp130, MAPKs, and NF-κB and the expression of E-selectin (data not shown), the results suggest that IL-6 may be able to exert direct effects on these signaling and gene expression in ECs. Using RT-PCR and enzyme-linked immunoabsorbent assay (ELISA) we have shown that the ECs used in the present study have constitutive expressions of IL-6R and sIL-6R, whereas the expressions of IL-6R and sIL-6R by SMCs were not detectable (Figure S4). It is probable that the constitutive expressions of IL-6R and sIL-6R by ECs can contribute to the SMC/IL-6 signaling in ECs.
Read et al21 have identified NF-κB and positive domain II (PDII), which contains a cAMP-responsive element/activating transcription factor (ATF)–like binding site, in the E-selectin promoter as responsive elements for expression of this gene induced by tumor necrosis factor-α (TNF-α). They showed that a heterodimer of transcription factors ATF-2 and c-JUN is constitutively bound to the PDII site, and that TNF-α stimulation of ECs induces marked activation of the JNK and p38 and their associations with c-JUN and ATF-2. Using immunoprecipitation assay, we have shown that coculture of ECs with SMCs for 30 minutes induces an increase in JNK/c-JUN and p38/ATF-2 associations (Figure S5), suggesting that the c-JUN and ATF-2 are downstream of JNK and p38 and may also be involved in SMC-induced E-selectin expression in ECs. Our results of EMSA using double-stranded oligonucleotides containing the PDII site (5′-GTACAATGATGTCAGAAACTCTGTC-3′)21 did not show specific DNA bindings of c-JUN/ATF-2 in the nucleus of ECs cocultured with SMCs. Thus, the c-JUN/ATF-2 responsive element in the E-selectin promoter in ECs in response to coculture with SMCs remains to be determined. In our present study, the specific inhibitors of JNK and p38 (ie, SP600125 and SB203580) did not inhibit the coculture-mediated increases in NF-κB–DNA binding activity and in vivo NF-κB–promoter binding, nor the decrease in the protein level of IκBα. This is in agreement with the findings by Read et al21 that NF-κB and JNK/p38 represent 2 separate signaling pathways, both of which are required for inflammatory cytokine responsiveness of E-selectin. It is possible that these 2 pathways are rapidly activated and converge on the E-selectin promoter to result in full activation of this gene in ECs by coculture with SMCs.
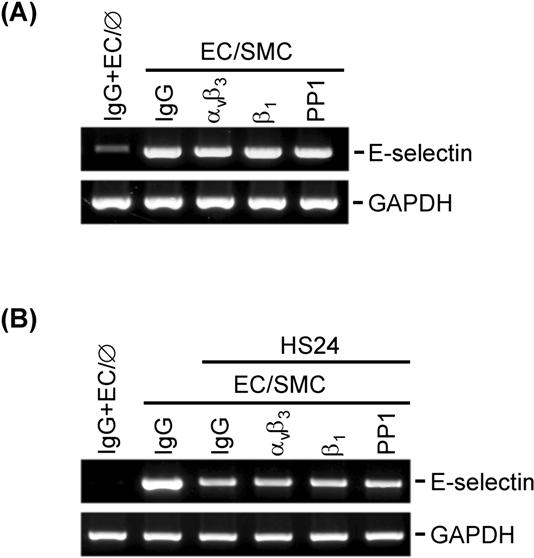
Our results indicate that the “preconditioning” of ECs by different levels of shear stress (HSS versus LSS) differentially modulates the response of EC gene expression to SMC coculture. The way in which HSS inhibits the EC E-selectin expression in cocultures is likely to be multifactorial. Blocking assays using antibodies against αvβ3 and β1 integrins, which are well-recognized mechanosenors in ECs in response to shear stress,22 did not eliminate the inhibitory effect of shear stress on SMC-induced E-selectin expression (Figure S6), suggesting that integrins may not be involved in the effect of shear stress on E-selectin expression in cocultured ECs. Because our results showed that HSS preshearing can inhibit the coculture-mediated activation of IRAK/gp130, JNK/p38, and NF-κB and reduction of IκBα, it is likely that the shear-mediated inhibition in coculture-induced E-selectin expression is attributable to the shear-mediated inhibition of the activation of these signaling pathways in cocultured ECs. In addition, our previous study showed that the SMC induction of proinflammatory genes in ECs was associated with their attenuation of endothelial nitric oxide synthase (eNOS) expression.8 Given that HSS can induce up-regulation of eNOS and production of nitric oxide (NO),23–26 the inhibitory effect of HSS on SMC-induced E-selectin expression in ECs could also be associated with the increased levels of NO in presheared ECs. Moreover, Krüppel-like factor 2 (KLF2) has been shown to be an important regulator of EC activation in response to proinflammatory stimuli and shear stress.27–30 Given that (1) the KLF2 expression in ECs can be inhibited by IL-1β and induced by shear stress and (2) overexpression of KLF2 can inhibit the IL-1β induction of E-selectin in ECs,28 it is likely that the KLF2 expression in ECs could be down-regulated by their coculture with SMCs and the inhibitory effect of preshearing on SMC-induced EC E-selectin expression could be due to, at least in part, the increase in the KLF2 expression in presheared ECs. Thus, KLF2 may serve as an additional mediator of the effects of SMCs and shear stress on E-selectin expression in ECs.
In summary, our present study has characterized the mechanisms by which (1) SMCs in close adjacency to ECs induce EC E-selectin expression and (2) shear stress inhibits this SMC-induced E-selectin expression (summarized in Figure 7). The cytokines IL-1β and IL-6 produced by EC/SMC coculture may interact with their corresponding receptors in ECs to induce their coupling with and activation of receptor-associated proteins IRAK and gp130 and the consequent activation of both JNK/p38 and NF-κB signaling pathways, and ultimately E-selectin expression. Shear stress may inhibit the SMC-induced E-selectin expression via the inhibition in SMC activation of IRAK/gp130, JNK/p38, and NF-κB. Our findings provide insights into the mechanisms underlying the interplays of SMCs with ECs and the protective homeostatic function of shear stress in modulating EC signaling and gene expression.
Figure 7.

Schematic representation of the signaling pathways regulating SMC-induced E-selectin expression in ECs and its inhibition by shear stress.
Supplementary Material
Acknowledgments
This work was supported by the National Health Research Institutes (Taiwan) (grant ME-095-PP-06) (J.-J.C.); the National Science Council (Taiwan) (grants 96-3112-B-400-009 and 95-2320-B-400-003) (J.-J.C.); and the National Heart, Lung, and Blood Institute (grants HL064382 and HL080518) (S.C.)
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: J.-J.C. and S.C. designed the research, wrote the paper, and provided financial support for the study; L.-J.C., C.-I.L., P.-L.L., M.-C.T., D.-Y.L., and C.-W.L. performed the experiments; S.U. contributed to the design of the EC/SMC coculture flow system.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Jeng-Jiann Chiu, Division of Medical Engineering Research, National Health Research Institutes, Miaoli 350, Taiwan, ROC; e-mail: jjchiu@nhri.org.tw.
References
- 1.Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev. 2004;84:767–801. doi: 10.1152/physrev.00041.2003. [DOI] [PubMed] [Google Scholar]
- 2.Zeiffer U, Schober A, Lietz M, et al. Neointimal smooth muscle cells display a proinflammatory phenotype resulting in increased leukocyte recruitment mediated by P-selectin and chemokines. Circ Res. 2004;94:776–784. doi: 10.1161/01.RES.0000121105.72718.5C. [DOI] [PubMed] [Google Scholar]
- 3.Rainger GE, Nash GB. Cellular pathology of atherosclerosis: smooth muscle cells prime co-cultured endothelial cells for enhanced leukocyte adhesion. Circ Res. 2001;88:615–622. doi: 10.1161/01.res.88.6.615. [DOI] [PubMed] [Google Scholar]
- 4.Chen CN, Chang SF, Lee PL, et al. Neutrophils, lymphocytes, and monocytes exhibit diverse behaviors in transendothelial and subendothelial migrations under co-culture with smooth muscle cells in disturbed flow. Blood. 2006;107:1933–1942. doi: 10.1182/blood-2005-08-3137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chiu JJ, Chen CN, Lee PL, et al. Analysis of the effect of disturbed flow on monocytic adhesion to endothelial cells. J Biomech. 2003;36:1883–1895. doi: 10.1016/s0021-9290(03)00210-0. [DOI] [PubMed] [Google Scholar]
- 6.Chappell DC, Varner SE, Nerem RM, Medford RM, Alexander RW. Oscillatory shear stress stimulates adhesion molecule expression in cultured human endothelium. Circ Res. 1998;82:532–539. doi: 10.1161/01.res.82.5.532. [DOI] [PubMed] [Google Scholar]
- 7.Morigi M, Zoja C, Figliuzzi M, et al. Fluid shear stress modulates surface expression of adhesion molecules by endothelial cells. Blood. 1995;85:1696–1703. [PubMed] [Google Scholar]
- 8.Chiu JJ, Chen LJ, Lee PL, et al. Shear stress inhibits adhesion molecule expression in vascular endothelial cells induced by coculture with smooth muscle cells. Blood. 2003;101:2667–2674. doi: 10.1182/blood-2002-08-2560. [DOI] [PubMed] [Google Scholar]
- 9.Wung BS, Cheng JJ, Chao YJ, Hsieh HJ, Wang DL. Modulation of Ras/Raf/extracellular signal-regulated kinase pathway by reactive oxygen species is involved in cyclic strain-induced early growth response-1 gene expression in endothelial cells. Circ Res. 1999;84:804–812. doi: 10.1161/01.res.84.7.804. [DOI] [PubMed] [Google Scholar]
- 10.Gimbrone MA., Jr . Culture of vascular endothelium. In: Spact TH, editor. Progress in Hemostasis and Thrombosis. Vol III. New York, NY: Grune and Stratton; 1976. pp. 1–28. [PubMed] [Google Scholar]
- 11.Johnson GL, Lapadat R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. doi: 10.1126/science.1072682. [DOI] [PubMed] [Google Scholar]
- 12.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296:1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 13.Barnes PJ. Nuclear factor-kappa B. Int J Biochem Cell Biol. 1997;29:867–870. doi: 10.1016/s1357-2725(96)00159-8. [DOI] [PubMed] [Google Scholar]
- 14.Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARalpha but not by PPARgamma activators. Nature. 1998;393:790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- 15.Hanada T, Yoshimura A. Regulation of cytokine signaling and inflammation. Cytokine Growth Factor Rev. 2002;13:413–421. doi: 10.1016/s1359-6101(02)00026-6. [DOI] [PubMed] [Google Scholar]
- 16.Kishimoto T, Akira S, Narazaki M, Taga T. Interleukin-6 family of cytokines and gp130. Blood. 1995;86:1243–1254. [PubMed] [Google Scholar]
- 17.Romano M, Sironi M, Toniatti C, et al. Role of IL-6 and its soluble receptor in induction of chemokines and leukocyte recruitment. Immunity. 1997;6:315–325. doi: 10.1016/s1074-7613(00)80334-9. [DOI] [PubMed] [Google Scholar]
- 18.Nilsson MB, Langley RR, Fidler IJ. Interleukin-6, secreted by human ovarian carcinoma cells, is a potent proangiogenic cytokine. Cancer Res. 2005;65:10794–10800. doi: 10.1158/0008-5472.CAN-05-0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ni CW, Hsieh HJ, Chao YJ, Wang DL. Interleukin-6-induced JAK2/STAT3 signaling pathway in endothelial cells is suppressed by hemodynamic flow. Am J Physiol Cell Physiol. 2004;287:C771–780. doi: 10.1152/ajpcell.00532.2003. [DOI] [PubMed] [Google Scholar]
- 20.Rajotte D, Arap W, Hagedorn M, Koivunen E, Pasqualini R, Ruoslahti E. Molecular heterogeneity of the vascular endothelium revealed by in vivo phage display. J Clin Invest. 1998;102:430–437. doi: 10.1172/JCI3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Read MA, Whitley MZ, Gupta S, et al. Tumor necrosis factor alpha-induced E-selectin expression is activated by the nuclear factor-kappaB and c-JUN N-terminal kinase/p38 mitogen-activated protein kinase pathways. J Biol Chem. 1997;272:2753–2761. doi: 10.1074/jbc.272.5.2753. [DOI] [PubMed] [Google Scholar]
- 22.Shyy JY, Chien S. Role of integrins in endothelial mechanosensing of shear stress. Circ Res. 2002;91:769–775. doi: 10.1161/01.res.0000038487.19924.18. [DOI] [PubMed] [Google Scholar]
- 23.Kuchan MJ, Jo H, Frangos JA. Role of G protein in shear stress-mediated nitric oxide production by endothelial cells. Am J Physiol. 1994;267:C753–C758. doi: 10.1152/ajpcell.1994.267.3.C753. [DOI] [PubMed] [Google Scholar]
- 24.Tsao PS, Lewis NP, Alpert A, Cooke JP. Exposure to shear stress alters endothelial adhesiveness: role of nitric oxide. Circulation. 1995;92:3513–3519. doi: 10.1161/01.cir.92.12.3513. [DOI] [PubMed] [Google Scholar]
- 25.Uematsu M, Ohara Y, Navas JP, et al. Regulation of endothelial cell nitric oxide synthase mRNA expression by shear stress. Am J Physiol. 1995;269:C1371–C1378. doi: 10.1152/ajpcell.1995.269.6.C1371. [DOI] [PubMed] [Google Scholar]
- 26.Ranjan V, Xiao Z, Diamond SL. Constitutive NOS expression in cultured endothelial cells is elevated by fluid shear stress. Am J Physiol. 1995;269:H550–H555. doi: 10.1152/ajpheart.1995.269.2.H550. [DOI] [PubMed] [Google Scholar]
- 27.Dekker RJ, van Soest S, Fontijn RD, et al. Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Kruppel-like factor (KLF2). Blood. 2002;100:1689–1698. doi: 10.1182/blood-2002-01-0046. [DOI] [PubMed] [Google Scholar]
- 28.SenBanerjee S, Lin Z, Atkins GB, et al. KLF2 is a novel transcriptional regulator of endothelial proinflammatory activation. J Exp Med. 2004;199:1305–1315. doi: 10.1084/jem.20031132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parmar KM, Larman HB, Dai G, et al. Integration of flow-dependent endothelial phenotypes by Kruppel-like factor 2. J Clin Invest. 2006;116:49–58. doi: 10.1172/JCI24787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang N, Miao H, Li YS, et al. Shear stress regulation of Kruppel-like factor 2 expression is flow pattern-specific. Biochem Biophys Res Commun. 2006;341:1244–1251. doi: 10.1016/j.bbrc.2006.01.089. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}