

Abstract
Chronic alcohol consumption induced liver injury in Cu, Zn-superoxide dismutase –deficient mice (Sod1−/−) with extensive centrilobular necrosis and inflammation and a reduction of hepatic ATP content. Mechanisms by which ethanol decreased ATP in these mice remain unclear. We investigated alterations in mitochondria of Sod1−/−mice produced by chronic ethanol treatment. These mitochondria had an increase in state 4 oxygen consumption with succinate and especially with glutamate plus malate compared to pair-fed Sod1−/−mice or mitochondria from wild type mice fed dextrose or ethanol. This uncoupling was associated with a decrease in ADP/O and respiratory control ratios, a decline in mitochondrial membrane potential, enhanced mitochondrial permeability transition and decreased aconitase activity. Total thiols and uncoupling protein 2 levels were elevated in the pair-fed Sod1−/−mitochondria, perhaps an adaptive response to oxidant stress. However, no such increases were found with the ethanol-fed Sod1−/−mitochondria suggesting a failure to develop these adaptations. The mitochondria from the ethanol-fed Sod1−/−mice had elevated levels of cleaved Bax, Bak, Bcl-xl and adenine nucleotide translocator. Immunoprecipitation studies revealed increased association of Bax and Bak with the adenine nucleotide translocator. ADP-ATP exchange was very low in the ethanol-fed Sod1−/−mitochondria. These results suggest that ethanol treatment of Sod1−/−mice, produces uncoupling and a decline in Δψ, swelling, increased association of proapoptotic proteins involved in the permeability transition and decreased adenine nucleotide translocator activity, which may be responsible for the decline in ATP levels and development of necrosis in this model of alcohol-induced liver injury.
Keywords: Alcohol induced liver disease, Cu, Zn- superoxide dismutase, knockout mice, mitochondria, oxidative stress
Introduction
Mitochondria play a key role in controlling the energy balance and in signaling of either a cell survival or death response, or whether mitochondrial injury results in apoptosis or necrosis. Mitochondria represent the major intracellular source of ROS especially superoxide radical, which is generated as a byproduct of oxygen consumption by the respiratory chain. Oxidative stress appears to play a central role in mechanisms by which ethanol is hepatotoxic (1). Damage to the mitochondria and increased production of ROS by mitochondria after ethanol treatment may contribute to ethanol-induced oxidative stress (2–5). Acute ethanol treatment of rat hepatocytes increased ROS production in mitochondria, produced depolarization of the mitochondrial membrane which resulted in a mitochondrial permeability transition (MPT) and caused Bax translocation from cytosol to mitochondria; these effects led to subsequent formation of Bax-VDAC complexes, release of cytochrome c, caspase 3 activation, and apoptosis (6–8). Chronic alcohol consumption induced inhibition of complexes I and III of the mitochondrial electron transport chain, impaired the rate of ATP synthesis, decreased levels of ATP and increased ROS production in rat liver (9, 10). Mitochondria isolated from rats chronically fed ethanol exhibited a markedly increased sensitivity to MPT induction caused by a variety of agents implicated in both necrotic (Ca 2+) and apoptotic (ceramide, GD3 ganglioside, and Bax) forms of cell death (11). Ethanol promotes oxidative stress in mitochondria, both by increasing ROS formation and by decreasing mitochondrial defense mechanisms. Mitochondrial GSH was depleted by an ethanol-induced impairment in mitochondrial GSH transport, due to decreased membrane fluidity (12). Patients and baboons with alcoholic hepatitis displayed mitochondria with morphological aberrations (13–15). While the role of oxidative stress in alcohol-induced cell injury is well documented (16), what alterations occur in mitochondria in in vivo models with alcohol-induced liver injury are still uncertain.
It has been difficult to study mechanisms responsible for alcohol-induced liver injury since oral administration of ethanol in liquid diets, while producing steatosis and oxidant stress in the liver, does not cause liver necrosis or fibrosis. There is a need for new oral models of ethanol treatment that results in the development of liver injury. Because alcoholic liver injury has been linked to oxidative stress, we reasoned that a compromised antioxidant defensive system in the liver could exaccerbate liver injury produced by ethanol. Several studies have shown a decrease in SOD1 activity in rat liver after alcohol treatment (17,18) and adenoviral delivery of SOD1 protected against liver injury caused by alcohol (19). A model of chronic alcohol-induced liver injury using Cu, Zn-superoxide dismutase –deficient mice (Sod1−/−) was developed (20). Feeding the homozygous Sod1−/−mice with ethanol (10% of total calories) for 3 weeks resulted in extensive centrilobular necrosis and inflammation and increased carbonyls, lipid peroxidation aldehydic products and 3-nitrotyrosine protein adducts, indicative of enhanced oxidative and nitrosative stress. Since liver injury was accompanied by reductions of hepatic ATP content, mitochondrial GSH levels and activity of the Mn-SOD, we suggest that the increased mitochondrial oxidative stress and subsequent alterations in mitochondria caused by chronic alcohol may be important in the mechanisms of this liver injury. The goal of this study was to evaluate mitochondrial function in livers of Sod1−/−mice chronically fed ethanol, especially as it may relate to MPT sensitivity.
Materials and Methods
Mice
Cu,Zn–superoxide dismutase deficient by targeted mutation mice (B6; 129S7-Sod1 tm1Leb) were purchased from Jackson Laboratories (Bar Harbor, Maine) and bred at the animal facility at Mount Sinai School of Medicine. All mice were cared for according to the NIH Guide for the Care and Use of Laboratory Animals. Mice were genotyped at 1month of age by polymerase chain reaction (PCR). Genomic DNA isolated from mouse tail was digested overnight in tail tip buffer containing 0.1 mM EDTA, 0.5 % SDS, Proteinase K (1 mg/ml), 50 mM Tris-HCl, pH 8.0. DNA was isolated by a chloroform/phenol procedure. The targeted allele was detected as a 250-bp fragment (HPRT) using primers IMR0781(5′-TGTTCTCCTCTTCCTCATCTCC-3′) mouse Pgk promoter forward primer for the PgkHPRT minigene and IMR0782 (5′-ACCCTTTCCAAATCCTCAGC-3′) as HPRT exon 2 reverse primer for the PgkHPRT minigene. The wild type allele was detected as a 123bp fragment using primers IMR0878 (5′-TGAACCAGTTGTGTTGTCAGG-3′) and IMR0888 (5′-TCCATCACTGGTCACTAGCC-3′). The 25 μl PCR reaction mixture contained Ready-To-Go™ PCR Beads, 300 ng DNA and 0.3 μM of each primer. The PCR conditions used were one cycle of denaturation at 94°C for 3min, followed by 12 cycles of denaturation at 94°C for 30 sec, annealing at 61°C for 30 sec and extension at 72°C for 35 sec, followed by 25 cycles at 94°C for 20 sec, at 58°C for 30 sec, at 72°C for 35 sec and a final cycle at 72°C for 5 min. Homozygous (Sod1−/−) and wild type (Sod1+/+) mice at age of 2 month were fed the ethanol diet at 10% of total calories for the first week and 20 % ethanol for the second week while control groups were pair-fed the control dextrose diet.
Mitochondrial isolation
After 2 weeks on the respective diets, the mice were sacrificed with an overdose of pentobarbital and livers were rapidly dissected in ice cold 0.25 mM sucrose. Two weeks of treatment were used in this study since severe liver injury occurs after 3 weeks of feeding alcohol to the SOD1 knockouts and we were interested in evaluating whether mitochondrial dysfunction occurs prior to the development of this severe injury. All procedures were carried out at 4°C. Livers were minced in fresh sucrose-based medium (250 mM sucrose, 1 mM MgCl2, 0.1 mM EGTA, 20 mM HEPES, pH 7.4) and gently homogenized manually (w/v) using a tissue grinder with loose-fitting pestle. The resulting homogenate was centrifuged at 800 × g for 5 min twice, and the supernatant fraction was centrifuged at 9,000 × g for 10 min to obtain the mitochondrial pellet. This pellet was washed with isolation medium (EGTA free for MPT measurements) and centrifuged at 10,000g for 10 min. The final pellet was homogenized in the above buffer. Protein concentration was measured by the DC protein Assay Kit II (Bio-Rad, Hercules, CA). To assure equivalent yield and purity of the mitochondria isolated from the 4 groups of mice, porin levels per mg mitochondrial protein were determined by Western blot analysis.
Determination of aconitase activity
The activity of aconitase in the mitochondrial fractions was measured by the conversion of citrate to α-ketoglutarate coupled to the reduction of NADP+. Aconitase activity was monitored for 3–5 min by determining the absorbance at 340nm in freshly prepared 50 mM Tris-HCl buffer containing 0.2 mM NADP+, 30 mM sodium citrate, 2 unit/ml of isocitrate dehydrogenase, and 0.6 mM MnCl2. Mitochondrial pellets were suspended in 2 mM sodium citrate buffer, 0.6 mM MnCl2, 1 mM sodium succinate and solubilized by repeating a freezing-thawing cycle 3 times. The reaction was started by adding the mitochondria samples. Aconitase activity is defined as the amount of enzyme necessary to catalyze the formation of 1 nmol of isocitrate/min at 37 °C.
Thiols and Sulfide
Total mitochondrial thiols were assayed using the Thiol and Sulfide Quantitation Kit (Molecular Probes, Carlsbad, CA), which provides an ultrasensitive colorimetric assay for quantitating both protein and nonprotein thiols. The nonprotein thiols essentially reflect GSH levels in the isolated mitochondria. In this assay thiols reduce a disulfide inhibited derivative of papain, stoichiometrically releasing the active enzyme. Proteins are denatured such that all protein thiols are accessible: the combination of nonprotein thiols plus protein thiols reduce the papain disulfide prosubstrate to the active reduced sulfhydryl form of papain and catalytic activity is proportional to the total free thiols available to reduce the papain disulfide prosubstrate. The activity of the papain is then measured using the chromogenic papain substrate, N-benzoyl-L-arginine, p-nitroanilide (L-BAPNA) according to kit instructions. Validation of this assay was shown by the lack of formation of p-nitroaniline with GSSG as compared to GSH, inhibition of p-nitroaniline formation when the mitochondrial extract was treated with diethylmaleate to bind free thiol groups and finding similar results when Ellman’s reagent was used to assay free thiol groups.
O2 consumption
Respiratory rates were assessed by measuring the oxygen consumption of mitochondria using Clark-type electrodes in 3 ml sealed Perspex chambers, which were continuously magnetically stirred at 30°C. Mice liver mitochondria (0.8mg/ml) were incubated in respiration buffer composed of 120 mM KCl, 3 mM HEPES, 1 mM EGTA, 25 mM sucrose, 5 mM MgCl2, 5 mM KH2PO4 at pH 7.4 in the presence of either 5 mM malate plus 5 mM glutamate or 15 mM succinate plus 2.7 uM rotenone as substrates to measure state 4 respiration. State 3 respiration was measured after the addition of ADP at a final concentration of 0.3 mM.
Spectrofluorometric measurements of Δψ
The electrical potential across the inner mitochondrial membrane (Δψ) was estimated by monitoring of fluorescence quenching of Rhodamine 123 (21). Fluorescence with excitation at 503 nm (2.5 nm slit) and emission at 527 nm (3 nm slit) was measured in a buffer containing 150 mM sucrose, 5 mM MgCl2, 2.7 μM rotenone; 0.2 μM Rhodamine 123; 5 mM KPi buffer; 20 mM potassium-HEPES buffer at pH 7.4 and mitochondria (0.167mg/ml). After incubation for 2–3 min, respiratory substrate disodium succinate (5 mM) was added to initiate state 4 respiration and then 300 μM ADP was added to initiate state 3 respiration. To de-energize the mitochondria, an uncoupler (1.3 μM CCCP) and respiratory inhibitor (1.3 μg/ml antimycin) were added. Rhodamine was dissolved in ethanol and its concentration was assayed spectrophotometrically at 507 nm (€507=101mM-1 cm−1). The ethanol concentration in all incubation mixtures was kept below 0.4% (v/v).
Measurement of mitochondrial permeability transition
Mitochondria (0.5mg/ml) were incubated in the buffer used above for oxygen consumption at 25° C. 1mM glutamate and 1mM malate were added as respiratory substrates. MPT was induced in mitochondria by addition of either 10, 50, or 150 μM CaCl2. 2 μM cyclosporin A was added before incubation as a control to prevent the MPT. Swelling was monitored every minute over a 30 min incubation by absorbance at 540 nm.
Detection of cytochrome c release
Mitochondria (1 mg of protein/ml) were incubated for 15 min at 25 °C in respiration buffer with the indicated additions. The reaction mixtures were then centrifuged at 13,000 × g for 10 min at 4 °C to obtain the mitochondrial pellets. The supernatant fractions were further spun at 100,000 × g for 15 min at 4 °C to eliminate mitochondrial membrane fragments and concentrated by ultrafiltration through Centricon 10 membranes (Millipore, Billerica, MA) at 4 °C. The concentrated supernatants were subjected to 18% SDS-PAGE, transferred to 0.2μm nitrocellulose membranes and analyzed by Western blotting using anti-cytochrome c antibody.
ADP/ATP translocase activity
Liver mitochondria were suspended (7mg/500μl) in 0.6 M mannitol, 0.2%BSA, 10 mM MOPS, and 0.1 mM EDTA at pH 6.8. Mitochondria were incubated with 15 μl of [3H] ATP (40Ci/mmol) for 45 min at 4°C in the presence or absence of 100 μM atractyloside. Mitochondria were washed twice to eliminate free [3H] ATP. The ADP-ATP exchange was initiated by addition of cold ADP (standard concentration of 400 μM) and stopped by addition of 100 μM atractyloside after 10 sec. The reaction mixtures were then centrifuged at 9,000 × g for 10 min at 4 °C to obtain the mitochondrial pellets. Supernatant and mitochondrial pellets were subjected to scintillation counting. ATP release into the 9000 × g supernatant fraction was measured and expressed as a percentage of [3H] counts compared to [3H] counts in control mitochondria.
Western blotting, protein cross-linking and immunoprecipitation
In vivo cross-linking for ANT interactions with proteins of the Bcl2 family and VDAC was performed with dimethyl 3,3′-dithio-bis(proprionate)x2HCl (DTBP) (Pierce Biotechnology, Inc., Rockford, IL). Mitochondria were treated with 2 mM of cross-linkers in PBS for 30 min at room temperature. After the reaction was quenched with 50 mM Tris-HCl for 10 min at 4°C, mitochondria were washed in PBS. Mitochondria were then lysed with lysis buffer : 10 mM Tris-HCl, pH 7.4, 142.5 mM KCl, 5 mM MgCl2, 1 mM EDTA, 1 mM PMSF, with 0.5% Nonidet P-40, and 20 μM leupeptin for 30 min on ice and centrifuged to remove insoluble debris. Immunoprecipitation was carried out as follows. Samples were precleared by mixing with control IgG protein and Protein G Plus-Agarose beads (Santa-Cruz Biotechnology, SantaCruz, CA) for 60 min at 4°C in a rocker, and the beads were removed by centrifugation. The resultant supernatants were incubated with anti-ANT antibodies (2 μg/ml) at 4°C for 2 h. Immunoprecipitates were collected by incubating with protein G-Sepharose for 60 min, followed by centrifugation for 2 min at 4°C. The pellets were washed with lysis buffer three times. After the final wash, the beads were suspended in SDS-sample buffer, and the samples were analyzed by SDS-PAGE and Western blotting.
Determination of caspase 3 activity
Caspase 3 activity in hepatic cytosols was assayed using the fluoregenic peptide 50 μM Ac-DEVD-AMC. The assay was carried out using 200 μg of cytosolic protein in a total volume of 100 μl. Following incubation in assay buffer at 37 °C for 30 min, the reaction was quenched by addition of 500 μl cold ethanol. A 2.0-ml aliquot of H2O was then added and the fluorescence of the samples was evaluated at 360/460 nm using a Bio-Tek Kontron SFM 25 spectrofluorimeter. Peptidase activities were measured in the absence and presence of the caspase 3 inhibitor Ac-DEVD-CHO (50μM); the difference between the two rates was attributed to the caspase -3 activity. The results were expressed as arbitrary units of fluorescence per milligram of protein.
Terminal Deoxynucleotidyl Transferase–Mediated dUTP Nick-End Labeling
DNA fragmentation was assessed via terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling (TUNEL) assay using the ApopTag in situ apoptosis detection kit (Serological Corp., Atlanta, GA). The quantitative analysis of positive nuclei with DNA fragmentation was performed by counting the average number of stained nuclei per visual field (original magnification × 400).
Statistics
Two-way analysis of variance (ANOVA) followed by Bonferroni post-hoc test was used for the determination of statistical significance of the experimental results.
Results
Oxidative stress in mitochondria of Sod1−/− mice
In order to evaluate oxidative stress in mitochondria of mice, we examined mitochondrial aconitase activity, a Fe-S cluster enzyme that is inactivated by superoxide and peroxynitrite. Inactivation of aconitase activity is believed to be a reliable marker for mitochondrial superoxide production (22,23). Deletion of the Sod1 gene caused 3.3- fold-decline in aconitase activity in liver of Sod1−/− mice fed dextrose (Fig. 1, A) Ethanol administration caused a 50% decrease in enzyme activity in mitochondria of the knockout mice and a 30% decrease in wild type mice. These results suggest that, the superoxide production in mitochondria of Sod1−/− mice is high and can be further increased with alcohol consumption. Because ROS oxidize thiol groups, we measured the concentration of protein-associated and low molecular weight thiols (mainly GSH) in mitochondria (Fig. 1B). The content of reduced thiols in mitochondria of Sod1 knockout mice was increased 2-fold compared with wild type mice, possibly an adaptive mechanism to oxidative stress. However, ethanol administration produced a decline in thiol content in mitochondria of Sod1−/− mice. No effect by ethanol was seen in mitochondria from the wild type mice (Fig. 1B). Interestingly, the pattern of changes in total thiol content in mitochondria of knockout mice produced by alcohol is similar to the changes in mitochondrial GSH content which were detected in our previous study i.e. an increase in the mitochondria from the Sod1−/− mice fed dextrose and a decrease after chronic ethanol treatment (20). One of the protective mechanisms against ROS production in the matrix of mitochondria is a mild uncoupling of respiration and phosphorylation by activation of uncoupling proteins (UCP)(24). UCP2 content was increased in mitochondria of Sod1−/− mice fed dextrose compared with the other groups (Fig. 1C and D), perhaps like the increase in GSH and thiols an adaptive mechanism to inhibit matrix ROS production. However, chronic ethanol treatment caused a decline in UCP2 content in mitochondria of Sod1−/− mice that probably reflects a failure to adapt protection factors with increasing levels of oxidative stress. Porin levels per mg mitochondrial protein were identical in the 4 groups of mice suggesting equivalent yield and purity of the isolated mitochondria.
Figure 1. Effect of chronic ethanol consumption on aconitase activity (A), levels of total thiols (B) and content of uncoupling proteins (C, D) in isolated liver mitochondria of Sod1+/+ and Sod1−/− mice.
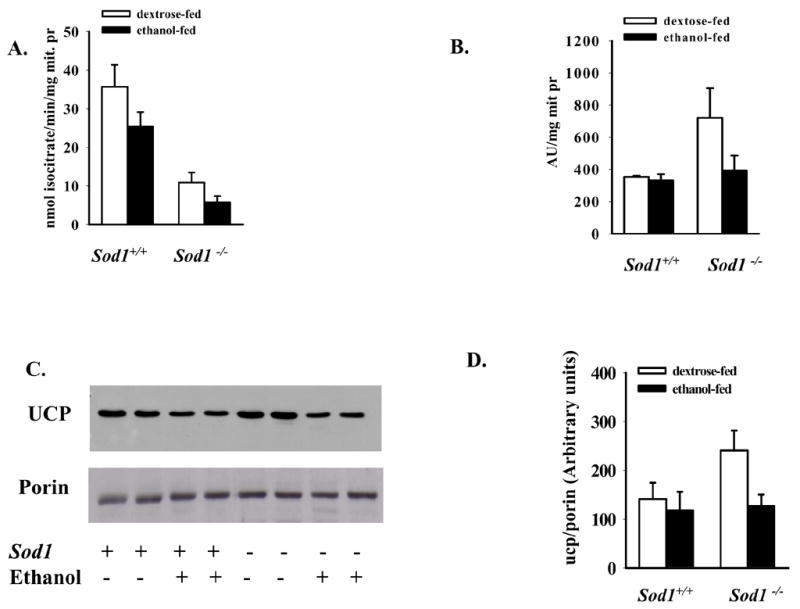
C. Representative Western blot of UCP2 content and porin levels in mitochondrial samples from 2 mice in each group, D. Quantification of the UCP2 content in mitochondria as detected by Western blots. The values represent the Mean ± SE of 4–6 mice. Effect of SOD deficiency and alcohol treatment were analyzed by two-way ANOVA.
A. Alcohol effect p=0.0037; SOD1 effect p=0.0012. B,D Alcohol effect p=0.033 and p=0.042 respectively.
O2 Consumption, Membrane potential
In the knockout mice, ethanol treatment increased the state 4 rate of oxygen consumption with glutamate plus malate as substrate, with no effect on state 3 rates of respiration (Tables 1). Respiratory control ratio values were decreased in Sod1−/− mice fed ethanol with either glutamate-malate or succinate as substrates due to the increase of state 4 respirations. A smaller increase in state 4 oxygen consumption was found with succinate as substrate (Table 1). Ethanol administration caused significant decreases in the ADP/O ratio for both glutamate-malate and succinate in Sod1−/− mice as compared with wild type mice fed ethanol (Table 1). The lower ADP/O ratio, and respiratory control ratio caused by ethanol in the Sod1 knockouts, can probably be attributed to uncoupling of oxidative phosphorylation. With respect to the wild type mice, chronic ethanol treatment had little or no significant effects on these parameters (Table 1, 1). The electrochemical potential of the proton gradient generated across the mitochondrial membrane (Δψ) was assessed by monitoring fluorescence quenching of Rh-123. Fig. 2 shows the effect of a series of additions on the dye fluorescence in isolated mitochondria of mice. Addition of succinate as a respiratory substrate to the medium containing mitochondria and Rh-123 decreased fluorescence, reflective of a high Δψ corresponding to state 4 of respiration. The decline in flourescence averaged 100,97, and 99 arbitrary units with mitochondria from the wild type mice fed dextrose or ethanol and the SOD1 knockout mice fed dextrose, respectively, whereas the decline averaged 49 arbitrary units with mitochondria from the SOD1 knockout mice fed ethanol. Quenching of Rh123 fluorescence by succinate in mitochondria of Sod1−/− mice fed alcohol was significantly less than it was in mitochondria of the other 3 groups of mice, indicating a reduction in Δψ. Addition of ADP induced an enhancement of steady-state fluorescence, which corresponds to state 3 respiration, as the proton gradient significantly decreases due to its utilization to promote ADP phosphorylation. This enlargement of Rh123 fluorescence was lower in mitochondria of Sod1−/− mice compared with the other experimental groups (Fig. 2). The increase in fluorescence caused by addition of ADP averaged 31,32 and 30 arbitrary units for mitochondria from wild type mice fed dextrose or ethanol or from the SOD1 knockout mice fed dextrose, respectively but averaged only 9 arbitrary units with the mitochondria from the SOD1 knockout mice fed ethanol. Quenching of fluorescence was reversed by deenergization with the uncoupler CCCP and respiratory inhibitor antimycin. Taken as a whole, these data suggest uncoupling and a decline in Δψ in mitochondria of Sod1−/− mice fed ethanol.
Table 1. Effect of Chronic Ethanol Feeding on Glutamate plus Malate and Succinate-Dependent Oxygen Consumption in Liver.
| Sod1+/+c | Sod1+/+e | Sod1−/−c | Sod1−/−e | |
|---|---|---|---|---|
| state 4 (glutamate-malate) | 0.017 ± 0.001 | 0.012 ± 0.000 | 0.013 ± 0.001 | 0.022 ± 0.001* |
| state 3 (glutamate-malate) | 0.085 ± 0.045 | 0.065 ± 0.010 | 0.064 ± 0.009 | 0.057 ± 0.005 |
| RCR (glutamate-malate) | 4.95 ± 0.45 | 4.40 ± 0.55 | 4.97 ± 0.58 | 2.63 ± 0.24** |
| P/O (glutamate-malate) | 2.36 ± 0.33 | 2.53 ± 0.15 | 2.71 ± 0.63 | 1.49 ± 0.03# |
| state 4 (succinate) | 0.034 ± 0.005 | 0.024 ± 0.004 | 0.025 ± 0.003 | 0.036 ± 0.003## |
| state 3 (succinate) | 0.110 ± 0.003 | 0.072 ± 0.004 | 0.080 ± 0.015 | 0.092 ± 0.008## |
| RCR (succinate) | 3.63 ± 0.50 | 3.06 ± 0.28 | 2.93 ± 0.29 | 2.56 ± 0.03 |
| P/O (succinate) | 1.88 ± 0.17 | 2.1 ± 0.12 | 1.62 ± 0.31 | 1.43 ± 0.32 |
Respiratory rates were assessed by measuring the oxygen consumption of mitochondria using Clark-type electrodes. Mouse liver mitochondria (0.8mg/ml) were incubated in respiration buffer at pH 7.4 at 30°C in the presence of 5 mM malate/5mM glutamate or succinate in the absence (State 4) or presence (State 3) of 0.3 mM ADP. Rates refer to uatoms O2 consumed per min per mg mitochondrial protein.
Effects of sod1 deficiency and alcohol treatment were analyzed by two-way ANOVA.
Alcohol effect p=0.04, sod1 effect p=0.015;interaction p=0.0029;
sod1 effect p=0.0075;
Interaction p=0.044
Interaction p=0.05
Figure 2. Decline in mitochondrial membrane potential in Sod1 −/− mice-fed ethanol.
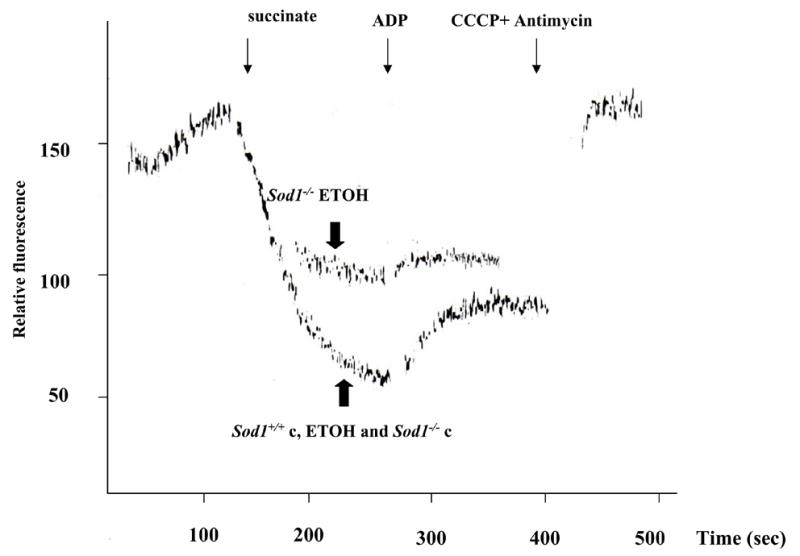
Mitochondial energization was monitored by fluorescence quenching of Rh-123. Fluorescence with excitation at 503 nm and emission at 527 nm was measured in a reaction buffer containing 150 mM sucrose; 5 mM MgCl2, 2.7 μM rotenone; 0.2 μM Rhodamine 123; 5 mM KPi buffer; 20 mM potassium-HEPES buffer, pH 7.4 and mitochondria (0.167mg/ml). Mitochondria were energized by addition of 5 mM disodium succinate (decrease in fluorescence) as respiratory substrate (State 4 respiration). Addition of 300 μM ADP causes an increase of fluorescence [quenching] (State 3 respiration). Mitochondria were deenergized with an uncoupler (1.3 μM CCCP) plus respiratory inhibitor (1.3 μg/ml antimycin). Each curve represents the Mean of 3–4 mice. Sod1+/+c and Sod1−/−c refer to the pair-fed wild type or Sod1 knockout mice. The traces for the mitochondria from the wild type mice fed dextrose or ethanol and from the SOD1 knockout mice fed dextrose are virtually identical and are overlaid on top of each other for all conditions (succinate, ADP, CCCP plus antimycin).
Permeabilization of the mitochondrial membrane, Cytochrome c release
Depolarization of the inner mitochondrial membrane would be expected to accompany a significant opening of the MPT pore. Mitochondria isolated from wild type mice fed either dextrose or ethanol tolerated concentrations of 10 μM Ca2+ without undergoing the MPT as assessed by mitochondrial swelling (Fig. 3). At 50 μM and 150 μM Ca2+, the mitochondria underwent the MPT and this was accompanied with cytochrome c release as presented in representative Western blots at the bottom of each figure. Ca2+-induced MPT opening and cytochrome c release from mitochondria were inhibited by cyclosporin A. Mitochondria from Sod1−/− mice fed dextrose demonstrated a small degree of swelling and cytochrome c release without Ca2+ addition, while 50 μM and 150 μM Ca2+ increased swelling of mitochondria resulting in cytochrome c release as was seen with the wild type mitochondria (Fig. 3C). Chronic ethanol consumption further increased induction of the MPT in mitochondria even without any addition of Ca2+causing significant cytochrome c release. In addition, alcohol sensitized the mitochondria to MPT induction at 10 μM Ca2+ (Fig. 3D), a calcium concentration which did not induce the MPT in the other 3 groups. Cyclosporin A partially inhibited this Ca 2+-induced MPT as well as cytochrome c release from mitochondria of Sod1−/− mice chronically fed ethanol.
Figure 3. Chronic ethanol consumption intensifies the mitochondrial permeability transition (MPT) and cytochrome C release from mitochondria of Sod1 −/− mice.
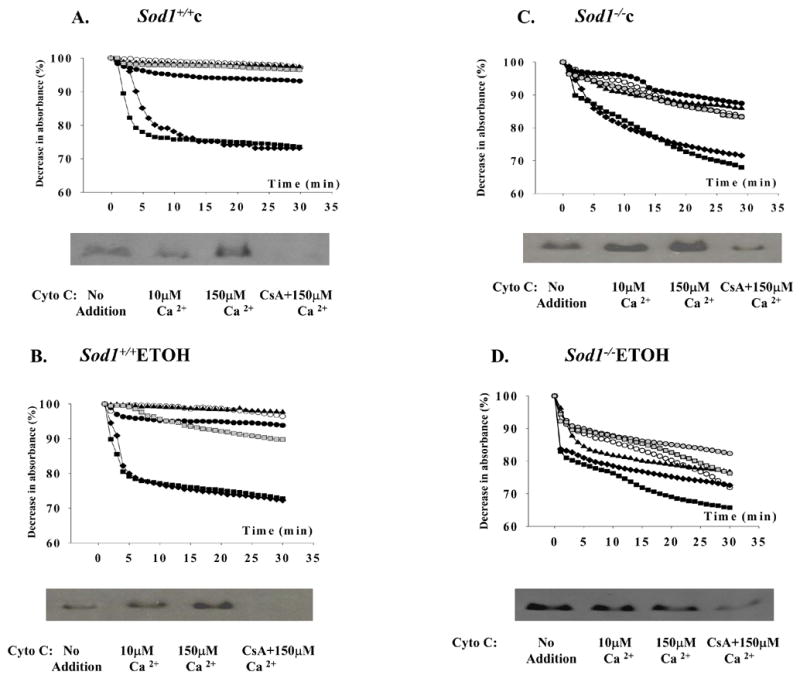
For MPT determination, mitochondria (0.5mg/ml) were incubated in respiration buffer at 25° C as described in Materials and Methods. Swelling was monitored for 30 min at 540 nm in mitochondria (○); mitochondria plus respiratory substrates 1 mM glutamate and 1mM malate without calcium (●); with 10 μM CaCl2 (▲); with 50 μM CaCl2 (◆), or with 150 μM CaCl2 (■). 2 μM cyclosporin A was added to mitochondria before incubation with (
 ) or without (
) or without (
 ) 150 μM Ca to prevent MPT. Each curve represents the Mean of 3–4 mice
) 150 μM Ca to prevent MPT. Each curve represents the Mean of 3–4 mice
For cytochrome C release to the incubation medium, mitochondria (1 mg/ml) were incubated for 15 min at 25 °C in respiration buffer with the indicated additions. The reaction mixtures were then centrifuged at 9,000 × g for 10 min at 4 °C and supernatant was concentrated by ultrafiltration through Centrikon 10 membranes (Amicon) at 4 °C. The concentrated supernatants were subjected to SDS-PAGE and analyzed by Western blotting using anti-cytochrome c antibody. Results show the cytochrome c present in the concentrated supernatants obtained after mitochondria from the 4 groups were incubated with no addition, or 10 uM calcium, or 150 uM calcium or 150 uM calcium plus 2 uM cyclosporin A.
The content of Bcl-2 family proteins in mitochondria and their association with ANT
For further evaluation of uncoupling of respiration and phosphorylation as well as the increase of nonspecific pore opening in mitochondria of Sod1−/− mice fed ethanol, the content of Bcl-2 family proteins were evaluated by SDS–PAGE and immunoblotting with the corresponding antibodies. Their possible association with ANT was also studied. We detected the 21kD form of Bax, a proapoptotic member of the Bcl-2 family in mitochondria of wild type mice fed dextrose or ethanol whereas the cleaved 19kD form of Bax (which is able to oligomerize) was observed in mitochondria of both knockout mice (Fig. 4A). Ethanol consumption produced a 3–fold increase in the content of cleaved Bax as compared with the Bax level in mitochondria of Sod1−/− mice fed dextrose (Fig. 4). Increased levels of another proapoptotic protein, Bak, were detected in mitochondria of Sod1−/− mice fed ethanol (Fig. 4A). Levels of the antiapoptotic Bcl-xl were also higher in the Sod1−/− mice fed ethanol, as were levels of ANT. The content of VDAC was not significantly changed in any of the groups. Chronic ethanol treatment had no effect on Bax, Bak Bcl-xl, VDAC or ANT protein levels in the wild type mice. Besides total levels of these proteins, their association with ANT was evaluated since ANT is one of the critical proteins regulating the MPT. After immunoprecipitating ANT from the mitochondria of the 4 groups, possible associated proteins were evaluated by Western blotting. Chronic ethanol feeding of the Sod1−/− mice caused an increase in levels of Bax and Bak but not Bcl-xl or VDAC in the ANT immunoprecipitate (Fig. 4 ANT-IP).
Figure 4. Expression of MPT pore components and their interactions in mitochondria of Sod1+/+ and Sod1−/− mice.
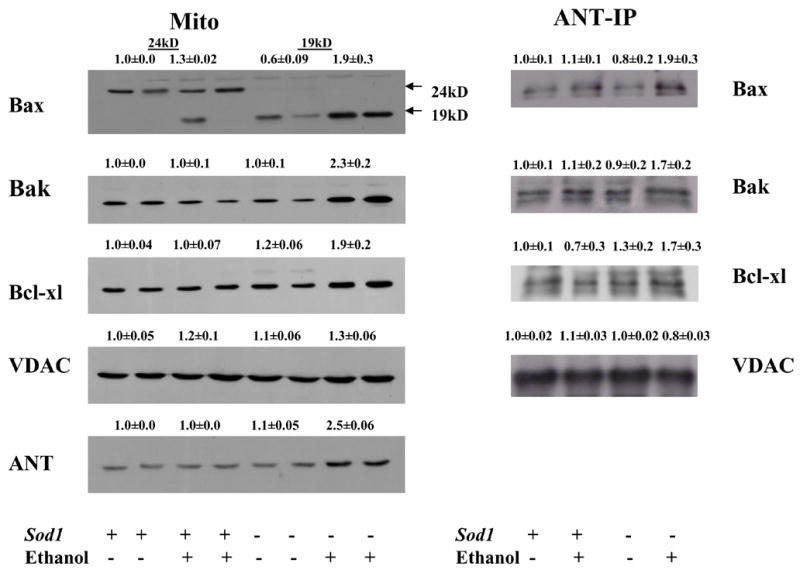
A. Western blotting was conducted with 100 μg mitochondrial protein using anti-Bax, anti-Bak, anti-Bcl-xl, anti-VDAC and anti-ANT antibodies. B. Isolated mitochondria were subjected to immunoprecipitation with an ANT specific antiserum as described in Materials and Methods. Equal amounts of precipitated ANT proteins, estimated by Western blot, were loaded for SDS-PAGE and protein interactions were detected by using the corresponding anti-Bax, anti-Bak, anti-Bcl-xl and anti-VDAC antibodies
Each immunoblot has been reproduced at least from three mice. Numbers above the blots refer to arbitrary densitometric units with results for the pair-fed wild type mice taken as 1.0.. Effects of SOD1 deficiency and alcohol treatment were analyzed by two-way ANOVA. Ethanol effect on ANT content p=0.003. Effect of SOD1 deficiency on Bax, Ant and ANT-Bax content p=0.0001;p=0.004, p=0.044 respectively; interaction p=0.0062; p=0.005; p=0.031 respectively. One way ANOVA followed by Student-Newman-Keul posthoc test indicated that the increase in Bax, Bak, Bclxl, ANT and the ANT-Bax and ANT-Bak comples was statistically significant (<0.05) as compared to the 3 other groups.
ANT activity
The content of ANT protein was elevated in the mitochondria of Sod1−/− mice fed ethanol (Fig. 4A). To investigate functional activity of ANT, we measured ADP/ATP exchange in the isolated mitochondria. Ethanol feeding significantly reduced ADP/ATP translocation through the mitochondrial membrane of Sod1−/− mice, as compared with the other 3 groups of mice (Fig. 5).
Figure 5. Decrease in ATP/ADP exchange in isolated mitochondria from Sod1 −/− mice.
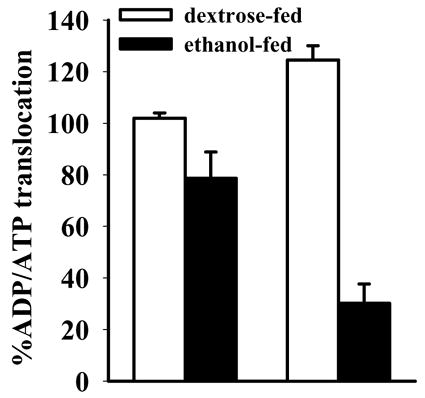
Isolated mitochondria (7 mg/500 μl buffer) were loaded with [3H]-labeled ATP. The exchange was initiated by addition of cold 400 μM ADP and stopped by addition of 100 μM atractyloside after 10 sec. The reaction mixture was centrifuged at 9,000 × g for 10 min at 4 °C and the supernatant and mitochondrial pellets were subjected to scintillation counting. ATP release into the mitochondrial supernatant was measured and expressed as a percentage of [3H] counts compared to [3H]counts in mitochondria with the results for the mitochondria from the Sod1+/+ mice-fed dextrose taken as the 100% value. Effects of SOD1 deficiency and alcohol treatment were analyzed by two-way ANOVA. Alcohol effect p=0.005; SOD1 effect p= 0.023; interaction p=0.0008.
DNA fragmentation and Caspase-3 activity
TUNEL results showed that there were more hepatocytes with positive staining nuclei in Sod1−/− mice fed ethanol (4.5 ± 0.9; per visual field of approximately 100 cells) than in the Sod1−/− mice fed dextrose (1.4 ± 0.3). No TUNEL staining was observed in the wild type mice fed ethanol or dextrose (Fig. 6A). Caspase-3 activity showed a significant increase in Sod1−/− mice fed dextrose compared to the alcohol-fed Sod1−/− mice and wild type mice fed dextrose or ethanol (Fig. 6B). These results with caspase-3 activity suggest that TUNEL reflects DNA fragmentation largely due to non-specific damage rather than apoptosis. Chronic ethanol feeding potentiates liver injury in the Sod1−/− mice however, this combined injury by ethanol in Sod1 knockout mice is primarily necrotic (20).
Figure 6. Effect of chronic ethanol consumption on DNA fragmentation and activity of caspase 3 in livers of Sod1 −/− mice.
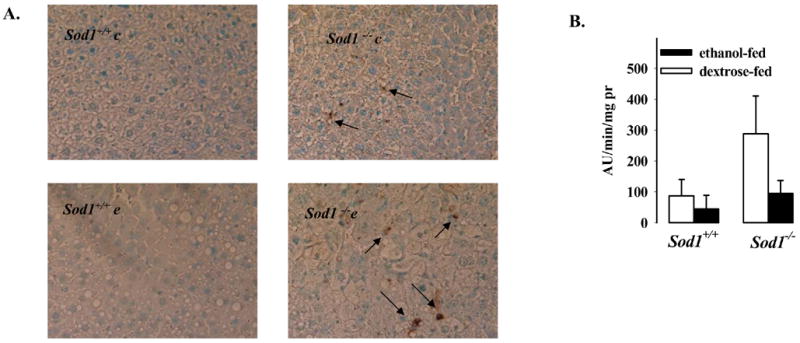
DNA fragmentation was determined by the TUNEL assay as described in Materials and Methods (A). Caspase 3 activity in hepatic cytosols was assayed using 50 μM Ac-DEVD-AMC fluorogenic peptide as substrate (B). The released AMC was determined fluorometrically and results expressed as arbitrary units of fluorescence per min per mg cytosolic protein. Effect of alcohol feeding on caspase 3 activity p=0.0039 by two-way ANOVA.
Discussion
Ethanol has long been known to inhibit mitochondrial oxygen consumption and ATP production (2,5, 8). While this is thought to play a role in alcohol-induced liver injury, most studies showing impairment of mitochondrial functions occur in models which do not exhibit significant liver injury beyond steatosis. A recent study reported that unlike results with rat liver mitochondria, chronic ethanol feeding had no significant effect on mouse liver mitochondrial oxygen uptake (25) a result confirmed in the current study with the wild type Sod1 positive mice fed ethanol. Thus the role of mitochondrial dysfunction, while likely and logically believed to be important in alcohol-induced liver injury, remains unclear and not proven.
We have previously shown that chronic ethanol feeding produced liver injury in mice lacking SOD1 (20). This model provides an oral alcohol feeding model which produces significant liver injury and lends some support for the overall hypothesis that oxidative stress plays a role in alcohol-induced toxicity to the liver. Hepatic ATP levels and mitochondrial GSH and SOD2 activity were lower in the ethanol-treated SOD1 knockout mice leading to the suggestion that mitochondrial damage may play an important role in the overall toxicity produced by ethanol in this model. The goal of the current study was to directly evaluate whether chronic alcohol treatment of SOD1 knockout mice indeed does produce mitochondrial injury. In this study, chronic ethanol administration was shown to intensify oxidative changes in mitochondria of Sod1−/− mice. There was an apparent increase in superoxide production in mitochondria of Sod1 knockout mice as reflected by inactivated aconitase activity. To cope with oxidative stress, mitochondria of Sod1−/− mice up-regulated some adaptive defensive responses such as an increase of total thiols, GSH and UCP2 protein content. Thiols, including glutathione are protective antioxidants, acting as free radical scavengers, therefore thiol-disulfide balance can be an indication of oxidative damage in tissues. The increase of UCP2 proteins is believed to occur during mild oxidative stress to cause some uncoupling due to lowering of the proton motive force, with a subsequent decrease in superoxide production (24). However, despite some adaptive mechanisms against oxidative stress, mitochondria of mice with deletion of the SOD1 enzyme and fed the dextrose diet still showed spontaneous swelling and MPT opening even in the absence of added Ca2+, some cytochrome c release, caspase-3 activation and apoptosis in liver tissue. These data confirm the important role of SOD1 in superoxide dismutation and protection of mitochondria from oxidative stress. Indeed, recent studies showed that SOD1 is localized in mitochondria (26, 27), and by protecting mitochondria from ROS-induced injury SOD1 could determine the fate of cells (28). A recent study showed that mitochondrial SOD1 was released from mitochondria in parallel to cytochrome c release, consequently increasing the susceptibility of mitochondria to oxidative stresses, induction of ANT oxidation, and the opening of MPT pores and eventually leading to the development of apoptotic reactions (29).
Chronic ethanol consumption increased oxidative stress as revealed by the further inactivation of aconitase activity and the decline in the adaptive enlargement of total thiol and UCP2 contents in mitochondria of Sod1−/− mice. In rats, chronic alcohol consumption induced inhibition of complexes I and III of the electron transport chain of the rat liver mitochondria and impaired the rate of ATP synthesis (9). We did not detect any significant decreases in the rate of respiration in mitochondria of wild type and knockout mice by ethanol as found with rat liver mitochondria. This possible species difference in sensitivity of mitochondrial respiration to ethanol has also been reported by others (30). However, ethanol enhanced the uncoupling of respiration and oxidative phosphorylation in mitochondria of Sod1−/− mice (but not in wild type mice) as detected by increased rates of respiration in state 4 and lowering of the respiratory control and the ADP/O ratio with glutamate plus malate as substrate. Quenching of Rh 123 fluorescence also showed that the mitochondria from the Sod1−/− mice did not utilize the Δψ efficiently as addition of ADP to these mitochondria did not elevate fluorescence, indicative of utilization of the Δψ to phosphorylate ADP to ATP, to the same extent as it did with mitochondria from the other 3 groups. The ethanol consumption increased MPT pore opening in mitochondria of Sod1−/− mice even in the absence of Ca2+resulting in significant swelling and cytochrome c release as well as sensitizing the mitochondria to MPT opening at 10μM Ca2+, a concentration without any effect in the other groups.
Since chronic alcohol consumption intensified swelling, uncoupled respiration and phosphorylation and produced a decline in membrane potential in mitochondria of Sod1−/− mice, the components of nonspecific pores (which regulate the MPT) were evaluated. The mitochondrial membrane permeability may be regulated by a so-called “megachannel,” whose composition is complex and controversial. The major proteins of these channels are the adenine nucleotide transporter (ANT) and the voltage dependent anion channel (VDAC) located at the contact sites of the inner and outer mitochondrial membrane (31, 32). While VDAC levels were not altered by ethanol feeding, ANT levels were increased 2.5 fold in mitochondria from the SOD1 knockout mice. The exact role of the ANT in MPT is not clear as mitochondria from knockout mice lacking ANTs could still be induced to undergo the MPT although higher concentrations of calcium were required to initiate the MPT (33). It was concluded that ANTs are non-essential structural components of the MPT although they do contribute to its regulation (33). The MPT is characterized by an increase in the inner membrane permeability to compounds with molecular mass less than 1,500 daltons. MPT function can be modulated by members of the Bcl 2 family (31), e.g. mitochondria undergo general permeabilization upon their interaction with Bax or Bak proteins (34). Levels of Bax and Bak were elevated in the mitochondria from the ethanol-treated SOD1 knockout mice. It is known that the activation of Bax is mediated by the translocation of Bax from the cytoplasm to the mitochondria, where it undergoes cleavages to lower molecular weight forms. When cleaved, Bax can oligomerize and oligomerized Bax inserts into the mitochondrial outer membrane (35, 36) where it can potentiate the MPT. A recent study using an in vivo model of apoptosis showed that there are apoptotic populations correspond to early, intermediate, and late apoptotic mitochondria, which are characterized by an increasing extent of permeabilization of their outer membrane and a gradual enrichment in oligomerized Bax proteins (37). We observed an increased level of Bax protein, and importantly of cleaved Bax in liver mitochondria of knockout mice fed ethanol. Bax has recently been reported to interact directly with the VDAC on the outer membrane to release cytochrome c, or with ANT on the inner membrane to initiate the permeability transition (31,32,38,39). During acute ethanol induced hepatocellular apoptosis, Bax, was shown to be translocated to the mitochondrial membrane before mitochondrial cytochrome c release, where it formed Bax/VDAC complexes (6–8). ROS generation in mitochondria was shown to oxidize critical thiol groups of ANT and to induce release of cytochrome c to initiate apoptosis (40). Thiol reductants (41,42) and oxidants (43,44) seem to regulate MPT by affecting the redox state of critical thiols relating to MPT pore formation. MPT occurs when thiol groups of inner membrane proteins including ANT are oxidized to induce conformational changes that form large nonselective pores. Indeed, cross-linking of inner membrane proteins was observed upon mitochondrial induction of the MPT (41,42,45). ANT is a bifunctional protein that, in physiological conditions, exchanges ATP and ADP on the inner mitochondrial membrane. Oxidation of Cys 160 and Cys 257 in ANT markedly increases the affinity of cyclophilin D to the Pro 62 residue of ANT, thereby inducing MPT (46). Pore formation by ANT would require a physical interaction with Bax. An increased association of Bax and Bak with ANT in mitochondria of Sod1−/− mice chronically fed with ethanol was detected, perhaps due, in part, to the overall increased content of these components in the mitochondria of the ethanol-fed SOD1 knockout mice.. A dramatic decline of ADP/ATP translocase activity in these mitochondria was observed in spite of an increase of ANT levels. This suggests that the ANT has lost functional activity in these mitochondria, perhaps as a result of oxidation of critical cysteine residues. We suggest that increases in proapoptotic proteins such as Bax and Bak and their increased association with ANT could play role in permeabilizing the mitochondrial membrane, leading to the decline in Δψ, RCR and ADP/O in mitochondria from Sod1−/− mice fed ethanol.
In tissue injuries due to ischemia/reperfusion, toxic chemicals, and viral infection, apoptotic and necrotic features often coexist, which can lead to cell killing either by apoptosis or necrosis (47–50). This process is described as “necrapoptosis”; and may begin with a common stress or death signal, proceed by shared pathways, but ultimately culminate in either cell lysis (necrosis) or programmed cellular resorption (apoptosis) depending on modifying factors; one important modifying factor is ATP (50). The liver injury produced by ethanol in Sod1−/− mice was mainly necrosis. We believe that the increase in TUNEL staining in the ethanol treated Sod1−/− mice reflects necrotic-induced DNA fragmentation rather than only apoptotic. Indeed, while some increase in caspase-3 activity was observed in the dextrose-fed Sod1−/− mice, compared to the wild type mice, this increase in caspase-3 activity was lower after the ethanol treatment which is not consistent with increased apoptosis. We previously reported that ATP levels were lowed by about 50% in livers of Sod1−/− mice fed ethanol (20). Hepatic ATP levels in the dextrose fed Sod1−/− mice were more then 80% wild type values (20). This switch of liver injury in Sod1−/− mice, from apoptosis to necrosis, by ethanol as reflected by lower caspase-3 activities may relate to the large decline in hepatic ATP level in the ethanol-treated knockout mice, a result of the uncoupling of respiration/oxidative phosphorylation and decline in mitochondrial membrane potential. The ANT interaction with proapoptoic proteins and its dysfunction as an ADP/ATP translocator in mitochondria of Sod1−/− mice as a consequence of chronic ethanol consumption may cause ATP depletion and necrosis, and thus contribute to the liver injury found in this model of ethanol toxicity.
Acknowledgments
These studies were supported by USPHS Grant AA06610 from The National Institute on Alcohol Abuse and Alcoholism.
Abbreviations
- ALD
alcoholic liver disease
- ANT
adenine nucleotide translocator
- GSH
reduced glutathione
- Δψ
mitochondrial membrane potential
- MPT
mitochondrial permeability transition
- ROS
reactive oxygen species
- SOD1
Cu, Zn-superoxide dismutase
- Sod1−/−c
Sod1 knockout mice fed dextrose
- Sod1−/−e
Sod1 knockout mice fed ethanol
- Sod1+/+c
-wild type mice fed dextrose
- Sod1+/+e
wild type mice fed ethanol
- TUNEL
terminal deoxynucleotidyl transferase–mediated dUTP nick-end labeling assay
- UCP2
uncoupling proteins 2
- VDAC
voltage-dependent anion channel protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Dey A, Cederbaum AI. Alcohol and oxidative liver injury. Hepatology. 2006;43:S63–74. doi: 10.1002/hep.20957. [DOI] [PubMed] [Google Scholar]
- 2.Bailey SM, Cunningham CC. Contribution of mitochondria to oxidative stress associated with alcoholic liver disease. Free Radic Biol Med. 2002;32:11–16. doi: 10.1016/s0891-5849(01)00769-9. [DOI] [PubMed] [Google Scholar]
- 3.Mansouri A, Gaou I, De Kerguenec C, Amsellem S, Haouzi D, Berson A, Moreau A, Feldmann G, Letteron P, Pessayre D, Fromenty B. An alcoholic binge causes massive degradation of hepatic mitochondrial DNA in mice. Gastroenterology. 1999;117:181–190. doi: 10.1016/s0016-5085(99)70566-4. [DOI] [PubMed] [Google Scholar]
- 4.Cahill A, Cunningham CC, Adachi M, Ishii H, Bailey SM, Fromenty B, Davies A. Effects of alcohol and oxidative stress on liver pathology: the role of the mitochondrion. Alcohol Clin Exp Res. 2002;26:907–915. [PMC free article] [PubMed] [Google Scholar]
- 5.Bailey SM. A review of the role of reactive oxygen and nitrogen species in alcohol-induced mitochondrial dysfunction. Free Radic Res. 2003;37:585–596. doi: 10.1080/1071576031000091711. [DOI] [PubMed] [Google Scholar]
- 6.Adachi M, Higuchi H, Miura S, Azuma T, Inokuchi S, Saito H, Kato S, Ishii H. Bax interacts with the voltage-dependent anion channel and mediates ethanol-induced apoptosis in rat hepatocytes. Am J Physiol Gastrointest Liver Physiol. 2004;287:G695–705. doi: 10.1152/ajpgi.00415.2003. [DOI] [PubMed] [Google Scholar]
- 7.Higuchi HAdachi M, Miura SGores GJ, Ishii H. The mitochondrial permeability transition contributes to acute ethanol-induced apoptosis in rat hepatocytes. Hepatology. 2001;34:320–328. doi: 10.1053/jhep.2001.26380. [DOI] [PubMed] [Google Scholar]
- 8.Adachi M, Ishii H. Role of mitochondria in alcoholic liver injury. Free Radic Biol Med. 2002;32:487–491. doi: 10.1016/s0891-5849(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 9.Bailey SM, Pietsch EC, Cunningham CC. Ethanol stimulates the production of reactive oxygen species at mitochondrial complexes I and III. Free Radic Biol Med. 1999;27:891–900. doi: 10.1016/s0891-5849(99)00138-0. [DOI] [PubMed] [Google Scholar]
- 10.Kukielka E, Dicker E, Cederbaum AI. Increased production of reactive oxygen species by rat liver mitochondria after chronic ethanol treatment. Arch Biochem Biophys. 1994;309:377–386. doi: 10.1006/abbi.1994.1127. [DOI] [PubMed] [Google Scholar]
- 11.Pastorino JG, Marcineviciute A, Cahill A, Hoek JB. Potentiation by chronic ethanol treatment of the mitochondrial permeability transition. Biochem Biophys Res Commun. 1999;265:405–409. doi: 10.1006/bbrc.1999.1696. [DOI] [PubMed] [Google Scholar]
- 12.Colell A, Garcia-Ruiz C, Morales A, Ballesta A, Ookhtens M, Rodes J, Kaplowitz N, Fernandez-Checa JC. Transport of reduced glutathione in hepatic mitochondria and mitoplasts from ethanol-treated rats: effect of membrane physical properties and S-adenosyl-L-methionine. Hepatology. 1997;26:699–708. doi: 10.1002/hep.510260323. [DOI] [PubMed] [Google Scholar]
- 13.Lieber CS, DeCarli L, Rubin E. Sequential production of fatty liver, hepatitis, and cirrhosis in sub-human primates fed ethanol with adequate diets. Proc Natl Acad Sci U S A. 1975;72:437–441. doi: 10.1073/pnas.72.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Inagaki T, Koike M, Ikuta K, Kobayashi S, Suzuki M, Kato K. Ultrastructural identification and clinical significance of light microscopic giant mitochondria in alcoholic liver injuries. Gastroenterol Jpn. 1989;24:46–53. doi: 10.1007/BF02774870. [DOI] [PubMed] [Google Scholar]
- 15.Bruguera M, Bertran A, Bombi JA, Rodes J. Giant mitochondria in hepatocytes: a diagnostic hint for alcoholic liver disease. Gastroenterology. 1977;73:1383–1387. [PubMed] [Google Scholar]
- 16.Cederbaum AI. Introduction-serial review: alcohol, oxidative stress and cell injury. Free Radic Biol Med. 2001;31:1524–1526. doi: 10.1016/s0891-5849(01)00741-9. [DOI] [PubMed] [Google Scholar]
- 17.Polavarapu R, Spitz DR, Sim JE, Follansbee MH, Oberley LW, Rahemtulla A, Nanji AA. Increased lipid peroxidation and impaired antioxidant enzyme function is associated with pathological liver injury in experimental alcoholic liver disease in rats fed diets high in corn oil and fish oil. Hepatology. 1998;27:1317–1323. doi: 10.1002/hep.510270518. [DOI] [PubMed] [Google Scholar]
- 18.Zidenberg-Cherr S, Olin KL, Villanueva J, Tang A, Phinney SD, Halsted CH, Keen CL. Ethanol-induced changes in hepatic free radical defense mechanisms and fatty-acid composition in the miniature pig. Hepatology. 1991;13:1185–1192. [PubMed] [Google Scholar]
- 19.Wheeler MD, Kono H, Yin M, Rusyn I, Froh M, Connor HD, Mason RP, Samulski RS, Thurman RG. Delivery of the Cu/Zn-superoxide dismutase gene with adenovirus reduces early alcohol-induced liver injury in rats. Gastroenterology. 2001;120:1241–1250. doi: 10.1053/gast.2001.23253. [DOI] [PubMed] [Google Scholar]
- 20.Kessova IG, Ho YS, Thung S, Cederbaum AI. Alcohol-induced liver injury in mice lacking Cu, Zn-superoxide dismutase. Hepatology. 2003;38:1136–1145. doi: 10.1053/jhep.2003.50450. [DOI] [PubMed] [Google Scholar]
- 21.Emaus RK, Grunwald R, Lemasters JJ. Rhodamine 123 as a probe of transmembrane potential in isolated rat-liver mitochondria: spectral and metabolic properties. Biochim Biophys Acta. 1986;850:436–448. doi: 10.1016/0005-2728(86)90112-x. [DOI] [PubMed] [Google Scholar]
- 22.Hausladen A, Fridovich I. Superoxide and peroxynitrite inactivate aconitases, but nitric oxide does not. J Biol Chem. 1994;269:29405–29408. [PubMed] [Google Scholar]
- 23.Hausladen A, Fridovich I. Measuring nitric oxide and superoxide: rate constants for aconitase reactivity. Methods Enzymol. 1996;269:37–41. doi: 10.1016/s0076-6879(96)69007-7. [DOI] [PubMed] [Google Scholar]
- 24.Echtay KS, Murphy MP, Smith RA, Talbot DA, Brand MD. Superoxide activates mitochondrial uncoupling protein 2 from the matrix side. Studies using targeted antioxidants. J Biol Chem. 2002;277:47129–47135. doi: 10.1074/jbc.M208262200. [DOI] [PubMed] [Google Scholar]
- 25.Venkatraman A, Shiva S, Wigley A, Ulasova E, Chhieng D, Bailey SM, Darley-Usmar V. The role of iNOS in alcohol-dependent hepatotoxicity and mitochondrial dysfunction in mice. Hepatology. 2004;40:565–573. doi: 10.1002/hep.20326. [DOI] [PubMed] [Google Scholar]
- 26.Kira Y, Sato EF; Inoue M. Association of Cu-Zn-type superoxide dismutase with mitochondria and peroxisomes. Arch Biochem Biophys. 2002;399:96–102. doi: 10.1006/abbi.2001.2738. [DOI] [PubMed] [Google Scholar]
- 27.Okado-Matsumoto A, Fridovich I. Subcellular distribution of superoxide dismutases (SOD) in rat liver: Cu,Zn-SOD in mitochondria. J Biol Chem. 2001;276:38388–38393. doi: 10.1074/jbc.M105395200. [DOI] [PubMed] [Google Scholar]
- 28.Mondola P, Santillo M, Seru R, Damiano S, Alvino C, Ruggiero G, Formisano P, Terrazzano G, Secondo A, Annunziato L. Cu,Zn superoxide dismutase increases intracellular calcium levels via a phospholipase C-protein kinase C pathway in SK-N-BE neuroblastoma cells. Biochem Biophys Res Commun. 2004;324:887–892. doi: 10.1016/j.bbrc.2004.09.131. [DOI] [PubMed] [Google Scholar]
- 29.Li Q, Sato EF, Kira Y, Nishikawa M, Utsumi K, Inoue M. A possible cooperation of SOD1 and cytochrome c in mitochondria-dependent apoptosis. Free Radic Biol Med. 2006;40:173–181. doi: 10.1016/j.freeradbiomed.2005.09.037. [DOI] [PubMed] [Google Scholar]
- 30.Venkatraman A, Shiva S, Davis AJ, Bailey SM, Brookes PS, Darley-Usmar VM. Chronic alcohol consumption increases the sensitivity of rat liver mitochondrial respiration to inhibition by nitric oxide. Hepatology. 2003;38:141–147. doi: 10.1053/jhep.2003.50293. [DOI] [PubMed] [Google Scholar]
- 31.Vyssokikh MY, Brdiczka D. The function of complexes between the outer mitochondrial membrane pore (VDAC) and the adenine nucleotide translocase in regulation of energy metabolism and apoptosis. Acta Biochim Pol. 2003;50:389–404. [PubMed] [Google Scholar]
- 32.Crompton M, Barksby E, Johnson N, Capano M. Mitochondrial intermembrane junctional complexes and their involvement in cell death. Biochimie. 2002;84:143–152. doi: 10.1016/s0300-9084(02)01368-8. [DOI] [PubMed] [Google Scholar]
- 33.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, MacGregor GR, Wallac DC. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature. 2004;427:461–465. doi: 10.1038/nature02229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Degli Esposti M, Dive C. Mitochondrial membrane permeabilisation by Bax/Bak. Biochem Biophys Res Commun. 2003;304:455–461. doi: 10.1016/s0006-291x(03)00617-x. [DOI] [PubMed] [Google Scholar]
- 35.Green DR, Kroemer G. The pathophysiology of mitochondrial cell death. Science. 2004;305:626–629. doi: 10.1126/science.1099320. [DOI] [PubMed] [Google Scholar]
- 36.Khosravi-Far R, Esposti MD. Death receptor signals to mitochondria. Cancer Biol Ther. 2004;3:1051–1057. doi: 10.4161/cbt.3.11.1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guihard G, Bellot G, Moreau C, Pradal G, Ferry N, Thomy R, Fichet P, Meflah K, Vallette FM. The mitochondrial apoptosis-induced channel (MAC) corresponds to a late apoptotic event. J Biol Chem. 2004;279:46542–46550. doi: 10.1074/jbc.M405153200. [DOI] [PubMed] [Google Scholar]
- 38.Verrier F, Deniaud A, Lebras M, Metivier D, Kroemer G, Mignotte B, Jan G, Brenner C. Dynamic evolution of the adenine nucleotide translocase interactome during chemotherapy-induced apoptosis. Oncogene. 2004;23:8049–8064. doi: 10.1038/sj.onc.1208001. [DOI] [PubMed] [Google Scholar]
- 39.Belzacq AS, Vieira HL, Verrier F, Vandecasteele G, Cohen I, Prevost MC, Larquet E, Pariselli F, Petit PX, Kahn A, Rizzuto R, Brenner C, Kroemer G. Bcl-2 and Bax modulate adenine nucleotide translocase activity. Cancer Res. 2003;63:541–546. [PubMed] [Google Scholar]
- 40.Kanno T, Sato EE, Muranaka S, Fujita H, Fujiwara T, Utsumi T, Inoue M, Utsumi K. Oxidative stress underlies the mechanism for Ca(2+)-induced permeability transition of mitochondria. Free Radic Res. 2004;38:27–35. doi: 10.1080/10715760310001626266. [DOI] [PubMed] [Google Scholar]
- 41.Fagian MM, Pereira-da-Silva L, Martins IS, Vercesi AE. Membrane protein thiol cross-linking associated with the permeabilization of the inner mitochondrial membrane by Ca2+ plus prooxidants. J Biol Chem. 1990;265:19955–19960. [PubMed] [Google Scholar]
- 42.Valle VG, Fagian MM, Parentoni LS, Meinicke AR, Vercesi AE. The participation of reactive oxygen species and protein thiols in the mechanism of mitochondrial inner membrane permeabilization by calcium plus prooxidants. Arch Biochem Biophys. 1993;307:1–7. doi: 10.1006/abbi.1993.1551. [DOI] [PubMed] [Google Scholar]
- 43.Bernardes CF, Meyer-Fernandes JR, Basseres DS, Castilho RF, Vercesi AE. Ca(2+)-dependent permeabilization of the inner mitochondrial membrane by 4,4′-diisothiocyanatostilbene-2,2′-disulfonic acid (DIDS) Biochim Biophys Acta. 1994;1188:93–100. doi: 10.1016/0005-2728(94)90026-4. [DOI] [PubMed] [Google Scholar]
- 44.Lenartowicz E, Bernardi P, Azzone GF. Phenylarsine oxide induces the cyclosporin A-sensitive membrane permeability transition in rat liver mitochondria. J Bioenerg Biomembr. 1991;23:679–688. doi: 10.1007/BF00785817. [DOI] [PubMed] [Google Scholar]
- 45.Castilho RF, Kowaltowski AJ, Vercesi AE. The irreversibility of inner mitochondrial membrane permeabilization by Ca2+ plus prooxidants is determined by the extent of membrane protein thiol cross-linking. J Bioenerg Biomembr. 1996;28:523–529. doi: 10.1007/BF02110442. [DOI] [PubMed] [Google Scholar]
- 46.McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore. Biochem J. 2002;367:541–548. doi: 10.1042/BJ20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Grasl-Kraupp B, Ruttkay-Nedecky B, Koudelka H, Bukowska K, Bursch W, Schulte-Hermann R. In situ detection of fragmented DNA (TUNEL assay) fails to discriminate among apoptosis, necrosis, and autolytic cell death: a cautionary note. Hepatology. 1995;21:1465–1468. doi: 10.1002/hep.1840210534. [DOI] [PubMed] [Google Scholar]
- 48.Ohno M, Takemura G, Ohno A, Misao J, Hayakawa Y, Minatoguchi S, Fujiwara T, Fujiwara H. “Apoptotic” myocytes in infarct area in rabbit hearts may be oncotic myocytes with DNA fragmentation: analysis by immunogold electron microscopy combined with In situ nick end-labeling. Circulation. 1998;98:1422–1430. doi: 10.1161/01.cir.98.14.1422. [DOI] [PubMed] [Google Scholar]
- 49.Kim JS, He L, Qian T, Lemasters JJ. Role of the mitochondrial permeability transition in apoptotic and necrotic death after ischemia/reperfusion injury to hepatocytes. Curr Mol Med. 2003;3:527–535. doi: 10.2174/1566524033479564. [DOI] [PubMed] [Google Scholar]
- 50.Lemasters JJ, Qian T, Bradham CA, Brenner DA, Cascio WE, Trost LC, Nishimura Y, Nieminen AL, Herman B. Mitochondrial dysfunction in the pathogenesis of necrotic and apoptotic cell death. J Bioenerg Biomembr. 1999;31:305–319. doi: 10.1023/a:1005419617371. [DOI] [PubMed] [Google Scholar]