

Abstract
The mitogen-activated protein kinases ERK (extracellular signal-regulated kinase), JNK (c-Jun N-terminal kinase), and p38 phosphorylate and activate transcription factors that promote proliferative and inflammatory responses, whereas glucocorticoid receptor (GR) activation inhibits cell growth and inflammation. We demonstrate that JNK and ERK but not p38 phosphorylate GR in vitro primarily at Ser-246. Selective activation of either ERK or JNK in vivo inhibits GR-mediated transcriptional activation, which depends on receptor phosphorylation at Ser-246 by JNK but not ERK. Thus, JNK inhibits GR transcriptional activation by direct receptor phosphorylation, whereas ERK does so indirectly. We propose that phosphorylation of GR by JNK or of a GR cofactor by ERK provides mechanisms to ensure the rapid inhibition of GR-dependent gene expression when it conflicts with mitogenic or proinflammatory signals.
The glucocorticoid receptor (GR) is a hormone-dependent transcription factor that induces differentiation, regulates metabolic processes, such as gluconeogenesis, and suppresses the inflammatory response (1–3). GR is expressed in virtually all tissues yet displays a remarkable capacity to regulate genes in a cell type-specific manner. Although steroid hormones, such as cortisol, act as the primary signal in activating the receptor’s transcriptional regulatory functions, GR-mediated transcriptional activation is also modulated both positively and negatively by phosphorylation (4). GR is phosphorylated at four major sites (Thr-171, Ser-224, Ser-232, and Ser-246) in its N-terminal transcriptional regulatory region, and we have recently shown that cyclin-dependent kinase (Cdk) and mitogen-activated protein kinase (MAPK) phosphorylate the GR in vitro at distinct residues that together correspond to the major receptor amino acids modified in vivo (5). In yeast, reduced Cdk activity results in decreased receptor-dependent transcriptional activation, indicating that Cdk function is necessary for full receptor-mediated transcriptional enhancement (5). In contrast, reduced MAPK activity in yeast, through the deletion of the MAPK homologues FUS3 and KSS1, increases transcriptional activation by the receptor, suggesting that MAPK inhibits receptor function (5). However, which mammalian MAPK family members are responsible for this inhibition and whether the reduction of GR-mediated transcriptional activation is mediated through receptor phosphorylation in vivo have yet to be determined.
Although the MAPK family members extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38 MAPK (p38) are thought to phosphorylate the same sequence motif (nonpolar-X-Ser/Thr-Pro), they are activated by different extracellular stimuli, respond to distinct sets of upstream activators, and phosphorylate specific substrates in vitro and in vivo (6, 7). The ERK pathway is stimulated in response to growth factors through Ras activation, which depends on translocation of the guanine nucleotide exchange factor Sos to the membrane (8–10). ERK substrates include transcription factors, such as Elk-1 (reviewed in ref. 11) and the estrogen receptor (12). JNK is activated by UV irradiation (13, 14), tumor necrosis factor α, proinflammatory cytokines, and the lipopolysaccharide of Gram-negative bacteria (15, 16). Activation of JNK is achieved primarily through the Rho family of GTPases, including Rac. JNK phosphorylates the N terminus of the transcription factor c-Jun (6) and ATF-2 (17), which increases their ability to activate transcription (7, 13, 18, 19). p38, like its yeast counterpart Hog1, is stimulated by osmotic stress, as well as by UV light, lipopolysaccharide, and inflammatory cytokines (20, 21). The substrates of p38 include transcription factors (e.g., ATF-2 and CHOP) and the translation factor PHAS-I (22, 23). Thus, the MAPKs are stimulated by a wide variety of extracellular stimuli and act upon a wide range of targets.
In this report, we analyze GR phosphorylation by MAPK family members ERK, JNK, and p38 and examine the consequences of selective activation of MAPK pathways on GR transcriptional activation in vivo.
MATERIALS AND METHODS
Purification of Receptor Derivatives, in Vitro Kinase Assays, and Phosphopeptide Mapping.
Wild-type rat GR derivatives containing N-terminal amino acids 106–318 or 106–414, or a receptor mutant with a single amino acid substitution (S246A), were expressed in Escherichia coli as glutathione S-transferase (GST)-fusion proteins (GST–GR) as previously described (5).
GST–GR substrate (2 μg) was bound to 100 μl of a 50% slurry of glutathione beads for 20 min on ice and washed twice with 1 ml of DK buffer (50 mM potassium phosphate, pH 7.15/10 mM MgCl2/5 mM NaF/5 mM DTT), supplemented with protease inhibitors [1 mM phenylmethylsulfonyl fluoride (Sigma) and 1 μg/ml each of aprotinin, pepstatin A, and leupeptin (Boehringer Mannheim)]. Reaction mixtures containing the immobilized receptor and recombinant purified ERK2 (New England Biolabs), bacterially expressed recombinant rat JNKβ, or murine p38 kinase (Stratagene) were set up according to the manufacturer’s instructions. All the kinase reactions were allowed to proceed for 30 min at room temperature (RT) with continuous shaking and stopped by washing the beads five times with 1 ml of PBS to remove unincorporated radioisotope. The labeled receptor protein was released by boiling for 2 min in an equal volume of 2× SDS sample buffer and run on SDS/polyacrylamide gels—10% (for JNK) or 12.5% (for ERK and p38)—along with the corresponding control substrates.
Myelin basic protein (MBP; Boehringer Mannheim, 2 μg per reaction), purified recombinant GST–c-Jun (Stratagene, 1 μg per reaction), and PHAS-I (Stratagene, 5 μg per reaction) were phosphorylated according to the manufacturer’s instructions and used as independent measures of kinase activity for ERK2, JNK, and p38 kinase, respectively. Equal amounts of GST–GR and the respective control protein were verified by Coomassie blue staining, and phosphorylation of substrates was visualized by autoradiography.
Two-dimensional phosphopeptide mapping of phosphorylated GST–GR was performed exactly as described (5, 24).
Cell Lines and Treatments.
Human osteosarcoma cell line SAOS2 ectopically expressing rat GR (25), U-2 OS human osteosarcoma cells (ATCC HTB 96), and human HeLa cervical carcinoma cells (ATCC, CCL 2) were maintained in DMEM (GIBCO/BRL) supplemented with 10% fetal bovine serum (FBS; HyClone), 50 units/ml of each penicillin, streptomycin, and 2 mM l-glutamine (GIBCO/BRL). Before metabolic labeling of SAOS2-GR cells with [32P]orthophosphate, cells were cultured in 150-mm dishes in serum-depleted medium (0.1% FBS/DMEM) for 48 h. All hormone treatments were done in DMEM/10% FBS containing either 100 nM dexamethasone (Dex) or an identical volume of 100% ethanol.
Metabolic Labeling of SAOS2-GR and HeLa Cells with [32P]Orthophosphate.
Serum-starved SAOS2-GR cells (25) were refed with 4 ml of phosphate-free medium supplemented with 100 nM Dex with or without 10% FBS and incubated for 4 h in the presence of 1 mCi/ml of [32P]orthophosphate per 150-mm dish. Transiently transfected HeLa cells were refed with 2.5 ml of phosphate-free medium supplemented with 100 nM Dex and incubated for 2 h with 1 mCi/ml of [32P]orthophosphate per 100-mm dish. Cells were washed twice with PBS and lysed in 0.5 ml of ice-cold lysis buffer (150 mM NaCl/50 mM Hepes, pH 7.5/1 mM EDTA/1 mM EGTA/10% glycerol/1% Triton X-100/1 mM NaF/25 μM ZnCl2), supplemented with protease inhibitors and phosphatase inhibitors. Metabolically labeled GR was immunoprecipitated from the cell extracts with BuGR2 mouse monoclonal antibodies (26) for 2 h on ice. Immune complexes were collected on protein A/G agarose beads (Santa Cruz Biotechnology) for 1 h at 4°C nutating, and washed four times with lysis buffer; the receptor was then released from the beads by boiling in 50 μl of 2× SDS sample buffer. The GR isolated from SAOS2-GR cells was purified on a 9% SDS/polyacrylamide gel and subjected to phosphopeptide mapping. The GR derivative isolated from HeLa cells was purified on a 9% SDS/polyacrylamide gel and transferred to Immobilon paper for autoradiography and Western blotting with the GR-specific rabbit polyclonal antibody, sc-1002 (Santa Cruz Biotechnology).
Plasmids.
pCMV-wt GR, pCMV-GR S246A, and pCMV-GR S224A S232A expression plasmids were used to produce rat GR, and ΔGTCO-CAT reporter plasmid, containing two consensus GREs upstream of chloramphenicol acetyltransferase (CAT) gene, was used to assess GR transcriptional activity. pCMV-LacZ plasmid produced β-galactosidase (β-Gal). pRK5-HA ERK1, pRK5-M2 JNK, pRK5-HA f-sos (27), and pCMV5-racQ61L (6) plasmids were used in transient transfection assays to selectively activate ERK and JNK pathways. Empty pRK5 expression vector was used to equalize the amount of DNA in all transient transfection mixes.
Transient Transfections and GR Activity Assays.
Subconfluent HeLa cells were washed once with serum-free medium and transfected with the indicated plasmids by using 25 μl of Lipofectamine reagent (GIBCO/BRL) in a total volume of 2.5 ml of serum-free medium per 60-mm dish according to the manufacturer’s instructions. At 3 h posttransfection, 2.5 ml of DMEM/10% FBS was added to each dish, and cells were incubated for another 12 h. The next day, cells were refed with fresh DMEM/10% FBS with or without 100 nM Dex and incubated for an additional 8 h.
For metabolic labeling, HeLa cells were washed once with serum-free medium and transfected with the indicated plasmids by using 60 μl of Lipofectamine reagent in a total volume of 5 ml of serum-free medium per 100-mm dish. At 3 h posttransfection, 5 ml of DMEM/20% FBS was added to each dish, and cells were incubated for another 12 h.
Human U-2 OS cells were refed with fresh DMEM/10% FBS 1 h before transfection and transfected with the indicated plasmids via the calcium phosphate precipitation method (28). After 8 h, cells were washed three times with PBS to remove calcium phosphate precipitates, allowed to recover overnight in DMEM/10% FBS, and then incubated with fresh medium with or without Dex for additional 8 h.
Transfected cells were washed twice with PBS and lysed directly on the dishes in 250 μl of 1× Reporter Lysis buffer (Promega) according to the manufacturer’s instructions. Nonchromatographic CAT assays and β-Gal assays were performed as described (29).
Western Blotting and JNK Assays.
To make protein extracts from transfected cells, HeLa and U-2 OS cells were washed twice with PBS and lysed directly on the dishes in 200 μl of ice-cold lysis buffer (described above). The lysates were collected and centrifuged at 10,000 × g for 10 min at 4°C. Protein concentrations in all samples were adjusted with the lysis buffer, and 30 μl of the whole cell extracts (WCE) was boiled in 30 μl of 2× SDS sample buffer. For Western blotting, protein extracts were fractionated on 10% SDS/polyacrylamide gels, transferred to Immobilon paper (Millipore), and probed with mouse monoclonal antibodies against hemagglutinin (HA) (Boehringer Mannheim), or FLAG (M2, Kodak), or with a rabbit polyclonal antiserum against GR (Santa Cruz Biotechnology). The blots were developed with horseradish peroxidase-coupled goat anti-mouse or goat anti-rabbit antibodies and enhanced chemiluminescence (Amersham).
To immunoprecipitate transfected JNK from the WCE, 200 μl of each extract was incubated with 1.5 μg of M2 antibodies for 1 h on ice, and the immune complexes were collected on 30 μl of the protein A/G agarose beads for 1 h at 4°C, nutating. The beads were washed three times in 750 μl of the lysis buffer and split in two. One half was boiled in 30 μl of 2× SDS sample buffer for immunoblotting, whereas the other half was washed twice in 600 μl of the kinase buffer (20 mM Hepes, pH 7.5/20 mM glycerophosphate/10 mM MgCl2/10 mM DTT/50 μM sodium orthovanadate), resuspended in 30 μl of the kinase buffer, and the kinase reaction was initiated by adding 1 μg of GST–c-Jun along with 20 μM ATP and 100 μCi of [γ-32P]ATP. The reaction proceeded for 30 min at RT with continuous shaking, and was terminated by boiling the proteins in 30 μl of the 2× SDS sample buffer.
RESULTS
JNK and ERK but Not p38 Kinase Phosphorylate GR in Vitro.
Using an in vitro kinase assay, we examined whether JNK, ERK, and p38 can phosphorylate a bacterially expressed and purified GST–GR fusion protein containing the receptor N-terminal residues 106–318, which harbor the four major receptor phosphorylation sites (GST–GR106–318). Fig. 1A demonstrates that both ERK and JNK use GST–GR106–318 derivative as a substrate in vitro (lanes 1 and 3), with efficiency comparable to the established ERK and JNK substrates, MBP and GST–c-Jun (lanes 2 and 4). In contrast, p38 failed to phosphorylate GR but displayed kinase activity toward the p38-specific substrate PHAS-I (lanes 5 and 6). These findings support the idea that GR is a substrate in vitro for ERK and JNK but not p38.
Figure 1.

Phosphorylation of GR by members of the MAPK family. (A) GR phosphorylation by ERK, JNK, and p38 in vitro. A GST–GR fusion protein containing receptor residues 106–318 (GST–GR106–318) was expressed in E. coli, purified by glutathione agarose affinity chromatography (5), and phosphorylated in vitro using purified recombinant ERK, JNK, and p38. MBP, purified recombinant GST– c-Jun, and PHAS-I were phosphorylated according to the manufacturer’s instructions and used as independent measure of kinase activity for ERK, JNK, and p38 kinase, respectively. The same amount of the GST–GR106–318 and the respective control proteins in each pair of lanes was verified by Coomassie blue staining (not shown), and phosphorylation of substrates was visualized by autoradiography. (B) Analysis of GR phosphorylation by ERK and JNK through two-dimensional phosphopeptide mapping. Polyacrylamide gels containing the receptor labeled by ERK or JNK were digested with V8 protease and subjected to phosphopeptide mapping as described (5). The two-dimensional map of the receptor metabolically labeled and isolated from Dex-treated rat hepatoma cells (in vivo) and the schematic representation of the GR phosphopeptides identified in vivo are shown on the left.
Two-dimensional peptide mapping was used to determine the amino acid residues phosphorylated in vitro by the different kinases, exploiting the known map positions of each in vivo phosphorylation site after the receptor is cleaved with V8 protease (Fig. 1B) (5, 24). Phosphorylation of GST–GR106–318 by ERK in vitro and subsequent V8 proteolysis result in the appearance of two major and one minor phosphopeptides that comigrate with phosphopeptides 5a, 5b, and 4 on the in vivo map (Fig. 1B). Peptides 5a and 5b result from incomplete digestion of a peptide containing a single phosphoserine at position Ser-246, whereas peptide 4 contains a single phosphothreonine at position Thr-171. JNK produced a phosphopeptide pattern strikingly similar to that obtained with ERK, suggesting that the same amino acid residues, Ser-246 and to a lesser extent Thr-171, are the likely targets of these protein kinases (Fig. 1B).
Serine 246 Is a Major GR Residue Phosphorylated by JNK in Vitro.
To verify that the major site of phosphate incorporation catalyzed by JNK is the residue Ser-246, we have constructed a GST–GR derivative with a single amino acid substitution of Ser-246 → Ala (S246A). GST–GR fusion proteins GST–GR106–318 (which has been used in Fig. 1), GST–GR106–414, and GST–GR106–414 S246A were expressed in E. coli and subjected to an in vitro kinase assay by using JNK. Fig. 2A demonstrates that both wild-type GR constructs are efficiently phosphorylated by JNK, whereas the S246A mutation decreases the amount of 32P incorporated into this receptor derivative by over 90%. Two-dimensional phosphopeptide analysis of the V8-digested receptor protein revealed that the S246A substitution completely eliminated phosphopeptides 5a and 5b (Fig. 2B). Thus, the receptor residue Ser-246 is the primary target for JNK phosphorylation in vitro.
Figure 2.

GR Ser-246 is the major site phosphorylated by JNK. (A) Phosphorylation of wild-type (wt) GR and GR S246A mutant by JNK in vitro. GST–GR106–318 (wt), as well as GST–GR derivatives containing residues 106–414 (GST–GR106–414), wt or with a single amino acid substitution S246A were expressed in E. coli, purified, and phosphorylated by JNK in vitro. Reaction products were separated by SDS/PAGE on 10% gels, stained with Coomassie blue to verify that equal amounts of the receptor derivatives were present in each lane, and autoradiography was performed. The densitometric measurement of the wt GR phosphorylation was arbitrarily set as 1. (B) JNK phosphorylates GR residue Ser-246 in vitro. GST–GR106–414 (wt GR) and GST-GR106–414 S246A (GR S246A) were phosphorylated by JNK in vitro and subjected to two-dimensional phosphopeptide mapping. Note the lack of peptide 5 on the map of the receptor S246A mutant.
Mitogen Stimulation Results in Increased GR Phosphorylation at Ser-246 in SAOS2 Cells in Vivo.
Our in vitro data suggest that in vivo receptor phosphorylation at Ser-246 should be catalyzed when the JNK and ERK pathways are activated, for example by serum stimulation, whereas in cells depleted of growth factors or serum, Ser-246 phosphorylation should be inhibited. SAOS2 cells stably expressing GR (25) were serum-starved and subjected to metabolic labeling with [32P]orthophosphate in the absence or presence of serum stimulation. Addition of serum to these cells results in a 10-fold induction of JNK activity but a less than 2-fold induction of ERK kinase potential (data not shown), suggesting that JNK is the major mitogen-induced kinase activity present in this cell type. The labeled GR was immunoprecipitated from stimulated or unstimulated cells, digested with V8 protease, and analyzed by two-dimensional phosphopeptide mapping. Fig. 3 illustrates that in serum-deprived cells, peptide 5, corresponding to GR phosphoserine-246, is not present, whereas peptide 4 containing phosphothreonine-171 is present, suggesting that Thr-171 might be a target of a different family of kinases, as MAPKs are largely inactive in serum-deprived cells. Our recent findings indicate that Thr-171 is a target for glycogen synthase kinase-3 (unpublished work). In contrast, receptor isolated from serum-stimulated cells gives rise to phosphopeptide 5, which reflects phosphorylation of Ser-246. In addition, in serum-stimulated but not serum-starved cells, we observe phosphopeptides 1 and 2 reflecting receptor phosphorylation at Ser-224 and Ser-232 by cyclin/Cdk complexes (5). This finding is consistent with the cells being in the G0 stage of the cell cycle before serum stimulation and reinitiation of the cell cycle when serum is repleted. Thus, serum stimulation of quiescent cells results in the phosphorylation of Ser-246.
Figure 3.
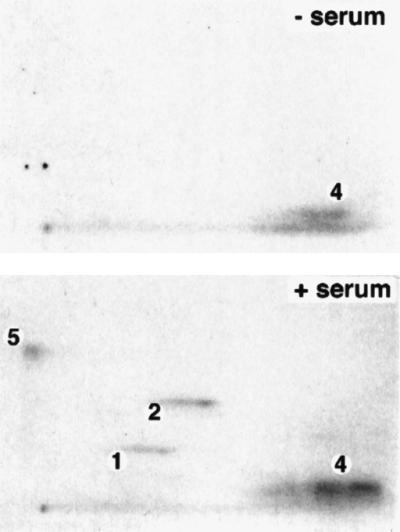
Mitogen stimulation results in Ser-246 phosphorylation in vivo. SAOS2 human osteosarcoma cells ectopically expressing the rat wt GR (25) were serum-starved for 48 h in 0.1% FBS/DMEM. The receptor was metabolically labeled for 4 h with 1 mCi/ml of [32P]orthophosphate and 100 nM Dex, in the absence (− serum) and presence (+ serum) of 10% FBS. In vivo labeled receptor was immunoprecipitated from cell lysates with the GR-specific monoclonal antibody, BuGR2 (26), and subjected to two-dimensional phosphopeptide mapping.
Selective Activation of ERK and JNK Pathway Results in Inhibition of GR Transcriptional Enhancement.
Before analyzing the consequences of ERK and JNK activation on GR hormone-dependent transcriptional activity, we developed conditions whereby either the ERK or JNK signaling pathways could be selectively activated in vivo. HeLa cells containing endogenous GR were transiently transfected with either ERK or JNK in the presence of constitutively active versions of their upstream activators, Sos and Rac, or an empty pRK5 expression vector. Sos is a nucleotide exchange factor that serves to activate Ras, and subsequently ERK, by catalyzing the exchange of the Ras-bound GDP for GTP (30). On the other hand, Rac, a member of the Rho protein family, is a small GTP-binding protein, capable of activating the JNK and p38, but not ERK, pathways (31). To induce activation of the ERK pathway, a constitutively active membrane-targeted HA-tagged-farnesylated Sos (HA-f-sos) (27) and an HA-tagged ERK (HA-ERK) were transfected into HeLa cells. WCE were prepared, and a Western blot was performed with an HA-specific monoclonal antibody that recognizes both transfected proteins. Fig. 4A demonstrates that co-expression of HA-f-sos induces a mobility shift in the HA-ERK as a result of a phosphorylation that is indicative of ERK activation. This active variant of Sos does not cause activation of the JNK pathway in HeLa cells as determined by an in vitro JNK kinase assay with recombinant GST–c-Jun as a substrate (data not shown). To activate the JNK pathway, a constitutively active Rac (racQ61L) mutant was used (31, 32). This active form of Rac does not cause activation of ERK, as judged by the lack of a mobility shift of ERK caused by activating phosphorylation (data not shown). Activation of the JNK pathway was confirmed via an in vitro kinase assay by immunoprecipitating JNK from HeLa cells in the presence or absence of racQ61L and using bacterially expressed and purified GST–c-Jun as the substrate. As illustrated in Fig. 4A (right panel), incorporation of 32P into GST–c-Jun in vitro is increased more than 10-fold when JNK is co-expressed with racQ61L, which reflects activation of the JNK pathway. This activation occurred to the same extent in the presence or absence of the GR agonist Dex. The level of JNK protein is not affected by racQ61L co-expression or by treatment of cells with Dex (data not shown). Thus, our transfection procedure ensured specific activation of either the ERK or JNK pathway.
Figure 4.

Activation of the ERK or the JNK pathway inhibits GR transcriptional activation. (A) Selective activation of ERK and JNK in HeLa cells by f-sos and racQ61L. pRK5-HA ERK (1.5 μg), pRK5-M2 JNK (1.5 μg), along with pRK5-HA f-sos (4 μg) or pCMV5-racQ61L (4 μg) or an empty pRK5 vector (4 μg), where indicated, was transfected into HeLa cells (2.5 × 105 cells/60-mm dish) using Lipofectamine. Transfected cells were treated with 100 nM Dex where indicated and harvested 8 h later. To visualize HA ERK and HA f-sos, WCE from ERK-transfected cells were immunoblotted with anti-HA mouse monoclonal antibody (left panel). FLAG-tagged JNK was immunoprecipitated from WCE by using the FLAG-specific mouse monoclonal antibody M2 and used in vitro to phosphorylate recombinant purified GST–c-Jun. Phosphorylated proteins were separated on 10% SDS/polyacrylamide gels, stained with Coomassie blue to verify equal amount of GST–c-Jun in each lane, and autoradiographed (right panel). (B) Inhibition of GR transcriptional enhancement by activated ERK and JNK. HeLa cells were transfected as described in A. In addition, each dish received 1.25 μg of the GR reporter plasmid ΔGTCO-CAT and 0.5 μg of pCMV-LacZ plasmid. Cells were harvested, and transcriptional activity of the endogenous GR was assessed via the CAT assay (29) and normalized to β-Gal activity. (C) Level of GR protein is unaffected by JNK or ERK activation. WCE were prepared from a parallel set of transfected cells. Equal amounts of total protein (50 μg/lane) were separated by SDS/PAGE, transferred to Immobilon paper, probed with a GR-specific polyclonal antibody, and visualized by enhanced chemiluminescence.
We next examined whether stimulation of the JNK and ERK pathways affected transcriptional activation by the endogenous GR in HeLa cells. As illustrated in Fig. 4B (left panel), GR transcriptional activity was induced over 7-fold by treatment with 100 nM Dex in the absence of activated ERK. In contrast, activation of ERK via f-sos resulted in more than a 4-fold reduction of hormone-dependent transcriptional enhancement by GR. Similarly, a 12-fold hormone-dependent GR transcriptional induction observed in the absence of activated JNK was inhibited 4-fold by co-expressing constitutively active racQ61L (Fig. 4B, right panel). This inhibition of GR transcriptional activity was not a result of a reduction in GR expression, as the level of receptor protein was unaffected by activation of the ERK or JNK pathways (Fig. 4C). Thus, activation of either the ERK or the JNK pathway results in a significant reduction in GR hormone-dependent transcriptional enhancement.
JNK Antagonizes GR Transcriptional Activity in Vivo by Phosphorylating Ser-246.
Because both ERK and JNK use the receptor residue Ser-246 as a target for their catalytic activity in vitro, we asked whether inhibition of GR transcriptional activity by these kinases is a direct consequence of phosphorylation event at Ser-246. Because HeLa cells contain endogenous GR, we used GR-deficient U-2 OS human osteosarcoma cells for studies with the mutant receptor. This allowed us to examine whether a point mutation that changes Ser-246 to a nonphosphorylatable Ala residue (S246A) will reverse the inhibition of GR transcriptional activity by the activated ERK or JNK. In the absence of ERK and JNK activation, the GR S246A mutant displays slightly higher transcriptional activity than the wild-type receptor (5–10%), consistent with the inhibitory role of Ser-246 phosphorylation, although the magnitude of this effect varies among experiments. As illustrated in Fig. 5A (left panel), transcriptional enhancement of both the wild-type GR and the S246A mutant were inhibited by ERK, indicating that additional factors play a role in down-regulating GR ability to activate transcription in response to ERK activation. In contrast, the S246A mutation largely restored the receptor transcriptional activity when co-expressed with the activated JNK (Fig. 5B). As in HeLa cells, expression of Sos or Rac in U-2 OS cells did not affect the expression of wild-type GR or GR S246A mutant (data not shown).
Figure 5.

Inhibition of GR transcriptional enhancement by JNK but not ERK is mediated through receptor phosphorylation of Ser-246. GR-deficient U-2 OS human osteosarcoma cells were transfected using the calcium phosphate precipitation method (28). Each 60-mm dish received 1.5 μg of pRK5-HA ERK with 4.0 μg of pRK5-HA f-sos or pRK5 plasmid (A) or 1.5 μg of pRK5-M2 JNK with 4.0 μg of pCMV5-racQ61L or pRK5 plasmid (B) as indicated. In addition, each dish received 0.7 μg of pCMV-GR (wt GR or GR S246A, as indicated), 1.25 μg of ΔGTCO-CAT, and 0.5 μg of pCMV-LacZ. Transfected cells were treated with 100 nM Dex for 8 h where indicated, harvested, and assayed for CAT and β-Gal activity. Each experiment was performed in duplicates, three times, with less than 5% variation.
Together, our findings suggest that JNK but not ERK-mediated inhibition of GR transcriptional activation occurs as a result of direct receptor phosphorylation at Ser-246. To determine whether JNK activation causes increased GR phosphorylation in vivo, we examined the amount of phosphate incorporated into GR under conditions where the JNK and ERK pathways are activated by Rac and Sos, respectively. We transiently co-expressed rat GR along with JNK, in the absence or presence of racQ61L, or ERK, in the absence or presence of f-sos, in HeLa cells and subjected these cells to metabolic labeling with [32P]orthophosphate for 2 h in the presence of Dex. For these experiments, we used a rat GR derivative that lacks the Cdk phosphorylation sites (GR S224A S232A) to eliminate any background phosphorylation by Cdk that could complicate the interpretation of our results. For each sample, the total amount of rat GR visualized by immunoblotting with a GR-specific antibody was used to standardize the amount of incorporated 32P. As shown in Fig. 6A, GR phosphorylation is increased 5-fold by activated JNK. In contrast, no detectable increase in the amount of phosphate incorporated into GR is observed in the presence of activated ERK (Fig. 6B). Thus, JNK but not ERK activation results in increased receptor phosphorylation in vivo.
Figure 6.

Activation of the JNK but not ERK pathway in vivo increases GR phosphorylation. HeLa cells (1 × 106) were transiently transfected in 100-mm dishes with pCMV-GR S224A S232A (2.3 μg) and (A) pRK5-M2 JNK (4.5 μg) with pCMV5-racQ61L (12 μg) or an empty pCMV5 vector (12 μg) or (B) pRK5-HA ERK (4.5 μg) with pRK5-HA f-sos (12 μg) or an empty pRK5 vector (12 μg) by using Lipofectamine. After 18 h, GR was metabolically labeled for 2 h with 1 mCi/ml of [32P]orthophosphate in the presence of 100 nM Dex. In vivo labeled receptor was immunoprecipitated from cell lysates with the rat GR-specific monoclonal antibody, BuGR2. Immunoprecipitates were separated on 9% SDS/PAGE gels, transferred to Immobilon membrane, and exposed to film to visualize the phosphorylated GR (Upper). To normalize to GR protein expression, Western blotting of the same membrane with anti-GR rabbit polyclonal antibody was performed (Lower). The densitometric value obtained in the absence of cotransfected racQ61L (A) or f-sos (B) is arbitrarily set as 1.
DISCUSSION
We have demonstrated that JNK phosphorylates GR primarily at Ser-246 in vitro and in vivo, which results in significant reduction of GR transcriptional activity in the cell types examined. Suppression of GR transcriptional activity by ERK, however, seems to be independent of Ser-246 phosphorylation and may reflect a modification of a GR cofactor by ERK. Although we cannot exclude the possibility that sites other than Ser-246 are phosphorylated by ERK in vivo, we think this is unlikely, as no increase in phosphate incorporated into GR is observed upon ERK activation. These findings also emphasize the complexity and specificity of mitogen- and stress-activated pathways, whereby biochemical evidence for ERK-mediated phosphorylation of Ser-246 in vitro is not sufficient to explain the inhibition of GR enhancement by ERKs in vivo, as the primary Ser-246 target site seems dispensable for this inhibition. Taken together, our in vitro phosphorylation and mapping studies, coupled with the in vivo labeling experiments, along with the activity assays using the GR phosphorylation site mutant indicate that JNK phosphorylates GR at Ser-246 and, as a consequence, reduces GR transcriptional activation.
Although the mechanism by which JNK phosphorylation inhibits GR transcriptional enhancement is currently unclear, phosphorylation may modulate GR activity by influencing its nuclear transport or affinity to factors involved in transcriptional regulation. The GR phosphorylation site Ser-246 and the sequences immediately surrounding it are highly conserved among species, suggesting functional conservation for this type of transcriptional antagonism between GR and JNK throughout evolution. In contrast, Thr-171 has diverged among species and is not conserved even between rat and human GR. Leucine-rich sequences flanking Ser-246 may be involved in nuclear export (33). It is tempting to speculate that phosphorylation of Ser-246 may increase GR nuclear export as the means of inactivating GR transcriptional enhancement, although this remains to be examined. Alternatively, the sequence surrounding Ser-246 may form a surface that interacts with factors involved in transcriptional activation, such that phosphorylation of Ser-246 disrupts this interaction or, conversely, recruits an inhibitory factor that reduces GR transcriptional activity.
We postulate that GR phosphorylation by JNK provides a novel mechanism of rapidly down-regulating GR transcriptional enhancement in response to mitogenic or proinflammatory signals (Fig. 7). This regulation would seem to be physiologically significant, as activated GR has potent anti-inflammatory and anti-proliferative effects in multiple cell types (34), and its action would be in conflict with the AP-1 signaling pathway. Thus, activation of JNK not only leads to the phosphorylation and increased transcriptional activity of mitogenic and proinflammatory regulators, including AP-1, but also ensures decreased transcriptional activity of anti-inflammatory and anti-proliferative factors such as GR (Fig. 7). Both actions of JNK are likely necessary for a robust inflammatory or proliferative response.
Figure 7.

Model for reciprocal regulation of GR and AP-1 transcriptional activation by JNK. Under conditions where JNK is inactive, AP-1-mediated transcription is low because c-Jun is not phosphorylated at sites required for full transcriptional activity, whereas hormone-induced GR-dependent transcription is high, as GR is not phosphorylated at the inhibitory site Ser-246. In the presence of growth factors or inflammatory cytokines, JNK is activated and phosphorylates both c-Jun and GR, which results in an increase in AP-1-mediated transcriptional activation and a decrease in GR-dependent transcriptional enhancement. The ability of activated JNK to affect the transcriptional activity of AP-1 and GR oppositely provides a mechanism to ensure the rapid inhibition of GR-dependent gene expression when it conflicts with mitogenic or proinflammatory signals.
Acknowledgments
We thank C. Basilico, N. Tanese, A. Wilson, and the members of the Garabedian laboratory for critically reading the manuscript. We acknowledge the generosity of J. Schlessinger for DNA constructs and K. Yamamoto for advice and support. We also thank the organizers of the GR Receptor Resource web site for cross-species GR amino acid sequence alignment. This work was supported by grants from the Army Breast Cancer Research Fund (DAMD17-94-J-4454, DAMD17-96-1-6032) and the Kaplan Cancer Center (to M.J.G.). S.K.L. is a Leukemia Society Special Fellow.
ABBREVIATIONS
- GR
glucocorticoid receptor
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- ERK
extracellular signal-regulated kinase
- p38
p38 MAP kinase
- Cdk
cyclin-dependent kinase
- GST
glutathione S-transferase
- MBP
myelin basic protein
- FBS
fetal bovine serum
- Dex
dexamethasone
- CAT
chloramphenicol acetyltransferase
- β-Gal
β-galactosidase
- WCE
whole cell extracts
- HA
hemagglutinin
References
- 1.Yamamoto K R. Annu Rev Genet. 1985;19:209–252. doi: 10.1146/annurev.ge.19.120185.001233. [DOI] [PubMed] [Google Scholar]
- 2.Didonato J A, Saatcioglu F, Karin M. Am J Respir Crit Care Med. 1996;154:11–15. doi: 10.1164/ajrccm/154.2_Pt_2.S11. [DOI] [PubMed] [Google Scholar]
- 3.Cato A C, Wade E. BioEssays. 1996;18:371–378. doi: 10.1002/bies.950180507. [DOI] [PubMed] [Google Scholar]
- 4.Webster J C, Jewell C M, Bodwell J E, Munck A, Sar M, Cidlowsky J A. J Biol Chem. 1997;272:9287–9293. doi: 10.1074/jbc.272.14.9287. [DOI] [PubMed] [Google Scholar]
- 5.Krstic M D, Rogatsky I, Yamamoto K R, Garabedian M J. Mol Cell Biol. 1997;17:3947–3954. doi: 10.1128/mcb.17.7.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Minden A, Lin A, Smeal T, Derijard B, Cobb M, Davis R, Karin M. Mol Cell Biol. 1994;14:6683–6688. doi: 10.1128/mcb.14.10.6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Karin M. Trans R Soc London. 1996;351:127–134. doi: 10.1098/rstb.1996.0008. [DOI] [PubMed] [Google Scholar]
- 8.Buday L, Downward J. Cell. 1993;73:611–620. doi: 10.1016/0092-8674(93)90146-h. [DOI] [PubMed] [Google Scholar]
- 9.Olivier J P, Raabe T, Henkemeyer M, Dickson B, Mbamalu G, Margolis B, Schlessinger J, Hafen E, Pawson T. Cell. 1993;73:179–191. doi: 10.1016/0092-8674(93)90170-u. [DOI] [PubMed] [Google Scholar]
- 10.Rozakis-Adcock M, Fernley R, Wade J, Pawson T, Bowtell D. Nature (London) 1993;363:83–85. doi: 10.1038/363083a0. [DOI] [PubMed] [Google Scholar]
- 11.Davis R J. J Biol Chem. 1993;268:14553–14556. [PubMed] [Google Scholar]
- 12.Kato S, Endoh H, Masuhiro Y, Kitamoto T, Uchiyama S, Sasaki H, Masushige S, Gotoh Y, Nishida E, Kawashima H, et al. Science. 1995;270:1491–1494. doi: 10.1126/science.270.5241.1491. [DOI] [PubMed] [Google Scholar]
- 13.Derijard B, Hibi M, Wu I H, Barrett T, Su B, Deng T, Karin M, Davis R J. Cell. 1994;76:1025–1037. doi: 10.1016/0092-8674(94)90380-8. [DOI] [PubMed] [Google Scholar]
- 14.Hibi M, Lin A, Smeal T, Minden A, Karin M. Genes Dev. 1993;7:2135–2148. doi: 10.1101/gad.7.11.2135. [DOI] [PubMed] [Google Scholar]
- 15.Westwick J K, Weitzel C, Minden A, Karin M, Brenner D A. J Biol Chem. 1994;269:26396–26401. [PubMed] [Google Scholar]
- 16.Sluss H K, Barrett T, Derijard B, Davis R J. Mol Cell Biol. 1994;14:8376–8384. doi: 10.1128/mcb.14.12.8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gupta S, Campbell D, Derijard B, Davis R J. Science. 1995;267:389–393. doi: 10.1126/science.7824938. [DOI] [PubMed] [Google Scholar]
- 18.Sanchez I, Hughes R T, Mayer B J, Yee K, Woodgett J R, Avruch J, Kyriakis J M, Zon L I. Nature (London) 1994;372:794–798. doi: 10.1038/372794a0. [DOI] [PubMed] [Google Scholar]
- 19.Livingstone C, Patel G, Jones N. EMBO J. 1995;14:1785–1797. doi: 10.1002/j.1460-2075.1995.tb07167.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Raingeaud J, Gupta S, Rogers J S, Dickens M, Han J, Ulevitch R J, Davis R J. J Biol Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 21.Han J, Lee J D, Bibbs L, Ulevitch R J. Science. 1994;265:808–811. doi: 10.1126/science.7914033. [DOI] [PubMed] [Google Scholar]
- 22.Raingeaud J, Whitmarsh A J, Barrett T, Derijard B, Davis R J. Mol Cell Biol. 1996;16:1247–1255. doi: 10.1128/mcb.16.3.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang X Z, Ron D. Science. 1996;272:1347–1349. doi: 10.1126/science.272.5266.1347. [DOI] [PubMed] [Google Scholar]
- 24.Krstic M D. Ph.D. thesis. San Francisco: Univ. of California; 1995. [Google Scholar]
- 25.Rogatsky I, Trowbridge J M, Garabedian M J. Mol Cell Biol. 1997;17:3181–3193. doi: 10.1128/mcb.17.6.3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gametchu B, Harrison R W. Endocrinology. 1984;114:274–279. doi: 10.1210/endo-114-1-274. [DOI] [PubMed] [Google Scholar]
- 27.Aronheim A, Engelberg D, Li N, al-Alawi N, Schlessinger J, Karin M. Cell. 1994;78:949–961. doi: 10.1016/0092-8674(94)90271-2. [DOI] [PubMed] [Google Scholar]
- 28.Ausubel F M, Brent R, Kingston R E, Moore D D, Seidman J G, Smith J A, Struhl K. Current Protocols in Molecular Biology. New York: Wiley; 1996. [Google Scholar]
- 29.Sleigh M J. Anal Biochem. 1986;156:251–256. doi: 10.1016/0003-2697(86)90180-6. [DOI] [PubMed] [Google Scholar]
- 30.Chardin P, Camonis J H, Gale N W, van Aelst L, Schlessinger J, Wigler M H, Bar-Sagi D. Science. 1993;260:1338–1343. doi: 10.1126/science.8493579. [DOI] [PubMed] [Google Scholar]
- 31.Minden A, Lin A, Claret F X, Abo A, Karin M. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- 32.Xu X, Barry D C, Settleman J, Schwartz M A, Bokoch G M. J Biol Chem. 1994;269:23569–23574. [PubMed] [Google Scholar]
- 33.Lee M S, Silver P A. Curr Opin Genet Dev. 1997;7:212–219. doi: 10.1016/s0959-437x(97)80131-1. [DOI] [PubMed] [Google Scholar]
- 34.Brattsand R, Linden M. Aliment Pharmacol Ther. 1996;2:81–90. doi: 10.1046/j.1365-2036.1996.22164025.x. [DOI] [PubMed] [Google Scholar]