

Abstract
The discovery of the receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin (OPG) system and its role in the regulation of bone resorption exemplifies how both serendipity and a logic-based approach can identify factors that regulate cell function. Before this discovery in the mid to late 1990s, it had long been recognized that osteoclast formation was regulated by factors expressed by osteoblast/stromal cells, but it had not been anticipated that members of the tumor necrosis factor superfamily of ligands and receptors would be involved or that the factors involved would have extensive functions beyond bone remodeling. RANKL/RANK signaling regulates the formation of multinucleated osteoclasts from their precursors as well as their activation and survival in normal bone remodeling and in a variety of pathologic conditions. OPG protects the skeleton from excessive bone resorption by binding to RANKL and preventing it from binding to its receptor, RANK. Thus, RANKL/OPG ratio is an important determinant of bone mass and skeletal integrity. Genetic studies in mice indicate that RANKL/RANK signaling is also required for lymph node formation and mammary gland lactational hyperplasia, and that OPG also protects arteries from medial calcification. Thus, these tumor necrosis factor superfamily members have important functions outside bone. Although our understanding of the mechanisms whereby they regulate osteoclast formation has advanced rapidly during the past 10 years, many questions remain about their roles in health and disease. Here we review our current understanding of the role of the RANKL/RANK/OPG system in bone and other tissues.
Introduction
Bone serves multiple functions in vertebrates, including support for muscles, protection of vital organs and hematopoietic marrow, and storage and release of vital ions, such as calcium. Unlike other durable structures, such as teeth, tendons, and cartilage, bone is continuously renewed by the process of bone remodeling in which pockets or trenches of bone are removed from the surfaces of trabecular and cortical bone by osteoclasts and subsequently replaced by new bone laid down by osteoblasts. There are at least one million of these microscopic remodeling foci at any one time in the adult skeleton, and the main function of this process is considered to be removal of effete or worn out parts of bones that have become damaged as part of normal wear and tear. It is a highly regulated process, but the molecular mechanisms that control its initiation, progression, and cessation at any given site remain poorly understood.
Bone remodeling becomes perturbed in a variety of pathologic conditions that affect the skeleton, including post-menopausal osteoporosis and rheumatoid arthritis, in which there is local and/or systemic alteration in the levels of hormones or proinflammatory cytokines that are known to stimulate or inhibit bone resorption in vitro and in vivo. It has been recognized since the early 1980s, when Rodan and Martin [1] postulated that osteoblasts regulate osteoclast formation, that factors expressed by osteoblasts within bone are produced in response to known stimulators of bone resorption, such as parathyroid hormone (PTH). Study of bones from genetically altered mice and from animal models of bone diseases during the past 10 years has greatly increased our knowledge of the factors that regulate the formation and activity of osteoclasts. In particular, identification in the mid to late 1990s of the receptor activator of nuclear factor-κB ligand (RANKL)/RANK/osteoprotegerin (OPG) signaling system provided a major breakthrough that clarified the role played by osteoblasts in these processes. More recently, it has become increasingly clear that osteoclasts are not simply trench digging cells, but that they have important regulatory functions as immunomodulators in pathologic states and that they may also regulate osteoblast function [2].
Regulation of osteoclast formation and activation
Osteoclasts are multinucleated bone resorbing cells formed by cytoplasmic fusion of their mononuclear precursors, which are in the myeloid lineage of hematopoietic cells that also give rise to macrophages. The switch to osteoclast differentiation requires expression in osteoclast precursors (OCPs) of c-Fos, a RANKL activated transcription factor [3]. To resorb bone effectively, osteoclasts attach themselves firmly to the bone surface using specialized actin-rich podosomes, which they use to form tightly sealed roughly circular extensions of their cytoplasm with the underlying bone matrix. Within these sealed zones they form ruffled membranes that increase the surface area of the cell membrane for secretion of hydrochloric acid and the proteolytic enzyme cathepsin K onto the bone surface [4]. They thereby simultaneously dissolve the mineral and degrade the matrix of bone, while protecting neighboring cells from harm by this sealing mechanism. They are activated by RANKL and by integrin-mediated signaling from bone matrix itself [4]. Osteoclasts work in packs within remodeling units under the control of osteoblast lineage cells expressing macrophage colony-stimulating factor (M-CSF) and RANKL.
Recent studies of the mechanisms by which PTH exerts its anabolic effects have suggested that osteoclasts are probably involved in the recruitment of packs of bone-forming osteoblasts to refill the trenches that they form on the bone surface [2]. This is based on studies showing that, after PTH injection, RANKL expression is increased by osteoblast/stromal cells, leading to activation of existing osteoclasts and release by them of a factor(s) that stimulates new bone formation. Also, antiresorptive treatment, at least in some studies, appears to reduce rather than enhance the anabolic action of PTH [5-7]. As is discussed below, osteoclasts also appear to regulate immune responses and their own production at sites of inflammation in bone, such as rheumatoid joints.
Osteoclasts are required during embryonic development for the removal of bone trabeculae formed under growth plates during endochondral ossification and thus for formation of the bone marrow cavity to facilitate normal hematopoiesis. Failure of osteoclast formation or activity results in osteopetrosis, some forms of which are lethal because of attendant immunodeficiency and increased risk for infection and recurrent fractures. Indeed, the development of osteopetrosis in a variety of knockout mice has identified necessary functions of genes in osteoclast biology that largely had not been anticipated [3,4].
Our understanding of the molecular mechanisms that regulate osteoclast formation and activation has advanced rapidly during the past 10 years since the discovery of the RANKL/RANK signaling system, and following the development in the late 1980s of in vitro assays that facilitated harvesting of large numbers of OCPs from bone marrow or spleen cells, which could then be cultured in the absence of osteoblast/stromal cells. The strategy for acquiring OCPs from these sources was developed in the knowledge that M-CSF expression by osteoblast/stromal cells was required for progenitor cells to differentiate into osteoclasts, but that M-CSF on its own was unable to complete this process. This requirement for M-CSF was based on the observation that op/op mice, which do not express functional M-CSF, have osteopetrosis because of a lack of osteoclasts [3]. Indeed, since 1981, when Rodan and Martin [1] proposed the novel hypothesis that osteoblast/stromal cells play a central role in the regulation of osteoclast formation and bone resorption, many investigators had attempted to identify the osteoclast-activating factor that completed the differentiation of precursors that had been exposed to M-CSF.
Discovery of osteoprotegerin, RANKL, and RANK
Between 1981 and the mid 1990s, the Rodan–Martin hypothesis was supported by many studies, but the factor(s) expressed by osteoblast/stromal or other cells remained undetermined until they were discovered independently by four groups using different approaches.
Boyle and coworkers [8] at Amgen Inc. (Thousand Oaks, CA, USA) discovered OPG unexpectedly in studies to identify tumor necrosis factor (TNF) receptor related molecules with possible therapeutic utility by generating transgenic mice that over-express various TNF receptor related cDNAs. Mice over-expressing one particular cDNA developed marked osteopetrosis because they did not have any osteoclasts in their bones. The protein encoded by the gene was named osteoprotegerin (the bone protector) [8], because it appeared to protect the skeleton from excessive bone resorption by limiting osteoclastic bone resorption. Independently, researchers at the Snow Brand Milk Products Co. (Sapporo, Hokkaido, Japan) reported their discovery of an identical molecule [9] using the standard approach to test the Rodan–Martin hypothesis of purifying a factor from human embryonic fibroblasts that inhibited osteoclastogenesis. They obtained a partial protein sequence and subsequently cloned the cDNA for OPG.
Using expression cloning and OPG as a probe, both groups quickly identified its ligand, which they called OPG ligand and osteoclast differentiation factor, respectively [10,11]. This ligand turned out to be identical to a member of the TNF ligand family, which had been identified in the preceding year as RANKL [12] and TNF-related activation induced cytokine [13]. Soon after OPG ligand/osteoclast differentiation factor was identified as a ligand for OPG, the cellular receptor was identified as being identical to the previously identified RANK, which Anderson and coworkers [12] at Immunex (Seattle, WA, USA) had discovered while they were sequencing cDNAs from a human bone marrow derived myeloid dendritic cell cDNA library. They found that RANK had partial homology to a portion of the extracellular domain of human CD40, a member of the TNF receptor superfamily, and that it was involved in the activation of T cells in the immune system. They then isolated RANKL by direct expression screening and found, like Wong and coworkers [13] did, that it increased dendritic cell stimulated naïve T cell proliferation and survival of RANK-expressing T cells. These discoveries that RANKL is involved in osteoclastogenesis and T cell activation have spawned the now growing field of osteoimmunology.
RANKL
RANKL is a type II homotrimeric transmembrane protein that is expressed as a membrane-bound and a secreted protein, which is derived from the membrane form as a result of either proteolytic cleavage or alternative splicing [14]. The proteo-lytic cleavage of RANKL requires ADAM (a disintegrin and metalloprotease domain) [15] and matrix metalloproteases [16]. RANKL expression is stimulated in osteoblast/stromal cells by most of the factors that are known to stimulate osteoclast formation and activity. It is highly expressed in lymph nodes, thymus and lung, and at low levels in a variety of other tissues including spleen and bone marrow [17]. In inflamed joints it is expressed by synovial cells and secreted by activated T cells. These sources of RANKL appear to be responsible, at least in part, for mediating the joint destruction in patients with rheumatoid arthritis [18]. TNF also mediates joint destruction in rheumatoid arthritis by systemically increasing the number of circulating OCPs, and by promoting their egress from the bone marrow into the peripheral blood and then to the inflamed joints, where it promotes fusion of these cells to osteoclasts along with RANKL and interleukin-1 [19].
RANKL, like TNF, stimulates the release of immature progenitors into the circulation. However, RANKL does not induce OCP mobilization in protein tyrosine phosphatase-ε knockout mice with osteoclasts that are defective in terms of bone adhesion and resorption [20]. Thus, RANKL-induced osteoclast activation may regulate progenitor recruitment as part of homeostasis and host defense, linking bone remodeling with regulation of hematopoiesis. Preclinical studies in mice have shown that RANKL is also expressed in mammary epithelial cells during pregnancy and is required for lactational hyperplasia of mammary epithelial cells and milk production [21]. It is also expressed by some malignant tumor cells that also express RANK, and thus it may play a role in inducing tumor cell proliferation [22] by an autocrine mechanism or in a paracrine manner if it is produced by accessory cells, such as activated T cells. However, production by T cells of RANKL also induces expression of interferon-β by activated osteoclasts through c-Fos to negatively regulate their formation [23]. This mechanism can be enhanced by T-cell produced interferon-γ, which degrades TNF receptor associated factor (TRAF)6, an essential adapter protein that is recruited to RANK to mediate RANK signaling (see below) [24].
RANK
RANK is a type I homotrimeric transmembrane protein whose expression was initially detected only on OCPs, mature osteoclasts, and dendritic cells. Like RANKL, however, it is expressed widely [17]. RANK protein expression has been reported in mammary gland [21] and some cancer cells, including breast and prostate cancers [22,25], two types of tumors with high bone metastasis potential. Although no humans have been identified to date with inactivating mutations or deletions of RANK, a deletion mutation occurred spontaneously in a line of transgenic mice, which consequently had all of the features of mice with targeted deletion of RANK, confirming the importance of RANK for osteoclast formation [26]. Activating mutations in exon 1 of RANK that cause an increase in RANK-mediated nuclear factor-κB (NF-κB) signaling and a resultant increase in osteoclast formation and activity account for the increased osteolysis seen in some patients with familial Paget's disease and have confirmed the importance of this system in humans [27]. A potential role for RANK in tumor cell proliferation [22] is being investigated and, if proven, could be a future target for anti-tumor therapy.
Osteoprotegerin
OPG is expressed in many tissues apart from osteoblasts, including heart, kidney, liver, spleen, and bone marrow [17]. Its expression is regulated by most of the factors that induce RANKL expression by osteoblasts. Although there are contradictory data, in general upregulation of RANKL is associated with downregulation of OPG, or at least lower induction of OPG, such that the ratio of RANKL to OPG changes in favor of osteoclastogenesis. Many reports have supported the assertion that the RANKL/OPG ratio is a major determinant of bone mass [28]. A osteoprotective role for OPG in humans is supported by the report of homozygous deletions of 100 kilobases of OPG in two patients with juvenile Paget's disease, an autosomal-recessive disorder characterized by increased bone remodeling, osteopenia, and fractures [29]. It is also supported by the identification of an inactivating deletion in exon 3 of OPG in three siblings with idiopathic hyperphosphatasia, which is an autosomal-recessive bone disease characterized by increased bone turnover associated with deformities of long bones, kyphosis, and acetabular protrusion in affected children [30]. A recent surprising finding is that OPG expression is regulated by Wnt/β-catenin signaling in osteoblasts, the same pathway that regulates osteoblastic bone formation [31]. Thus, bone mass is determined by the combined efforts of osteoblasts and osteoclasts, and is regulated in osteoblasts by two major signaling pathways: RANKL/RANK and Wnt/β-catenin.
OPG also appears to protect large blood vessels from medial calcification, based on the observation of renal and aortic calcification occurring in OPG knockout mice [32]. Furthermore, the absence of OPG in OPG/apolipoprotein E double knockout mice accelerates the calcific atherosclerosis that develops in apolipoprotein E knockout mice, suggesting that OPG protects against this complication of atherosclerosis [33]. Whether OPG and RANKL signaling plays important roles in cardiovascular disease remains to be determined and is controversial [34]. For example, there is also an association between high levels of OPG in serum and cardiovascular disease, diabetes, and chronic renal failure in humans [34]. However, OPG in this latter setting does not appear to protect the skeleton against the increased bone resorption of secondary hyperparathyroidism mediated by PTH in patients with renal osteodystrophy or against vascular calcification. It is possible that OPG in the serum of such patients is bound to a plasma protein(s) and thus rendered inactive, but further studies will be required to determine the significance of these observations, which question whether the RANKL/OPG ratio in serum is indicative of bone mass/bone resorption in these settings [35].
Transcription factor activation by RANKL/RANK in osteoclasts
With the knowledge that RANKL/RANK signaling is essential for osteoclast formation, major efforts have been made to identify the signaling pathways that are activated downstream and to determine the full extent of the involvement of RANKL in osteoclast biology and common bone diseases. After RANKL binds to RANK, a key preliminary step in downstream signaling is binding of TRAFs to specific sites within the cytoplasmic domain of RANK, which is a transmembrane protein that – like the TNF receptors – has no intrinsic ability to activate protein kinases to mediate signaling. TRAF2, -5, and -6 all bind to RANK, but of these only TRAF6 appears to be essential in osteoclasts, because only TRAF6 knockout mice develop osteopetrosis. Interestingly, although two independently produced mutant TRAF6 mice have osteopetrosis, surprisingly one has normal numbers of osteoclasts (but they are inactive) [36] and the other has no osteoclasts [37]. At least seven signaling pathways are activated by RANK-mediated protein kinase signaling; four of them directly mediate osteoclastogenesis (inhibitor of NF-κB kinase/NF-κB, c-Jun amino-terminal kinase/activator protein-1, c-myc, and calcineurin/nuclear factor of activated T cells [NFAT]c1) and three mediate osteoclast activation (src and MKK6/p38/MITF) and survival (src and extracellular signal-regulated kinase). It remains unexplained how inactivation of TRAF6 resulted in two different osteoclast phenotypes, however.
Several adapter molecules bind to RANK along with TRAFs to mediate signaling. Among these is Grb-2 associated binder protein 2, a member of a family of adapter molecules that are phosphorylated at tyrosine residues and recruit a variety of signaling molecules that contain Src homology 2 domains. Loss of Grb-2 associated binder protein 2 results in reduced RANKL/RANK-induced osteoclast differentiation, decreased bone resorption, and mild osteopetrosis. This indicates that it plays a significant role in RANKL-induced osteoclastogenesis [38].
The essential role played by NF-κB/activator protein-1/NFATc1 signaling for osteoclast formation was discovered after the generation of mice with targeted deletion of both p50 and p52 subunits of NF-κB and of c-Fos [3], and following elegant molecular rescue experiments in which adoptive transfer of Nfatc1-/- hematopoietic stem cells to Fos-/- mice induced osteoclast formation [39]. Over-expression of a constitutively active form of NFATc1 induces osteoclast formation by M-CSF treated Fos-/- or NF-κB p50/p52-/- osteoclast precursors in the absence of RANKL [40], indicating that it is downstream from NF-κB and c-Fos (Figure 1). On the basis of all of these studies, NFATc1 has been described as the master regulator of osteoclastogenesis [39]. It is activated by calcium-dependent calcineurin dephosphorylation. Cyclosporine A, a calcineurin inhibitor, inhibits NFATc1 activation, and thus it was surprising that treatment of patients with this immunosuppressant was associated with bone loss [41]. The likely explanation for this effect of cyclosporine A is that NFATc1 also positively regulates expression of osterix, an essential transcription factor that regulates osteoblast function [42], and the net effect is reduced bone formation and osteoporosis [43].
Figure 1.
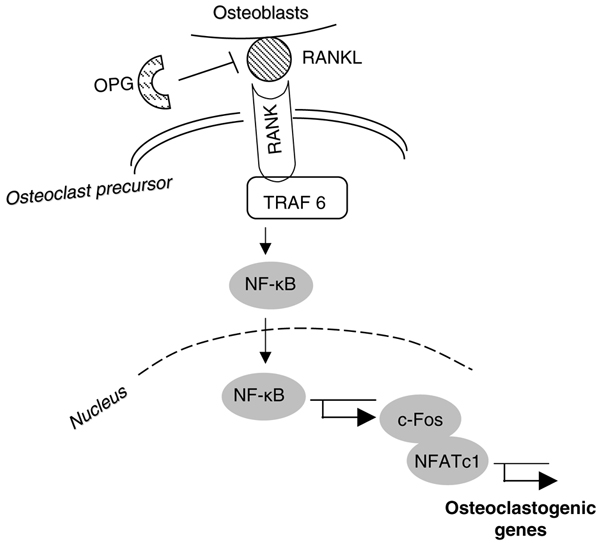
The essential signaling pathway for normal osteoclastogenesis. Under physiologic conditions, RANKL produced by osteoblasts binds to RANK on the surface of osteoclast precursors and recruits the adaptor protein TRAF6, leading to NF-κB activation and translocation to the nucleus. NF-κB increases c-Fos expression and c-Fos interacts with NFATc1 to trigger the transcription of osteoclastogenic genes. OPG inhibits the initiation of the process by binding to RANKL. NFAT, nuclear factor of activated T cells; NF-κB, nuclear factor-κB; OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor-κB ligand; TRAF, tumor necrosis factor receptor associated factor.
Immunoreceptors, osteoimmunology, and RANKL
It is not yet clear how calcium signaling is activated during osteoclast formation, but it appears to involve the Fc receptor common γ subunit (FcRγ) immunoreceptor expressed by osteoclasts and the adapter protein DNAX-activating protein 12, which associates with an immunoreceptor tyrosine-based activation motif [44]. DNAX-activating protein 12/FcRγ double knockout mice have impaired RANKL-induced NFATc1 activation and osteoclast formation, and are severely osteopetrotic [45]. However, this receptor-mediated signaling pathway cannot induce osteoclast formation on its own. Thus, like M-CSF, it is necessary but not sufficient for osteoclastogenesis. FcRγ-associating receptors include osteoclast-associated receptor, whose expression is regulated by NFATc1. Thus, signaling through RANKL/RANK and these receptors in inflammatory disorders provides a NFATc1-mediated amplifying mechanism that potentially can increase osteoclast formation beyond that of RANKL alone. This mechanism for enhanced osteoclast formation in inflammatory bone disease can be augmented also by TNF, which not only induces c-fms expression by OCPs [46] but also increases proliferation and survival of these cells as well as enhancing their egress from bone marrow into the bloodstream, from where they can alight in increased numbers at sites of inflammation. These OCPs also increase their production of TNF in response to TNF and RANKL, and thereby induce an auto-amplifying autocrine vicious cycle to increase osteoclast numbers. It is likely that osteoclasts and their precursors secrete many more factors that interact with immune and other cells to affect bone volume and turnover in a variety of bone disorders (Figure 2).
Figure 2.
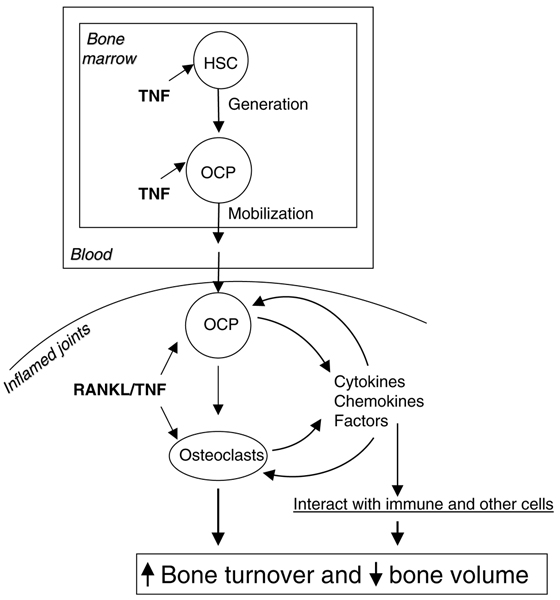
Osteoclasts and precursors are secretory cells. In inflammatory bone disease, systemic elevated TNF stimulates the generation of OCPs in the bone marrow and also enhances their egress into the bloodstream, from where they can alight in increased numbers at sites of inflammation. These OCPs differentiate to osteoclasts and increase their production of many factors in response to TNF and RANKL and thereby induce an auto-amplifying autocrine vicious cycle to increase osteoclast numbers and interact with immune and other cells to affect bone volume and turnover. OCP, osteoclast precursor; RANKL, receptor activator of nuclear factor-κB ligand; TNF, tumor necrosis factor.
Conclusion
The RANKL/RANK/OPG system is one of the most important discoveries in bone biology in the past decade. This system is critical for skeletal health, and disruption of it leads to or causes numerous bone diseases. Recent studies have indicated that RANKL/RANK signaling also plays an important role in other tissues (Figure 3).
Figure 3.
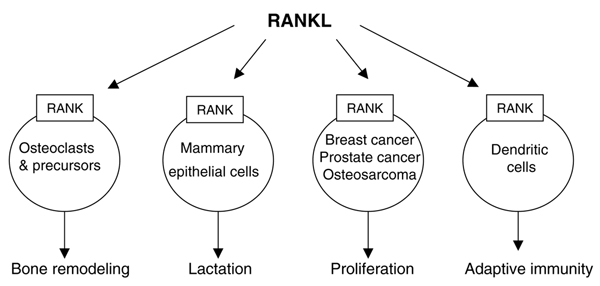
The role of the RANKL/RANK system in bone and other tissues. RANKL is produced by a variety of cell types and its expression is regulated by many physiologic and pathologic factors. Preclinical studies in mice and studies of human tissues have revealed functions for RANKL/RANK signaling in normal and pathologic states. OPG can bind to RANKL and prevent its interaction with RANK to inhibit osteoclast formation, but its effects on other cellular functions of RANKL have yet to be determined. OPG, osteoprotegerin; RANKL, receptor activator of nuclear factor-κB ligand.
However, several important issues remain unresolved. For example, how is this system activated and then inactivated during normal bone remodeling? Which cells specifically in the osteoblast/stromal cell lineage regulate normal and pathologic bone remodeling? Are they all immature, non-matrix-producing stromal cells or do osteoid synthesizing osteoblasts play a major regulatory role in addition to keeping osteoclasts away from them while they are filling in resorption trenches? Do stromal cells circulate in sufficient numbers, like OCPs, to play important roles in RANKL/RANK-mediated normal and pathologic bone remodeling? How does RANKL/RANK signaling regulate osteoblast function and the anabolic effects of PTH or other potential anabolic agents? Do serum levels of RANKL or OPG reflect what is happening in bone and joints in patients? Does RANKL/RANK signaling play an important role in cancer cell growth and interaction with other cells in bone? How exactly do immune cells influence osteoclast and osteoblast/stromal cell function in normal and disease states? It is becoming recognized increasingly that the osteoclast per se – as a secretory cell – is an immune cell. How important are osteoclasts and their precursors in regulating their own formation and function relative to macrophages and other immune cells? Elucidation of the specific roles played by RANKL/RANK in these various types of cells will probably link bone remodeling with regulation of the function of other organ systems in health and disease.
Abbreviations
FcRγ = Fc receptor common γ subunit; M-CSF = macrophage colony-stimulating factor; NFAT = nuclear factor of activated T cells; NF-κB = nuclear factor-κB; OCP = osteoclast precursor; OPG = osteoprotegerin; PTH = parathyroid hormone; RANK = receptor activator of NF-κB; RANKL = receptor activator of nuclear factor-κB ligand; TNF = tumor necrosis factor; TRAF = TNF receptor associated factor.
Competing interests
BFB has received payment for consulting for Amgen as a member of their International Advisory Panel in 2005. LX has no competing interests to declare.
Acknowledgments
Acknowledgements
This article is published as part of Arthritis Research & Therapy Volume 9 Supplement 1, 2007: Basic science, rationale, background and future of denosumab: a RANK ligand inhibitor. The full contents of the supplement are available online at http://arthritis-research.com/supplements/9/S1.
Publication of the supplement has been supported by an unrestricted grant from Amgen Inc.
References
- Rodan GA, Martin TJ. Role of osteoblasts in hormonal control of bone resorption: a hypothesis. Calcif Tissue Int. 1981;33:349–351. doi: 10.1007/BF02409454. [DOI] [PubMed] [Google Scholar]
- Martin TJ, Sims NA. Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med. 2005;11:76–81. doi: 10.1016/j.molmed.2004.12.004. [DOI] [PubMed] [Google Scholar]
- Karsenty G, Wagner EF. Reaching a genetic and molecular understanding of skeletal development. Dev Cell. 2002;2:389–406. doi: 10.1016/S1534-5807(02)00157-0. [DOI] [PubMed] [Google Scholar]
- Teitelbaum SL, Ross FP. Genetic regulation of osteoclast development and function. Nat Rev Genet. 2003;4:638–649. doi: 10.1038/nrg1122. [DOI] [PubMed] [Google Scholar]
- Cosman F, Nieves J, Zion M, Woelfert L, Luckey M, Lindsay R. Daily and cyclic parathyroid hormone in women receiving alendronate. N Engl J Med. 2005;353:566–575. doi: 10.1056/NEJMoa050157. [DOI] [PubMed] [Google Scholar]
- Black DM, Greenspan SL, Ensrud KE, Palermo L, McGowan JA, Lang TF, Garnero P, Bouxsein ML, Bilezikian JP, Rosen CJ, PaTH Study Investigators The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N Engl J Med. 2003;349:1207–1215. doi: 10.1056/NEJMoa031975. [DOI] [PubMed] [Google Scholar]
- Ma YL, Bryant HU, Zeng Q, Schmidt A, Hoover J, Cole HW, Yao W, Jee WS, Sato M. New bone formation with teriparatide [human parathyroid hormone-(1-34)] is not retarded by long-term pretreatment with alendronate, estrogen, or raloxifene in ovariectomized rats. Endocrinology. 2003;144:2008–2015. doi: 10.1210/en.2002-221061. [DOI] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Shima N, Nakagawa N, Mochizuki SI, Yano K, Fujise N, Sato Y, Goto M, Yamaguchi K, Kuriyama M, et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): a mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology. 1998;139:1329–1337. doi: 10.1210/en.139.3.1329. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/S0092-8674(00)81569-X. [DOI] [PubMed] [Google Scholar]
- Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Wong BR, Rho J, Arron J, Robinson E, Orlinick J, Chao M, Kalachikov S, Cayani E, Bartlett FS, III, Frankel WN, et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J Biol Chem. 1997;272:25190–25194. doi: 10.1074/jbc.272.40.25190. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Kasai M, Utsuyama M, Hirokawa K. Determination of three isoforms of the receptor activator of nuclear factor-[kappa]B ligand and their differential expression in bone and thymus. Endocrinology. 2001;142:1419–1426. doi: 10.1210/en.142.4.1419. [DOI] [PubMed] [Google Scholar]
- Hikita A, Yana I, Wakeyama H, Nakamura M, Kadono Y, Oshima Y, Nakamura K, Seiki M, Tanaka S. Negative regulation of osteoclastogenesis by ectodomain shedding of receptor activator of NF-kappa B ligand. J Biol Chem. 2006;281:36846–36855. doi: 10.1074/jbc.M606656200. [DOI] [PubMed] [Google Scholar]
- Lynch CC, Hikosaka A, Acuff HB, Martin MD, Kawai N, Singh RK, Vargo-Gogola TC, Begtrup JL, Peterson TE, Fingleton B, et al. MMP-7 promotes prostate cancer-induced osteolysis via the solubilization of RANKL. Cancer Cell. 2005;7:485–496. doi: 10.1016/j.ccr.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Wada T, Nakashima T, Hiroshi N, Penninger JM. RANKL-RANK signaling in osteoclastogenesis and bone disease. Trends Mol Med. 2006;12:17–25. doi: 10.1016/j.molmed.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Schett G, Hayer S, Zwerina J, Redlich K, Smolen JS. Mechanisms of disease: the link between RANKL and arthritic bone disease. Nat Clin Pract Rheumatol. 2005;1:47–54. doi: 10.1038/ncprheum0036. [DOI] [PubMed] [Google Scholar]
- Li P, Schwarz EM, O'Keefe RJ, Ma L, Looney RJ, Ritchlin CT, Boyce BF, Xing L. Systemic tumor necrosis factor alpha mediates an increase in peripheral CD11bhigh osteoclast precursors in tumor necrosis factor alpha-transgenic mice. Arthritis Rheum. 2004;50:265–276. doi: 10.1002/art.11419. [DOI] [PubMed] [Google Scholar]
- Kollet O, Dar A, Shivtiel S, Kalinkovich A, Lapid K, Sztainberg Y, Tesio M, Samstein RM, Goichberg P, Spiegel A, et al. Osteoclasts degrade endosteal components and promote mobilization of hematopoietic progenitor cells. Nat Med. 2006;12:657–664. doi: 10.1038/nm1417. [DOI] [PubMed] [Google Scholar]
- Fata JE, Kong YY, Li J, Sasaki T, Irie-Sasaki J, Moorehead RA, Elliott R, Scully S, Voura EB, Lacey DL, et al. The osteoclast differentiation factor osteoprotegerin-ligand is essential for mammary gland development. Cell. 2000;103:41–50. doi: 10.1016/S0092-8674(00)00103-3. [DOI] [PubMed] [Google Scholar]
- Kim NS, Kim HJ, Koo BK, Kwon MC, Kim YW, Cho Y, Yokota Y, Penninger JM, Kong YY. Receptor activator of NF-kappaB ligand regulates the proliferation of mammary epithelial cells via Id2. Mol Cell Biol. 2006;26:1002–1013. doi: 10.1128/MCB.26.3.1002-1013.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Ogasawara K, Hida S, Chiba T, Murata S, Sato K, Takaoka A, Yokochi T, Oda H, Tanaka K, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature. 2000;408:600–605. doi: 10.1038/35046102. [DOI] [PubMed] [Google Scholar]
- Chen G, Sircar K, Aprikian A, Potti A, Goltzman D, Rabbani SA. Expression of RANKL/RANK/OPG in primary and metastatic human prostate cancer as markers of disease stage and functional regulation. Cancer. 2006;107:289–298. doi: 10.1002/cncr.21978. [DOI] [PubMed] [Google Scholar]
- Kapur RP, Yao Z, Iida MH, Clarke CM, Doggett B, Xing L, Boyce BF. Malignant autosomal recessive osteopetrosis caused by spontaneous mutation of murine Rank. J Bone Miner Res. 2004;19:1689–1697. doi: 10.1359/JBMR.040713. [DOI] [PubMed] [Google Scholar]
- Hughes AE, Ralston SH, Marken J, Bell C, MacPherson H, Wallace RG, van Hul W, Whyte MP, Nakatsuka K, Hovy L, et al. Mutations in TNFRSF11A, affecting the signal peptide of RANK, cause familial expansile osteolysis. Nat Genet. 2000;24:45–48. doi: 10.1038/71667. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Schoppet M. Clinical implications of the osteoprotegerin/RANKL/RANK system for bone and vascular diseases. JAMA. 2004;292:490–495. doi: 10.1001/jama.292.4.490. [DOI] [PubMed] [Google Scholar]
- Whyte MP, Obrecht SE, Finnegan PM, Jones JL, Podgornik MN, McAlister WH, Mumm S. Osteoprotegerin deficiency and juvenile Paget's disease. N Engl J Med. 2002;347:175–184. doi: 10.1056/NEJMoa013096. [DOI] [PubMed] [Google Scholar]
- Cundy T, Hegde M, Naot D, Chong B, King A, Wallace R, Mulley J, Love DR, Seidel J, Fawkner M, et al. A mutation in the gene TNFRSF11B encoding osteoprotegerin causes an idiopathic hyperphosphatasia phenotype. Hum Mol Genet. 2002;11:2119–2127. doi: 10.1093/hmg/11.18.2119. [DOI] [PubMed] [Google Scholar]
- Boyce BF, Xing L, Chen D. Osteoprotegerin, the bone protector, is a surprising target for beta-catenin signaling. Cell Metab. 2005;2:344–345. doi: 10.1016/j.cmet.2005.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucay N, Sarosi I, Dunstan CR, Morony S, Tarpley J, Capparelli C, Scully S, Tan HL, Xu W, Lacey DL, et al. Osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998;12:1260–1268. doi: 10.1101/gad.12.9.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett BJ, Scatena M, Kirk EA, Rattazzi M, Varon RM, Averill M, Schwartz SM, Giachelli CM, Rosenfeld ME. Osteoprotegerin inactivation accelerates advanced atherosclerotic lesion progression and calcification in older ApoE-/- mice. Arterioscler Thromb Vasc Biol. 2006;26:2117–2124. doi: 10.1161/01.ATV.0000236428.91125.e6. [DOI] [PubMed] [Google Scholar]
- Collin-Osdoby P. Regulation of vascular calcification by osteoclast regulatory factors RANKL and osteoprotegerin. Circ Res. 2004;95:1046–1057. doi: 10.1161/01.RES.0000149165.99974.12. [DOI] [PubMed] [Google Scholar]
- Rogers A, Eastell R. Circulating osteoprotegerin and receptor activator for nuclear factor kappaB ligand: clinical utility in metabolic bone disease assessment. J Clin Endocrinol Metab. 2005;90:6323–6331. doi: 10.1210/jc.2005-0794. [DOI] [PubMed] [Google Scholar]
- Lomaga MA, Yeh WC, Sarosi I, Duncan GS, Furlonger C, Ho A, Morony S, Capparelli C, Van G, Kaufman S, et al. TRAF6 deficiency results in osteopetrosis and defective interleukin-1, CD40, and LPS signaling. Genes Dev. 1999;13:1015–1024. doi: 10.1101/gad.13.8.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito A, Azuma S, Tanaka S, Miyazaki T, Takaki S, Takatsu K, Nakao K, Nakamura K, Katsuki M, Yamamoto T, et al. Severe osteopetrosis, defective interleukin-1 signalling and lymph node organogenesis in TRAF6-deficient mice. Genes Cells. 1999;4:353–362. doi: 10.1046/j.1365-2443.1999.00265.x. [DOI] [PubMed] [Google Scholar]
- Wada T, Nakashima T, Oliveira-dos-Santos AJ, Gasser J, Hara H, Schett G, Penninger JM. The molecular scaffold Gab2 is a crucial component of RANK signaling and osteoclastogenesis. Nat Med. 2005;11:394–399. doi: 10.1038/nm1203. [DOI] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Koga T, Nishina H, Isshiki M, Yoshida H, Saiura A, Isobe M, Yokochi T, Inoue J, et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev Cell. 2002;3:889–901. doi: 10.1016/S1534-5807(02)00369-6. [DOI] [PubMed] [Google Scholar]
- Yao Z, Matsuo K, Nishimura R, Xing L, Boyce BF. c-Fos/NFAT1-or 2-mediated osteoclastogenesis requires NF-kB p50/p52 expression. J Bone Miner Res. 2005. p. S145.
- Thiebaud D, Krieg MA, Gillard-Berguer D, Jacquet AF, Goy JJ, Burckhardt P. Cyclosporine induces high bone turnover and may contribute to bone loss after heart transplantation. Eur J Clin Invest. 1996;26:549–555. doi: 10.1046/j.1365-2362.1996.00170.x. [DOI] [PubMed] [Google Scholar]
- Nakashima K, Zhou X, Kunkel G, Zhang Z, Deng JM, Behringer RR, de Crombrugghe B. The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell. 2002;108:17–29. doi: 10.1016/S0092-8674(01)00622-5. [DOI] [PubMed] [Google Scholar]
- Koga T, Matsui Y, Asagiri M, Kodama T, de Crombrugghe B, Nakashima K, Takayanagi H. NFAT and Osterix cooperatively regulate bone formation. Nat Med. 2005;11:880–885. doi: 10.1038/nm1270. [DOI] [PubMed] [Google Scholar]
- Koga T, Inui M, Inoue K, Kim S, Suematsu A, Kobayashi E, Iwata T, Ohnishi H, Matozaki T, Kodama T, et al. Costimulatory signals mediated by the ITAM motif cooperate with RANKL for bone homeostasis. Nature. 2004;428:758–763. doi: 10.1038/nature02444. [DOI] [PubMed] [Google Scholar]
- Takayanagi H. Mechanistic insight into osteoclast differentiation in osteoimmunology. J Mol Med. 2005;83:170–179. doi: 10.1007/s00109-004-0612-6. [DOI] [PubMed] [Google Scholar]
- Yao Z, Li P, Zhang Q, Schwarz EM, Keng P, Arbini A, Boyce BF, Xing L. Tumor necrosis factor-α increases circulating osteoclast precursor numbers by promoting their proliferation and differentiation in the bone marrow through up-regulation of c-Fms expression. J Biol Chem. 2006;281:11846–11855. doi: 10.1074/jbc.M512624200. [DOI] [PubMed] [Google Scholar]