

Abstract
Inflammation and degradation of bone are two closely linked processes. Chronic inflammatory arthritis not only leads to inflammatory bone loss but it also involves local erosion of articular bone. This osteo-destructive feature of chronic inflammatory arthritis is a major cause of disability in patients with rheumatoid arthritis. Osteoclasts are essential for the resorption of mineralized cartilage and subchondral bone in chronic arthritis. The observed up-regulation of osteoclast differentiation factors (receptor activator of nuclear factor-κB ligand [RANKL]) in the synovial membrane of chronically inflamed joints indicates that osteoclasts are abundant in this setting, leading to rapid degradation of mineralized tissue. Blockade of osteoclast formation is thus a key strategy in preventing structural damage in arthritis. Denosumab, a humanized antibody that neutralizes RANKL, is an attractive candidate agent to inhibit inflammatory bone loss.
Introduction
Pathologic changes in bone mass and bone quality can be the manifestation of an intrinsic, often genetically based dysfunction of the skeletal system, which can lead to fragile or otherwise altered bone. Osteogenesis imperfecta or Paget's disease are among the best known examples of primary genetically based bone disease; the former leads to fragile bone and the latter to increased bone mass. More frequently, however, bone becomes the target of extraskeletal processes, and changes in the exogenous or endogenous environment can affect skeletal tissue, accelerate bone loss, and decrease bone quality. Lack of mobility, smoking, and drug therapy are well established exogenous environmental factors that affect bone quality. Endogenous factors include age-induced loss of muscular strength and endocrine changes, such as reduction in sex hormones during the menopause or dysfunction of the thyroid or adrenal hormone axis.
Systemic inflammation is a key example of a condition that profoundly affects the skeleton but is not a primary disorder of bone itself. Recent data suggest that even a minor and subclinical increase in systemic inflammation precipitates bone disease and increases fracture risk [1]. This tight interaction between inflammation and bone is highlighted by the observation that virtually all chronic inflammatory diseases, particularly rheumatic disease and chronic inflammatory bowel disease, are associated with a high prevalence of osteoporosis and increased fracture risk [2-6]. In the case of a more localized inflammatory process, these systemic effects on bone are accompanied by local bone damage at the skeletal sites closest to the inflammatory focus. Similar to destruction mediated by an earthquake, bone damage is most common and most severe in the vicinity of the (inflammatory) epicenter, whereas the effects are less severe at sites distant from the epicenter, although they can still be detected (for instance, bone density measurements in the case of bone loss). Local resorption of alveolar bone in periodontal disease is a good example of local bone loss; another clinically important example is bone erosion in inflammatory arthritis.
Local bone erosion in arthritis
Only few diseases lead to local resorption of bone. Apart from tumor metastases, various granulomatous diseases (including tuberculosis, sarcoidosis, and histiocytosis) can precipitate local bone erosion. One of the most frequent causes for local bone erosion, however, is arthritis, which involves destruction of juxta-articular bone. This process is a hallmark of rheumatoid arthritis (RA) but it also occurs within the context of other chronic forms of arthritis, particularly in psoriatic arthritis (Figure 1). Determination of bone erosion is almost exclusively based on radiographic findings, because direct assessment of these lesions through biopsy is only rarely practical. The term 'bone erosion' describes loss of mineralized tissue at juxta-articular sites, which is commonly associated with a break in the cortical lining.
Figure 1.
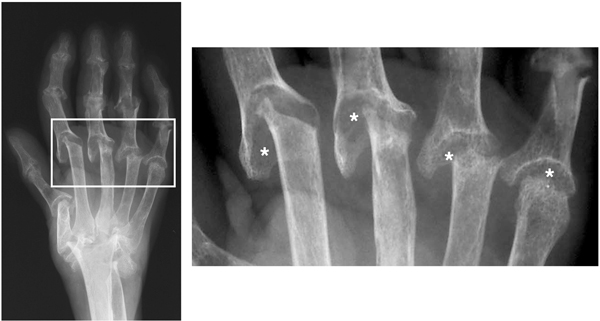
Hand radiograph of a patient with rheumatoid arthritis. Imaged is the hand of a patient with advanced rheumatoid arthritis with severe destruction of the joint architecture. Asterisks indicate bone erosion.
Signs of inflammatory bone erosion have been found in skeletal remains of individuals from several Indian tribes in Northern America [7]. Destruction of bone by arthritis was first described more than 100 years ago. These earliest studies used histopathologic examination to investigate areas of structural joint damage [8]. Later, with the development of radiographic imaging, bone erosions could be visualized directly and became not only part of the diagnostic criteria for RA but also a valuable tool for monitoring disease [9,10]. Virtually all clinical studies of anti-inflammatory and immuno-modulatory drugs for the treatment of RA have employed clinical end-points as efficacy measures and radiologic endpoints to define drug effects on structural damage. Importantly, the resorption of mineralized tissue that is evident on conventional radiographs is strongly associated with poor functional outcome in patients with chronic arthritis [11-13]. With the ultimate aim being to preserve functional status and joint architecture in RA patients, characterization of early disease has been an important goal in RA research over the past 10 years. It soon became evident that bone erosion starts early in disease and progresses most rapidly during the first year [14]. These findings have fostered the concepts that retardation, arrest, or even repair of structural damage should be considered central goals in the treatment of RA.
Cartilage and bone erosion are different
The early and rapid occurrence of structural damage in RA originates from a close interaction of the synovial membrane of the joint cavity and of the tendon sheaths with cartilage and subchondral bone. By definition, synovial tissue becomes inflamed during the course of arthritis and simultaneously involves adjacent cartilage and bone. In particular, synovial tissue close to the joint ends plays a central role in initiating structural damage [15]. Studies of joints from animals and humans have shown that this layer of mineralized cartilage is rapidly resorbed, whereas the unmineralized surface cartilage remains intact for a certain period of time.
Invasion into mineralized cartilage is a multicellular process, involving inflammatory cells such as fibroblasts, lymphocytes, and monocytes (Figure 2). The two former cell types trigger differentiation of monocytes to osteoclasts, which can remove mineral as well as matrix (either dominated by collagen type II in the case of mineralized cartilage or rich in collagen type I in the case of subchondral bone; Figure 3) [16,17]. The particular susceptibility of mineralized cartilage to osteoclast-mediated bone resorption is intuitive because the most abundant pathway of ossification, namely enchondral ossification, involves removal of mineralized cartilage and its remodeling into bone. Resorption of the subchondral bone layer also requires osteoclasts, which depend on the presence of minerals to exert their resorptive actions. In contrast to these mineralized tissues, resorption of unmineralized surface cartilage is not an osteoclast-dependent process. Although the molecular mechanisms of degradation of surface cartilage in the inflamed joint are not fully understood, invasion of synovial fibroblasts into the cartilage and expression of matrix-degrading enzymes (for instance, aggrecanases and matrix metalloproteinases) by synovial fibroblasts, neutrophils, and chondrocytes are key processes in cartilage damage [18]. Joint destruction can thus be considered a process that combines several different events: destruction of surface cartilage, resorption of mineralized cartilage, and erosion of bone. The radiologic image of structural damage represents a picture in which these processes are fused.
Figure 2.
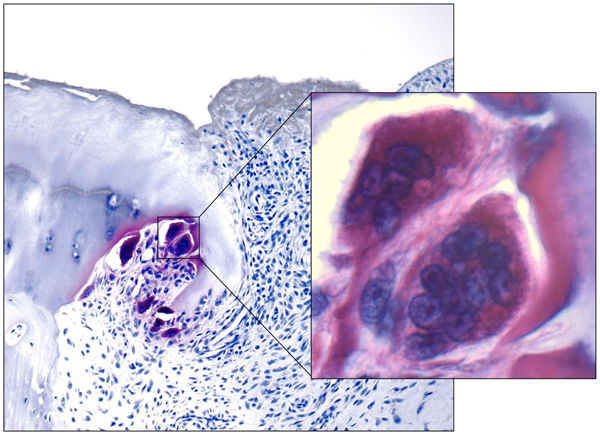
Microphotograph of human bone erosion. Tartrate acid phosphatase staining through a finger joint of patient with rheumatoid arthritis shows osteoclasts (inset) resorbing the mineralized cartilage.
Figure 3.
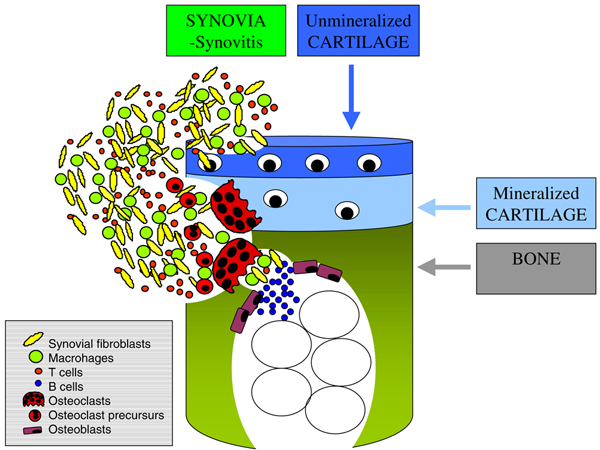
Scheme of arthritic bone erosion. In synovial inflammatory tissue the interaction of fibroblasts, T cells, and monocytes leads to differentiation of osteoclast precursor cells and mature osteoclasts; this results in destruction of mineralized tissue, including cartilage and cortical bone.
RANKL in arthritis
The observations that osteoclasts are an integral part of the mixed cellular infiltrate of inflammatory arthritis and that accumulation of these cells at sites of structural damage suggest that molecules involved in osteoclast formation are important players in the destructive processes of the disease. Receptor activator of nuclear factor-κB ligand (RANKL) exists in two different isoforms: a transmembrane cell-bound form and a soluble from, which shares structural similarities with tumor necrosis factor (TNF) but which binds to a different receptor, termed receptor antagonist of nuclear factor-κB (RANK) [19]. Interaction of lower affinity with another member of the TNF receptor family, namely TNF-related apoptosis-inducing ligand, has also been described. The interaction between RANKL and RANK is crucial for osteoclast formation as well as lymph node organogenesis [19].
Mice that are unable to express either RANKL or RANK are osteopetrotic because of a complete lack of osteoclasts. These animals also exhibit complete developmental arrest of the lymph node anlagen during embryogenesis [20,21]. Inhibition rather than absence of RANKL is achieved by over-expression of the natural inhibitor of RANK/RANK, namely osteoprotegerin (OPG). Whereas lymph node anlagen develop normally in mice that over-express OPG, the bone phenotype is very similar to that in RANKL and RANK knockout mice, because these animals have few osteoclasts and are osteopetrotic [22]. Also, preclical studies of RANKL inhibition demonstrated normal immune responses [23].
RANKL is expressed in synovial tissue in chronic inflammatory arthritides such as RA and psoriatic arthritis [24-27]. Expression can be found on T and B cells and on synovial fibroblasts. Importantly, many proinflammatory cytokines, including TNF, IL-1, IL-6, and IL-17, are important inducers of RANKL expression, as is prostaglandin E2 [28]. This plethora of inflammatory mediators in arthritis facilitates expression of RANKL, which engages its ligand RANK on monocytic cells, a population that is abundant in inflammatory synovial tissue. Formation of osteoclasts and their precursor cells is thus rapid, early, and triggered by continuous cytokine production and cell influx. In accordance with this, kinetic analyses of osteoclast formation in animal models of arthritis have revealed that cells form only a few days after the onset of inflammation [29].
Effects of RANKL in experimental arthritis
The role played by RANKL in inflammatory bone destruction has been intensively characterized in experimental models of arthritis. In particular, mouse models with a genetic block in osteoclast formation have permitted new insights into the mechanisms of arthritis. For example, osteopetrotic mice such as c-fos-/- mice, RANKL-/- mice, and RANK-/- mice develop arthritis but do not exhibit bone erosion [30-32]. This switch from a destructive to a nondestructive phenotype not only proves that osteoclasts play a role in destruction of the joint architecture, but it also hints that blockade of RANKL might serve as an interesting approach to preventing inflammatory bone erosion.
OPG has been used as a neutralizing agent to achieve pharmacologic blockade of RANKL in various models. Destruction of mineralized tissue was effectively blocked by this form of RANKL inhibition in models including adjuvant-induced arthritis, TNF-mediated arthritis, and collagen-induced arthritis [33-36]. In contrast, OPG did not ameliorate the inflammatory signs of disease, and histologic examination of inflamed joints from these models revealed full-blown development of synovial inflammation, suggesting that the interaction between RANKL and RANK is not critically involved in joint inflammation. Despite the presence of inflammation, however, structural damage did not occur to a significant degree in these models, indicating that RANKL-mediated osteoclast formation is a key player in structural damage in arthritis. Treatment with OPG completely blocked osteoclast formation in inflammatory tissue and prevented the formation of osteoclast precursor cells (Figure 4). This arrest of osteoclast differentiation elicited by OPG, together with the effect on function of mature osteoclasts, is crucial to appreciating the effective bone-sparing properties of RANKL blockade. Given the abundance of monocytes as potential osteoclast precursors in the inflammatory tissue and the large number of cells that actually differentiate from this precursor pool to the osteoclast lineage, this dual inhibitory effect on both differentiation and activation of osteoclasts can be considered an important feature of RANKL blockade. This may provide an explanation as to why relatively high amounts of bisphosphonates are necessary to block inflammatory bone erosions in animal models of arthritis [37]. Also, the uptake and deposition of bisphosphonates at cortical bone sites affected by arthritis might be insufficient to allow complete protection of bone.
Figure 4.
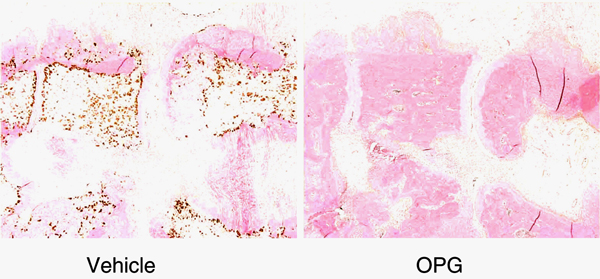
Effect of RANKL blockade on experimental arthritis. Adjuvant-induced arthritis of rats treated with vehicle or osteoprotegerin (OPG). Note the protection of bone and the absence of (stained) osteoclasts. RANKL, receptor activator of nuclear factor-κB ligand.
It is not entirely clear whether RANKL blockade also affects proteoglycan loss in superficial articular cartilage. There is no doubt that targeting of osteoclasts would protect mineralized cartilage, an area that is highly sensitive to osteoclast-mediated resorption. Nonmineralized cartilage is not affected by osteoclasts, and it is therefore unlikely that RANKL blockade would affect this compartment directly. In some of the experimental arthritis models, however, protection against proteoglycan loss was observed following RANKL blockade, whereas it has not been observed in others. There are two potential explanations for the protection of surface cartilage after RANKL blockade. The first of these is a direct effect of RANKL blockade on cartilage by engagement of RANKL and RANK, which are expressed on articular chondrocytes [38]. However, a specific function of RANK/RANKL on chondrocytes remains to be defined. A second possibility is an indirect protective effect on cartilage, based on prevention of resorption of mineralized cartilage attached to surface cartilage. Replacement of this interaction by an inflammatory tissue that attacks unmineralized cartilage not only from the surface (joint cavity) but also at the base may create a forceps-like destruction pattern that could dramatically affect the structure of this remaining surface cartilage. In this regard, inflammatory tissue not only creates aggrecanases and matrix metalloproteinases, which destroy the cartilage matrix, but it also leads to internal production of these enzymes by chondrocytes through the expression of proinflammatory cytokines. Thus, RANKL blockade probably affects the microenvironment of articular cartilage in inflammatory arthritis by preserving surrounding mineralized cartilage and bone.
RANKL inhibition in human arthritis
Blockade of RANKL is considered a powerful future anti-resorptive strategy for the treatment of human bone disease. First proof of principal for this concept was derived from use of Fc:OPG as a blocker of RANKL; a single injection of this protein in humans resulted in increased bone mass and rapid decline in bone resorption parameters [39]. This study was conducted in postmenopausal women with low bone mass. Recently, a fully human neutralizing antibody directed against RANKL, namely denosumab (formerly known as AMG 162), was developed that has sustained antiresorptive activity, lasting for up to 6 months after a single injection [40]. Repeated injections of this antibody have resulted in profound and significant increases in bone mass at both axial and peripheral skeletal sites, as well as in trabecular and cortical areas of bone [41]. Thus far, no major adverse effects of denosumab have been reported; this is reassuring, but further careful and long-term surveillance of infection rate and cardiovascular events are needed before conclusions regarding the long-term safety of this drug can be drawn. These findings make denosumab an attractive therapeutic tool for treating patients with RA. It is conceivable that the effects of denosumab extend not only to the prevention and treatment of generalized bone loss in RA but also to the prevention and treatment of joint destruction. A phase II study is currently underway that aims to investigate the role of RANKL blockade in inhibiting structural damage in RA, and preliminary data suggest that denosumab can indeed inhibit the process of bone erosion, as detected by magnetic resonance imaging of joints [42].
Although RANKL blockade does not inhibit inflammation, necessitating its combination with anti-inflammatory therapy, there are several rationales for targeting RANKL in human RA. First, structural damage often progresses despite use of disease-modifying antirheumatic drugs, especially in those patients who are not treated with TNF blockers. Second, structural damage is a leading cause of decreased joint function and disability in RA patients, emphasizing the need for effective drug therapy to preserve joint architecture. Finally, targeting of RANKL specifically and directly affects the mechanism responsible for the loss of mineralized cartilage and bone in chronic arthritis.
Conclusion
Chronic arthritis is an osteodestructive process, which leads to an accumulation of joint damage over time and prevents a restitutio ad integrum, as observed in spurious forms of arthritis. In addition to controlling inflammation, prevention of structural damage is a key objective of antirheumatic therapy. Because structural damage in RA is based on formation of osteoclasts in and around the joint, which resorb mineralized cartilage and subchondral bone, and because generation of these cells depends on RANKL, inhibition of this molecule is an interesting way to improve therapeutic outcomes in RA.
Abbreviations
IL = interleukin; OPG = osteoprotegerin; RA = rheumatoid arthritis; RANK = receptor activator of nuclear factor-κB; RANKL = receptor activator of nuclear factor-κB ligand; TNF = tumor necrosis factor.
Competing interests
GS occasionally serves as a consultant for Amgen Inc.
Acknowledgments
Acknowledgements
This article is published as part of Arthritis Research & Therapy Volume 9 Supplement 1, 2007: Basic science, rationale, background and future of denosumab: a RANK ligand inhibitor. The full contents of the supplement are available online at http://arthritis-research.com/supplements/9/S1.
Publication of the supplement has been supported by an unrestricted grant from Amgen Inc.
References
- Schett G, Kiechl S, Weger S, Pederiva A, Mayr A, Petrangeli M, Oberhollenzer F, Lorenzini R, Redlich K, Axmann R, et al. High-sensitivity C-reactive protein and risk of nontraumatic fractures in the Bruneck study. Arch Intern Med. 2006;166:2495–2501. doi: 10.1001/archinte.166.22.2495. [DOI] [PubMed] [Google Scholar]
- Gough AK, Lilley J, Eyre S, Holder RL, Emery P. Generalized bone loss in patients with early rheumatoid arthritis. Lancet. 1994;344:23–27. doi: 10.1016/S0140-6736(94)91049-9. [DOI] [PubMed] [Google Scholar]
- Spector TD, Hall GM, McCloskey EV, Kanis JA. Risk of vertebral fracture in women with rheumatoid arthritis. BMJ. 1993;306:558. doi: 10.1136/bmj.306.6877.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooyman JR, Melton LJ, III, Nelson AM, O'Fallon WM, Riggs BL. Fractures after rheumatoid arthritis. A population-based study. Arthritis Rheum. 1984;27:1353–1361. doi: 10.1002/art.1780271205. [DOI] [PubMed] [Google Scholar]
- Bultink IE, Lems WF, Kostense PJ, Dijkmans BA, Voskuyl AE. Prevalence of and risk factors for low bone mineral density and vertebral fractures in patients with systemic lupus erythematosus. Arthritis Rheum. 2005;52:2044–2050. doi: 10.1002/art.21110. [DOI] [PubMed] [Google Scholar]
- Bernstein CN, Blanchard JF, Leslie W, Wajda A, Yu BN. The incidence of fracture among patients with inflammatory bowel disease. A population based cohort study. Ann Intern Med. 2000;133:795–799. doi: 10.7326/0003-4819-133-10-200011210-00012. [DOI] [PubMed] [Google Scholar]
- Lougheed T. The Vector Theory. Did rheumatoid arthritis originate in the New World? Arthritis News Magazine. 1989;7(2) [Google Scholar]
- Weichselbaum A. The finer changes of cartilage with fungus synovitis and caries of the joint ends [in German] Archiv Pathol Anat Physiol Klin Med. 1878;73:461–475. doi: 10.1007/BF01995720. [DOI] [Google Scholar]
- Arnett FC, Edworthy SM, Bloch DA, McShane DJ, Fries JF, Cooper NS, Healey LA, Kaplan SR, Liang MH, Luthra HS, et al. The American Rheumatism Association 1987 revised criteria for the classification of rheumatoid arthritis. Arthritis Rheum. 1988;31:315–324. doi: 10.1002/art.1780310302. [DOI] [PubMed] [Google Scholar]
- Sharp JT, Young DY, Bluhm GB, Brook A, Brower AC, Corbett M, Decker JL, Genant HK, Gofton JP, Goodman N, et al. How many joints in the hands and wrists should be included in a score of radiologic abnormalities used to assess rheumatoid arthritis? Arthritis Rheum. 1985;28:1326–1335. doi: 10.1002/art.1780281203. [DOI] [PubMed] [Google Scholar]
- Scott DL, Pugner K, Kaarela K, Doyle DV, Woolf A, Holmes J, Hieke K. The links between joint damage and disability in rheumatoid arthritis. Rheumatology (Oxford) 2000;39:122–132. doi: 10.1093/rheumatology/39.2.122. [DOI] [PubMed] [Google Scholar]
- Pincus T. Rheumatoid arthritis: disappointing long-term outcomes despite successful short-trem clinical trials. J Clin Epidemiol. 1988;41:1037–1041. doi: 10.1016/0895-4356(88)90072-8. [DOI] [PubMed] [Google Scholar]
- Welsing PM, van Gestel AM, Swinkels HL, Kiemeney LA, van Riel PL. The relationship between disease activity, joint destruction, and functional capacity over the course of rheumatoid arthritis. Arthritis Rheum. 2001;44:2009–2017. doi: 10.1002/1529-0131(200109)44:9<2009::AID-ART349>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Van der Heijde DM. Joint erosions and patients with early rheumatoid arthritis. Br J Rheumatol. 1995;34:74–78. [PubMed] [Google Scholar]
- Hayer S, Redlich K, Korb A, Hermmann S, Smolen JS, Schett G. Tendosynovitis and osteoclast formation as the initial changes of preclinical inflammatory arthritis. Arthritis Rheum. 2007;56:79–88. doi: 10.1002/art.22313. [DOI] [PubMed] [Google Scholar]
- Bromley M, Woolley DE. Chondroclasts and osteoclasts at subchondral sites of erosion in the rheumatoid joint. Arthritis Rheum. 1984;27:968–975. doi: 10.1002/art.1780270902. [DOI] [PubMed] [Google Scholar]
- Gravallese EM, Harada Y, Wang JT, Gorn AH, Thornhill TS, Goldring SR. Identification of cell types responsible for bone resorption in rheumatoid arthritis and juvenile rheumatoid arthritis. Am J Pathol. 1998;152:943–951. [PMC free article] [PubMed] [Google Scholar]
- Pap T, Aupperle KR, Gay S, Firestein GS, Gay RE. Invasiveness of synovial fibroblasts is regulated by p53 in the SCID mouse in vivo model of cartilage invasion. Arthritis Rheum. 2001;44:676–681. doi: 10.1002/1529-0131(200103)44:3<676::AID-ANR117>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/S0092-8674(00)81569-X. [DOI] [PubMed] [Google Scholar]
- Kong YY, Yoshida H, Sarosi I, Tan HL, Timms E, Capparelli C, Morony S, Oliveira-dos-Santos AJ, Van G, Itie A, et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature. 1999;397:315–323. doi: 10.1038/16852. [DOI] [PubMed] [Google Scholar]
- Dougall WC, Glaccum M, Charrier K, Rohrbach K, Brasel K, De Smedt T, Daro E, Smith J, Tometsko ME, Maliszewski CR, et al. RANK is essential for osteoclast and lymph node development. Genes Dev. 1999;13:2412–2424. doi: 10.1101/gad.13.18.2412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Stolina M, Guo J, Faggioni R, Brown H, Senaldi G. Regulatory effects of osteoprotegerin on cellular and humoral immune responses. Clin Immunol. 2003;109:347–354. doi: 10.1016/j.clim.2003.09.001. [DOI] [PubMed] [Google Scholar]
- Shigeyama Y, Pap T, Kunzler P, Simmen BR, Gay RE, Gay S. Expression of osteoclast differentiation factor in rheumatoid arthritis. Arthritis Rheum. 2000;43:2523–2530. doi: 10.1002/1529-0131(200011)43:11<2523::AID-ANR20>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Gravallese EM, Manning C, Tsay A, Naito A, Pan C, Amento E, Goldring SR. Synovial tissue in rheumatoid arthritis is a source of osteoclast differentiation factor. Arthritis Rheum. 2000;43:250–258. doi: 10.1002/1529-0131(200002)43:2<250::AID-ANR3>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Ritchlin CT, Haas-Smith SA, Li P, Hicks DG, Schwarz EM. Mechanisms of TNF-alpha- and RANKL-mediated osteoclastogenesis and bone resorption in psoriatic arthritis. J Clin Invest. 2003;111:821–831. doi: 10.1172/JCI200316069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stolina M, Adamu S, Ominsky M, Dwyer D, Asuncion F, Geng Z, Middleton S, Brown H, Pretorius J, Schett G, et al. RANKL is a marker and mediator of local and systemic bone loss in two rat models of inflammatory arthritis. J Bone Miner Res. 2005;20:1756–1765. doi: 10.1359/JBMR.050601. [DOI] [PubMed] [Google Scholar]
- Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schett G, Stolina M, Bolon B, Middleton S, Adlam M, Brown H, Zhu L, Feige U, Zack DJ. Analysis of the kinetics of osteoclastogenesis in arthritic rats. Arthritis Rheum. 2005;52:3192–3201. doi: 10.1002/art.21343. [DOI] [PubMed] [Google Scholar]
- Redlich K, Hayer S, Ricci R, David JP, Tohidast-Akrad M, Kollias G, Steiner G, Smolen JS, Wagner EF, Schett G. Osteoclasts are essential for TNF-alpha-mediated joint destruction. J Clin Invest. 2002;110:1419–1427. doi: 10.1172/JCI200215582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit AR, Ji H, von Stechow D, Muller R, Goldring SR, Choi Y, Benoist C, Gravallese EM. TRANCE/RANKL knockout mice are protected from bone erosion in a serum transfer model of arthritis. Am J Pathol. 2001;159:1689–1699. doi: 10.1016/S0002-9440(10)63016-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Schwarz EM, O'Keefe , Ma L, Boyce BF, Xing L. RANK signaling is not required for TNF-mediated increase in CD11(hi) osteoclast precursors but is essential for mature osteoclast formation in TNFalpha-mediated inflammatory arthritis. J Bone Miner Res. 2004;19:207–213. doi: 10.1359/JBMR.0301233. [DOI] [PubMed] [Google Scholar]
- Kong YY, Feige U, Sarosi I, Bolon B, Tafuri A, Morony S, Capparelli C, Li J, Elliott R, McCabe S, et al. Activated T cells regulate bone loss and joint destruction in adjuvant arthritis through osteoprotegerin ligand. Nature. 1999;402:304–309. doi: 10.1038/46303. [DOI] [PubMed] [Google Scholar]
- Redlich K, Hayer S, Maier A, Dunstan CR, Tohidast-Akrad M, Lang S, Turk B, Pietschmann P, Woloszczuk W, Haralambous S, et al. Tumor necrosis factor α-mediated joint destruction is inhibited by targeting osteoclasts with osteoprotegerin. Arthritis Rheum. 2002;46:785–792. doi: 10.1002/art.10097. [DOI] [PubMed] [Google Scholar]
- Romas E, Gillespie MT, Martin TJ. Involvement of receptor activator of NFκB ligand and tumor necrosis factor-a in bone destruction in rheumatoid arthritis. Bone. 2002;30:340–346. doi: 10.1016/S8756-3282(01)00682-2. [DOI] [PubMed] [Google Scholar]
- Lubberts E, van den Bersselaar L, Oppers-Walgreen B, Schwarzenberger P, Coenen-de Roo CJ, Kolls JK, Joosten LA, van den Berg WB. IL-17 promotes bone erosion in murine collagen-induced arthritis through loss of the receptor activator of NF-kappa B ligand/osteoprotegerin balance. J Immunol. 2003;170:2655–2662. doi: 10.4049/jimmunol.170.5.2655. [DOI] [PubMed] [Google Scholar]
- Herrak P, Gortz B, Hayer S, Redlich K, Reiter E, Gasser J, Bergmeister H, Kollias G, Smolen JS, Schett G. Zoledronic acid protects against local and systemic bone loss in tumor necrosis factor-mediated arthritis. Arthritis Rheum. 2004;50:2327–2337. doi: 10.1002/art.20384. [DOI] [PubMed] [Google Scholar]
- Komuro H, Olee T, Kuhn K, Quach J, Brinson DC, Shikhman A, Valbracht J, Creighton-Achermann L, Lotz M. The osteoprotegerin/receptor activity of nuclear factor kappaB/receptor activator of nuclear factor kappaB ligand system in cartilage. Arthritis Rheum. 2001;44:2768–2776. doi: 10.1002/1529-0131(200112)44:12<2768::AID-ART464>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- Bekker PJ, Holloway D, Nakanishi A, Arrighi M, Leese PT, Dunstan CR. The effect of a single dose of osteoprotegerin in postmenopausal women. J Bone Miner Res. 2001;16:348–360. doi: 10.1359/jbmr.2001.16.2.348. [DOI] [PubMed] [Google Scholar]
- Bekker PJ, Holloway DL, Rasmussen AS, Murphy R, Martin SW, Leese PT, Holmes GB, Dunstan CR, DePaoli AM. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner Res. 2004;19:1059–1066. doi: 10.1359/JBMR.040305. [DOI] [PubMed] [Google Scholar]
- McClung MR, Lewiecki EM, Cohen SB, Bolognese MA, Woodson GC, Moffett AH, Peacock M, Miller PD, Lederman SN, Chesnut CH, AMG 162 Bone Loss Study Group et al. Denosumab in postmenopausal women with low bone mineral density. N Engl J Med. 2006;354:821–831. doi: 10.1056/NEJMoa044459. [DOI] [PubMed] [Google Scholar]
- Cohen SB, Valen P, Ritchlin C, Schechtman J, Peterfy C, van der Heijde D, Zhou L, Newmark R, Tsuji W. RANKL inhibition with denosumab reduces progression of bone erosions in patients with rheumatoid arthritis: month 6 MRI results [abstract 2120] Arthritis Rheum. 2006;54(Suppl 9):S831. [Google Scholar]