

Abstract
Multiple myeloma, a cancer of plasma cells, is associated with excessive tumor-induced, osteoclast-mediated bone destruction. Hypercalcemia remains the most frequent metabolic complication of myeloma in patients, and excessive osteolysis plays a major contributory role in its pathogenesis. The clinical presentation of hypercalcemia in patients varies depending on the level of ionized calcium; it can be life threatening, as in the case of hypercalcemic crisis, requiring immediate medical treatment to prevent death. During the past few years there have been exciting developments in our understanding of the pathogenesis of myeloma bone disease; in particular, key mediators of the osteoclastic bone resorption in myeloma have been identified, including receptor activator of nuclear factor-κB ligand (RANKL) and macrophage inflammatory protein-1α. There is also increasing evidence that Dickkopf 1, which has been shown to be over-expressed in myeloma patients, is also a potent stimulator of osteoclast formation and activity. Importantly, the available data suggest that RANKL is the final common mediator of osteoclastic bone resorption, irrespective of the upstream initiator molecule. This brief review presents an overview of the roles played by these mediators in inducing osteolysis in myeloma bone disease, and it discusses targeting RANKL as a potential new treatment strategy in myeloma bone disease and myeloma-associated hypercalcemia.
Introduction
Multiple myeloma, a clonal neoplasm of plasma cells, is the second most common adult hematologic malignancy, and it is the most common cancer with skeleton as its primary site. Multiple myeloma, which affects 70,000 people in the USA, with 15,000 new cases accruing yearly, accounts for 1% to 2% of cancer-related deaths. Myeloma is unique in its propensity to cause osteolysis, with 80% of patients suffering from devastating and progressive bone destruction; this results in the complications that are responsible for the high morbidity and mortality rates associated with the disease. These complications, which include severe and unremitting bone pain, pathologic fractures (Figure 1), spinal cord compression, and hypercalcemia, are a significant clinical problem for which there is no effective cure. Hypercalcemia, which can range in presentation from mild to severe and life threatening, is the most common metabolic complication of myeloma and occurs in approximately one-third of patients.
Figure 1.
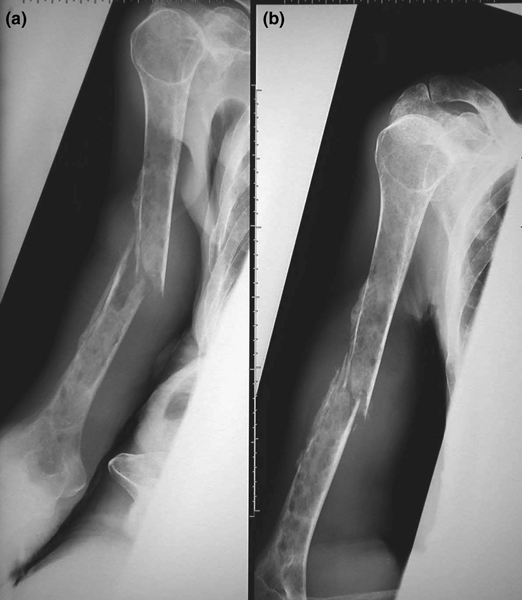
Osteolytic lesions and pathologic fracture in a 76-year-old female myeloma patient. With permission from Nair and Pearson [45]. Copyright © 2004 Massachusetts Medical Society. All rights reserved.
Hypercalcemia in myeloma
In myeloma patients, the primary cause of the hypercalcemia is widespread tumor-induced bone destruction. This is primarily due to increased osteoclastic bone resorption caused by potent cytokines expressed or secreted locally by the myeloma cells (receptor activator of nuclear factor-κB ligand [RANKL], macrophage inflammatory protein [MIP]-1α, and tumor necrosis factors [TNFs]; see below) or over-expressed by other cells in the local microenvironment [1]. This bone resorption in turn leads to efflux of calcium into the extracellular fluid. However, the pathogenesis of hypercalcemia in myeloma is probably more complex than this because not all patients with significant myeloma bone disease develop hypercalcemia and, even in the patients who do develop it, hypercalcemia is often a prominent feature only late in the course of disease.
Hypercalcemia is most common in those myeloma patients who have the greatest tumor volume, irrespective of serum parathyroid hormone-related protein (PTHrP) status. The reasons for this are still unclear but they may be related to the amount of bone-resorbing activity produced by myeloma cells as well as glomerular filtration status. Because myeloma patients often have irreversible impairment in renal function and increased renal tubular calcium reabsorption, the capacity of the kidneys to clear excess calcium load from the circulation effectively is overwhelmed, resulting in elevated serum calcium levels. Elevated serum PTHrP is not a consistent finding in myeloma patients, and it remains unclear why myeloma patients present with increased renal tubular calcium reabsorption. Consistent with this, hypercalcemia is more frequent in patients with plasma cell leukemia, a late stage complication of myeloma, than in overt myeloma per se [2], despite the fact that plasma cell leukemias induce comparatively weaker bone-resorbing activity. This is a consequence of the severe renal insufficiency associated with plasma cell leukemia. Measurements of total body myeloma cell burden together with production of bone-resorbing activity by cultured bone marrow myeloma cells in vitro do not correlate closely with hypercalcemia, although they do correlate somewhat with the extent of osteolytic bone lesions [3]. Thus, other factors are probably involved in the pathogenesis of hypercalcemia, in addition to those that promote osteoclast formation and induce osteoclast activation.
There are other differences between the hypercalcemia that occurs in myeloma and classical humoral hypercalcemia of malignancy in patients with solid tumors. First, in contrast to myeloma patients with hypercalcemia, in which PTHrP is implicated only sporadically, humoral hypercalcemia of malignancy is almost always due to excessive secretion of PTHrP by the tumor, resulting in elevated circulating levels. Second, in patients with hypercalcemia due to myeloma, there is almost always impaired renal function and increased serum phosphate that is associated with decreased glomerular filtration rate. Third, markers of bone formation such as serum alkaline phosphatase are commonly not elevated in patients with myeloma, because bone formation is often not altered and may in fact be suppressed for reasons that are not entirely clear but may involve Dickkopf (DKK)1 (see below). Finally, patients with hypercalcemia due to myeloma usually respond very rapidly to treatment with corticosteroids, mainly because of their rapid suppression of myeloma tumor growth, unlike patients with humoral hypercalcemia caused by solid tumors [4].
Myeloma bone disease
The excessive bone destruction that is characteristic of multiple myeloma is dependent on osteoclast stimulation. Although it has been known for more than three decades that osteoclasts are hyperstimulated by cytokines in myeloma, the osteoclast-activating factors (OAFs) responsible proved elusive until the late 1990s. For some time the erstwhile approach of identifying such factors by use of cultured tumor cell lines, followed by protein purification, was unsuccessful. It is now apparent that cell-cell interactions involving myeloma cells and other cells in the marrow microenvironment, including stromal cells, cells of the osteoblast lineage, and possibly osteoclasts themselves, are required for production of OAFs in myeloma. There is now abundant evidence that the OAFs that are probably responsible for bone resorption in myeloma are the cytokine RANKL and the chemokine MIP-1α. However, a number of other cytokines, including TNF-α, lymphotoxin (TNF-β), and PTHrP, have also been implicated in the disease. There is still considerable debate as to whether in myeloma these bone-resorbing cytokines are produced by the tumor cells or accessory cells involved in cell-cell interactions, or both; indeed, there is evidence in support of both notions.
RANKL
RANKL is part of a system of TNF ligand and receptor superfamily members that include its cognate signaling receptor RANK (receptor activator of nuclear factor-κB) and the decoy receptor antagonist osteoprotegerin (OPG), which regulate osteoclastogenesis and bone resorption in vivo. Both RANKL and OPG are expressed by bone marrow stromal cells (BMSCs) and osteoblasts. It is now well established that RANKL is increased and OPG is markedly decreased in bone marrow in multiple myeloma. Early studies demonstrated that co-culture of myeloma cells and BMSCs leads to marked induction of RANKL expression by both cell types, and a concomitant downregulation of OPG expression by BMSCs [5]; this illustrates the importance of interactions between myeloma cells and stromal cells. This induction of RANKL and downregulation of OPG in myeloma cell-BMSC co-cultures is abrogated by neutralizing antibody to α4 integrin receptor subunit, and treatment of myeloma tumor cells with a recombinant soluble form of vascular cell adhesion molecule (VCAM)-1 (a ligand for the α4 adhesion molecule) was sufficient to induce RANKL expression [5]. These data suggest that cell-cell interactions, mediated by stromal cell expressed VCAM-1 binding to very late antigen-4 (α4β1 integrin) on myeloma cells, are necessary for tumor cells to express RANKL.
These findings of RANKL induction and concomitant OPG downregulation in bone-derived cells in myeloma were subsequently replicated in other studies involving myeloma cell-BMSC and myeloma cell-osteoblast co-cultures [6,7], although these latter studies and others did not observe an increase in RANKL expression in tumor cells [8]. However, evidence is accumulating that supports the notion that myeloma cells themselves also express RANKL, thus providing a mechanism for direct stimulation of osteoclast formation by myeloma cells [9-13]. Nevertheless, irrespective of whether myeloma cells themselves are capable of driving osteoclastogenesis in myeloma directly, it is incontrovertible that RANKL is over-expressed in the marrow micro-environment in myeloma patients, with the ratio of RANKL to OPG markedly upregulated as compared with that in normal control individuals [14].
Data from a number of studies have shown that neutralizing the biologic effects of this over-expressed RANKL in vivo, using either a genetically engineered soluble form of RANK (RANK-Fc) or OPG-Fc, reduces bone destruction and has a beneficial effect in murine models of myeloma bone disease [6,15-17]. These chimeric molecules act as decoys by binding to and sequestering RANKL, thus preventing it from interacting with its native receptor on hematopoietic osteoclast precursors and mature osteoclasts. As proof of principle that targeting the RANKL/RANK system is beneficial in hypercalcemia, RANK-Fc (constructed by fusing the extracellular domain of murine RANK to the Fc region of human lgG1) was found to inhibit normal and pathologic osteoclastic bone resorption in a mouse model of tumor-induced hypercalcemia [18]. Importantly, neutralizing monoclonal antibodies raised against the mouse α4 integrin subunit also decreased osteolysis and overall tumor burden in a mouse model of myeloma bone disease [19]. The latter finding is consistent with the notion that RANKL levels are modulated by stromal cell-expressed VCAM-1 binding to very late antigen-4 on myeloma tumor cells [5]. Taken together, the available data strongly suggest that RANKL is not only the final common mediator of osteoclast activation but also that it is a myeloma cell product, and that aberrant expression of this cytokine within the myeloma marrow millieu is likely to be pivotal in initiating and maintaining the characteristic bone destruction as well as promoting hypercalcemia.
Unlike most of the other mediators previously implicated in myeloma bone disease, RANKL is expressed in various cell types as a transmembrane protein, although it can be cleaved, as occurs in activated T cells, to yield a soluble form that remains biologically active. Unlike for T cells, however, there is no unequivocal evidence to date that 'soluble RANKL' in myeloma is either naturally secreted or cleaved from the cell surface by a proteinase. Nevertheless, whether the predominant source of RANKL in the myeloma bone microenvironment is tumor cells or BMSCs, or both, the consequent exaggerated osteoclast formation and activation is likely to be exacerbated by a concomitant decrease in locally available OPG. The reduced levels of OPG in bone marrow of patients with myeloma is likely to be further exarcebated by binding, internalizing, and degrading of OPG by tumor cells [20]. Consistent with this, serum OPG levels in myeloma patients are significantly reduced compared with those in healthy control individuals [21].
Macrophage inflammatory protein-1α
MIP-1α is a member of the C-C chemokine family produced by myeloma cells and has been implicated as an important mediator in myeloma bone disease that stimulates osteoclast formation and differentiation, and bone resorption [22-27]. Consistent with the notion that it plays an important role in the pathogenesis of myeloma-associated osteolysis in vivo, there is now convincing evidence, in RANK knockout mice, that MIP-1α also stimulates osteoclast differentiation and bone resorption in vivo in a RANKL/RANK-dependent manner when it is expressed adjacent to bone surfaces [22].
MIP-1α is present in bone marrow and marrow supernatants of patients with myeloma bone disease at much higher levels than in patients with other hematologic malignancies and in normal control individuals [23]. Furthermore, its levels correlate with presence of lytic lesions as well as disease stage and activity [24,25]. MIP-1α was also found to be elevated in the serum of myeloma patients with severe bone disease and correlated positively with bone resorption markers, providing evidence for a causal role of the chemokine in the development of lytic bone lesions in myeloma [26,27]. Interestingly, MIP-1α is also produced by osteoblast-like cells in response to stimulation by interleukin-1 and TNF-α [28]. Because myeloma cells secrete high levels of both cytokines constitutively [29], this would serve to amplify further the concentrations of the chemokine locally in the bone micro-environment. In addition, MIP-1α signaling through cell surface receptors for MIP-1α (C-C chemokine receptor [CCR]-1 and CCR5) on bone cells enhances adhesive interactions between myeloma and marrow stromal cells. This, in turn, upregulates levels of expression of RANKL and interleukin-6, which further exacerbates bone destruction and increases tumor burden [30]. Systemic administration of neutralizing antibodies to MIP-1α or small molecule CCR-1-specific and CCR-5-specific antagonists inhibited tumor-induced osteolysis and limited disease progression in mouse models of myeloma bone disease [22,31]. Therefore, the MIP-1α and its cognate receptors may serve as targets for the development of novel therapies to combat hypercalcemia. In this regard, MIP-1α has been reported to induce severe hypercalcemia in other hematopoietic malignancies [32].
Dickkopf 1
DKK1, the prototypic member of a family of proteins that act as extracellular antagonists of canonical Wnt signaling, is expressed in bone in osteoblasts, osteocytes, and BMSCs. It was identified on cDNA microarrays as being preferentially and abundantly expressed in bone marrow aspirates from patients with myeloma. Specifically, DKK1 over-expression was reported to be associated with the presence of focal lesions detected on magnetic resonance imaging [33]. These data have led to the notion that DKK1 is one of the major mediators of the osteolysis that occurs in myeloma bone disease. Interestingly, canonical Wnt signaling was recently shown to modulate RANKL and OPG expression in osteoblasts in vitro, resulting in inhibition of osteoclastogenesis [34]. Consistent with these observations, it was recently reported that DKK1 enhances osteoclast formation and bone resorption in vitro [35], and systemic administration of anti-DKK1 antibodies was shown to block osteoclast formation in vivo in a murine model of myeloma bone disease [36].
Diagnosis and treatment of hypercalcemia
The diagnosis of hypercalcemia is based on concentration of ionized calcium, because the serum calcium level may be erroneously low due to binding of circulating calcium to albumin. The clinical presentation is frequently dependent on the calcium level; patients may be asymptomatic (≤ 3 mmol/l) or they may present with symptoms such as dry mouth, anorexia and vomiting, polyuria, polydipsia, depression, or confusion (3 to 4 mmol/l). Occasionally, patients may develop a life-threatening 'hypercalcemic crisis' (≥ 4 mmol/l) and present in coma.
The best therapy for myeloma-associated hypercalcemia is, by and large, successful elimination of the primary tumor, but myeloma remains an incurable cancer and its unique propensity to cause bone destruction is, in part, responsible for the associated hypercalcemia observed in some patients. Over the past decade, saline replenishment and bisphosphonates, which are potent inhibitors of osteoclastic bone resorption, have become the mainstay of treatment for cancer-induced osteolytic bone disease and management of PTHrP-mediated humoral hypercalcemia of malignancy, as well as hypercalcemia induced by myeloma [37]. In myeloma patients with hypercalcemia, these agents are usually used in combination regimens that include corticosteroids and/or calcitonin, the latter being effective in patients at high risk for developing a hypercalcemic crisis. The available bisphosphonates, including clodronate (a first-generation bisphosphonate), pamidronate (a second-generation bisphosphonate), and zoledronic acid and ibandronate (third-generation bisphosphonates), have exhibited varying degrees of efficacy in multiple clinical trials, undoubtedly influenced by tolerance or toxicity issues. Orally administered clodronate has exhibited little ability to slow the development of skeletal complications in patients. In contrast, oral administration of the more potent ibandronate or intravenous infusions of either pamidronate or zoledronic acid have reduced skeletal complications in patients with cancer-induced bone diseases, with zoledronic acid being the most effective to date [38-40].
The exciting discovery that the RANKL/RANK/OPG pathway is indispensable not only for normal bone resorption but also for pathologic bone resorption and hypercalcemia induced by myeloma and other cancers that metastasize to the skeleton has spurred the clinical development of a number of other types of new antiresorptive agents. Available data from preclinical and clinical studies strongly suggesting that RANKL is the final common mediator in tumor-induced bone destruction that precedes malignancy-associated hypercalcemia, irrespective of the upstream initiator, provide a rationale for pursuing this therapeutic strategy.
Interestingly, data from preclinical studies in murine models of humoral hypercalcemia of malignancy suggest that inhibition of RANKL, using OPG-Fc, causes greater suppression of bone resorption and hypercalcemia than do bisphosphonates [41]. Although its development has been discontinued, AMGN-0007, a recombinant OPG construct, was shown in early studies to be efficacious in patients with multiple myeloma or breast cancer related skeletal metastases. At present, the most advanced anti-RANKL approach in clinical development is a fully human monoclonal antibody to RANKL, namely denosumab (Amgen Inc., Thousand Oaks, CA, USA). Clinical trials with denosumab are ongoing in patients with multiple myeloma and recent data suggest that it is efficacious [42]. In addition, studies are underway in bone metastases from breast cancer, prostate cancer, and other solid tumors [43].
The observation that an intact RANK signaling pathway is indispensable for the osteoclastogenic effect of MIP-1α in vivo [22] lends further support to the notion that RANKL is indeed the final common mediator of the excessive osteoclast number and activity in myeloma. It also suggests that denosumab may be effective in treating hypercalcemia, irrespective of its cause. With regard to targeting MIP-1α or its receptors, available preclinical data provide a basis for further studies to determine the efficacy of orally bioavailable small molecule synthetic antagonists of the receptors that mediate the effects of MIP-1α (CCR-1 and CCR-5), as adjuncts to current standard therapeutic approaches in myeloma-induced hypercalcemia. Although it is presently unclear which of the MIP-1α signaling receptors mediates its osteoclastogenic activity, small molecule antagonists of both CCR-1 and CCR-5 are currently under development for other disease conditions [44].
Finally, a small molecule (IIC3; Enzo Biochem) that is capable of binding to and thereby inhibiting the biologic activity of DKK1 is also currently under development. Given the recently discovered role of canonical Wnt signaling in osteoclast formation and the osteoclastogenic effect of DKK1, further insights into the mechanisms involved in modulating the effects of DKK1 in myeloma bone disease will help to define other novel molecular targets, facilitating development of new therapies for the treatment of associated hypercalcemia.
Conclusion
Hypercalcemia is the most common metabolic complication of multiple myeloma but its pathogenesis remains unclear.
Recently available data support a role for several cytokines such as RANKL, MIP-1α and DKK1 in the exaggerated osteoclastic bone resorption that characterizes myeloma bone disease, with compelling evidence for RANKL being the final common mediator. Given that the increased osteolysis contributes to the development and progression of hypercalcemia in patients, strategies targeting these molecules and the signaling pathways involved may result in novel therapies to manage this distressing and often life-threatening complication of myeloma bone disease.
Abbreviations
BMSC = bone marrow stromal cell; CCR = C-C chemokine receptor; DKK = Dickkopf; MIP = macrophage inflammatory protein; OAF = osteoclast-activating factor; OPG = osteoprotegerin; PTHrP = parathyroid hormone-related protein; RANK = receptor activator of nuclear factor-κB; RANKL = receptor activator of nuclear factor-κB ligand; TNF = tumor necrosis factor; VCAM = vascular cell adhesion molecule.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
This work was supported by grants from the NIH/NCI (KO1 CA104180 and PO1 CA40035).
This article is published as part of Arthritis Research & Therapy Volume 9 Supplement 1, 2007: Basic science, rationale, background and future of denosumab: a RANK ligand inhibitor. The full contents of the supplement are available online at http://arthritis-research.com/supplements/9/S1.
Publication of the supplement has been supported by an unrestricted grant from Amgen Inc.
References
- Oyajobi BO, Mundy GR. Pathophysiology of myeloma bone disease. In: Gahrton G, Durie BGM, Samson DS, editor. Multiple Myeloma and Related Disorders. 2. London, UK: Arnold; 2004. pp. 74–88. [Google Scholar]
- Garcia-Sanz R, Orfão A, Gonzalez M, Tabernero MD, Bladé J, Moro MJ, Fernandez-Calvo J, Sanz MA, Perez-Simon JA, Rasillo A, et al. Primary plasma cell leukemia: clinical, immunophenotypic, DNA ploidy, and cytogenetic characteristics. Blood. 1999;93:1032–1037. [PubMed] [Google Scholar]
- Tuttle KR, Kunau RT, Loveridge N, Mundy GR. Altered renal calcium handling in hypercalcemia of malignancy. J Am Soc Nephrol. 1991;2:191–199. doi: 10.1681/ASN.V22191. [DOI] [PubMed] [Google Scholar]
- Durie BGM, Salmon SE, Mundy GR. Relation of osteoclast activating factor production to the extent of bone disease in multiple myeloma. Br J Haematol. 1981;47:21–30. doi: 10.1111/j.1365-2141.1981.tb02758.x. [DOI] [PubMed] [Google Scholar]
- Oyajobi BO, Traianedes K, Yoneda T, Mundy GR. Expression of RANK ligand (RANKL) by myeloma cells requires binding to bone marrow stromal cells via an α4β1-VCAM-1 interaction. Bone. 1998. p. S180.
- Pearse RN, Sordillo EM, Yaccoby S, Wong BR, Liau DF, Colman N, Michaeli J, Epstein J, Choi Y. Multiple myeloma disrupts the TRANCE/osteoprotegerin cytokine axis to trigger bone destruction and promote tumor progression. Proc Natl Acad Sci USA. 2001;98:11581–11586. doi: 10.1073/pnas.201394498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giuliani N, Bataille R, Mancini C, Lazarretti M, Barille S. Myeloma cells induce imbalance in the osteoprotegerin/osteoprotegerin ligand system in the human bone marrow environment. Blood. 2001;98:3527–3533. doi: 10.1182/blood.V98.13.3527. [DOI] [PubMed] [Google Scholar]
- Roux S, Meignin V, Quillard J, Meduri G, Guiochon-Mantel A, Fermand J-P, Milgrom E, Mariette X. RANK (receptor activator of nuclear factor-κB) and RANKL expression in multiple myeloma. Br J Haematol. 2002;117:86–92. doi: 10.1046/j.1365-2141.2002.03417.x. [DOI] [PubMed] [Google Scholar]
- Farrugia AN, Atkins GJ, To LB, Pan B, Horvath N, Findlay DM, Bardy A, Zannettino AC. Receptor activator of nuclear factor-κB ligand expression by human myeloma cells mediates osteoclast formation in vitro and correlates with bone destruction in vivo. Cancer Res. 2003;63:5438–5445. [PubMed] [Google Scholar]
- Croucher PI, Shipman CM, Lippitt J, Perry M, Asosingh K, Hijzen A, Brabbs AC, van Beek EJR, Holen I, Skerry TM, et al. Osteoprotegerin inhibits the development of osteolytic bone disease in multiple myeloma. Blood. 2001;98:3534–3540. doi: 10.1182/blood.V98.13.3534. [DOI] [PubMed] [Google Scholar]
- Sezer O, Heider U, Jakob C, Zavrski I, Eucker J, Possinger K, Sers C, Krenn V. Immunocytochemistry reveals RANKL expression of myeloma cells. Blood. 2002;99:4646–4647. doi: 10.1182/blood-2002-01-0148. [DOI] [PubMed] [Google Scholar]
- Sezer O, Heider U, Jakob C, Eucker J, Possinger K. Human bone marrow myeloma cells express RANKL. J Clin Oncol. 2002;20:353–354. doi: 10.1200/JCO.2002.20.1.353. [DOI] [PubMed] [Google Scholar]
- Lai FP, Cole-Sinclair M, Cheng WJ, Quinn JM, Gillespie MT, Sentry JW, Schneider HG. Myeloma cells can directly contribute to the pool of RANKL in bone bypassing the classic stromal and osteoblast pathway of osteoclast stimulation. Br J Haematol. 2004;126:192–201. doi: 10.1111/j.1365-2141.2004.05018.x. [DOI] [PubMed] [Google Scholar]
- Roux S, Mariette X. The high rate of bone resorption in multiple myeloma is due to RANK (receptor activator of nuclear factor-kappaB) and RANK ligand expression. Leuk Lymphoma. 2004;45:1111–1118. doi: 10.1080/10428194310001593193. [DOI] [PubMed] [Google Scholar]
- Oyajobi BO, Williams PJ, Traianedes K, Yoneda T, Anderson DM, Mundy GR. Efficacy of a genetically-engineered soluble Receptor Activator of NF-κB (RANK) fusion protein on bone resorption in vitro and in vivo. Cancer. 2000. p. 3093.
- Yaccoby S, Pearse RN, Johnson CL, Barlogie B, Choi Y, Epstein J. Myeloma interacts with the bone marrow microenvironment to induce osteoclastogenesis and is dependent on osteoclast activity. Br J Haematol. 2002;116:278–290. doi: 10.1046/j.1365-2141.2002.03257.x. [DOI] [PubMed] [Google Scholar]
- Vanderkerken K, De Leenheer E, Shipman CM, Asosingh K, Willems A, Van Camp B, Croucher P. Recombinant osteoprotegerin decreases tumor burden and increases survival in a mouse model of multiple myeloma. Cancer Res. 2003;63:287–289. [PubMed] [Google Scholar]
- Oyajobi BO, Anderson DM, Traianedes K, Williams PJ, Yoneda T, Mundy GR. Therapeutic efficacy of a soluble receptor activator of nuclear factor kappaB-IgG Fc fusion protein in suppressing bone resorption and hypercalcemia in a model of humoral hypercalcemia of malignancy. Cancer Res. 2001;61:2572–2578. [PubMed] [Google Scholar]
- Mori Y, Shimizu N, Dallas M, Niewolna M, Williams PJ, Mundy GR, Yoneda T. Anti-α4 integrin antibody suppresses the development of myeloma and associated osteolysis. Blood. 2004;104:2149–2154. doi: 10.1182/blood-2004-01-0236. [DOI] [PubMed] [Google Scholar]
- Standal T, Seidel C, Hjertner O, Plesner T, Sanderson RD, Waage A, Borset M, Sundan A. Osteoprotegerin is bound, internalized and degraded by multiple myeloma cells. Blood. 2002;100:3002–3007. doi: 10.1182/blood-2002-04-1190. [DOI] [PubMed] [Google Scholar]
- Seidel C, Hjertner O, Abildgaard N, Heickendorf L, Hjorth M, Westin J, Nielsen JL, Hjorth-Hansen H, Waagew A, Sundan A, et al. Serum osteoprotegerin levels are reduced in patients with multiple myeloma with lytic bone disease. Blood. 2001;98:2269–2271. doi: 10.1182/blood.V98.7.2269. [DOI] [PubMed] [Google Scholar]
- Oyajobi BO, Franchin G, Williams PJ, Pulkrabek D, Gupta A, Munoz S, Grubbs B, Zhao M, Chen D, Sherry B, Mundy GR. Dual effects of macrophage inflammatory protein-1α on osteolysis and tumor burden in the murine 5TGM1 model of myeloma bone disease. Blood. 2003;102:311–319. doi: 10.1182/blood-2002-12-3905. [DOI] [PubMed] [Google Scholar]
- Choi SJ, Cruz JC, Craig F, Chung H, Devlin RD, Roodman GD, Alsina M. Macrophage inflammatory protein-1α is a potential osteoclast stimulatory factor in multiple myeloma. Blood. 2000;96:671–675. [PubMed] [Google Scholar]
- Abe M, Hiura K, Wilde J, Moriyama K, Hashimoto T, Ozaki S, Wakatsuki S, Kosaka M, Kido S, Inoue D, et al. Role for macrophage inflammatory protein (MIP)-1α and MIP-1β in the development of osteolytic lesions in multiple myeloma. Blood. 2002;100:2195–2202. [PubMed] [Google Scholar]
- Hashimoto T, Abe M, Oshima T, Shibata H, Ozaki S, Inoue D, Matsumoto T. Ability of myeloma cells to secrete macrophage inflammatory protein (MIP)-1alpha and MIP-1beta correlates with lytic bone lesions in patients with multiple myeloma. Br J Haematol. 2004;125:38–41. doi: 10.1111/j.1365-2141.2004.04864.x. [DOI] [PubMed] [Google Scholar]
- Uneda S, Hata H, Matsuno F, Harada N, Mitsuya Y, Kawano F, Mitsuya H. Macrophage inflammatory protein-1α is produced by human multiple myeloma (MM) cells and its expression correlates with bone lesions in patients with MM. Br J Haematol. 2003;120:53–55. doi: 10.1046/j.1365-2141.2003.04040.x. [DOI] [PubMed] [Google Scholar]
- Terpos E, Politou M, Szydlo R, Goldman JM, Apperley JF, Rahemtulla A. Serum levels of macrophage inflammatory protein-1 alpha (MIP-1 alpha) correlate with the extent of bone disease and survival in patients with multiple myeloma. Br J Haematol. 2003;123:106–109. doi: 10.1046/j.1365-2141.2003.04561.x. [DOI] [PubMed] [Google Scholar]
- Taichman RS, Reilly MJ, Matthew LS. Human osteoblast-like cells and osteosarcoma cell lines synthesize macrophage inhibitory protein 1α in response to interleukin 1β and tumour necrosis factor-α stimulation in vitro. Br J Haematol. 2000;108:275–283. doi: 10.1046/j.1365-2141.2000.01873.x. [DOI] [PubMed] [Google Scholar]
- Lichtenstein A, Berenson J, Norman D, Chang MP, Carlile A. Production of cytokines by bone marrow cells obtained from patients with multiple myeloma. Blood. 1989;74:1266–1273. [PubMed] [Google Scholar]
- Oba Y, Lee JW, Ehrlich LA, Chung HY, Jelinek DF, Callander NS, Horuk R, Choi SJ, Roodman GD. MIP-1α utilizes both CCR1 and CCR5 to induce osteoclast formation and increase adhesion of myeloma cells to marrow stromal cells. Exp Hematol. 2005;33:272–278. doi: 10.1016/j.exphem.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Menu E, De Leenheer E, De Raeve H, Coulton L, Imanishi T, Miyashita K, Van Valckenborgh E, Van Riet I, Van Camp B, Horuk R, et al. Role of CCR1 and CCR5 in homing and growth of multiple myeloma and in the development of osteolytic lesions: a study in the 5TMM model. Clin Exp Metastasis. 2006;23:291–300. doi: 10.1007/s10585-006-9038-6. [DOI] [PubMed] [Google Scholar]
- Okada Y, Tsukada J, Nakano K, Tonai S, Mine S, Tanaka Y. Macrophage inflammatory protein-1α induces hypercalcemia in adult T-cell leukemia. J Bone Miner Res. 2004;19:1105–1111. doi: 10.1359/JBMR.040314. [DOI] [PubMed] [Google Scholar]
- Tian E, Zhan F, Walker R, Rasmussen E, Ma Y, Barlogie B, Shaughnessy JD., Jr The role of the Wnt-signaling antagonist DKK1 in the development of osteolytic lesions in multiple myeloma. N Engl J Med. 2003;349:2483–2494. doi: 10.1056/NEJMoa030847. [DOI] [PubMed] [Google Scholar]
- Spencer GJ, Utting JC, Etheridge SL, Arnett TR, Genever PG. Wnt signalling in osteoblasts regulates expression of the receptor activator of NF-κB ligand and inhibits osteoclastogenesis in vitro. J Cell Sci. 2006;119:1283–1296. doi: 10.1242/jcs.02883. [DOI] [PubMed] [Google Scholar]
- Oyajobi BO, Gupta A, Flores A, Wideman C, Shipman C, Mundy GR, Garrett IR. Dkk1 modulates osteoclastogenesis and bone resorption: Implications for myeloma bone disease. J Bone Miner Res. 2005. p. S32.
- Yaccoby S, Ling W, Zhan F, Walker R, Barlogie B, Shaughnessy JD., Jr Antibody-based inhibition of DKK1 suppresses tumor-induced bone resorption and multiple myeloma growth in vivo. Blood. 2007;109:2106–2011. doi: 10.1182/blood-2006-09-047712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart AF. Hypercalcemia associated with cancer. N Engl J Med. 2005;352:373–379. doi: 10.1056/NEJMcp042806. [DOI] [PubMed] [Google Scholar]
- Body JJ. Bisphosphonates for malignancy-related bone disease: current status, future developments. Support Care Cancer. 2006;14:408–418. doi: 10.1007/s00520-005-0913-5. [DOI] [PubMed] [Google Scholar]
- Bell R. Efficacy of ibandronate in metastatic bone disease: review of clinical data. Oncologist. 2005;(Suppl 1):8–13. doi: 10.1634/theoncologist.10-90001-8. [DOI] [PubMed] [Google Scholar]
- Rosen LS, Gordon D, Antonio BS, Kaminski M, Howell A, Belch A, Mackey JA, Apffelstaedt J, Hussein M, Coleman RE, et al. Zoledronic acid versus pamidronate in the treatment of skeletal metastases in patients with breast cancer or osteolytic lesions of multiple myeloma: a phase III, double-blind, comparative trial. Cancer J. 2001;7:377–387. [PubMed] [Google Scholar]
- Morony S, Warmington K, Adamu S, Asuncion F, Geng Z, Grisanti M, Tan HL, Capparelli C, Starnes C, Weimann B, et al. The inhibition of RANKL causes greater suppression of bone resorption and hypercalcemia compared with bisphosphonates in two models of humoral hypercalcemia of malignancy. Endocrinology. 2005;146:3235–3243. doi: 10.1210/en.2004-1583. [DOI] [PubMed] [Google Scholar]
- Body JJ, Facon T, Coleman RE, Lipton A, Geurs F, Fan M, Holloway D, Peterson MC, Bekker PJ. A study of the biological receptor activator of nuclear factor-κB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res. 2006;12:1221–1228. doi: 10.1158/1078-0432.CCR-05-1933. [DOI] [PubMed] [Google Scholar]
- ClinicalTrials.gov http://www.clinicaltrials.gov
- Liang M, Mallari C, Rosser M, Ng HP, May K, Monahan S, Bauman JG, Islam I, Ghannam A, Buckman B, et al. Identification and characterization of a potent, selective and orally active antagonist of the CC chemokine receptor-1. J Biol Chem. 2000;275:19000–19008. doi: 10.1074/jbc.M001222200. [DOI] [PubMed] [Google Scholar]
- Nair SR, Pearson SB. Images in clinical medicine. Pathologic fracture and lytic lesions in multiple myeloma. N Engl J Med. 2004;351:1874. doi: 10.1056/NEJMicm031146. [DOI] [PubMed] [Google Scholar]