

Abstract
Establishment of skeletal metastasis involves bidirectional interactions between the tumor cell and the cellular elements in the bone microenvironment. A better understanding of the pathophysiology of bone metastasis will be critical in developing the means to prevent bone metastasis or inhibit its progression. The receptor activator of nuclear factor-κB (RANK)/RANK ligand pathway has emerged as the key pathway regulating osteolysis in skeletal metastasis. A number of candidate factors, including the Wnt (wingless int) proteins, endothelin-1, and bone morphogenetic proteins, have been implicated in the establishment of osteoblastic metastasis. The complex nature of tumor-bone microenvironment interactions and the presence of multiple pathways that lead to bone metastasis suggests that simultaneous targeting of these pathways in the metastatic cascade are required for effective treatment. This review discusses current understanding of the pathophysiologic mechanisms that underlie the establishment of bone metastasis and potential molecular therapeutic strategies for prevention and treatment of bone metastasis.
Introduction
Metastases from carcinomas are the most common malignant tumors involving bone. It is estimated that there will be 1,444,920 new cases of cancer and 559,650 cancer deaths in USA in 2007 [1]. Prostate, breast, and lung cancers are the most common malignancies in adults and are the most common tumors that metastasize to bone [1,2]. Moreover, bone metastasis affects more than 60% of advanced stage breast and prostate cancer patients [2]. Carcinomas of lung, kidney and thyroid, and melanomas are other common tumors that metastasize to bone [2].
Bone metastasis is associated with increased morbidity and portends a poor outcome, with decreased survival, in cancer patients [3,4]. Bone metastases are classified as osteolytic, osteoblastic, or mixed, based on their radiographic appearance [4]. Bone metastases from prostate cancer are predominantly osteoblastic, whereas metastatic lesions in bone from breast cancer can be osteoblastic, osteolytic, or mixed. Irrespective of the mechanisms that are involved in the generation of these radiographic phenotypes, the end result is a change in bone architecture, which predisposes the patient to a variety of skeletal complications [3,5].
Molecular mechanism of bone metastasis
Sir Stephen Paget enunciated the 'soil and seed hypothesis' more than a century ago, stating that certain tumor cells (seeds) will selectively colonize distant organs (soil) because of the presence of a favorable environment for their localization and growth [6]. Metastasis to bone is a complex multistep event, which involves a bidirectional interaction of the tumor cells with cellular elements in three different micro-environments [7]: the site of primary neoplasm, the circulation, and the bone microenvironment. The metastatic tumor cells must escape from the primary tumor into the circulation and reach the skeletal sites, where they establish themselves, proliferate, and then induce metastatic lesions [7]. The preferential skeletal localization of tumor cells is attributed to the biologic and molecular characteristics of tumor cells as well as the bone microenvironment [4,7-10]. The pathophysiology of bone metastasis is poorly understood. According to the traditional model of metastasis, the potential to metastasize resides in a small subset of tumor cells that have acquired this property through a set of mutations that occur during the later stages of tumor progression [11]. An emerging concept has recently challenged this existing model of metastasis by demonstrating that the potential to metastasize is encoded in the bulk of the tumor and is present early in tumor pathogenesis [11-14].
Recent work by Kang and coworkers [15] in a mouse intracardiac model of bone metastasis has demonstrated the presence of a tissue-specific metastatic phenotype that is associated with and promoted by a specific set of genes that pre-exist in the primary tumor. Furthermore, these distinctive bone metastasis gene signatures are different from the poor prognosis gene expression signatures [13,14]. Kang and coworkers hypothesized that the poor prognosis gene signature facilitates the emergence of metastatic cells in the primary tumor, but the specific set of genes associated with bone metastasis is responsible for the cellular activities necessary to form bone metastasis [13-15]. Over-expression of the bone metastasis specific gene set (namely the genes encoding C-X-C chemokine receptor [CXCR]4, IL-11, connective tissue growth factor, and matrix metalloproteinase [MMP]-1), along with the osteopontin gene, in various combinations considerably enhanced the metastatic potential of breast cancer cells to bone in this model. In addition, each of these genes, when expressed individually, failed to confer high skeletal tropism [15]. The bone metastasis specific genes encode mostly cell surface and secretory proteins, which participate in multiple steps involved in homing, invasion, angiogenesis, and proliferation of tumor cells in the bone microenvironment [15].
Smid and coworkers [16] analyzed 107 primary human breast tumors in order to identify genes associated with breast cancer metastasis to bone. These investigators also identified distinct poor prognosis and bone metastasis signatures in primary breast tumors. Again, ability to metastasize is distinct from the potential to home to bone and form a bone metastasis.
The data reported by Kang [15] and Smid [16] and their coworkers demonstrate that ability to form a bone metastasis is associated with a bone metastatic phenotype. The goal is therefore to develop specific therapies that can inhibit these genes in primary tumors and prevent or reduce metastasis to bone.
Bone as a unique environment for metastasis
Bone metastases are usually multifocal and have a predilection for the hematopoietic marrow sites in the proximal long bones and axial skeleton (vertebrae, pelvis, ribs, and cranium) [9]. Continuous and dynamic turnover of the bone matrix and bone marrow provides a fertile ground for tumor cells to utilize the vast available resources (cells, growth factors, cytokines, and receptors) for their homing and subsequent proliferation [17]. Both anatomic and molecular characteristics of bone make it a favorable site for metastasis [9]. Abundant sinusoids and sluggish blood flow in the metaphysis facilitate an intimate interaction between endothelium and tumor cells, which is necessary for their initial colonization in the bone marrow [9,18]. Moreover, it appears that a subset of bone marrow cells (vascular endothelial growth factor receptor-1 expressing hematopoietic progenitor cells and fibroblasts) may form a 'premetastatic niche' in response to the humoral factors secreted by the primary neoplasm. The cells comprising the premetastatic niche express cell surface ligands and receptors (integrin and fibronectin), which provide a permissive environment for the migrating tumor cells [19]. In addition, various growth factors and cytokines in the bone marrow such as endothelin (ET)-1, basic fibroblast growth factor, transforming growth factor (TGF)-β, IL-6, and IL-8 serve as paracrine regulators of the initial growth of metastatic tumor cells [20]. The interaction of receptor molecules in the bone marrow stroma (urokinase receptor, vascular cell adhesion molecule-1, and fibronectin) with the ligands that are over-expressed on the tumor cells (β1, α4β1 and α5β1 integrins, cadherin-11, connective tissue growth factor, and CXCR4) promotes colonization of circulating malignant cells in the bone marrow [9,21]. The extracellular matrix proteins (especially type I collagen, type IV collagen, vitronectin, fibronectin, osteopontin, osteocalcin, bone sialoprotein, osteonectin, and stromal cell derived factor-1) are chemotactic for tumor cells and promote colonization of circulating tumor cells in the bone marrow [9,22,23].
The bone matrix is a vast storehouse of latent growth factors such as insulin-like growth factor, TGF-β, bone morphogenetic protein (BMP), platelet-derived growth factor, and vascular endothelial growth factor. The release of these factors during bone remodeling may promote cell homing and appears to promote colonization and initial proliferation of tumor cells [24,25]. Release of these factors during the formation of both osteolytic and osteoblastic lesions stimulates a vicious cycle of tumor growth that leads to tumor cell proliferation and progression of bone lesions [26,27].
Mechanism of osteolytic and osteoblastic metastasis
Bone metastases are classified as osteolytic, osteoblastic, or mixed osteolytic and osteoblastic, based on their radiographic appearance [4]. These phenotypes are a reflection of the interactions between tumor cells and cellular elements (osteoclasts and osteoblasts) of the bone microenvironment. Previous studies conducted in our laboratory and others have demonstrated that human cancer cells that metastasize to bone are characterized by a distinct cytokine profile, which dictates the final phenotype of the skeletal lesion [28,29]. We demonstrated that human prostate cancer cells that produce osteolytic lesions over-expressed factors such as IL-1, receptor activator of nuclear factor-κB ligand (RANKL), and tumor necrosis factor (TNF)-α, which are involved in the stimulation of osteoclast differentiation and activation [28]. In addition, we also showed that human prostate cancer cell lines forming osteoblastic lesions produced factors such as BMP, osteoprotegerin (OPG), and TGF-β, which are associated with bone formation [28].
Osteolytic metastasis
The most common manifestation of bone metastasis is osteolysis [26,27]. Numerous in vivo studies in animals suggest the existence of a vicious cycle in the pathogenesis and progression of an osteolytic metastasis [4,26,27]. This complex vicious cycle includes reciprocal interactions between tumor cells, bone cells (osteoclasts and osteoblasts), and the bone matrix (Figure 1). The tumor cells secrete various soluble factors that promote osteoclast differentiation, proliferation, and activation, which leads to increased osteolysis (Table 1). Growth factors (TGF-β, insulin-like growth factor, basic fibroblast growth factor, and BMP) mobilized from bone following osteolysis support the growth and survival of the tumor cells [4,24,26,27]. In return, the growing tumor secretes more pro-osteolytic factors, which results in further osteolysis and perpetuation of the vicious cycle. A better understanding of the complex tumor cell-host cell interactions in the bone microenvironment, and of the autocrine and the paracrine effects of the secreted factors (tumor cells) and released factors (from bone matrix) will facilitate development of effective strategies to inhibit disease progression [26].
Figure 1.
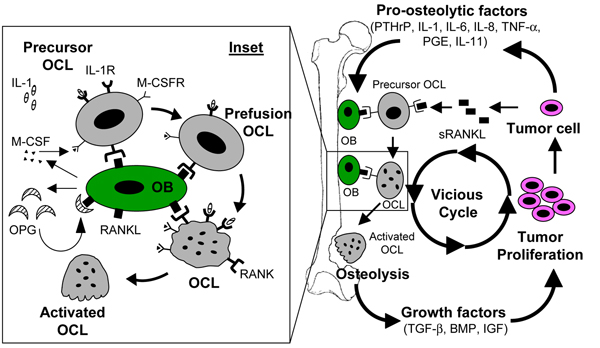
Vicious cycle in osteolytic bone metastasis. The pro-osteolytic factors secreted by the tumor cells (PTHrP, IL-1, IL-8, IL-11, soluble RANKL, TNF-α, and PGE) promote osteolysis by stimulating osteoclast formation and maturation. The growth factors secreted following osteolysis (BMP, IGF, and TGF-β) are stimulatory for tumor growth, which results in increased tumor burden and eventually more osteolysis. The inset delineates the regulation of osteoclast formation and activation. RANKL on the osteoblast/stromal cells interacts with the RANK on the osteoclast precursors in the presence of M-CSF to stimulate their differentiation into mature osteoclasts. An alternate pathway (RANKL independent) of osteoclast differentiation (mediated by IL-1 and its receptor IL-1R on the osteoclast) is also shown. BMP, bone morphogenetic protein; IGF, insulin-like growth factor; M-CSF, macrophage colony-stimulating factor; OB, osteoblast; OCL, osteoclast; PG, prostaglandin; PTHrP, parathyroid hormone related peptide; IL, interleukin; RANKL, receptor activator of nuclear factor-κB ligand; TGF, transforming growth factor; TNF, tumor necrosis factor.
Table 1.
Pro-osteolytic tumor-secreted factors and their described role in the pathogenesis of osteolytic metastasis
| Tumor-secreted factors | Role in the pathogenesis of osteolytic metastasis |
| PTHrP | Upregulates RANKL expression and decreases OPG expression [26,41] |
| Soluble RANKL | Stimulates osteoclastogenesis by binding directly to RANK [43] |
| IL-6 | Increases osteoclastogenesis via gp130 signal transduction pathway; enhances the effect of PTHrP [48] |
| IL-1 | Increases osteoclastogenesis (RANKL dependent and independent pathway); promotes osteoclast activation and survival [78,79] |
| TNF-α | Increases osteoclastogenesis and osteoclast activation (via gp130 signal transduction pathway as well as RANKL primed pathway) [80,81] |
| IL-8 | Increases osteoclastogenesis by direct stimulation of CXCR1 receptors on the osteoclast precursor [47] |
| IL-11 | Increases osteoclastogenesis via gp130 signal transduction pathway [48,82] |
| M-CSF | Upregulates RANKL expression on stromal cells; chemotactic role for attracting osteoclasts to resorptive sites and prolongs survival of the mature osteoclast by inhibiting apoptosis [83] |
| TGF-β | Inhibits osteoclast formation but can also directly stimulate osteoclast formation (in absence of RANKL) [49] |
| Prostaglandin | Upregulates RANKL expression and enhances the effect of soluble RANKL [26,84] |
| VEGF | Induces angiogenesis and promotes osteoclastogenesis [85] |
| MMPs | Assist osteoclast mediated bone resorption [86] |
CXCR, C-X-C chemokine receptor; IL, interleukin; M-CSF, macrophage colony-stimulating factor; MMP, matrix metalloproteinase; OPG, osteoprotegerin; PTHrP, parathyroid hormone-related peptide; RANKL, receptor activator of nuclear factor-κB ligand; TGF, transforming growth factor; TNF, tumor necrosis factor; VEGF, vascular endothelial growth factor.
The osteoclasts, which are derivatives of the pluripotent hematopoietic precursors in the marrow, are the primary cells involved in tumor-mediated osteolysis [30]. Osteoclast differentiation and maturation is the critical cellular process involved in the pathophysiology of osteolytic metastasis [31,32]. The OPG/receptor activator of nuclear factor-κB (RANK)/RANKL pathway has emerged as the key pathway regulating osteoclast formation and survival in physiologic and pathologic states, including skeletal metastases [33,34].
RANKL is a member of the TNF ligand superfamily and is expressed on numerous cell types, including osteoblast/stromal cells [35]. RANKL exists either as a membrane bound or a soluble isoform, and it is the principal ligand for RANK [36]. RANK is a TNF receptor that is expressed on osteoclasts and dendritic cells [37]. RANK-RANKL interaction in the presence of macrophage colony-stimulating factor activates multiple intracellular signaling cascades (nuclear factor-κB, p38 mitogen-activated protein kinase, cellular Src kinases, and Jun amino-terminal kinases) in the precursor osteoclasts [31,32,38]. Stimulation of these pathways eventually results in increased formation, maturation, and survival of osteoclasts [31,32]. OPG – the third member of the triad – is a secreted TNF receptor that acts as a soluble decoy receptor for RANKL and TNF-related apoptosis-inducing ligand (TRAIL) [39,40]. OPG is a negative regulator of the RANK/RANKL pathway and prevents RANK-RANKL interaction by sequestering RANKL. Hence, OPG decreases osteoclastogenesis and promotes osteoclast apoptosis [35,39,40].
Laboratory studies have demonstrated the integral involvement of the OPG/RANK/RANKL axis in the pathogenesis of osteolytic skeletal metastasis. The tumor cells secrete numerous cytokines and growth factors, including parathyroid hormone-related peptide (PTHrP), IL-1, IL-6, IL-8, IL-11, and TNF-α, which upregulate expression of RANKL on osteoblast/stromal cells (Table 1) [26,34]. PTHrP is one of the major mediators of breast cancer related hypercalcemia and osteolytic bone metastasis [41]. PTHrP contributes to osteolysis by upregulating expression of RANKL on osteoblasts [26,41]. Experiments in our laboratory and others have demonstrated increased secretion of soluble RANKL by human prostate cancer cells, forming osteolytic lesions [28,42,43]. A prior study in our laboratory using a mouse intratibial injection model of metastasis demonstrated that blocking the RANK-RANKL interaction by RANK-Fc (a recombinant soluble fusion protein consisting of extracellular domain of RANK coupled with the Fc domain of human IgG) limits the formation of osteolytic bone lesions [44]. In another study, conducted in the same animal model, exogenous RANK-Fc administration was effective at preventing osteolysis and reducing tumor volume in mixed osteolytic and osteoblastic bone lesions [45]. However, blocking the OPG/RANK/RANKL pathway with RANK-Fc had no effect on tumor cells in nonosseous sites; the decrease in tumor burden following inhibition of the tumor-mediated osteolysis is an indirect effect secondary to interruption of the vicious cycle of osteolytic metastasis [43-45].
Recently, we noted that simultaneous blocking of the osteolytic and osteoblastic pathways in a metastatic mixed lung cancer lesion of bone is superior to inhibition of either pathway alone [45]. Because many metastatic lesions in bone are considered mixed (although the lytic or the blastic phase may be predominant), therapeutic strategies that target both the osteolytic and osteoblastic components may be necessary to inhibit progression of a skeletal metastatic lesion effectively.
These studies clearly highlight the pivotal role of the OPG/RANKL axis in the pathogenesis of tumor-mediated osteolysis in a pure lytic as well as a mixed metastatic skeletal lesion. The ratio of RANKL to OPG has also been postulated as one of the factors that determines the final phenotype (osteolytic versus osteoblastic) of the skeletal metastasis [33,46,47]. Osteolysis is characterized by an increase in the ratio of RANKL to OPG; tumor-secreted factors that have been implicated in osteolysis increase this ratio by up-regulating the RANKL expression on osteoblasts/stromal cells or by downregulating OPG secretion [26,34,41].
Cytokines such as IL-6, TNF-α, IL-1, and IL-8 can stimulate osteoclastogenesis independent of RANKL [47,48]. Bendre and coworkers [47] demonstrated that IL-8 binds to CXCR1 present on osteoclast precursors and enhances osteoclast differentiation independent of RANKL. The contribution of these RANKL independent pathways to the osteoclastogenesis associated with osteolytic metastasis is not clear at present [47-49].
Osteoblastic metastasis
Osteoblasts are the key cells involved in forming the woven bone seen in osteoblastic metastasis [4,5,27]. Tumor cells forming osteoblastic metastases secrete numerous pro-osteoblastic factors (cytokines, transcription factors, and growth factors) that switch normal bone remodeling toward a predominant bone-forming state (Table 2). These factors stimulate various steps that are involved in the differentiation, proliferation, and maturation of osteoblasts or inhibition of osteoclast pathways [4,27] (Table 2). Activated osteoblasts secrete numerous growth factors during the formation of woven bone (TGF-β, BMP, and vascular cell endothelial growth factor), which are used by tumor cells to potentiate their survival and growth. The growing tumor secretes more pro-osteoblastic factors, thereby amplifying the formation of woven bone and perpetuating the vicious cycle [4,50].
Table 2.
Pro-osteoblastic tumor-secreted factors and their described role in the establishment of osteoblastic metastasis
| Tumor-secreted factors | Probable mechanisms underlying new bone formation in osteoblastic metastases |
| Wnt | Stimulates differentiation and activation, and promotes survival and activity of osteoblasts; inhibits osteoclast activity [52] |
| ET-1 | Stimulates proliferation of osteoblasts, promotes mineralization, inhibits osteoclast motility, and potentiates the pro-osteogenic effects of other growth factors [59,60] |
| BMP | Stimulates osteoblast proliferation, activity, and survival; increases OPG production [62-66] |
| IGF-1, IGF-2 | Stimulate osteoblast proliferation and survival [87] |
| IL-6 | Regulates osteoblast function [88] |
| OPG | Inhibits osteoclastic activity by binding to RANKL [89] |
| TGF-β | Stimulates osteoblast proliferation [90] |
| Urokinase (uPA) | Stimulates osteoblast proliferation [91] |
| PDGF-BB | Promotes angiogenesis and osteoblast activity [92] |
| FGF-1, FGF-2, and FGF-8 | Promote differentiation and proliferation of osteoblasts [93] |
| PSA | Inactivation of PTHrP and stimulation of latent growth factors (TGF-β) [94] |
| VEGF | Promotes osteoblast differentiation [95] |
| MDA-BF-1 | Stimulates osteoblast formation and activation [96] |
BMP, bone morphogenetic protein; ET, endothelin; FGF, fibroblast growth factor; IGF, insulin-like growth factor; OPG, osteoprotegerin; PDGF, platelet-derived growth factor; TGF, transforming growth factor; PSA, prostate-specific antigen; PTHrP, parathyroid hormone-related peptide; uPA, urokinase plasminogen activator; Wnt, wingless int; VEGF, vascular endothelial growth factor.
In 1958, Roland [51] introduced the theory that every primary or metastatic cancer in bone (including osteoblastic prostate cancers) begins with osteolysis. Whether initial osteolysis is required for development of osteoblastic metastasis is unclear [28,43,44,52]. Bisphosphonates and other agents that inhibit osteoclastic activity have failed to prevent formation of osteoblastic lesions in murine models of metastasis, which is indirect evidence that osteoblastic lesions can form in the absence of initial osteoclastic activity [28,43,53]. In a mouse tibial injection model using human prostate cancer cells that produce osteoblastic metastasis, Lee and coworkers [54] demonstrated that zoledronate failed to halt the formation of osteoblastic lesions, indicating that osteoclasts may not be essential in the establishment of osteoblastic metastasis. Using a similar animal model, Whang and coworkers [44] administered RANK-Fc to block osteolysis in an attempt to prevent the establishment of osteoblastic lesions by human prostate cancer cells. RANK-Fc treatment failed to prevent or delay the establishment of osteoblastic lesions, but overall tumor growth was limited. These findings suggest that blocking osteolytic activity is important even when treating osteoblastic lesions because it slows the release of growth factors from bone matrix that may enhance tumor proliferation.
The Wnt (wingless int) pathway, the ET axis, and the BMP pathway have emerged as key regulators of the establishment of osteoblastic skeletal metastasis [25,52,55]. Wnt proteins are soluble glycoproteins that promote embryonic and postnatal bone formation [56]. These proteins bind to a membrane receptor complex comprised of frizzled (FZD) G-protein-coupled receptor and a low-density lipoprotein receptor-related protein [56]. The formation of this ligand-receptor complex initiates a number of intracellular signaling cascades that modulate differentiation, survival, and activity of the osteoblasts. Prostate cancer cells forming osteoblastic and mixed osteoblastic and osteolytic metastases express a variety of Wnt proteins [52,57]. Hall and coworkers [52] recently explored the roles played by the autocrine and paracrine effects of Wnt proteins in the establishment of osteoblastic metastatic prostate cancer. Limiting the activity of Wnt proteins by their natural antagonist Dickkopf-1 decreased the osteoblastic component of mixed osteolytic and osteoblastic bone lesions produced by human prostate cancer cells in an intratibial injection model [52].
Nelson and coworkers [55] first hypothesized that there is an association between ET-1 and osteoblastic metastasis in patients with advanced prostate cancer. ET-1 promotes osteogenic differentiation, stimulates bone matrix formation, and inhibits osteoclast formation and motility [58]. The paracrine effects of ET-1 in the bone microenvironment are predominantly mediated by ET receptor subtype A (ETA) receptors [55,58-60]. Yin and coworkers [58] demonstrated that both human prostate and breast cancer cell lines that form osteoblastic lesions in the bone produce ET-1. Blocking the ET axis by exogenous administration of an ETA receptor antagonist decreased the number of osteoblastic lesions formed in nude mice following intracardiac injection of human breast cancer cells.
Considering the integral role played by BMP in skeletal development and postnatal bone repair, BMP was hypothesized to be involved in the new bone formation observed in bone metastasis [61,62]. The BMPs, especially BMP-2, BMP-6 and BMP-7, are known to play a critical role in formation of osteoblastic bone metastasis by virtue of their osteoinductive and cell type specific proliferative activity [25,45,61-65]. Studies conducted in our laboratory and others have shown that human prostate, lung, or breast cancer cells forming osteoblastic bone metastasis secrete a variety of BMPs and express BMP receptors [25,45,64-66]. In addition, BMPs stimulate invasion, migration, and proliferation of human prostate and lung cancer cells, which suggests participation of BMPs in an autocrine loop to regulate tumor growth [25,45]. Finally, blocking the effect of BMPs with noggin (an endogenous antagonist of BMP) leads to a decrease in the formation of osteoblastic lesions in a mouse intratibial injection model [45].
Although the animal models that are currently available provide useful data, they do not truly simulate metastatic disease. Transgenic mouse models and syngeneic models of breast and prostate cancer exhibit a low incidence of metastasis to bone and lack reproducibility [67]. The injection of tumor cells into a metastatic site (long bones) does not allow one to evaluate the factors associated with tumor cell migration, invasion, and preferential localization to a particular metastatic site. Intracardiac injection of tumor cells does not truly simulate metastasis from prostate, breast, or lung tumors because it bypasses the early steps in the metastatic process. In addition, it may take months for lesions to form, limiting the feasibility of these experiments. It is essential to recognize these limitations when using these animal data to develop a therapeutic strategy in humans [67].
Therapeutic implications and molecular treatment strategies
The ultimate goal of treating metastatic bone disease is either to prevent a bone lesion from developing or to limit the progression of an established bone metastasis. At present our understanding of the development of bone metastasis is limited. Therefore, the currently available therapies (bisphosphonates, radiotherapy, radiopharmaceuticals, and surgery) for bone metastasis focus on symptomatic management and limiting the progression of established metastatic disease [2,4]. Considering the complex nature of the tumor-bone microenvironment interactions and the existence of multiple pathways that are involved in the development of bone metastasis, it is plausible that simultaneous targeting of multiple steps in metastasis formation will be superior to inhibiting one specific target [45,68].
Identification of pathways and molecular checkpoints in the pathogenesis of skeletal metastasis has led to discovery of novel molecular targets for therapeutic intervention (Table 3). Following their success in preclinical animal studies, most of these agents are being tested in clinical trials to assess their safety profile and efficacy in debulking established bone metastases (Table 3). It may be more appropriate to develop combination therapeutic strategies to prevent metastasis rather than attempting to limit progression of established disease. Patients with established metastatic disease may benefit from a combination of agents that not only affect bone turnover but also inhibit or kill tumor cells. For example, direct injection of agents into bony metastatic sites to kill tumor cells and limit bone destruction may confer relief from pain and improve quality of life, as well as limiting the progression of metastatic disease. The availability of computed tomography guided injection techniques would permit precise targeting of anatomic regions that are difficult to reach without extensive surgical procedures, such as pelvis, spine, and proximal femur.
Table 3.
Therapeutic strategies for targeting molecules/pathways involved in the pathogenesis of bone metastasis
| Therapeutic strategy | Target and rationale for therapy |
| Recombinant OPG construct | Blocks RANKL and TRAIL pathway [69] |
| Soluble RANK-Fc | Blocks the effect of RANKL without any effect on the TRAIL pathway [43-45] |
| Human monoclonal antibody to RANKL (denosumab) | Blocks the effect of RANKL without any effect on the TRAIL pathway [72] |
| Oligonucleotides to NF-κB, P2X7 receptor antagonists | Block the effect of NF-κB activation [97] |
| Humanized anti-PTHrP monoclonal antibody | Inhibits PTHrP-mediated osteolysis via the RANKL pathway [41] |
| PDGFR antagonist (ST1571, Imatinib mesylate/Gleevec) | Inhibits tumor growth and angiogenesis by inhibiting PDGFR tyrosine kinase signaling [98] |
| ETA receptor inhibitor (atrasentan) | Blocks ET-1 mediated bone formation in prostate skeletal metastasis [55,58,59] |
| EMD121974 (cilengitide) | Inhibits tumor-ECM interactions involved in tumor metastasis, growth, and angiogenesis [76] |
| MMP inhibitors | Inhibit MMP mediated tumor growth, metastasis, and angiogenesis [99] |
ECM, extracellular matrix; ET, endothelin; ETA, endothelin receptor subtype A; MMP, matrix metalloproteinase; NF-κB, nuclear factor-κB; OPG, osteoprotegerin; PDGF, platelet-derived growth factor; PTHrP, parathyroid hormone related peptide; RANKL, receptor activator of nuclear factor-κB ligand; TRAIL, TNF-related apoptosis-inducing ligand.
The OPG/RANK/RANKL pathway offers multiple molecular checkpoints for therapeutic targeting in osteolytic metastases [34]. Abrogation of this axis has demonstrated therapeutic efficacy in restricting tumor-mediated osteolysis in vitro as well as in animal models of metastasis [31,34,43-45]. Recombinant OPG construct was one of the first RANKL inhibitors to be used in clinical trials for osteolytic metastatic disease [69]. In a randomized, double-blind, phase I clinical trial, a single subcutaneous dose of recombinant OPG construct was well tolerated and effective in suppressing bone resorption in multiple myeloma and breast cancer patients with established skeletal metastasis [69]. However, the ability of OPG to block the TRAIL apoptosis pathway in cancer cells was noted, and there were concerns that this could lead to a flare of tumor growth [70]. Anti-RANKL antibodies (for instance, denosumab) represent a new class of RANKL inhibitors that are characterized by high affinity and specificity for RANKL [71,72]. In addition, they do not inhibit the TRAIL apoptosis pathway [71]. In a recent randomized, double-blind, double-dummy, active-controlled multicenter phase I clinical study, Body and coworkers [72] demonstrated the effectiveness of denosumab. A single subcutaneous dose of denosumab (AMG 162) given to patients with multiple myeloma or bone metastasis from breast cancer yielded a dose dependent and sustained reduction in bone resorption.
Atrasentan (an ETA receptor antagonist) exhibited efficacy in treatment of osteoblastic lesions in preclinical animal models of prostate cancer bone metastasis and in phase I and II clinical trials [55,59]. However, the results of recent phase III clinical trials of atrasentan in men with metastatic hormone-refractory prostate cancer were inconclusive [73,74]. In a randomized, double-blind, placebo-controlled, phase III clinical trial, an intent-to-treat analysis failed to demonstrate a significant delay in time to clinical and radiographic progression with atrasentan as compared with placebo in men with metastatic hormone-refractory prostate cancer [59,73,74].
Advances in structural deciphering of biomolecules that are involved in the pathogenesis of bone metastasis, such as OPG, integrins, and MMP, have allowed scientists to design molecules that mimic these critical targets [75]. These artificial, designed molecules can be used to block or increase the activity of a particular therapeutic target, and have significant potential in the treatment of bone metastasis [75,76]. The use of RGD-based peptidomimetics (which contain sequence of arginine-glycine-aspartic acid) and MMP inhibitor peptidomimetics in animal models of bone metastasis is associated with smaller size and decreased number of osteolytic lesions [76].
Recent discovery of gene signatures and organ-specific metastasis genes that are predictive of metastatic potential and organ of metastasis, respectively, is a promising step in designing strategies to prevent and treat bone metastasis [15,16]. Further development and validation of bone metastasis specific genes would theoretically allow stratification of cancer patients into good or poor metastatic prognosis and permit patient-tailored therapeutic strategies. However, these types of therapies may not be feasible from an economic standpoint [12,77].
Conclusion
Prevention or limitation of established bone metastasis would significantly improve the quality of lives of patients diagnosed with advanced lung, prostate, or breast cancer. This is especially true for prostate cancer patients, in whom progression of bone metastasis occurs over many years. We must focus our research efforts on elucidating the factors that allow the tumor cells to escape into the circulation, deposit in the bone, and then proliferate to form skeletal lesions. Such work will enable investigators to develop effective treatment modalities to prevent as well as treat bone metastasis.
Abbreviations
BMP = bone morphogenetic protein; CXCR = C-X-C chemokine receptor; ET = endothelin; ETA = endothelin receptor subtype A; IL = interleukin; MMP = matrix metalloproteinase; OPG = osteoprotegerin; PTHrP = parathyroid hormone-related peptide; RANK = receptor activator of nuclear factor-κB; RANKL = receptor activator of nuclear factor-κB ligand; TGF = transforming growth factor; TNF = tumor necrosis factor; TRAIL = TNF-related apoptosis-inducing ligand.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
The authors thank Christine O Kang, Staff Research Associate, University of California of Los Angeles, for developing the figures, list of references, and abbreviations, and her useful suggestions in the drafting, revision, and preparation of the review content.
This article is published as part of Arthritis Research & Therapy Volume 9 Supplement 1, 2007: Basic science, rationale, background and future of denosumab: a RANK ligand inhibitor. The full contents of the supplement are available online at http://arthritis-research.com/supplements/9/S1.
Publication of the supplement has been supported by an unrestricted grant from Amgen Inc.
References
- American Cancer Society . Cancer Facts and Figures, 2007. Atlanta: American Cancer Society; 2007. http://www.cancer.org/downloads/STT/CAFF2007PWSecured.pdf [Google Scholar]
- Coleman RE. Metastatic bone disease: clinical features, pathophysiology and treatment strategies. Cancer Treat Rev. 2001;27:165–176. doi: 10.1053/ctrv.2000.0210. [DOI] [PubMed] [Google Scholar]
- Coleman RE. Skeletal complications of malignancy. Cancer. 1997;(Suppl):1588–1594. doi: 10.1002/(SICI)1097-0142(19971015)80:8+<1588::AID-CNCR9>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Roodman GD. Mechanisms of bone metastasis. N Engl J Med. 2004;350:1655–1664. doi: 10.1056/NEJMra030831. [DOI] [PubMed] [Google Scholar]
- Mundy GR. Metastasis to bone: causes, consequences and therapeutic opportunities. Nat Rev Cancer. 2002;2:584–593. doi: 10.1038/nrc867. [DOI] [PubMed] [Google Scholar]
- Paget S. The distribution of secondary growths in cancer of the breast. 1889. Cancer Metastasis Rev. 1989;8:98–101. [PubMed] [Google Scholar]
- Steeg PS. Tumor metastasis: mechanistic insights and clinical challenges. Nat Med. 2006;12:895–904. doi: 10.1038/nm1469. [DOI] [PubMed] [Google Scholar]
- Hoon DS, Kitago M, Kim J, Mori T, Piris A, Szyfelbein K, Mihm MC, Jr, Nathanson SD, Padera TP, Chambers AF, et al. Molecular mechanisms of metastasis. Cancer Metastasis Rev. 2006;25:203–220. doi: 10.1007/s10555-006-8500-x. [DOI] [PubMed] [Google Scholar]
- Mastro AM, Gay CV, Welch DR. The skeleton as a unique environment for breast cancer cells. Clin Exp Metastasis. 2003;20:275–284. doi: 10.1023/A:1022995403081. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Bernards R, Weinberg RA. A progression puzzle. Nature. 2002;418:823. doi: 10.1038/418823a. [DOI] [PubMed] [Google Scholar]
- Ramaswamy S, Ross KN, Lander ES, Golub TR. A molecular signature of metastasis in primary solid tumors. Nat Genet. 2003;33:49–54. doi: 10.1038/ng1060. [DOI] [PubMed] [Google Scholar]
- Kang Y, Siegel PM, Shu W, Drobnjak M, Kakonen SM, Cordon-Cardo C, Guise TA, Massague J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell. 2003;3:537–549. doi: 10.1016/S1535-6108(03)00132-6. [DOI] [PubMed] [Google Scholar]
- Smid M, Wang Y, Klijn JG, Sieuwerts AM, Zhang Y, Atkins D, Martens JW, Foekens JA. Genes associated with breast cancer metastatic to bone. J Clin Oncol. 2006;24:2261–2267. doi: 10.1200/JCO.2005.03.8802. [DOI] [PubMed] [Google Scholar]
- Schneider A, Kalikin LM, Mattos AC, Keller ET, Allen MJ, Pienta KJ, McCauley LK. Bone turnover mediates preferential localization of prostate cancer in the skeleton. Endocrinology. 2005;146:1727–1736. doi: 10.1210/en.2004-1211. [DOI] [PubMed] [Google Scholar]
- Phadke PA, Mercer RR, Harms JF, Jia Y, Frost AR, Jewell JL, Bussard KM, Nelson S, Moore C, Kappes JC, et al. Kinetics of metastatic breast cancer cell trafficking in bone. Clin Cancer Res. 2006;12:1431–1440. doi: 10.1158/1078-0432.CCR-05-1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan RN, Riba RD, Zacharoulis S, Bramley AH, Vincent L, Costa C, MacDonald DD, Jin DK, Shido K, Kerns SA, et al. VEGFR1-positive haematopoietic bone marrow progenitors initiate the premetastatic niche. Nature. 2005;438:820–827. doi: 10.1038/nature04186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pirtskhalaishvili G, Nelson JB. Endothelium-derived factors as paracrine mediators of prostate cancer progression. Prostate. 2000;44:77–87. doi: 10.1002/1097-0045(20000615)44:1<77::AID-PROS10>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Hiraga T. Crosstalk between cancer cells and bone microenvironment in bone metastasis. Biochem Biophys Res Commun. 2005;328:679–687. doi: 10.1016/j.bbrc.2004.11.070. [DOI] [PubMed] [Google Scholar]
- Zhang JH, Wang J, Tang J, Barnett B, Dickson J, Hahsimoto N, Williams P, Ma W, Zheng W, Yoneda T, et al. Bone sialoprotein promotes bone metastasis of a non-bone-seeking clone of human breast cancer cells. Anticancer Res. 2004;24:1361–1368. [PubMed] [Google Scholar]
- Stewart DA, Cooper CR, Sikes RA. Changes in extracellular matrix (ECM) and ECM-associated proteins in the metastatic progression of prostate cancer. Reprod Biol Endocrinol. 2004;2:2. doi: 10.1186/1477-7827-2-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hauschka PV, Mavrakos AE, Iafrati MD, Doleman SE, Klagsbrun M. Growth factors in bone matrix. Isolation of multiple types by affinity chromatography on heparin-sepharose. J Biol Chem. 1986;261:12665–12674. [PubMed] [Google Scholar]
- Feeley BT, Gamradt SC, Hsu WK, Liu N, Krenek L, Robbins P, Huard J, Lieberman JR. Influence of BMPs on the formation of osteoblastic lesions in metastatic prostate cancer. J Bone Miner Res. 2005;20:2189–2199. doi: 10.1359/JBMR.050802. [DOI] [PubMed] [Google Scholar]
- Guise TA, Kozlow WM, Heras-Herzig A, Padalecki SS, Yin JJ, Chirgwin JM. Molecular mechanisms of breast cancer metastases to bone. Clin Breast Cancer. 2005. pp. S46–S53. [DOI] [PubMed]
- Yin JJ, Pollock CB, Kelly K. Mechanisms of cancer metastasis to the bone. Cell Res. 2005;15:57–62. doi: 10.1038/sj.cr.7290266. [DOI] [PubMed] [Google Scholar]
- Lee Y, Schwarz E, Davies M, Jo M, Gates J, Wu J, Zhang X, Lieberman JR. Differences in the cytokine profiles associated with prostate cancer cell induced osteoblastic and osteolytic lesions in bone. J Orthop Res. 2003;21:62–72. doi: 10.1016/S0736-0266(02)00095-5. [DOI] [PubMed] [Google Scholar]
- Pederson L, Winding B, Foged NT, Spelsberg TC, Oursler MJ. Identification of breast cancer cell line-derived paracrine factors that stimulate osteoclast activity. Cancer Res. 1999;59:5849–5855. [PubMed] [Google Scholar]
- Shimamura T, Amizuka N, Li M, Freitas PH, White JH, Henderson JE, Shingaki S, Nakajima T, Ozawa H. Histological observations on the microenvironment of osteolytic bone metastasis by breast carcinoma cell line. Biomed Res. 2005;26:159–172. doi: 10.2220/biomedres.26.159. [DOI] [PubMed] [Google Scholar]
- Roodman GD. Regulation of osteoclast differentiation. Ann N Y Acad Sci. 2006;1068:100–109. doi: 10.1196/annals.1346.013. [DOI] [PubMed] [Google Scholar]
- Boyle WJ, Simonet WS, Lacey DL. Osteoclast differentiation and activation. Nature. 2003;423:337–342. doi: 10.1038/nature01658. [DOI] [PubMed] [Google Scholar]
- Hofbauer LC, Neubauer A, Heufelder AE. Receptor activator of nuclear factor-kappaB ligand and osteoprotegerin: potential implications for the pathogenesis and treatment of malignant bone diseases. Cancer. 2001;92:460–470. doi: 10.1002/1097-0142(20010801)92:3<460::AID-CNCR1344>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wittrant Y, Theoleyre S, Chipoy C, Padrines M, Blanchard F, Heymann D, Redini F. RANKL/RANK/OPG: new therapeutic targets in bone tumours and associated osteolysis. Biochim Biophys Acta. 2004;1704:49–57. doi: 10.1016/j.bbcan.2004.05.002. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/S0092-8674(00)81569-X. [DOI] [PubMed] [Google Scholar]
- Ikeda T, Kasai M, Utsuyama M, Hirokawa K. Determination of three isoforms of the receptor activator of nuclear factor-kappaB ligand and their differential expression in bone and thymus. Endocrinology. 2001;142:1419–1426. doi: 10.1210/en.142.4.1419. [DOI] [PubMed] [Google Scholar]
- Anderson DM, Maraskovsky E, Billingsley WL, Dougall WC, Tometsko ME, Roux ER, Teepe MC, DuBose RF, Cosman D, Galibert L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature. 1997;390:175–179. doi: 10.1038/36593. [DOI] [PubMed] [Google Scholar]
- Boyce BF, Yamashita T, Yao Z, Zhang Q, Li F, Xing L. Roles for NF-kappaB and c-Fos in osteoclasts. J Bone Miner Metab. 2005. pp. 11–15. [DOI] [PubMed]
- Simonet WS, Lacey DL, Dunstan CR, Kelley M, Chang MS, Luthy R, Nguyen HQ, Wooden S, Bennett L, Boone T, et al. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 1997;89:309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Yasuda H, Shima N, Nakagawa N, Yamaguchi K, Kinosaki M, Mochizuki S, Tomoyasu A, Yano K, Goto M, Murakami A, et al. Osteoclast differentiation factor is a ligand for osteoprote-gerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc Natl Acad Sci USA. 1998;95:3597–3602. doi: 10.1073/pnas.95.7.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guise TA, Yin JJ, Taylor SD, Kumagai Y, Dallas M, Boyce BF, Yoneda T, Mundy GR. Evidence for a causal role of parathyroid hormone-related protein in the pathogenesis of human breast cancer-mediated osteolysis. J Clin Invest. 1996;98:1544–1549. doi: 10.1172/JCI118947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Cheng YY, Chow LT, Zheng MH, Kumta SM. Tumour cells produce receptor activator of NF-kappaB ligand (RANKL) in skeletal metastases. J Clin Pathol. 2002;55:877–878. doi: 10.1136/jcp.55.11.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Dai J, Yao Z, Lu Y, Dougall W, Keller ET. Soluble receptor activator of nuclear factor kappaB Fc diminishes prostate cancer progression in bone. Cancer Res. 2003;63:7883–7890. [PubMed] [Google Scholar]
- Whang PG, Schwarz EM, Gamradt SC, Dougall WC, Lieberman JR. The effects of RANK blockade and osteoclast depletion in a model of pure osteoblastic prostate cancer metastasis in bone. J Orthop Res. 2005;23:1475–1483. doi: 10.1016/j.orthres.2005.05.004.1100230634. [DOI] [PubMed] [Google Scholar]
- Feeley BT, Liu NQ, Conduah AH, Krenek L, Roth K, Dougall WC, Huard J, Dubinett S, Lieberman JR. Mixed metastatic lung cancer lesions in bone are inhibited by noggin overexpression and RANK:Fc administration. J Bone Miner Res. 2006;21:1571–1580. doi: 10.1359/jbmr.060706. [DOI] [PubMed] [Google Scholar]
- Grimaud E, Soubigou L, Couillaud S, Coipeau P, Moreau A, Passuti N, Gouin F, Redini F, Heymann D. Receptor activator of nuclear factor kappaB ligand (RANKL)/osteoprotegerin (OPG) ratio is increased in severe osteolysis. Am J Pathol. 2003;163:2021–2031. doi: 10.1016/s0002-9440(10)63560-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendre MS, Margulies AG, Walser B, Akel NS, Bhattacharrya S, Skinner RA, Swain F, Ramani V, Mohammad KS, Wessner LL, et al. Tumor-derived interleukin-8 stimulates osteolysis independent of the receptor activator of nuclear factor-kappaB ligand pathway. Cancer Res. 2005;65:11001–11009. doi: 10.1158/0008-5472.CAN-05-2630. [DOI] [PubMed] [Google Scholar]
- Kudo O, Sabokbar A, Pocock A, Itonaga I, Fujikawa Y, Athanasou NA. Interleukin-6 and interleukin-11 support human osteoclast formation by a RANKL-independent mechanism. Bone. 2003;32:1–7. doi: 10.1016/S8756-3282(02)00915-8. [DOI] [PubMed] [Google Scholar]
- Itonaga I, Sabokbar A, Sun SG, Kudo O, Danks L, Ferguson D, Fujikawa Y, Athanasou NA. Transforming growth factor-beta induces osteoclast formation in the absence of RANKL. Bone. 2004;34:57–64. doi: 10.1016/j.bone.2003.08.008. [DOI] [PubMed] [Google Scholar]
- Loberg RD, Logothetis CJ, Keller ET, Pienta KJ. Pathogenesis and treatment of prostate cancer bone metastases: targeting the lethal phenotype. J Clin Oncol. 2005;23:8232–8241. doi: 10.1200/JCO.2005.03.0841. [DOI] [PubMed] [Google Scholar]
- Roland SI. Calcium studies in ten cases of osteoblastic prostatic metastasis. J Urol. 1958;79:339–342. doi: 10.1016/S0022-5347(17)66278-5. [DOI] [PubMed] [Google Scholar]
- Hall CL, Kang S, MacDougald OA, Keller ET. Role of Wnts in prostate cancer bone metastases. J Cell Biochem. 2006;97:661–672. doi: 10.1002/jcb.20735. [DOI] [PubMed] [Google Scholar]
- Kiefer JA, Vessella RL, Quinn JE, Odman AM, Zhang J, Keller ET, Kostenuik PJ, Dunstan CR, Corey E. The effect of osteoprote-gerin administration on the intra-tibial growth of the osteoblastic LuCaP 23.1 prostate cancer xenograft. Clin Exp Metastasis. 2004;21:381–387. doi: 10.1007/s10585-004-2869-0. [DOI] [PubMed] [Google Scholar]
- Lee YP, Schwarz EM, Davies M, Jo M, Gates J, Zhang X, Wu J, Lieberman JR. Use of zoledronate to treat osteoblastic versus osteolytic lesions in a severe-combined-immunodeficient mouse model. Cancer Res. 2002;62:5564–5570. [PubMed] [Google Scholar]
- Nelson JB, Nguyen SH, Wu-Wong JR, Opgenorth TJ, Dixon DB, Chung LW, Inoue N. New bone formation in an osteoblastic tumor model is increased by endothelin-1 overexpression and decreased by endothelin A receptor blockade. Urology. 1999;53:1063–1069. doi: 10.1016/S0090-4295(98)00658-X. [DOI] [PubMed] [Google Scholar]
- Bodine PV, Komm BS. Wnt signaling and osteoblastogenesis. Rev Endocr Metab Disord. 2006;7:33–39. doi: 10.1007/s11154-006-9002-4. [DOI] [PubMed] [Google Scholar]
- Chen G, Shukeir N, Potti A, Sircar K, Aprikian A, Goltzman D, Rabbani SA. Up-regulation of Wnt-1 and beta-catenin production in patients with advanced metastatic prostate carcinoma: potential pathogenetic and prognostic implications. Cancer. 2004;101:1345–1356. doi: 10.1002/cncr.20518. [DOI] [PubMed] [Google Scholar]
- Yin JJ, Mohammad KS, Kakonen SM, Harris S, Wu-Wong JR, Wessale JL, Padley RJ, Garrett IR, Chirgwin JM, Guise TA. A causal role for endothelin-1 in the pathogenesis of osteoblastic bone metastases. Proc Natl Acad Sci USA. 2003;100:10954–10959. doi: 10.1073/pnas.1830978100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JB. Endothelin receptor antagonists. World J Urol. 2005;23:19–27. doi: 10.1007/s00345-004-0478-9. [DOI] [PubMed] [Google Scholar]
- Stern PH, Tatrai A, Semler DE, Lee SK, Lakatos P, Strieleman PJ, Tarjan G, Sanders JL. Endothelin receptors, second messengers, and actions in bone. J Nutr. 1995. pp. 2028S–2032S. [DOI] [PubMed]
- Lieberman JR, Daluiski A, Einhorn TA. The role of growth factors in the repair of bone. Biology and clinical applications. J Bone Joint Surg Am. 2002;84-A:1032–1044. doi: 10.2106/00004623-200206000-00022. [DOI] [PubMed] [Google Scholar]
- Bentley H, Hamdy FC, Hart KA, Seid JM, Williams JL, Johnstone D, Russell RG. Expression of bone morphogenetic proteins in human prostatic adenocarcinoma and benign prostatic hyperplasia. Br J Cancer. 1992;66:1159–1163. doi: 10.1038/bjc.1992.427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker KD, Corey E, Brown LG, Vessella RL. Bone morphogenetic protein signaling in prostate cancer cell lines. J Cell Biochem. 2004;91:151–160. doi: 10.1002/jcb.10679. [DOI] [PubMed] [Google Scholar]
- Masuda H, Fukabori Y, Nakano K, Takezawa Y, T CS, Yamanaka H. Increased expression of bone morphogenetic protein-7 in bone metastatic prostate cancer. Prostate. 2003;54:268–274. doi: 10.1002/pros.10193. [DOI] [PubMed] [Google Scholar]
- Bobinac D, Maric I, Zoricic S, Spanjol J, Dordevic G, Mustac E, Fuckar Z. Expression of bone morphogenetic proteins in human metastatic prostate and breast cancer. Croat Med J. 2005;46:389–396. [PubMed] [Google Scholar]
- Dai J, Keller J, Zhang J, Lu Y, Yao Z, Keller ET. Bone morphogenetic protein-6 promotes osteoblastic prostate cancer bone metastases through a dual mechanism. Cancer Res. 2005;65:8274–8285. doi: 10.1158/0008-5472.CAN-05-1891. [DOI] [PubMed] [Google Scholar]
- Rosol TJ, Tannehill-Gregg SH, LeRoy BE, Mandl S, Contag CH. Animal models of bone metastasis. Cancer. 2003;(Suppl):748–757. doi: 10.1002/cncr.11150. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Sasaki A, Dunstan C, Williams PJ, Bauss F, De Clerck YA, Mundy GR. Inhibition of osteolytic bone metastasis of breast cancer by combined treatment with the bisphosphonate ibandronate and tissue inhibitor of the matrix metalloproteinase-2. J Clin Invest. 1997;99:2509–2517. doi: 10.1172/JCI119435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Body JJ, Greipp P, Coleman RE, Facon T, Geurs F, Fermand JP, Harousseau JL, Lipton A, Mariette X, Williams CD, et al. A phase I study of AMGN-0007 a recombinant osteoprotegerin construct, in patients with multiple myeloma or breast carcinoma related bone metastases. Cancer. 2003;(Suppl):887–892. doi: 10.1002/cncr.11138. [DOI] [PubMed] [Google Scholar]
- Neville-Webbe HL, Cross NA, Eaton CL, Nyambo R, Evans CA, Coleman RE, Holen I. Osteoprotegerin (OPG) produced by bone marrow stromal cells protects breast cancer cells from TRAIL-induced apoptosis. Breast Cancer Res Treat. 2004;86:269–279. doi: 10.1023/B:BREA.0000036900.48763.b3. [DOI] [PubMed] [Google Scholar]
- Abrahamsen B, Teng AY. Technology evaluation: denosumab, Amgen. Curr Opin Mol Ther. 2005;7:604–610. [PubMed] [Google Scholar]
- Body JJ, Facon T, Coleman RE, Lipton A, Geurs F, Fan M, Holloway D, Peterson MC, Bekker PJ. A study of the biological receptor activator of nuclear factor-kappaB ligand inhibitor, denosumab, in patients with multiple myeloma or bone metastases from breast cancer. Clin Cancer Res. 2006;12:1221–1228. doi: 10.1158/1078-0432.CCR-05-1933. [DOI] [PubMed] [Google Scholar]
- Carducci MA, Jimeno A. Targeting bone metastasis in prostate cancer with endothelin receptor antagonists. Clin Cancer Res. 2006;12:6296s–6300s. doi: 10.1158/1078-0432.CCR-06-0929. [DOI] [PubMed] [Google Scholar]
- Murphy G. Atrasentan for metastatic hormone refractory prostate cancer. Issues Emerg Health Technol. 2005;77:1–4. [PubMed] [Google Scholar]
- Dunehoo AL, Anderson M, Majumdar S, Kobayashi N, Berkland C, Siahaan TJ. Cell adhesion molecules for targeted drug delivery. J Pharm Sci. 2006;95:1856–1872. doi: 10.1002/jps.20676. [DOI] [PubMed] [Google Scholar]
- Harms JF, Welch DR, Samant RS, Shevde LA, Miele ME, Babu GR, Goldberg SF, Gilman VR, Sosnowski DM, Campo DA, et al. A small molecule antagonist of the alpha(v)beta3 integrin suppresses MDA-MB-435 skeletal metastasis. Clin Exp Metastasis. 2004;21:119–128. doi: 10.1023/B:CLIN.0000024763.69809.64. [DOI] [PubMed] [Google Scholar]
- Kominsky SL, Davidson NE. A 'bone' fide predictor of metastasis? Predicting breast cancer metastasis to bone. J Clin Oncol. 2006;24:2227–2229. doi: 10.1200/JCO.2005.05.5319. [DOI] [PubMed] [Google Scholar]
- Lee SK, Gardner AE, Kalinowski JF, Jastrzebski SL, Lorenzo JA. RANKL-stimulated osteoclast-like cell formation in vitro is partially dependent on endogenous interleukin-1 production. Bone. 2006;38:678–685. doi: 10.1016/j.bone.2005.10.011. [DOI] [PubMed] [Google Scholar]
- Jimi E, Nakamura I, Duong LT, Ikebe T, Takahashi N, Rodan GA, Suda T. Interleukin 1 induces multinucleation and bone-resorbing activity of osteoclasts in the absence of osteoblasts/stromal cells. Exp Cell Res. 1999;247:84–93. doi: 10.1006/excr.1998.4320. [DOI] [PubMed] [Google Scholar]
- Fuller K, Murphy C, Kirstein B, Fox SW, Chambers TJ. TNFalpha potently activates osteoclasts, through a direct action independent of and strongly synergistic with RANKL. Endocrinology. 2002;143:1108–1118. doi: 10.1210/en.143.3.1108. [DOI] [PubMed] [Google Scholar]
- Lam J, Takeshita S, Barker JE, Kanagawa O, Ross FP, Teitelbaum SL. TNF-alpha induces osteoclastogenesis by direct stimulation of macrophages exposed to permissive levels of RANK ligand. J Clin Invest. 2000;106:1481–1488. doi: 10.1172/JCI11176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan H, Tumber A, Hill PA. Breast cancer cells induce osteoclast formation by stimulating host IL-11 production and downregulating granulocyte/macrophage colony-stimulating factor. Int J Cancer. 2004;109:653–660. doi: 10.1002/ijc.20056. [DOI] [PubMed] [Google Scholar]
- Mancino AT, Klimberg VS, Yamamoto M, Manolagas SC, Abe E. Breast cancer increases osteoclastogenesis by secreting M-CSF and upregulating RANKL in stromal cells. J Surg Res. 2001;100:18–24. doi: 10.1006/jsre.2001.6204. [DOI] [PubMed] [Google Scholar]
- Wani MR, Fuller K, Kim NS, Choi Y, Chambers T. Prostaglandin E2 cooperates with TRANCE in osteoclast induction from hemopoietic precursors: synergistic activation of differentiation, cell spreading, and fusion. Endocrinology. 1999;140:1927–1935. doi: 10.1210/en.140.4.1927. [DOI] [PubMed] [Google Scholar]
- Peyruchaud O, Serre CM, NicAmhlaoibh R, Fournier P, Clezardin P. Angiostatin inhibits bone metastasis formation in nude mice through a direct anti-osteoclastic activity. J Biol Chem. 2003;278:45826–45832. doi: 10.1074/jbc.M309024200. [DOI] [PubMed] [Google Scholar]
- Nemeth JA, Yousif R, Herzog M, Che M, Upadhyay J, Shekarriz B, Bhagat S, Mullins C, Fridman R, Cher ML. Matrix metalloproteinase activity, bone matrix turnover, and tumor cell proliferation in prostate cancer bone metastasis. J Natl Cancer Inst. 2002;94:17–25. doi: 10.1093/jnci/94.1.17. [DOI] [PubMed] [Google Scholar]
- Fizazi K, Yang J, Peleg S, Sikes CR, Kreimann EL, Daliani D, Olive M, Raymond KA, Janus TJ, Logothetis CJ, et al. Prostate cancer cells-osteoblast interaction shifts expression of growth/survival-related genes in prostate cancer and reduces expression of osteoprotegerin in osteoblasts. Clin Cancer Res. 2003;9:2587–2597. [PubMed] [Google Scholar]
- Shariat SF, Andrews B, Kattan MW, Kim J, Wheeler TM, Slawin KM. Plasma levels of interleukin-6 and its soluble receptor are associated with prostate cancer progression and metastasis. Urology. 2001;58:1008–1015. doi: 10.1016/S0090-4295(01)01405-4. [DOI] [PubMed] [Google Scholar]
- Corey E, Brown LG, Kiefer JA, Quinn JE, Pitts TE, Blair JM, Vessella RL. Osteoprotegerin in prostate cancer bone metastasis. Cancer Res. 2005;65:1710–1718. doi: 10.1158/0008-5472.CAN-04-2033. [DOI] [PubMed] [Google Scholar]
- Matuo Y, Nishi N, Takasuka H, Masuda Y, Nishikawa K, Isaacs JT, Adams PS, McKeehan WL, Sato GH. Production and significance of TGF-beta in AT-3 metastatic cell line established from the Dunning rat prostatic adenocarcinoma. Biochem Biophys Res Commun. 1990;166:840–847. doi: 10.1016/0006-291X(90)90886-R. [DOI] [PubMed] [Google Scholar]
- Achbarou A, Kaiser S, Tremblay G, Ste-Marie LG, Brodt P, Goltzman D, Rabbani SA. Urokinase overproduction results in increased skeletal metastasis by prostate cancer cells in vivo. Cancer Res. 1994;54:2372–2377. [PubMed] [Google Scholar]
- Yi B, Williams PJ, Niewolna M, Wang Y, Yoneda T. Tumor-derived platelet-derived growth factor-BB plays a critical role in osteosclerotic bone metastasis in an animal model of human breast cancer. Cancer Res. 2002;62:917–923. [PubMed] [Google Scholar]
- Valta MP, Hentunen T, Qu Q, Valve EM, Harjula A, Seppanen JA, Vaananen HK, Harkonen PL. Regulation of osteoblast differentiation: a novel function for fibroblast growth factor 8. Endocrinology. 2006;147:2171–2182. doi: 10.1210/en.2005-1502. [DOI] [PubMed] [Google Scholar]
- Cramer SD, Chen Z, Peehl DM. Prostate specific antigen cleaves parathyroid hormone-related protein in the PTH-like domain: inactivation of PTHrP-stimulated cAMP accumulation in mouse osteoblasts. J Urol. 1996;156:526–531. doi: 10.1016/S0022-5347(01)65919-6. [DOI] [PubMed] [Google Scholar]
- Kitagawa Y, Dai J, Zhang J, Keller JM, Nor J, Yao Z, Keller ET. Vascular endothelial growth factor contributes to prostate cancer-mediated osteoblastic activity. Cancer Res. 2005;65:10921–10929. doi: 10.1158/0008-5472.CAN-05-1809. [DOI] [PubMed] [Google Scholar]
- Vakar-Lopez F, Cheng CJ, Kim J, Shi GG, Troncoso P, Tu SM, YuLee LY, Lin SH. Up-regulation of MDA-BF-1, a secreted isoform of ErbB3, in metastatic prostate cancer cells and activated osteoblasts in bone marrow. J Pathol. 2004;203:688–695. doi: 10.1002/path.1568. [DOI] [PubMed] [Google Scholar]
- Penolazzi L, Magri E, Lambertini E, Calo G, Cozzani M, Siciliani G, Piva R, Gambari R. Local in vivo administration of a decoy oligonucleotide targeting NF-kappaB induces apoptosis of osteoclasts after application of orthodontic forces to rat teeth. Int J Mol Med. 2006;18:807–811. [PubMed] [Google Scholar]
- Lev DC, Kim SJ, Onn A, Stone V, Nam DH, Yazici S, Fidler IJ, Price JE. Inhibition of platelet-derived growth factor receptor signaling restricts the growth of human breast cancer in the bone of nude mice. Clin Cancer Res. 2005;11:306–314. [PubMed] [Google Scholar]
- Lara PN, Jr, Stadler WM, Longmate J, Quinn DI, Wexler J, Van Loan M, Twardowski P, Gumerlock PH, Vogelzang NJ, Vokes EE, et al. A randomized phase II trial of the matrix metalloproteinase inhibitor BMS-275291 in hormone-refractory prostate cancer patients with bone metastases. Clin Cancer Res. 2006;12:1556–1563. doi: 10.1158/1078-0432.CCR-05-2074. [DOI] [PubMed] [Google Scholar]