

Abstract
Total joint replacement, although considered an excellent surgical procedure, can be complicated by osteolysis induced by implant particles and subsequent aseptic loosening of the implant. The pathogenesis of implant-associated osteolysis includes inflammatory and osteolytic processes. The sustained chronic inflammatory response initiated by particulate debris at the implant-bone interface is manifested by recruitment of a wide array of cell types. These cells include macrophages, fibroblasts, giant cells, neutrophils, lymphocytes, and – most importantly – osteoclasts, which are the principal bone resorbing cells. The 'cellular response' entails secretion of osteoclastogenic and inflammatory cytokines that favor exacerbated osteoclast activity and enhanced osteolysis. An appreciation of the complex network that leads to these cellular and inflammatory responses will form a foundation on which to develop therapeutic interventions to combat inflammatory periprosthetic bone loss.
Introduction
Aseptic loosening of joint implants is a disabling condition that can affect patients 10 to 20 years after joint replacement surgery (Figure 1). Total joint replacement for end-stage joint diseases such as osteoarthritis and inflammatory rheumatoid arthritis is an effective surgical intervention [1]. Unfortunately, wear debris, primarily generated from the prosthetic joint articular surface, remains the major factor limiting the survival of joint implants. Subtle progression of tissue destruction around the implant imposes a major challenge, because signs and symptoms may not be clinically apparent until late stages of failure.
Figure 1.
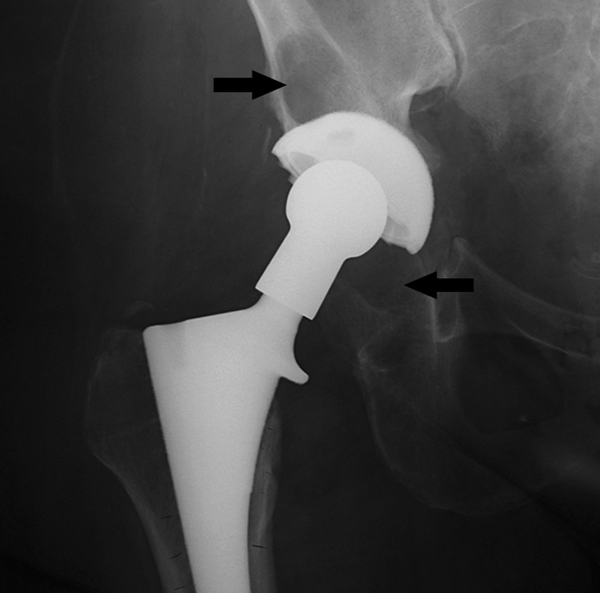
Radiographic appearance of osteolysis. Radiograph of the right hip of a 68-year-old woman 10 years after undergoing cementless primary total hip replacement shows inflammatory osteolysis; the periarticular bone loss is indicated by arrows.
The basis of periprosthetic tissue destruction, the so-called 'biologic response' to implant debris, is complex. However, this response is the outcome of multiple factors, including physical and biologic components [2,3]. At the core of the biologic response that leads to osteolysis is activation of the receptor activator of nuclear factor-κB (RANK)/RANK ligand (RANKL) axis, which is indicated by expression of RANK, RANKL, and osteoprotegerin (OPG) in periprosthetic membranes [4,5]. This activation culminates in enhanced osteoclast recruitment and activity adjacent to bone-implant interfaces, leading to osteolysis [6,7].
Aseptic loosening and osteolysis: etiology, pathogenesis, and cellular responses
Wear debris is formed at prosthetic joint articulations, modular interfaces, and nonarticulating interfaces [1,8]. Although a wide range of particles has been found, the majority of particles formed are less than 5 μm in diameter and are randomly shaped. Studies have suggested that the cellular response to particles may vary with size, shape, composition, charge, and number of particles [9,10]. Furthermore, it was proposed that particle phagocytosis represents an important component of the cellular response to implants; hence, the size of these particles is significant. In this regard, several reports have estimated that particles ranging from 0.2 to 10 μm in diameter undergo phagocytosis by macrophages [11]. In vitro studies of macrophage cultures clearly indicated that smaller polymethylmethacrylate (PMMA) and polyethylene particles (< 20 μm) elicited a significantly greater inflammatory cytokine response, as indicated by increased release of tumor necrosis factor (TNF), IL-1, IL-6, prostaglandin (PG)E2, matrix metalloproteinases, and other factors [9,12-14]. Although particle phagocytosis has been identified as a critical component of this biologic response, recent studies in human macrophages indicate that direct interactions between particle and cell surface are sufficient to activate osteoclastogenic signaling pathways [9,15].
Aseptic loosening can be the result of inadequate initial fixation, mechanical loss of fixation over time, or biologic loss of fixation caused by particle-induced osteolysis around the implant. The causes of particle accumulation vary from implant interface wear, micromotion occurring in response to corrosion, oxidative reactions, and minor pathogen contaminations [2,3]. In general, the initial response is a localized anti-inflammatory response that is characterized by formation of fibrous tissue that encapsulates the implant. Often, synovial fluid and synovial lining membranes are also formed, and granulomatous tissue is established. Immunohistochemical studies of these tissues have revealed an abundance of macrophages, fibroblasts, giant cells, neutrophils, and lymphocytes [16-18]. However, aseptic loosening is characterized by poorly vascularized connective tissue dominated by fibroblasts and macrophages [15]. Subsequently, secretion of proinflammatory factors, gelatinases, and proteases contributes to periprosthetic osteolysis and to failure of the joint implant [19]. Perhaps the most important aspect of the response to particles is its sustained duration, caused by its resistance to enzymatic degradation and digestion [20,21]. This characteristic fuels and continuously builds up the pathologic response, leading to aggressive osteolysis.
The rate at which particles accumulate is also considered an important factor in the occurrence of osteolysis. Various studies found osteolysis to be associated with increased wear rates [3,12,20,22]. Specifically, areas of increased lysis were found to contain significantly greater concentrations of particles (of similar size and shape) as compared with nonosteolytic regions of the loosened implant at revision [10,20]. These observations were further supported in vitro studies in which induction of transcriptional activity and cytokine release were found to be particle dose dependent. An effect of particle concentration on secretions by various types of cells has also been reported [12,21-24]. In primary human monocytes, release of cytokines, PGE2, and hexosaminidase was found to depend on size, concentration, and surface area of particles [12,21-24]. Other studies using macrophage cell lines found dose-dependent apoptosis of the cells when they were treated with ceramic and polyethylene particles [21]. In studies of other cells such as synovial fibroblasts and chondrocytes, treatment with increasing doses of cobalt and vanadium was found to have toxic effects [25].
Clearly, particulate debris load and particle composition are important factors in the osteolytic process. Therefore, ongoing investigations into and evaluations of alternative weight-bearing surfaces are critical if we are to identify materials that are optimal in terms of minimizing particle generation over time [15,26]. Ceramics, and use of highly cross-linked polyethylene and of metal-on-metal articulations show promise as approaches that may markedly reduce the production of wear particles.
Biologic and mechanical factors have been incriminated in the early and late stages of the development of osteolysis following joint replacement. Inflammatory reactions develop at early stages as part of the resolution process. These reactions primarily include increased circulation and elevated fluid levels in the vicinity of the affected tissue [26,27]. In addition, apart from massive recruitment of macrophages to the site of injury, some studies identified recruitment of lymphocytes, indicating that the immune system may be involved in this inflammatory response [15,26,28-32]. Perhaps the most complex aspect of this reaction is secretion of various cytokines and factors by these lymphocytes and other cells, leading over time to adverse effects [7,33,34]. The cellular and associated inflammatory response is not restricted to the initial healing process, but rather it appears to take center stage at progression and in the late stages of periprosthetic osteolysis.
Several co-factors have been described as facilitating propagation of inflammatory and eventual osteolytic events. One major factor relates to poor implant fixation and subsequent motion. In this regard, ample evidence indicates that loosening of implants is associated with excessive motion and physical stress, which result in accelerated release of particulate debris [24]. This is apparent from the progressive nature of bone resorption around loose screws associated with wear debris. In recent years compelling evidence has been reported indicating that the release of such debris leads to inflammatory responses that ultimately result in bone loss. Evidence implicating particulate debris as a major component in osteolysis is derived from studies conducted in animal models exposed to particulate debris, studies with macrophages [35,36] and osteoclasts conducted in vitro and in vivo, and evaluations of osteolysis and co-factors in clinically successful and failed total joint prostheses [21,37-41]. The amounts of particulate debris around implants in most cases exhibit a fair correlation with the degree of aseptic loosening, although certain patients have an exaggerated biologic response to particulate debris. However, particles have been documented in remote tissues as well. Factors that contribute to particle distribution include (but are not limited to) particle number and size, fluid flow, implant design, and joint space [10,26].
Biology of the osteolytic response
The cellular response is dominated by phagocytes and macrophages [26,35,42,43]. However, the osteolytic response includes various cell types, such as osteoclasts, fibroblasts, and osteoblasts/stromal cells. This is supported by the wide range of factors that are secreted by various cells, including cytokines, growth factors, metalloproteinases, prostanoids and lysosomal enzymes, among others [44,45]. The mechanisms that underlie induction by particles of cellular responses and osteolysis are largely unclear and remain subject to vigorous investigation. It is believed that recognition of particles relies on phagocytosis of small-sized particles by macrophages and unidentified cell surface interactions. The latter interactions may include nonspecific physical induction of transmembrane proteins or recognition of cell surface molecules by particles or proteins/factors that are adherent to the surface. However, the precise nature of stimulation of cells by particles remains unknown. Furthermore, it is well accepted that host defense cells, which comprise the core of the inflammatory reaction, recognize particles and release large quantities of proinflammatory cytokines and factors [15,26,28-30,32], including TNF, IL-1α and IL-1β, IL-6, RANKL and PGE2, among others [34,37,38,42,46-48]. These factors stimulate bone loss by targeting osteoclasts and their monocytic precursors. In this regard, RANK/RANKL is the principal axis that regulates the osteoclastogenic event.
The RANK/RANKL pathway was discovered during the late 1990s and resolved the mystery regarding osteoclast differentiation from hematopoetic monocyte/macrophage precursors [7,38]. Osteoclast differentiation is primarily controlled by the stromal/osteoblast-derived proteins RANKL (also known as OPG ligand and osteoclast differentiation factor) and macrophage colony-stimulating factor [49]. A major breakthrough in our understanding of the regulation of osteoclastogenesis was achieved with the identification of OPG, a soluble protein of the TNF receptor family [50] that binds RANKL and acts as a decoy molecule (for review [6,7,51]). Several studies have demonstrated that OPG is a potent inhibitor of bone loss, thus regulating bone density and mass in mouse and humans. This secreted cytokine was also shown to be effective in blocking metabolic, pathologic (including periprosthetic osteolysis), and metastatic bone loss. A similar approach using soluble extracellular RANK protein, which is similar to OPG, neutralizes RANKL by sequestering it in an inactive complex [52]. This approach was further proven to be effective in vitro and in vivo by studies in which Fc-RANK fusion protein was found to block bone loss pathologies. Also important to regulation of osteoclastogenesis and bone loss is that many proinflammatory and anti-inflammatory cytokines act directly to enhance or inhibit the RANK/RANKL axis. For example, the effect of TNF as a stimulator and activator of osteoclastogenesis appears to be entirely dependent on an active RANK/RANKL pathway [53]. In this regard, signaling by TNF appears to be superimposed over pathways initiated by RANKL in osteoclast precursors. A similar synergistic pattern has been observed for other pathologic insults such as implant particles and pathogens. These observations are in discord with studies indicating that TNF, in the absence of RANK, is sufficient to induce osteoclastogenesis [54].
Studies conducted in animal models and in vitro cell cultures have shown that TNF plays a crucial role as a mediator of particle-induced osteoclastogenic and osteolytic events [15,42,47]. In this regard, PMMA particles fail to elicit aggressive osteolysis in TNF receptor null animals or in the presence of TNF neutralizing agents, for instance various soluble TNF binding proteins. Likewise, animals that lack RANK or RANKL resist induction by particles of osteolysis, which is similar to their protection from arthritic bone erosion [34,40,47]. This is not unexpected, considering that TNF family members, especially RANKL, are prerequisites for osteoclast formation.
Further downstream signaling by wear particles, unsurprisingly, overlaps with that of TNF and RANKL. In this regard, activation of kinases and recruitment of molecules that are essential for osteoclast differentiation and activation have been documented [13,15,42,47,55]. Notably, particle-induced pathways lead to activation of kinases and transcription factors that are essential for osteoclastogenesis. Among these are activation of the tyrosine kinase c-src, mitogen-activated protein kinases, and the nuclear factor-κB (NF-κB) cascade [7,13,34]. Although activation of these pathways might be secondary to other events, selective blockade of these downstream pathways reduces particle transmitted effects.
Potential therapeutic intervention in inflammatory osteolysis: molecular approaches to arrest osteoclast activity
The biologic mechanisms of debris-mediated osteolysis are complex and involve various cell types and inflammatory cytokines of the periprosthetic membrane. Particles inhibit bone formation by targeting osteoblasts and activating macrophages and osteoclasts, which leads to significant periprosthetic bone loss [15,56]. Therefore, in combination with improvements in implant integration, strategies to target the cellular components (osteoblasts and osteoclasts) that contribute to implant failure represent potentially effective therapeutic interventions. In this regard, it should be noted that differentiation of bone marrow macrophages (osteoclast precursors) into mature osteoclasts requires recognition and binding of the osteoblast, fibroblast, and T cell secreted factor RANKL by its cognate receptor, RANK, which is expressed on the surface of osteoclast precursors [6,38,57]. This process is regulated by another factor secreted by osteoblasts, namely OPG, which acts as a decoy receptor by binding to RANKL and reducing its bioavailability. On the other hand, binding of RANKL to RANK prompts induction of several intracellular pathways by this receptor, leading to activation of key transcription factors, most notably NF-κB.
It has been reported that the NF-κB family of transcription factors is central to pathologic responses and is essential for osteoclast differentiation (for review [7,51]). It consists of several family members that primarily form heterodimers. These dimers are found bound to an inhibitory protein (inhibitor of NF-κB [IκB]) that, in the absence of stimulation, retains the complex in the cytoplasm. Stimulation by RANKL or other specific stimuli leads to activation of upstream IκB kinases (IKKs), which in turn phosphorylate IκB, leading to its dissociation from the NF-κB complex and eventually to its degradation by the proteosome system. IκB-liberated NF-κB then translocates to the nucleus, binds to specific DNA sites, and induces basal transcription (for review [7,58]).
Knowledge has accumulated of the NF-κB activation mechanisms that eventually lead to osteoclast formation and, when induced by factors such as TNF and PMMA particles, exacerbate osteoclastogenesis and inflammatory responses. Several approaches that exploit this knowledge have been designed to perturb this pathway and hence alleviate the deleterious effects of inflammatory osteolysis. A number of approaches to osteoclast-based therapy may be considered. The first involves targeting osteoclast precursor cells, which are recruited to inflammatory sites by circulating cytokines. The second entails targeting precursors that are stimulated by the particle-mediated cellular response to differentiate and form bone resorbing osteoclasts. The third approach involves targeting activation mechanisms of mature osteoclasts.
There are various approaches to inhibiting differentiation of precursor cells to osteoclasts. First and foremost, direct inhibition of osteoclast differentiation may be achieved by application of RANKL decoy molecules such as OPG and the soluble fusion protein RANK-Fc [52,59]. Indeed, studies in animal models and in vitro osteoclast cultures have identified significant inhibition of osteoclastogenesis and reduced hallmarks of osteolysis [16,52]. Other approaches target key intracellular signal transduction pathways that are essential for osteoclast differentiation and inflammatory exacerbation of this process. The transcription factor NF-κB is at the center of this process, and is essential for both inflammatory and osteolytic responses; inhibition of NF-κB hinges on disrupting its translocation to the nuclei, as noted above. Transduction of a dominant-negative form of the NF-κB inhibitory protein, IκB, which retains NF-κB in the cytoplasm, was indeed sufficient to block osteoclast formation and activity [39,55,60]. Another viable approach is to block activation of the upstream IKK complex, which is responsible for phosphorylation of IκB and subsequent activation of NF-κB. This was possible by introducing a small peptide that perturbs assembly of the IKK complex and attenuates activation of NF-κB [61]. More importantly, administration of the dominant-negative IκB protein or the IKK inhibitory small peptide to arthritic mice significantly was found to block bone erosion associated with inflammatory arthritis and particle-induced osteolysis of calvaria in mice (for review [7]). Because NF-κB is central to most host immune and inflammatory responses and induces expression of a large number of genes encoding proteins that are associated with bone pathology, efforts have been directed toward identifying specific NF-κB-mediated genes and their products. Steps in this direction should lead to the design of a future generation of selective inhibitors that might be effective in alleviating inflammatory bone loss.
Several genes have been shown to be critical for osteoclast differentiation. Among these are c-fms, c-fos, RANKL, NF-κB, c-src, and the proton ATPase [51]. Recent studies have revealed that proinflammatory cytokines such as TNF act directly on some of these genes and their products, in particular c-src and NF-κB, to accelerate osteoclast formation and cause a potent osteoclastic response (for review [7,62]). Selective inhibitors of the c-src tyrosine kinase have shown great promise in halting osteoclast activity, and future studies should be directed toward testing the effect of such inhibitors on inflammatory osteolysis [63,64]. Another promising approach involves the use of bisphosphonates [65]. These compounds inhibit osteoclast function and induce their apoptosis. Animal models have been used to study the efficacy of an oral bisphosphonate in inhibiting bone resorption mediated by wear debris [26,66,67]. The findings indicate reduced radiographic periprosthetic radiolucency, although the levels of PGE2 and IL-1 remained elevated in tissue cultures from these implants. These studies have served as the basis for clinical trials using alendronate in patients with radiographically evident osteolytic lesions. In other studies, bone loss around implant caused by intra-articular injection of particles was prevented and treated by alendronate [68-71]. Thus, bisphosphonates may be useful in preventing particle-induced osteolysis around total joint implants.
Inhibiting recruitment of osteoclast precursor and other cells to the inflammatory site entails use of anti-inflammatory approaches to neutralize mediators such as TNF, IL-1, IL-6, and others. It has been proposed that pharmacologic intervention targeted at the macrophage may provide the means to slow the response to wear debris. Indeed, local cytokine inhibition may reduce inflammation in the periprosthetic tissue, and several biologic mediators have been identified as useful for clinical application. In particular, the IL-1 receptor antagonist protein has been reported to reduce inflammation, and the anti-inflammatory cytokine IL-10 appears able to reduce cell mediated reactions in inflammation [28,72]. Using particle-stimulated murine air pouch and calvarial models to evaluate the potential of gene therapy to treat inflammation induced by orthopedic wear debris, recent studies showed that retroviral vectors encoding human IL-1 receptor antagonist, human TNF receptor, and viral IL-10 led to marked decreases in inflammation, decreased pouch fluid accumulation, and reduced macrophage influx [40,48]. In the murine air pouch study, histologic assessments revealed that pouches transduced with viral IL-10 or IL-1 receptor antagonist exhibited a 40% reduction in inflammatory cell infiltration when compared with nonviral controls or LacZ transduced membranes. The calvaria study corroborated these findings by independently demonstrating that gene delivery of viral IL-10 inhibits three processes that are critically involved in periprosthetic osteolysis: proinflammatory cytokine production induced by wear debris, osteoclastogenesis, and osteolysis.
Anti-inflammatory cytokines secreted by T lymphocytes such as IFN-γ, IL-4, and IL-10 have also been shown to be effective in antagonizing the actions of proinflammatory cytokines (for review [7,40]). These cytokines are being now exploited in erosive inflammatory responses because they arrest osteoclast formation. The finding that activated T-helper-1 cells (which secrete RANKL and proinflammatory cytokines) and T-helper-2 cells (which secrete IL-4) are present in the synovium strongly implicates the immune system as a key regulator of inflammatory bone disease. Of significance is a recent finding that IL-4 mRNA was more frequently identified in nonerosive than in erosive disease (38% versus 15%). These findings provide indirect evidence that IL-4 has bone sparing effects in vivo. Additionally, IL-4 adenoviral gene therapy has been shown to be effective in reducing inflammation, inhibiting proinflammatory cytokine secretion, and sparing bone destruction in a model of adjuvant-induced arthritis [73].
Clarifying the molecular mechanisms that underlie the anti-osteoclastogenic actions of these anti-inflammatory cytokines should unveil useful molecular targets. In this regard, recent progress toward elucidating the anti-osteoclastogenic action of IL-4 and IFN-γ was reported. Specifically, IL-4 binds to membrane receptors expressed on the surface of osteoclast precursors, and activates key transcription factors, most notably the signal transducer and activator of transcription (STAT)6. The mechanism responsible entails activation by IL-4 of Janus kinases, which phosphorylate STAT6, prompting its dimerization and translocation to the nucleus, where it binds to DNA and regulates target genes [74]. In osteoclasts and their precursors, STAT6 was found to be crucial for IL-4 inhibition of osteoclastogenesis and inhibits inflammatory bone erosion. Although the precise mechanism is unknown, STAT6 is presumed to inhibit NF-κB and mitogen-activated protein kinase activation and thus inhibits transcription of genes, including those encoding inflammatory cytokines such as TNF, IL-6, NF-κB family members, and more [75]. These observations indicate that STAT6 is a potentially useful target for anti-erosive drug design.
IFN-γ is another major product of immune cells that potently inhibits bone resorption (for review [7,58]). Recent reports illustrated that IFN-γ interferes with the RANK\RANKL signal transduction in osteoclasts and their precursors. It induces rapid degradation of TNF receptor associated factor 6, a RANK adaptor protein. This action results in arrest of RANK downstream signals, such as NF-κB and c-Jun\Janus kinase pathways. In another study [76] it was reported that RANKL-induced secretion of IFN-γ by osteoclast precursors counterbalances bone resorption by blocking osteoclastogenesis in an autoregulatory manner. Although a direct role for IFNs in particle-induced inflammatory osteolysis has not yet been established, the possibility that these cytokines will block the bone loss associated with this disease is worthy of further investigation.
Conclusion
Aseptic loosening secondary to wear debris mediated inflammatory osteolysis is a serious health problem and will have an impact on increasing numbers of patients over the coming decades. The initial response to particulate debris includes a subtle inflammatory response, which becomes more pronounced as osteolysis progresses. The inflammatory environment provokes a cellular response characterized by elevated levels of secreted factors such as TNF, RANKL, IL-6, IL-1, and IL-11. Most of these cytokines affect osteoclast differentiation and activity directly, and result in enhanced osteolysis. Clarifying the molecular details of these signaling cascades and cellular responses represents, and will continue to represent, an enormous therapeutic challenge.
Abbreviations
IFN = interferon; IκB = inhibitor of NF-κB; IKK = IκB kinase; IL = interleukin; NF-κB = nuclear factor-κB; OPG = osteoprotegerin; PG = prostaglandin; PMMA = polymethylmethacrylate; RANK = receptor activator of nuclear factor-κB; RANKL = receptor activator of nuclear factor-κB ligand; STAT = signal transducer and activator of transcription; TNF = tumor necrosis factor.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
The authors acknowledge the generous support by the National Institute of Health (NIH; grants # DE13754, AR47443, AR49192, and AR47096) and by the Shriners Hospital for Children. These funds were instrumental for completing studies by the authors cited in this review.
This article is published as part of Arthritis Research & Therapy Volume 9 Supplement 1, 2007: Basic science, rationale, background and future of denosumab: a RANK ligand inhibitor. The full contents of the supplement are available online at http://arthritis-research.com/supplements/9/S1.
Publication of the supplement has been supported by an unrestricted grant from Amgen Inc.
References
- Harris WH. The problem is osteolysis. Clin Orthop Rel Res. 1995;311:46–53. [PubMed] [Google Scholar]
- Kadoya Y, Kobayashi A, Ohashi H. Wear and osteolysis in total joint replacements. Acta Orthop Scand. 1998;69:1–16. [PubMed] [Google Scholar]
- Maloney W, Smith R. Periprosthetic osteolysis in total hip arthroplasty: the role of particulate waer debris. J Bone Joint Surg. 1995;77A:1448–1461. [PubMed] [Google Scholar]
- Crotti TN, Smith MD, Findlay DM, Zreiqat H, Ahern MJ, Weedon H, Hatzinikolous G, Capone M, Holding C, Haynes DR. Factors regulating osteoclast formation in human tissues adjacent to peri-implant bone loss: expression of receptor activator NFkappaB, RANK ligand and osteoprotegerin. Biomaterials. 2004;25:565–573. doi: 10.1016/S0142-9612(03)00556-8. [DOI] [PubMed] [Google Scholar]
- Gehrke T, Sers C, Morawietz L, Fernahl G, Neidel J, Frommelt L, Krenn V. Receptor activator of nuclear factor kappaB ligand is expressed in resident and inflammatory cells in aseptic and septic prosthesis loosening. Scand J Rheumatol. 2003;32:287–294. doi: 10.1080/03009740310003929. [DOI] [PubMed] [Google Scholar]
- Khosla S. Minireview: the OPG/RANKL/RANK system. Endocrinology. 2001;142:5050–5055. doi: 10.1210/en.142.12.5050. [DOI] [PubMed] [Google Scholar]
- Abu-Amer Y. Advances in osteoclast differentiation and function. Curr Drug Targets Immune Endocr Metabol Disord. 2005;5:347–355. doi: 10.2174/1568008054863808. [DOI] [PubMed] [Google Scholar]
- Goldring SR, Clark CR, Wright TM. The problem in total joint arthroplasty: aseptic loosening. J Bone Joint Surg. 1993;75A:799–801. doi: 10.2106/00004623-199306000-00001. [DOI] [PubMed] [Google Scholar]
- Gonzalez O, Smith RL, Goodman SB. Effect of size, concentration, surface area, and volume of polymethylmethacrylate particles on human macrophages in vitro. J Biomed Mater Res. 1996;30:463–473. doi: 10.1002/(SICI)1097-4636(199604)30:4<463::AID-JBM4>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Sabokbar A, Pandey R, Athanasou NA. The effect of particle size and electrical charge on macrophage-osteoclast differentiation and bone resorption. J Mater Sci Mater Med. 2003;14:731–738. doi: 10.1023/A:1025088418878. [DOI] [PubMed] [Google Scholar]
- Gelb H, Schumacher HR, Cuckler J, Baker DG. In vivo inflammatory response to polymethylmethacrylate particulate debris: effect of size, morphology, and surface area. J Ortho Res. 1994;12:83–92. doi: 10.1002/jor.1100120111. [DOI] [PubMed] [Google Scholar]
- Shanbhag AS, Jacobs JJ, Glant TT, Gilbert JL, Black J, Galante JO. Composition and morphology of wear debris in failed uncemented total hip replacement. J Bone Joint Surg Br. 1994;76:60–67. [PubMed] [Google Scholar]
- Abbas S, Clohisy JC, Abu-Amer Y. Mitogen-activated protein (MAP) kinases mediate PMMA-induction of osteoclasts. J Orthop Res. 2003;21:1041–1048. doi: 10.1016/S0736-0266(03)00081-0. [DOI] [PubMed] [Google Scholar]
- O'Keefe RJ, Rosier RN, Teot LA, Stewart JM, Hicks DG. Cytokine and matrix metalloproteinase expression in pigmented villonodular synovitis may mediate bone and cartilage destruction. Iowa Orthop J. 1998;18:26–34. [PMC free article] [PubMed] [Google Scholar]
- Gallo J, Kamâinek P, Tichâa V, Rihâakovâa P, Ditmar R. Particle disease. A comprehensive theory of periprosthetic osteolysis: a review. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2002;146:21–28. doi: 10.5507/bp.2002.004. [DOI] [PubMed] [Google Scholar]
- Ulrich-Vinther M, Carmody EE, Goater JJ, Sballe K, O'Keefe RJ, Schwarz EM. Recombinant adeno-associated virus-mediated osteoprotegerin gene therapy inhibits wear debris-induced osteolysis. J Bone Joint Surg. 2002;84:1405. doi: 10.2106/00004623-200208000-00016. [DOI] [PubMed] [Google Scholar]
- Goldring SR. Bone and joint destruction in rheumatoid arthritis: what is really happening? J Rheumatol Suppl. 2002;65:44–48. [PubMed] [Google Scholar]
- Clohisy JC, Calvert G, Tull F, McDonald D, Maloney WJ. Reasons for revision hip surgery: a retrospective review. Clin Orthop Relat Res. 2004;429:188–192. doi: 10.1097/01.blo.0000150126.73024.42. [DOI] [PubMed] [Google Scholar]
- Voloshin I, Gelinas J, Maloney MD, O'Keefe RJ, Bigliani LU, Blaine TA. Proinflammatory cytokines and metalloproteases are expressed in the subacromial bursa in patients with rotator cuff disease. Arthroscopy. 2005;21:1076. doi: 10.1016/j.arthro.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Shanbhag AS, Bailey HO, Hwang DS, Cha CW, Eror NG, Rubash HE. Quantitative analysis of ultrahigh molecular weight polyethylene (UHMWPE) wear debris associated with total knee replacements. J Biomed Mater Res. 2000;53:100–110. doi: 10.1002/(SICI)1097-4636(2000)53:1<100::AID-JBM14>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- Jacobs JJ, Hallab NJ, Urban RM, Wimmer MA. Wear particles. J Bone Joint Surg Am. 2006;(Suppl 2):99–102. doi: 10.2106/JBJS.F.00102. [DOI] [PubMed] [Google Scholar]
- Shanbhag AS, Jacobs JJ, Glant TT, Gilbert JL, Black J, Galante JO. Composition and morphology of wear debris in failed uncemented total hip replacement. J Bone Joint Surg Br. 1994;76:60–67. [PubMed] [Google Scholar]
- Shanbhag AS, Jacobs JJ, Black J, Galante JO, Glant TT. Cellular mediators secreted by interfacial membranes obtained at revision total hip arthroplasty. J Arthroplasty. 1995;10:498–506. doi: 10.1016/S0883-5403(05)80152-4. [DOI] [PubMed] [Google Scholar]
- Hirakawa K, Jacobs JJ, Urban R, Saito T. Mechanisms of failure of total hip replacements: lessons learned from retrieval studies. Clin Orthop Relat Res. 2004;420:10–17. doi: 10.1097/00003086-200403000-00003. [DOI] [PubMed] [Google Scholar]
- Shanbhag AS, Jacobs JJ, Black J, Galante JO, Glant TT. Effects of particles on fibroblast proliferation and bone resorption in vitro. Clin Orthop Relat Res. 1997;342:205–217. doi: 10.1097/00003086-199709000-00028. [DOI] [PubMed] [Google Scholar]
- Purdue PE, Koulouvaris P, Potter HG, Nestor BJ, Sculco TP. The cellular and molecular biology of periprosthetic osteolysis. Clin Orthop Relat Res. 2007;454:251–261. doi: 10.1097/01.blo.0000238813.95035.1b. [DOI] [PubMed] [Google Scholar]
- Chambers TJ. The cellular basis of bone resorption. Clin Orthop. 1980;151:283–293. [PubMed] [Google Scholar]
- Arora A, Song Y, Chun L, Huie P, Trindade M, Smith RL, Goodman S. The role of the TH1 and TH2 immune responses in loosening and osteolysis of cemented total hip replacements. J Biomed Mater Res A. 2003;64:693–697. doi: 10.1002/jbm.a.10200. [DOI] [PubMed] [Google Scholar]
- Farber A, Chin R, Song Y, Huie P, Goodman S. Chronic antigen-specific immune-system activation may potentially be involved in the loosening of cemented acetabular components. J Biomed Mater Res. 2001;55:433–441. doi: 10.1002/1097-4636(20010605)55:3<433::AID-JBM1033>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Goodman SB. Does the immune system play a role in loosening and osteolysis of total joint replacements? J Long Term Eff Med Implants. 1996;6:91–101. [PubMed] [Google Scholar]
- Groot D, Gosens T, Leeuwen NC, Rhee MV, Teepen HJ. Wear-induced osteolysis and synovial swelling in a patient with a metal-polyethylene wrist prosthesis. J Hand Surg. 2006;31:1615–1618. doi: 10.1016/j.jhsa.2006.09.009. [DOI] [PubMed] [Google Scholar]
- Hallab NJ, Anderson S, Stafford T, Glant T, Jacobs JJ. Lymphocyte responses in patients with total hip arthroplasty. J Orthop Res. 2005;23:384–391. doi: 10.1016/j.orthres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Hallab NJ, Anderson S, Stafford T, Glant T, Jacobs JJ. Lymphocyte responses in patients with total hip arthroplasty. J Orthop Res. 2005;23:384–391. doi: 10.1016/j.orthres.2004.09.001. [DOI] [PubMed] [Google Scholar]
- Lam J, Abu-Amer Y, Nelson C, Fermont D, Ross F, Teitelbaum S. Tumor necrosis factor superfamily cytokines and the pathogenesis of inflammatory osteolysis. Ann Rheum Dis. 2002;61:82–83. doi: 10.1136/ard.61.1.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn J, Joyner C, Triffitt JT, Athanasou NA. Polymethylmethacrylate-induced inflammatory macrophages resorb bone. J Bone Joint Surg Br. 1992;74:652–658. doi: 10.1302/0301-620X.74B5.1527108. [DOI] [PubMed] [Google Scholar]
- Sabokbar A, Itonaga I, Sun SG, Kudo O, Athanasou NA. Arthroplasty membrane-derived fibroblasts directly induce osteoclast formation and osteolysis in aseptic loosening. J Orthop Res. 2005;23:511–519. doi: 10.1016/j.orthres.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Schwarz EM, O'Keefe RJ, Looney RJ. Bone implant interface, osteolysis and potential therapies. J Musculoskelet Neuronal Interact. 2004;4:390–392. [PubMed] [Google Scholar]
- Ritchlin CT, Schwarz EM, O'Keefe RJ, Looney RJ. RANK, RANKL and OPG in inflammatory arthritis and periprosthetic osteolysis. J Musculoskelet Neuronal Interact. 2004;4:276–284. [PubMed] [Google Scholar]
- Clohisy J, Hirayama T, Frazier E, Han S, Abu-Amer Y. NF-κB signaing blockade abolishes implant particle-induced osteoclastogenesis. J Orthop Res. 2004;22:13–20. doi: 10.1016/S0736-0266(03)00156-6. [DOI] [PubMed] [Google Scholar]
- Abu-Amer Y. Mechanisms of inflammatory mediators in bone loss diseases. In: Rosier RN, Evans CH, editor. Molecular Biology in Orthopedics. Rosemont, IL: American Academy of Orthopedic Surgeons; 2003. pp. 229–239. [Google Scholar]
- Clohisy J, Teitelbaum S, Chen S, Erdmann J, Abu-Amer Y. TNF mediates PMMA particle-induced NF-κB activation in osteoclast precursor cells. J Orthop Res. 2002;20:174–181. doi: 10.1016/S0736-0266(01)00088-2. [DOI] [PubMed] [Google Scholar]
- Archibeck MJ, Jacobs JJ, Roebuck KA, Glant TT. The basic science of periprosthetic osteolysis. Instr Course Lect. 2001;50:185–195. [PubMed] [Google Scholar]
- Neale SD, Athanasou NA. Cytokine receptor profile of arthroplasty macrophages, foreign body giant cells and mature osteoclasts. Acta Orthop Scand. 1999;70:452–458. doi: 10.3109/17453679909000980. [DOI] [PubMed] [Google Scholar]
- Perry MJ, Mortuza FY, Ponsford FM, Elson CJ, Atkins RM. Analysis of cell types and mediator production from tissues around loosening joint implants. Br J Rheumatol. 1995;34:1127–1134. doi: 10.1093/rheumatology/34.12.1127. [DOI] [PubMed] [Google Scholar]
- Shanbhag AS, Jacobs JJ, Black J, Galante JO, Glant TT. Cellular mediators secreted by interfacial membranes obtained at revision total hip arthroplasty. J Arthroplasty. 1995;10:498–506. doi: 10.1016/S0883-5403(05)80152-4. [DOI] [PubMed] [Google Scholar]
- Mundy GR. Mechanisms of osteolytic bone destruction. Bone. 1991;(Suppl 1):S1–S6. doi: 10.1016/8756-3282(91)90057-P. [DOI] [PubMed] [Google Scholar]
- Schwarz E, Lu P, Goater J, Benz E, Kollias G, Rosier R, Puzas J, O'Keefe R. TNF-α/NF-κB signaling in periprosthetic osteolysis. J Orthop Res. 2000;18:472–480. doi: 10.1002/jor.1100180321. [DOI] [PubMed] [Google Scholar]
- Looney RJ, Schwarz EM, Boyd A, O'Keefe RJ. Periprosthetic osteolysis: an immunologist's update. Curr Opin Rheumatol. 2006;18:80–87. doi: 10.1097/01.bor.0000198004.88568.96. [DOI] [PubMed] [Google Scholar]
- Lacey DL, Timms E, Tan HL, Kelley MJ, Dunstan CR, Burgess T, Elliott R, Colombero A, Elliott G, Scully S, et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell. 1998;93:165–176. doi: 10.1016/S0092-8674(00)81569-X. [DOI] [PubMed] [Google Scholar]
- Simonet W, Lacey DL, Dunstan CR, Kelly M, Chang M, Nguyen S, Wooden S, Bennett L, Boone G. Osteoprotegerin: a novel secreted protein involved in the regulation of bone density. Cell. 2001;89:309–319. doi: 10.1016/S0092-8674(00)80209-3. [DOI] [PubMed] [Google Scholar]
- Teitelbaum S. Bone resorption by osteoclasts. Science. 2000;289:1504–1508. doi: 10.1126/science.289.5484.1504. [DOI] [PubMed] [Google Scholar]
- Childs L, Paschalis E, Xing L, Dougall W, Anderson D, Bosky A, Puzas J, Rosier R, O'Keefe R, Boyce B, et al. In vivo RANK signaling blockade using the receptor activator of NF-κB:Fc effectively prevents and ameliorates wear debris-induced osteolysis via osteoclast depletion without inhibiting osteogenesis. J Bone Miner Res. 2002;17:192–199. doi: 10.1359/jbmr.2002.17.2.192. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Heulsmann A, Tondravi M, Mukherjee A, Abu-Amer Y. Tumor necrosis factor-alpha (TNF) stimulates RANKL-induced osteoclastogenesis via coupling of TNF type I receptor and RANK signaling pathways. J Biol Chem. 2000;276:563–568. doi: 10.1074/jbc.M008198200. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Takahashi N, Jimi E, Udagawa N, Takami M, Kotake S, Nakagawa N, Shima N, Yasuda H, Higashio K, et al. Tumor necrosis factor alpha stimulates osteoclast differentiation by a mechanism independent of the ODF/RANKL-RANK interaction. J Exp Med. 2000;191:275–285. doi: 10.1084/jem.191.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abbas S, Abu-Amer Y. Dominant-negative IkB facilitates apoptosis of osteoclasts by tumor necrosis factor-α. J Biol Chem. 2003;278:20077–20082. doi: 10.1074/jbc.M208619200. [DOI] [PubMed] [Google Scholar]
- Zreiqat H, Crotti TN, Howlett CR, Capone M, Markovic B, Haynes DR. Prosthetic particles modify the expression of bone-related proteins by human osteoblastic cells in vitro. Biomaterials. 2003;24:337–346. doi: 10.1016/S0142-9612(02)00324-1. [DOI] [PubMed] [Google Scholar]
- Wei X, Zhang X, Zuscik MJ, Drissi MH, Schwarz EM, O'Keefe RJ. Fibroblasts express RANKL and support osteoclastogenesis in a COX-2-dependent manner after stimulation with titanium particles. J Bone Miner Res. 2005;20:1136–1148. doi: 10.1359/JBMR.050206. [DOI] [PubMed] [Google Scholar]
- Abu-Amer Y, Faccio R. Therapeutic approaches in bone pathogeneses: targeting the IKK/NF-κB axis. Future Rheumatol. 2006;1:133–146. doi: 10.2217/17460816.1.1.133. [DOI] [Google Scholar]
- Ulrich-Vinther M, Carmody EE, Goater JJ, Sballe K, O'Keefe RJ, Schwarz EM. Recombinant adeno-associated virus-mediated osteoprotegerin gene therapy inhibits wear debris-induced osteolysis. J Bone Joint Surg Am. 2002;84-A:1405–1412. doi: 10.2106/00004623-200208000-00016. [DOI] [PubMed] [Google Scholar]
- Clohisy J, Roy B, Biondo C, Frazier E, Willis D, Teitelbaum S, Abu-Amer Y. Direct inhibition of NF-κB blocks bone erosion associated with inflammatory arthritis. J Immunol. 2003;171:5547–5553. doi: 10.4049/jimmunol.171.10.5547. [DOI] [PubMed] [Google Scholar]
- Dai S, Hirayama T, Abbas S, Abu-Amer Y. The IκB kinase (IKK) inhibitor, NEMO-binding domain peptide, blocks osteoclastogenesis and bone erosion in inflammatory arthritis. J Biol Chem. 2004;279:37219–37222. doi: 10.1074/jbc.C400258200. [DOI] [PubMed] [Google Scholar]
- Abu-Amer Y, Faccio R. Therapeutic approaches in bone pathogeneses: targeting the IKK/NF-κB axis. Future Med. 2006;1:133–146. [Google Scholar]
- Susva M, Missbach M, Green J. Src inhibitors: drugs for the treatment of osteoporosis, cancer or both? Trends Pharmacol Sci. 2000;21:489–495. doi: 10.1016/S0165-6147(00)01567-4. [DOI] [PubMed] [Google Scholar]
- Violette S, Guan W, Bartlett C, Smith J, Bardelay C, Antoine E, Rickles R, Mandine E, Adams S, Lynch B, et al. Bone-targeted src SH2 inhibitors block src cellular activity and osteoclast-mediated resorption. Bone. 2001;28:54–64. doi: 10.1016/S8756-3282(00)00427-0. [DOI] [PubMed] [Google Scholar]
- Goldring SR, Gravallese EM. Bisphosphonates: environmental protection for the joint? Arthritis Rheum. 2004;50:2044–2047. doi: 10.1002/art.20383. [DOI] [PubMed] [Google Scholar]
- Sato M, Grasser W. Effects of bisphosphonates on isolated rat osteoclasts as examined by reflected light microscopy. J Bone Miner Res. 1990;5:31–40. doi: 10.1002/jbmr.5650050107. [DOI] [PubMed] [Google Scholar]
- Carano A, Teitelbaum SL, Konsek JD, Schlesinger PH, Blair HC. Bisphosphonates directly inhibit the bone resorption activity of isolated avian osteoclasts in vitro. J Clin Invest. 1990;85:456–461. doi: 10.1172/JCI114459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Grasser W, Endo N, Akins R, Simmons H, Thompson DD, Golub E, Rodan GA. Bisphosphonate action. Alendronate localization in rat bone and effects on osteoclast ultrastructure. J Clin Invest. 1991;88:2095–2105. doi: 10.1172/JCI115539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seedor JG, Quartuccio HA, Thompson DD. The bisphosphonate alendronate (MK-217) inhibits bone loss due to ovariectomy in rats. J Bone Miner Res. 1991;6:339–346. doi: 10.1002/jbmr.5650060405. [DOI] [PubMed] [Google Scholar]
- Balena R, Toolan BC, Shea M, Markatos A, Myers ER, Lee SC, Opas EE, Seedor JG, Klein H, Frankenfield D, et al. The effects of 2-year treatment with the aminobisphosphonate alen-dronate on bone metabolism, bone histomorphometry, and bone strength in ovariectomized nonhuman primates. J Clin Invest. 1993;93:2577–2586. doi: 10.1172/JCI116872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millett PJ, Allen MJ, Bostrom MPG. Effects of alendronate on particle-induced osteolysis in a rat model. J Bone Joint Surg. 2002;84:236. doi: 10.2106/00004623-200202000-00011. [DOI] [PubMed] [Google Scholar]
- van de Loo FA, van den Berg WB. Gene therapy for rheumatoid arthritis. Lessons from animal models, including studies on interleukin-4, interleukin-10, and interleukin-1 receptor antagonist as potential disease modulators. Rheum Dis Clin North Am. 2002;28:127–149. doi: 10.1016/S0889-857X(03)00073-5. [DOI] [PubMed] [Google Scholar]
- Woods J, Katschke JK, Volin M, Ruth J, Woodruff D, Amin M, Connors M, Kurata H, Arai K, Haines IG. IL-4 adenoviral gene therapy reduces inflammation, proinflammatory cytokines, vascularization, and bony destruction in rat adjuvant-induced arthritis. J Immunol. 2001;166:1214–1222. doi: 10.4049/jimmunol.166.2.1214. [DOI] [PubMed] [Google Scholar]
- Takeda K, Tanaka T, Shi W, Matsumoto M, Minami M, Kashiwamura S, Nakanishi K, Yoshida N, Kishimoto T, Akira S. Essential role of Stat6 in IL-4 signalling. Nature. 1996;380:627–630. doi: 10.1038/380627a0. [DOI] [PubMed] [Google Scholar]
- Abu-Amer Y. IL-4 abrogates osteoclastogenesis through STAT6-dependent inhibition of NF-κB. J Clin Invest. 2001;107:1375–1385. doi: 10.1172/JCI10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takayanagi H, Kim S, Matsuo K, Suzuki H, Suzuki T, Sato K, Yokochi T, Oda H, Nakamura K, Ida N, et al. RANKL maintains bone homeostasis through c-Fos-dependent induction of interferon-beta. Nature. 2002;416:744–749. doi: 10.1038/416744a. [DOI] [PubMed] [Google Scholar]