

Abstract
Embryogenesis involves two distinct processes. On the one hand, cells must specialize, acquiring fates appropriate to their positions (differentiation); on the other hand, they must physically construct the embryo through coordinated mechanical activity (morphogenesis). In early vertebrate development, fibroblast growth factor (FGF) regulates multiple embryonic events, including germ layer differentiation and morphogenesis; the cellular components that direct FGF signaling to evoke these different responses remain largely unknown. We show here that the copper transporter 1 (Ctr1) protein is a critical router of FGF signals during early embryogenesis. Ctr1 both promotes the differentiation and inhibits the morphogenesis of mesoderm and neurectoderm in embryos of the frog Xenopus laevis, thereby coordinating normal development. Signal sorting by Ctr1 involves the activation of the Ras–MAP kinase cascade and appears to be independent of its role in copper transport. Mouse embryonic stem (ES) cells deficient for Ctr1 (Ctr1−/−) retain characteristics of pluripotency under conditions that favor differentiation in wild-type ES cells, indicating a conserved role for Ctr1 during amphibian and mammalian cell fate determination. Our studies support a model in which vertebrate Ctr1 functions as a key regulator of the differentiation capacity of both stem and progenitor cell populations.
Keywords: Xenopus, FGF, pluripotent, ERK, Laloo
A recurrent theme in animal development is the prominent role held by a small number of signaling molecules in the initiation of events with a wide variety of biological outcomes. A dramatic example of this phenomenon occurs during early vertebrate embryogenesis, when fibroblast growth factor (FGF) mediates multiple developmental processes. During mesoderm induction, FGF triggers activation of a Ras–MAPK cascade (1–12). In contrast, subsequent morphogenesis of the dorsal mesoderm is regulated by a noncanonical FGF pathway that is independent of Ras–MAPK signaling and may involve stimulation of Ca2+ release and/or activation of phospholipase Cγ (PLCγ) (13, 14). FGF, then, regulates cell determination and morphogenesis via divergent signaling cascades.
The mechanisms underlying the distinct responses to FGF during embryogenesis remain largely mysterious. FGF-mediated activation of the Ras–MAPK cascade during mesoderm development in the frog Xenopus laevis is triggered by the coordinate phosphorylation of the FRS2α/SNT-1 docking protein by the FGF receptor and by the Src family of nonreceptor tyrosine kinases (10–12). We have identified the high-affinity copper transporter 1 (Ctr1) protein as a factor that physically interacts with the Src-related kinase, Laloo. Analysis of Ctr1 in frog embryos and in mouse embryonic stem (ES) cells demonstrates that Ctr1 is a critical regulator of tissue morphogenesis and of both stem and progenitor cell fate determination. Our studies suggest that Ctr1 is required for the proper interpretation and coordination of differentiation and morphogenetic cues during early vertebrate development.
Results and Discussion
Protein interaction screens were performed to isolate novel components involved in interpreting FGF-stimulated functions. Xenopus copper transporter-1 (Xctr1) was identified as a factor that physically associates with the tyrosine kinase Laloo, required for FGF-mediated mesoderm induction [supporting information (SI) Fig. 5] (9, 15). Microinjection of Xctr1 cRNA at early cleavage stages gave rise to embryos with shortened, kinked axes and reduced or absent anterior structures, including eyes and cement glands (93%; n = 46) (Fig. 1A). Xctr1-injected embryos underwent at least some mesodermal differentiation, as demonstrated by staining with an antibody (12/101) against a somite-specific epitope (96%; n = 46) (16) (Fig. 1A Left). To better define the effects of Xctr1 misexpression, ectodermal (animal cap) explant assays were performed. Untreated animal caps normally “round up” and develop as epidermis (Fig. 1B Upper Left); animal caps injected with Xctr1 RNA appeared indistinguishable from controls (data not shown). As expected, animal caps treated with high doses of the TGFβ ligand, activin, formed dorsal mesoderm (notochord and somites) and elongated in a process known as convergent extension (17, 18); injection of a control RNA (GFP) had no effect on elongation or gene expression (Fig. 1B Upper Center, Fig. 1C, and data not shown). Injection of Xctr1 RNA, however, markedly inhibited activin-induced elongation (Fig. 1B Lower Left). This effect was not secondary to a block of dorsal mesodermal differentiation because expression of the notochord marker collagen type II (19) was not affected in these explants; activin-mediated induction of the somite marker muscle actin (20) was modestly reduced or unaffected by Xctr1 misexpression (Fig. 1C, lanes 2 and 4, and data not shown). Xctr1 misexpression in animal caps cultured in saline induced the expression of the neural-specific marker NRP-1 (21) (Fig. 1D). This expression appears to reflect primary, rather than secondary, neural induction, in that neural induction by Xctr1, assayed at several time points, was not accompanied by mesoderm induction (SI Fig. 6). Thus, Xctr1 misexpression neuralizes ectodermal explants and disrupts gastrulation movements without inhibiting mesodermal differentiation.
Fig. 1.

Xctr1 misexpression inhibits morphogenesis in explants and embryos. (A) Whole-mount immunohistochemistry of stage 32 Xenopus embryos injected in the dorsal marginal zone at early cleavage stages with 1 ng of either Xctr1 or β-gal RNA. The 12/101 antibody is directed against a somite-specific epitope at this stage. (B) Effects of injection of RNA from wild-type and mutant Xctr1 on activin-mediated elongation of stage 20 animal caps; 1 ng of GFP RNA-injected animal caps were used as an injection control. Animal caps were explanted at stage 8; immediately after dissection, activin was added at a concentration of 0.5 ng/ml, and CuSO4 was added to achieve a concentration of 10 μM as indicated. (C–E) RT-PCR analysis of the effects of copper, wild-type, and mutant Xctr1 constructs on dorsal mesoderm induction by activin (stage 20) (C), neural induction (stage 20) (D), and metallothionein expression (stage 11) (E). (F) Xctr1 synergizes with FGF, but not activin, to induce Xbra expression (stage 13). (G) Copper-binding Xctr1 mutants, but not copper, synergize with FGF to induce Xbra expression (stage 13). Explants were treated with 0.5 ng/ml (C) or 0.1 ng/ml (F) activin protein or 10 ng/ml FGF protein (F and G) as listed; 1 ng of wild-type or mutant Xctr1 RNA was injected at early cleavage stages as listed.
Ctr1, shown to mediate the uptake of cisplatin and other platinum chemotherapy agents, encodes a putative three-pass transmembrane protein thought to function in vivo as a high-affinity homotrimeric transporter for reduced, monovalent copper in an energy-independent process (22–24). To determine the role of copper transport in the regulation of cell movement, Xctr1 mutants were generated based on mouse Ctr1 mutants shown to be defective in copper transport (25). The corresponding cRNAs (M2, M149) were effective in blocking elongation of activin-treated animal caps at a range of concentrations similar to that of wild-type Xctr1 (Fig. 1B Lower). Moreover, like wild-type Xctr1, expression of the mutant constructs induced NRP-1 expression and had little or no effect on the induction of dorsal mesoderm by activin (Fig. 1 C and D, compare lanes 4, 5, and 6). Consistent with the observation that high levels of intracellular copper induce the expression of the metallothionein gene (26), treatment of animal caps with solutions of copper sulfate that had no effect on either cell fate or morphogenesis resulted in strong metallothionein expression (Fig. 1 B–E). In contrast, expression of wild-type and mutant Xctr1 RNA at levels that block morphogenesis in these assays did not induce metallothionein expression (Fig. 1E). Although we cannot rule out the possibility that the Ctr1 mutant constructs retain the ability to affect development via the uptake of other metals, these studies suggest a prominent role for Ctr1 during embryogenesis that is independent of its role in copper transport.
FGF signaling, like TGFβ signaling, is required for mesoderm formation during Xenopus embryogenesis. Although FGF is not thought to function as a primary mesoderm inducer in vivo, exogenous FGF can induce mesoderm in ectodermal explants (27). The Xctr1-interacting protein, Laloo, is required for FGF-mediated mesoderm induction and induces expression of the early panmesodermal gene Xbrachyury (Xbra) in gastrula-stage animal cap explants (9). Xctr1 RNA injection alone does not induce Xbra expression in this assay (Fig. 1F and SI Fig. 6). However, Xctr1 misexpression enhances Xbra expression in the presence of low levels of FGF, but not activin, protein (Fig. 1F and data not shown). As observed in the context of Xctr1-mediated morphogenesis and neural induction, copper-binding Xctr1 mutants functioned like wild-type Xctr1 in the mesoderm induction assay; moreover, exogenous copper sulfate did not enhance mesoderm induction by FGF (Fig. 1G). These studies indicate that Xctr1 can synergize with FGF, but not activin, during mesoderm induction.
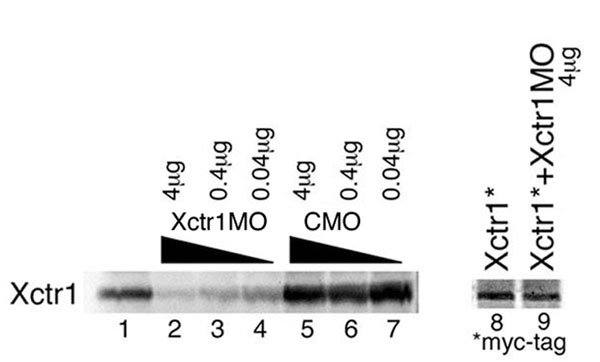
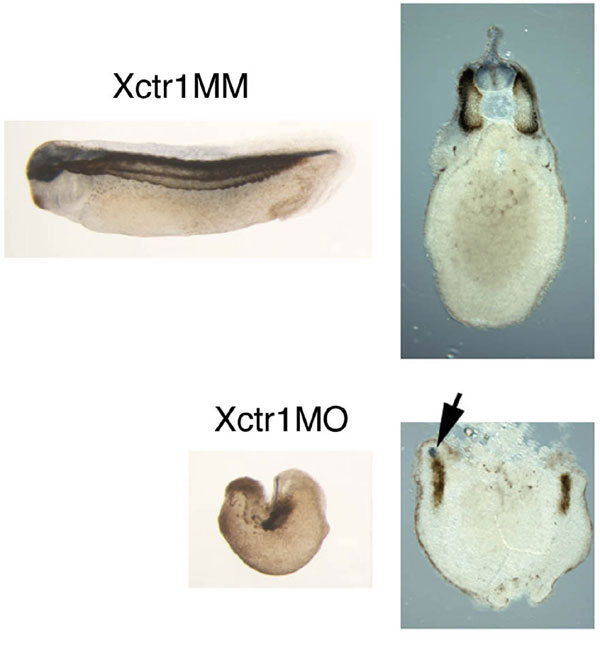
To establish the requirement for Xctr1 during embryogenesis, we performed knockdown studies with a translation-blocking Xctr1 morpholino oligonucleotide (Xctr1MO; SI Fig. 7). Dorsal marginal zone injection of Xctr1MO resulted in a phenotype similar to that seen in Xctr1 gain-of-function studies (compare Figs. 1A Left and 2A Left). Few, if any, defects were seen after injection of a control morpholino, Xctr1 mismatch (Xctr1MM), that differs from Xctr1 by five base-pair substitutions (Fig. 2A). Strikingly, more than half of the Xctr1MO-injected embryos did not form somites, as demonstrated by the absence of 12/101 staining (56%; n = 68) (Fig. 2A and SI Fig. 8). Differentiation was partially rescued, in a dose-dependent manner, by the coexpression of an Xctr1MO-insensitive Xctr1 (Xctr1*) construct (Fig. 2A).
Fig. 2.

Xctr1 knockdown inhibits differentiation and enhances morphogenesis. (A) Whole-mount immunohistochemistry of stage 32 embryos injected in the dorsal marginal zone at early cleavage stages with 250 ng of either Xctr1MO or Xctr1 5 base-pair mismatch (MM) morpholinos. Rescue experiments were performed by coexpression of a morpholino-insensitive Xctr1 construct (Xctr1*) in which the Xctr1MO-binding region was separated from the remainder of Xctr1 by 6 Myc epitope tags; somite formation was scored on a scale of 0 to 5 (0, complete, bilateral absence of 12/101 stain; 5, normal staining and somite morphology). Xctr1MO embryos were scored as 1.49 ± 0.3 (n = 41), and Xctr1MO + 500-pg Xctr1* embryos were scored as 2.01 ± 0.07 (n = 39). Xctr1* RNA (250 and 500 pg) were coinjected with Xctr1MO as indicated. (B) Effect of Xctr1 knockdown on FGF-mediated induction of Xbrachyury (Xbra) expression and its rescue by coexpression of 500 pg of Xctr1* RNA. Xctr1*, like Xctr1, induces expression of the neural marker NRP-1, albeit at higher doses than wild-type (data not shown). (C) Xctr1 knockdown inhibits Xbra expression in vivo. Whole-mount in situ hybridization analysis of gastrula-stage embryos using an antisense Xbra probe. Red-gal staining used as a lineage trace is demarcated with brackets; Xbra stain in Xctr1MO-injected embryos is indicated with arrows. (Upper) Marginal views. (Lower) Vegetal views. (D) Effects of Xctr1 knockdown on FGF- and activin-mediated morphogenesis of stage 20 animal caps. (E) Expression of dominant-negative Dishevelled (Xdd1) RNA inhibits Xctr1 knockdown-mediated elongation by FGF; 10 ng/ml FGF and 0.5 ng/ml (high) or 0.1 ng/ml (low) activin was added as indicated.
Consistent with the observed loss of somites in the Xctr1MO-injected embryos, Xctr1 knockdown inhibited the induction of the mesodermal early response gene Xbra by FGF at gastrula stages; this effect was partially rescued by coexpression of Xctr1* (Fig. 2B). Gastrula-stage Xbra expression was also potently inhibited by Xctr1 knockdown in vivo (Fig. 2C). At later stages of development, Xctr1 knockdown inhibited markers of dorsal mesoderm induced by either FGF or activin; in contrast, induction of ventrolateral mesoderm markers, including hoxb9, was only modestly inhibited by Xctr1MO injection (SI Fig. 9 A and B). No effects on gene expression or morphology were observed in animal cap explants injected with Xctr1MO or Xctr1MM and cultured without exogenous growth factors (SI Fig. 9C). These data indicate that Xctr1 is required during or before gastrulation for subsequent development of the mesoderm in Xenopus. We cannot at present determine whether Xctr1 knockdown results in a failure of mesoderm differentiation and/or a conversion of affected cells into distinct fates for which we have not yet assayed.
Misexpression studies suggested a role for Xctr1 during both differentiation and morphogenesis. Despite a requirement for Xctr1 in the development of the dorsal mesoderm, Xctr1 loss of function enhanced elongation of caps with FGF; this effect was partially rescued by coexpression of Xctr1* (Fig. 2D Top). The period during which FGF stimulated the elongation of Xctr1 morpholino-treated explants parallels the window of competence for FGF-mediated mesoderm induction, suggesting that FGF (or activin) in these studies at least partially mesodermalized explants, which then elongated in the absence of Xctr1 (SI Fig. 10A) (28). These data are consistent with the finding that Xbra expression is required for differentiation-independent morphogenesis by an activated form of the Shp2 phosphatase (29). Similar enhancement of elongation was seen in animal caps treated with low doses of activin (Fig. 2D Middle). Xctr1 knockdown did not markedly alter the elongation of animal caps treated with high doses of activin (Fig. 2D Bottom) and did not promote elongation of animal caps cultured in the absence of growth factors (data not shown). Elongation of FGF-treated explants by Xctr1MO was inhibited by a dominant-negative form of Dishevelled, which has been shown to regulate vertebrate convergent extension movements through a noncanonical Wnt “Planar Cell Polarity” pathway (Fig. 2E) (18). Therefore, Xctr1 inhibits morphogenesis upstream of or parallel to Dishevelled. Finally, we found that Xctr1 knockdown similarly promotes elongation of caudal neurectoderm, which undergoes extensive convergent extension during early development (30) (SI Fig. 10 B and C). Xctr1MO injection inhibited differentiation in this context as well: Markers of caudal neural fate, as well as the general neural markers NCAM and NRP-1, were all strongly inhibited by Xctr1 knockdown in explant cultures (SI Fig. 10 B and D). Inhibition of neural fate was also observed in the context of the intact embryo (SI Fig. 8). Xctr1 knockdown thus enhances morphogenetic movements and inhibits differentiation in both presumptive mesoderm and neurectoderm.
Mesoderm formation in Xenopus is dependent on the FGF-mediated activation of the ERK MAP kinase (27); phosphorylation of the docking protein SNT-1 by the tyrosine kinase Laloo is a critical intermediate in this process (10–12). Exogenous Xctr1, Laloo, and SNT-1 formed a trimeric complex in Xenopus embryos (Fig. 3A). Tyrosine phosphorylation of SNT-1 by Laloo was partially inhibited after Xctr1 knockdown, suggesting that Xctr1 regulates SNT-1 activity in vivo (Fig. 3B). Injection of Xctr1MO also inhibited ERK phosphorylation by FGF in gastrula-stage animal caps, demonstrating a key role for Xctr1 in FGF-dependent ERK activation (Fig. 3C). Ras activation transmits FGF receptor stimulation upstream of ERK and is essential for mesoderm formation in Xenopus, including the Smad pathway-mediated induction of the dorsal mesodermal genes collagen type II and muscle actin by activin (Fig. 3D) (1, 3, 27). Although expression of an activated form of Ras (v-Ras) was insufficient to induce dorsal mesoderm, coexpression of v-Ras rescued the block to activin-mediated dorsal mesoderm induction by Xctr1MO (Fig. 3E). Taken together these data suggest that Xctr1 is required upstream of Ras, as a component of the canonical FGF signaling cascade, during mesoderm formation. Expression of a dominant inhibitory form of Ras does not enhance FGF-mediated elongation, however, indicating that antagonism of canonical FGF signaling is not sufficient to promote morphogenesis (Fig. 3F).
Fig. 3.

Xctr1 regulates Ras-ERK signaling. (A) Coprecipitation of Xctr1, Laloo, and the docking protein, SNT-1/FRS2α. (B) Xctr1 knockdown inhibits Laloo-mediated SNT-1 phosphorylation (0.37 ± 0.11-fold reduction by Xctr1MO; n = 3). Signal represents the phosphorylation of up to six tyrosine residues on SNT-1 (10–12). (C) Xctr1 knockdown inhibits ERK phosphorylation (0.62 ± 0.27-fold reduction by MO; n = 4). Effects of injection of Xctr1MO and Xctr1MM on the FGF-mediated dual phosphorylation (dp) of the ERK MAP kinase in animal cap explants. Explants were cultured in the presence of FGF for 2 h before Western blot analysis; inhibition of ERK phosphorylation by Xctr1MO was not apparent for the first hour of FGF stimulation (data not shown). (D) Inhibition of activin-mediated dorsal mesoderm induction in animal cap explants by 1 ng of dominant inhibitory Ras (dnRas) RNA (1). (E) Rescue of Xctr1 morpholino-mediated dorsal mesoderm inhibition by coinjection of 40-pg constitutively active Ras (v-Ras) RNA (1). (F) Injection of 1 ng dnRas RNA does not enhance elongation of animal caps by FGF. Ras inhibition also inhibits elongation in FGF-treated caps from embryos injected with Xctr1MO (data not shown). (G) RT-PCR analysis of Xctr1 expression in early stage Xenopus embryos. (H) Whole-mount in situ hybridization analysis of Xctr1 expression. Stages (Upper) 4, 8 (animal views), 18 (dorsal view), and (Lower) 32 (lateral view) are shown. Pronephric tubules are indicated by arrowhead; 250-ng morpholinos were injected at early cleavage stages as listed, and 10 ng/ml FGF and 0.5 ng/ml activin were added to animal cap explants at stage 8 as listed.
Maternal Xctr1 RNA is expressed ubiquitously and at high levels through blastula stages and at much lower levels during gastrula and neurula stages (Fig. 3 G and H and data not shown). Zygotic Xctr1 transcripts were detected in the brain and pronephros (Fig. 3H). This expression pattern is consistent with an early role for Xctr1 in stimulating mesoderm differentiation and inhibiting premature convergent extension movements during Xenopus embryogenesis. The presumably high levels of maternal Xctr1 protein may explain the inability of Xctr1MO to fully block early FGF-mediated SNT-1 and ERK phosphorylation (Fig. 3 B and C).
Having demonstrated a requirement for Xctr1 in amphibian development, we next asked whether Ctr1 has a similar function in higher vertebrates. Mouse embryos lacking the Ctr1 gene display clear defects in gastrulation and mesoderm formation and die in utero (31, 32). Unfortunately, the early embryonic lethality of the Ctr1-null mutants precludes in-depth analysis of the function of this gene in cellular differentiation during murine embryogenesis. To define the requirement for Ctr1 in mammalian cell differentiation, we derived multiple ES cell lines from blastocysts of Ctr1+/− mouse crosses. The differentiation potential of Ctr1−/− ES cells was then assessed by culture in medium lacking leukemia inhibitory factor (LIF).
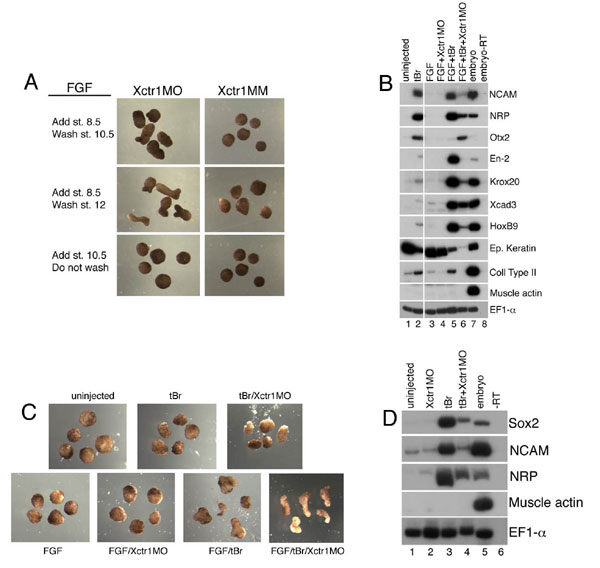
Cultures of Ctr1−/− cells proliferated less and maintained an undifferentiated phenotype compared with wild-type ES cells, which rapidly differentiated and generated cell types with distinct morphologies (Fig. 4A and data not shown). Because Xctr1 expression has profound effects on mesodermal development in Xenopus, we first asked whether cells expressing the general mesodermal marker, Flk-1 (33), were generated from Ctr1-deficient mouse ES cells. When allowed to differentiate as monolayers in vitro, Ctr1−/− ES cells failed to give rise to Flk-1(+) cells by day 4, the peak of mesodermal development in the wild-type ES cells (Fig. 4B). Ctr1+/− ES cells also showed a reduction in formation of Flk-1(+) cells (Fig. 4B). However, Ctr1+/− mice are viable and fertile, suggesting that any alterations in mesoderm development that may occur in the heterozygotes are not detrimental to embryonic development in vivo (31, 32). To further elucidate the defect in Ctr1−/− ES differentiation, we cultured the ES cells as embryoid bodies (EBs) and assessed the expression of stem cell and differentiation markers. Ctr1 expression was observed in wild-type ES cells and differentiating EBs at all stages examined. Ctr1 transcripts were not present in Ctr1−/− cells, confirming the deletion of the Ctr1 gene in these clones (Fig. 4C Top Left). Robust expression of the panmesodermal markers Flk-1 and Brachyury and the anterior streak and definitive endoderm marker Foxa2 was observed in wild-type cells; in contrast, in the Ctr1−/− EBs, induction was much weaker and was delayed by at least 1 day (Fig. 4C Middle and data not shown) (34–36). Consistent with the dramatic reduction in expression of mesodermal marker genes, Ctr1−/− EBs did not develop red pigmentation (i.e., they did not form erythroid cells) or develop into beating cardiomyocytes (data not shown). Although expression of Nanog, Oct3/4, and Rex1 (genes that maintain ES cell pluripotency) was rapidly down-regulated in wild-type cultures, expression of these genes was dramatically extended in Ctr1−/−EBs (Fig. 4C Bottom) (37). Ctr1 thus appears to regulate whether ES cells self-renew or begin to differentiate. Together these studies demonstrate that Ctr1 is essential for normal differentiation in both lower and higher vertebrates.
Fig. 4.

Ctr1−/− ES cells exhibit prolonged pluripotency and defects in differentiation. (A) Morphology of wild-type, Ctr1+/−, and Ctr1−/− monolayers cultured for 4 days in the absence of LIF. Two lines each of Ctr1+/− and Ctr1−/− ES cells were assayed with similar results. One line derived from a wild-type blastocyst served as a control. (B) Flow-cytometric analysis of Flk-1 protein expression in wild-type, Ctr1+/−, and Ctr1−/− monolayers cultured for 4 days in the absence of LIF. Levels of c-Kit, a protein expressed in undifferentiated ES cells, hematopoietic stem cells, and germ cells, were not altered in Ctr1+/− or Ctr1−/− monolayers (41). Plots correspond to cells in A, as denoted by arrows. (C) RT-PCR analysis of wild-type (+/+), Ctr1+/−, and Ctr1−/− EB cultures. Differentiation is inhibited in the Ctr1−/− EBs, whereas markers of pluripoptent ES and/or germ cell progenitors are enriched in these cultures. Morphology and expression of ES cell markers in Ctr1+/− lines generally resembled that seen in wild-type lines. Expression of markers of differentiated fates was often slightly delayed and/or diminished relative to that seen in wild-type cells, suggesting a modest Ctr1 haploinsufficient phenotype (this figure and data not shown). In this regard, we note that Ctr1+/− mice are viable and fertile (31, 32). β-actin was used as a loading control. (D) Proposed function of Ctr1 during early vertebrate development. (Upper) Model depicting a role for Xctr1 in the interpretation of FGF–receptor interaction as either a differentiation (Left) or a morphogenesis (Right) cue. (Lower) Comparison of Ctr1 function in amphibian embryonic and mammalian ES cell development. Red bars signify levels of Ctr1 activity.
Understanding the mechanisms that coordinate cell fate and movement remains a central issue in developmental biology. A necessary first step in unraveling these mechanisms is identifying the key cellular components that direct signals to induce differentiation versus morphogenetic responses. Our data indicate that Ctr1 plays an essential role in this process before, during, and shortly after gastrulation. During embryonic cell fate determination, Ctr1 links FGF signaling to ERK activation via Laloo-mediated SNT-1 phosphorylation. Down-regulation of Ctr1, however, not only inhibits canonical FGF signaling (and thus differentiation), but also promotes a distinct biological outcome (coordinated tissue movements) via stimulation of an alternate signaling cascade (Fig. 4D Upper). This latter network requires active Dishevelled and may stimulate Ca2+ release and/or PKCδ activation (13, 14).
This study also defines a conserved role for Ctr1 in the differentiation of Xenopus embryos and mouse ES cells and in the inhibition of tissue morphogenesis and stem cell pluripotency (Fig. 4D Lower). The observation that Oct4 expression is prolonged in embryoid bodies after treatment with the MEK (MAPK/ERK kinase) inhibitor PD098059 indicates that Ctr1 may promote differentiation through ERK pathway activation in mammalian ES cells as well as in the frog (38). The requirement for Ctr1 during ES cell development further suggests that Ctr1 is a key component of a conserved signal transduction mechanism by which both stem and progenitor cells establish and/or maintain the competence to differentiate. The elucidation of a role for Ctr1 in ES cell differentiation should facilitate characterization of the signaling networks that govern mammalian stem cell pluripotency.
Materials and Methods
Bacterial Two-Hybrid Screen.
Two-hybrid screening was performed as recommended (BacterioMatch II two-hybrid system; Stratagene, La Jolla, CA). The full coding sequence of Laloo was amplified by PCR (gggaattccGGCTGCATCAAGTCAAAGG, ccctcgagTTAAGGTTGTGCCTGGTAC) and ligated into the EcoRI and XhoI sites of pBT for use as bait to screen a Xenopus cDNA library (stages 38–40; Stratagene).
Xctr1 Cloning and Mutant Construction.
An image clone containing the Xenopus homolog Ctr1 was obtained from Open Biosystems (Huntsville, AL) (IMAGE ID: 6877868; accession no. AAH53785). The full coding sequence of Xctr1 was amplified by PCR and cloned into the BamHI and XhoI sites of pCS2++. For Myc-Xctr1 (also referred to as Xctr1*), the coding sequence of Xctr1, minus the start codon, was subcloned downstream of six Myc epitopes. All Xctr1-deletion constructs were fused downstream of either one V5 (GKPIPNPLLGLDST) or six Myc (EQKLISEEDLNEM) epitopes.
Mutations of Xctr1 amino acids 35–40, corresponding to the M2 region of human Ctr1 (MHMMAM→AAAAAAM, Xctr1-M2), and amino acid 149, corresponding to methionine-154 of human Ctr1 (M→A, Xctr1-M149) (39), were introduced by PCR with cloned Pfu (Stratagene) (M2, tgctgctgcCCCAGACCCTCCCCCat, p-gcagcagcaACCTTTTACTTTGGATA; M149, p-gcaACGTACAACGCTTACCTGTGC, GAAGATCAACATAAGGAAATAACT).
RT-PCR, Whole-Mount in Situ Hybridization and Immunohistochemistry, and Protein Interaction Assays.
Primers designed for this study were as follows: Xctr1, F-GTGTGTCTGCTGGAAACTAAG, R-TTCCCGCGAAATCTTCAG; Xcad3, F-agttcagccagtccaaac, R-gatggttgcagtgaatcc; Xenopus metallothionein, F-TCTTGCTCCTGTGGTACTAC, R-AGTTAGTCCTCTGCCCACTG; and mCtr1 genotyping primers (used as a mix of three primers), CT57- GCTATGGTATTTGGGATCCTGCAAA, CT2′-CTACAGCAATTAATAAGGCCATATG, and Neo3730-CGATTCGCAGCGCATCGCCTTCTAT. All other primer sequences are as described (40). Whole-mount antibody staining and in situ hybridization was performed as described (9). The 12/101 and Xen-1 antibodies (Developmental Studies Hybridoma Bank, Iowa City, IA) were used at a 1:1 dilution for somite and neural staining, respectively. The secondary antibody, a donkey anti-mouse IgG coupled to horseradish peroxidase (Jackson ImmunoResearch Laboratories, West Grove, PA), was used at 1:1,000 dilution. Color reactions were performed by using the Vector SG and DAB kits (Vector Laboratories, Burlingame, CA). Coimmunoprecipitation assays were performed as described (11). SNT-Myc and Laloo-Flag constructs were described previously (10).
Morpholinos.
Morpholino oligonucleotides (Gene Tools, Philomath, OR) were heated for 5 min at 65°C and then quenched on ice before injection. Sequences were as follows: Xctr1-MO, ATATGGTGAGAGTGATTCATGTCCA; mismatch/MM, ATATcGTGAcAGTcATTgATcTCCA; and generic control morpholino, 5′-CCTCTTACCTCAGTTACAATTTATA.
ES Cell Generation.
ES cell lines were generated from flushed blastocysts obtained by mating of Ctr1+/− heterozygous mice. Cells from microdissected blastocysts were initially grown on STO feeder cells in the presence of LIF (ESGRO; Chemicon International, Temecula, CA) and expanded to generate stable ES cells lines, which were genotyped by PCR. Cells were subsequently passaged in the presence of LIF without STO cells.
ES Cell Culture, EB Culture, and FACS Analysis.
ES cells were differentiated as EBs for ≤15 days as described (41). For monolayer analysis of ES differentiation, 50,000 ES cells were plated onto 10-cm2 dishes precoated with 0.1% gelatin [protocol modified from Nishikawa et al. (42)]. At day 4, cells were pelleted and incubated with anti-mouse Flk1 Ab (eBiosciences, San Diego, CA) conjugated to biotin for 20 min on ice. After washes with PBS/10% FCS, cells were incubated with streptavidin-APC (BD Biosciences, San Jose, CA) for 20 min, washed, and resuspended in PBS/10% FCS with 0.01% propidium iodide for dead cell exclusion. Flow cytometry was performed by using a FACSCalibur (BD Biosciences) at the Flow Cytometry Core Facility at the Mount Sinai School of Medicine. Data were analyzed by using the Flowjo software package (TreeStar, Ashland, OR).
Supplementary Material
Acknowledgments
We thank E. Amaya, A. Brivanlou, J. Gitschier, S. Sokol, D. Thiele, and P. Wilson for gifts of reagents. This work was supported by Public Health Service Grants R01-GM61671 (to D.C.W.) and R01-DK52191, R01-HL62248, and R01-EB02209 (to M.H.B.), and an Exploratory Research Award from The Black Family Stem Cell Institute (to D.C.W.). Y.-M.K. was supported by the Howard Hughes Medical Institute.
Abbreviations
- EB
embryoid body
- LIF
leukemia inhibitory factor.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/cgi/content/full/0701413104/DC1.
References
- 1.Whitman M, Melton DA. Nature. 1992;357:252–254. doi: 10.1038/357252a0. [DOI] [PubMed] [Google Scholar]
- 2.MacNicol AM, Muslin AJ, Williams LT. Cell. 1993;73:571–583. doi: 10.1016/0092-8674(93)90143-e. [DOI] [PubMed] [Google Scholar]
- 3.LaBonne C, Burke B, Whitman M. Development (Cambridge, UK) 1995;121:1475–1486. doi: 10.1242/dev.121.5.1475. [DOI] [PubMed] [Google Scholar]
- 4.Gotoh Y, Masuyama N, Suzuki A, Ueno N, Nishida E. EMBO J. 1995;14:2491–2498. doi: 10.1002/j.1460-2075.1995.tb07246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Umbhauer M, Marshall CJ, Mason CS, Old RW, Smith JC. Nature. 1995;376:58–62. doi: 10.1038/376058a0. [DOI] [PubMed] [Google Scholar]
- 6.Tang TL, Freeman RM, Jr, O'Reilly AM, Neel BG, Sokol SY. Cell. 1995;80:473–483. doi: 10.1016/0092-8674(95)90498-0. [DOI] [PubMed] [Google Scholar]
- 7.Gupta RW, Mayer BJ. Oncogene. 1998;17:2155–2165. doi: 10.1038/sj.onc.1202158. [DOI] [PubMed] [Google Scholar]
- 8.Carballada R, Yasuo H, Lemaire P. Development (Cambridge, UK) 2001;128:35–44. doi: 10.1242/dev.128.1.35. [DOI] [PubMed] [Google Scholar]
- 9.Weinstein DC, Marden J, Carnevali F, Hemmati-Brivanlou A. Nature. 1998;394:904–908. doi: 10.1038/29808. [DOI] [PubMed] [Google Scholar]
- 10.Hama J, Xu H, Goldfarb M, Weinstein DC. Mech Dev. 2001;109:195–204. doi: 10.1016/s0925-4773(01)00524-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hama J, Suri C, Haremaki T, Weinstein DC. J Biol Chem. 2002;277:19806–19810. doi: 10.1074/jbc.M110637200. [DOI] [PubMed] [Google Scholar]
- 12.Kusakabe M, Masuyama N, Hanafusa H, Nishida E. EMBO Rep. 2001;2:727–735. doi: 10.1093/embo-reports/kve152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nutt SL, Dingwell KS, Holt CE, Amaya E. Genes Dev. 2001;15:1152–1166. doi: 10.1101/gad.191301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sivak JM, Petersen LF, Amaya E. Dev Cell. 2005;8:689–701. doi: 10.1016/j.devcel.2005.02.011. [DOI] [PubMed] [Google Scholar]
- 15.Dancis A, Yuan DS, Haile D, Askwith C, Eide D, Moehle C, Kaplan J, Klausner RD. Cell. 1994;76:393–402. doi: 10.1016/0092-8674(94)90345-x. [DOI] [PubMed] [Google Scholar]
- 16.Kintner CR, Brockes JP. Nature. 1984;308:67–69. doi: 10.1038/308067a0. [DOI] [PubMed] [Google Scholar]
- 17.Klein PS, Melton DA. Endocr Rev. 1994;15:326–341. doi: 10.1210/edrv-15-3-326. [DOI] [PubMed] [Google Scholar]
- 18.Keller R. Science. 2002;298:1950–1954. doi: 10.1126/science.1079478. [DOI] [PubMed] [Google Scholar]
- 19.Bieker JJ, Yazdani-Buicky M. J Histochem Cytochem. 1992;40:1117–1120. doi: 10.1177/40.8.1619277. [DOI] [PubMed] [Google Scholar]
- 20.Mohun TJ, Brennan S, Dathan N, Fairman S, Gurdon JB. Nature. 1984;311:716–721. doi: 10.1038/311716a0. [DOI] [PubMed] [Google Scholar]
- 21.Richter K, Good PJ, Dawid IB. New Biol. 1990;2:556–565. [PubMed] [Google Scholar]
- 22.Puig S, Thiele DJ. Curr Opin Chem Biol. 2002;6:171–180. doi: 10.1016/s1367-5931(02)00298-3. [DOI] [PubMed] [Google Scholar]
- 23.Aller SG, Unger VM. Proc Natl Acad Sci USA. 2006;103:3627–3632. doi: 10.1073/pnas.0509929103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Safaei R, Howell SB. Crit Rev Oncol Hematol. 2005;53:13–23. doi: 10.1016/j.critrevonc.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 25.Lee J, Petris MJ, Thiele DJ. J Biol Chem. 2002;277:40253–40259. doi: 10.1074/jbc.M208002200. [DOI] [PubMed] [Google Scholar]
- 26.Dameron CT, Harrison MD. Am J Clin Nutr. 1998;67:1091S–1097S. doi: 10.1093/ajcn/67.5.1091S. [DOI] [PubMed] [Google Scholar]
- 27.Heasman J. Development (Cambridge, UK) 2006;133:1205–1217. doi: 10.1242/dev.02304. [DOI] [PubMed] [Google Scholar]
- 28.Green JB, Howes G, Symes K, Cooke J, Smith JC. Development (Cambridge, UK) 1990;108:173–183. doi: 10.1242/dev.108.1.173. [DOI] [PubMed] [Google Scholar]
- 29.O'Reilly AM, Pluskey S, Shoelson SE, Neel BG. Mol Cell Biol. 2000;20:299–311. doi: 10.1128/mcb.20.1.299-311.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wallingford JB, Harland RM. Development (Cambridge, UK) 2001;128:2581–2592. doi: 10.1242/dev.128.13.2581. [DOI] [PubMed] [Google Scholar]
- 31.Lee J, Prohaska JR, Thiele DJ. Proc Natl Acad Sci USA. 2001;98:6842–6847. doi: 10.1073/pnas.111058698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuo YM, Zhou B, Cosco D, Gitschier J. Proc Natl Acad Sci USA. 2001;98:6836–6841. doi: 10.1073/pnas.111057298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ema M, Takahashi S, Rossant J. Blood. 2006;107:111–117. doi: 10.1182/blood-2005-05-1970. [DOI] [PubMed] [Google Scholar]
- 34.Robertson SM, Kennedy M, Shannon JM, Keller G. Development (Cambridge, UK) 2000;127:2447–2459. doi: 10.1242/dev.127.11.2447. [DOI] [PubMed] [Google Scholar]
- 35.Kabrun N, Buhring HJ, Choi K, Ullrich A, Risau W, Keller G. Development (Cambridge, UK) 1997;124:2039–2048. doi: 10.1242/dev.124.10.2039. [DOI] [PubMed] [Google Scholar]
- 36.Kubo A, Shinozaki K, Shannon JM, Kouskoff V, Kennedy M, Woo S, Fehling HJ, Keller G. Development (Cambridge, UK) 2004;131:1651–1662. doi: 10.1242/dev.01044. [DOI] [PubMed] [Google Scholar]
- 37.Boiani M, Scholer HR. Nat Rev Mol Cell Biol. 2005;6:872–884. doi: 10.1038/nrm1744. [DOI] [PubMed] [Google Scholar]
- 38.Burdon T, Stracey C, Chambers I, Nichols J, Smith A. Dev Biol. 1999;210:30–43. doi: 10.1006/dbio.1999.9265. [DOI] [PubMed] [Google Scholar]
- 39.Puig S, Lee J, Lau M, Thiele DJ. J Biol Chem. 2002;277:26021–26030. doi: 10.1074/jbc.M202547200. [DOI] [PubMed] [Google Scholar]
- 40.Suri C, Haremaki T, Weinstein DC. Dev Biol. 2004;265:90–104. doi: 10.1016/j.ydbio.2003.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Keller G, Kennedy M, Papayannopoulou T, Wiles MV. Mol Cell Biol. 1993;13:473–486. doi: 10.1128/mcb.13.1.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Nishikawa SI, Nishikawa S, Hirashima M, Matsuyoshi N, Kodama H. Development (Cambridge, UK) 1998;125:1747–1757. doi: 10.1242/dev.125.9.1747. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}