

Abstract
Objective
Interleukin-10 (IL-10) exerts potent anti-inflammatory actions and modulates matrix metalloproteinase expression. We hypothesized that endogenous IL-10 may regulate infarct healing and left ventricular remodeling by promoting resolution of the post-infarction inflammatory response and by modulating extracellular matrix metabolism.
Methods
IL-10 null and wildtype (WT) mice underwent reperfused infarction protocols. We compared the healing response and remodeling-associated parameters between IL-10 -/- and WT infarcts. In addition, we studied the effects of IL-10 on inflammatory gene synthesis by stimulated murine cardiac fibroblasts.
Results
Infarcted IL-10 -/- mice exhibited comparable mortality rates with WT animals. Although IL-10 -/-mice had higher peak tumor necrosis factor (TNF)-α and monocyte chemoattractant protein (MCP)-1/CCL2 mRNA levels in the infarcted heart than WT mice, both groups demonstrated timely repression of pro-inflammatory cytokine and chemokine mRNA synthesis after 24h of reperfusion and exhibited a similar time course of resolution of the neutrophil infiltrate. IL-10 gene disruption did not alter fibrous tissue deposition and dilative remodeling of the infarcted heart. Pre-incubation with IL-10 did not modulate the pro-inflammatory phenotype of TNF-α-stimulated cardiac fibroblasts, failing to inhibit chemokine mRNA synthesis. In contrast, transforming growth factor (TGF)-β1 pre-incubation suppressed interferon-γ-inducible protein (IP)-10/CXCL10 synthesis by cardiac fibroblasts exposed to TNF-α.
Conclusions
IL-10 signaling plays a non-critical role in suppression of inflammatory mediators, resolution of the inflammatory response, and fibrous tissue deposition following myocardial infarction. This may be due to the relative selectivity of IL-10-mediated anti-inflammatory actions, with respect to cell type and stimulus. Resolution of postinfarction inflammation is likely to involve multiple overlapping regulatory mechanisms controlling various pro-inflammatory pathways activated in the infarcted myocardium.
INTRODUCTION
Myocardial infarction triggers an intense inflammatory reaction associated with induction of pro-inflammatory cytokines and chemokines, and recruitment of neutrophils and mononuclear cells in the infarcted area [1], [2]. Release of pro-inflammatory mediators and leukocyte infiltration play an important role in clearance of the wound from dead cells and matrix debris. Pro-inflammatory signals are crucial in mediating the response to injury, regulate debridement of the infarcted myocardium and may initiate the cellular events necessary for scar formation. However, optimal healing requires activation of inhibitory mechanisms suppressing cytokine and chemokine synthesis and mediating resolution of the inflammatory infiltrate. These “stop signals” may prevent persistent inflammation, inhibiting tissue injury and promoting the healing process [3].
Few studies have explored the significance of pathways inhibiting inflammation in healing myocardial infarcts. Timely suppression of the inflammatory response appears to be important for containment of injury and optimal infarct healing. However, the mechanisms responsible for repression of inflammatory gene synthesis and resolution of the leukocytic infiltrate remain poorly understood. We have recently demonstrated the critical role of endogenous thrombospondin (TSP)-1, a crucial TGF-β activator [4], in resolution and containment of the inflammatory response following myocardial infarction [5]. TSP-1 null mice demonstrated prolonged cytokine and chemokine expression and expansion of the inflammatory infiltrate into the non-infarcted area, resulting in enhanced adverse remodeling [5]. These findings suggested that activation of TGF-β signaling pathways may be important in resolution and spatial containment of the inflammatory response following myocardial infarction.
Interleukin (IL)-10 is another candidate gene with a potential role in repression of inflammatory gene synthesis. IL-10 is a mononuclear cell product that potently inhibits production of pro-inflammatory cytokines (such as IL-1, IL-6 and TNF-α) by activated monocyte/macrophages [6], [7] and suppresses synthesis of both CC and CXC chemokines by stimulated monocytes [7], [8]. We have previously reported marked IL-10 upregulation in both canine [9] and mouse [10] myocardial infarcts, and suggested that IL-10 induction may suppress expression of proinflammatory cytokines in the infarcted myocardium. Beyond its suppressive effects on inflammatory gene synthesis, IL-10 also regulates extracellular matrix metabolism [11] and angiogenesis [12]. IL-10-mediated tissue inhibitor of metalloproteinases (TIMP)-1 upregulation [11], [9] may play an important role in promoting extracellular matrix deposition in the infarct [9].
Investigations using IL-10 -/- animals have suggested an important role for endogenous IL-10 in suppressing peak inflammation following myocardial infarction. IL-10 null mice exhibited markedly increased mortality, larger infarct size and enhanced peak serum TNF-α levels [13], [14] compared with wildtype (WT) animals. Although these investigations suggested cardioprotective effects of IL-10 in the early stages of myocardial infarction, the potential role of IL-10 in resolution of inflammation and in regulation of the cellular and molecular events associated with infarct healing and post-infarction remodeling has not been investigated. The current study was designed to examine the potential effects of IL-10 in resolution of inflammation and regulation of fibrous tissue deposition and cardiac remodeling following reperfused myocardial infarction. In addition, we investigated the in vitro effects of IL-10 in modulating pro-inflammatory activation of isolated murine cardiac fibroblasts.
METHODS
1. Murine model of reperfused infarction
The investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No 85-23, revised 1996). IL-10 null and WT C57/BL/6 mice were purchased from Jackson laboratories. Female WT and IL-10 -/-mice, 8-12 weeks of age (18.0-22.0 g body weight), were anesthetized by an intraperitoneal injection of sodium pentobarbital (60 μg/g). A closed-chest mouse model of reperfused myocardial infarction was utilized as previously described [15], [10], in order to avoid the confounding effects of surgical trauma and inflammation, which may influence the baseline levels of chemokines and cytokines. The left anterior descending (LAD) coronary artery was occluded for 1h then reperfused for 6h to 7 days. At the end of the experiment, the chest was opened and the heart was immediately excised, fixed in zinc-formalin, and embedded in paraffin for histological studies, or snap frozen and stored at –80°C for RNA isolation. Sham animals were prepared identically without undergoing coronary occlusion/reperfusion. Animals used for histology underwent 24 h, 72 h, and 7 day reperfusion protocols (8 animals per group). Mice used for RNA extraction underwent 6 h, 24 h, and 72 h of reperfusion (8 animals per group). Additional animals (IL-10 null mice, n=16; WT mice, n=14) were used for perfusion-fixation after 7 days of reperfusion in order to assess remodeling-associated parameters.
2. Immunohistochemistry and quantitative histology
Murine hearts were fixed in zinc-formalin (Z-fix; Anatech, Battle Creek, MI), and embedded in paraffin. Sections were cut at 3 μm and stained immunohistochemically with the following antibodies: monoclonal anti-α smooth muscle actin (α-SMA) antibody (Sigma, St. Louis, MO), rat anti-mouse macrophage antibody Mac-2 (Cedarlane), and rat anti-neutrophil antibody (Serotec, Raleigh NC). Staining was performed using a peroxidase-based technique with the Vectastain ELITE rat, or goat kit (Vector Labs, Burlingame, CA) and developed with diaminobenzidine + nickel (Vector). The Mouse on Mouse (MOM) kit (Vector) was used for α-SMA immunohistochemistry. Quantitative assessment of neutrophil and macrophage density was performed by counting the number of neutrophils and Mac-2-immunoreactive cells respectively in the infarcted area as previously described [16]. Myofibroblasts were identified as extravascular α-SMA positive cells and counted in the infarcted myocardium [5]. Macrophage, neutrophil and myofibroblast density was expressed as cells/mm2. The collagen network was assessed using picrosirius red staining as previously described [16].
3. Echocardiography
Echocardiographic studies were performed prior to instrumentation and after 7 days of reperfusion (WT: n=7, IL-10 KO: n=8) using an 8 MHz probe (Sequoia C256; Acuson, Mountain View, CA). Short axis M-mode was used for measurement of systolic and diastolic ventricular and anterior wall diameters. The following echocardiographic parameters were measured as indicators of function and remodeling: Left Ventricular End-Diastolic Diameter (LVEDD), Left Ventricular End-Systolic Diameter (LVESD), Fractional shortening (FS=[LVEDD-LVESD]x100/LVEDD). The percent change in these parameters after infarction was quantitatively assessed using the following formulas: ΔLVEDD= (LVEDD 7 days – LVEDD pre)X100/LVEDD pre, ΔLVESD= (LVESD 7 days – LVESD pre)X100/LVESD pre, ΔFS= (FS pre – FS 7 days)X100/FS pre.
4. Perfusion fixation and assessment of ventricular volumes
For assessment of post-infarction remodeling, infarcted hearts after 7 days of reperfusion were used for perfusion-fixation (n=14 for WT and n=16 for IL-10 null animals) as previously described [16]. A cardioplegic solution was perfused through the jugular vein to promote relaxation. After excision and rinsing in cold cardioplegic solution, the aorta was cannulated, a PE-50 catheter was pushed into the left ventricle and secured in placed. Hearts were fixed for 10 min with 10% zinc-buffered formalin by aortic perfusion. After paraffin embedding, the entire heart was cross-sectioned from base to apex at 250μ intervals. 10 serial 5μ sections were obtained at each interval. The Left Ventricular End-Diastolic Volume (LVEDV) and LVEDD were assessed with ImagePro software using methods developed in our laboratory [17], [16], [9]. The size of the infarct was expressed as a percentage of the left ventricular volume.
5. RNA extraction and Ribonuclease protection assay
Inflammatory gene expression in murine hearts was assessed using Ribonuclease Protection Assay (RPA) as previously described [10]. The mRNA expression level of the chemokines Macrophage Inflammatory Protein (MIP)-1α, MIP-1β, MIP-2, MCP-1, and IP-10, the cytokines TNF-α, IL-1β, IL-6, TGF-β1, 2, and 3, was determined using a ribonuclease protection assay (RPA) (RiboQuant; Pharmingen) according to the manufacturer’s protocol. Phosphorimaging of the gels was performed (Storm 860; Molecular Dynamics, Sunnyvale, CA) and signals were quantified using Image QuaNT software and normalized to the ribosomal protein L32 mRNA.
6. Isolation and stimulation of murine cardiac fibroblasts
Mouse cardiac fibroblasts were isolated and cultured according to the method of Egbalhi-Webb [18], [19] as modified by Lafontant et al. [20]. Briefly, mouse hearts were obtained from three to five 12 -week-old C57BL/6J mice, were minced, and digested in 100 U/ml collagenase CLS 2 (Worthington Biochemical, Lakewood, NJ) at 37° C. Supernatants were filtered through a 70 um filter. Cells passed through the filter were pelleted, washed to remove the collagenase, then resuspended in Dulbecco’s modified Eagles medium (DMEM) containing 10% fetal bovine serum (FBS), and antibiotics (PenG 100U/ml, Streptomycin sulfate 100 ug/ml, Amphotericin B 0.25 ug/ml, GIBCO, Grand Island, NY), plated on 25 or 75-cm2flasks, and incubated at 37° C. Adherent cells were characterized at passage 1 by immunofluorescence microscopy and found to be positive for vimentin and α-SMA, but negative for desmin, and CD31 (CD31/PECAM-1; a leukocyte and endothelial cell marker). Fibroblasts were used up to passage 4 for cytokine stimulation studies. 16 hours prior to each experiment, cells were incubated in DMEM with 2% serum. Cells were washed 3 times in Hank’s Balanced Salt Solution (HBSS) prior to treatment with cytokines. All experiments were performed in serum-free DMEM. In order to study the effects of the inhibitory cytokines IL-10 and TGF-β1 in modulating inflammatory chemokine synthesis, cardiac fibroblasts were stimulated with IL-10 (100 ng/ml), or TGF-β1(10 ng/nl) (both from R&D Systems, San Diego, CA), or pre-incubated with IL-10 (100 ng/ml) and TGF-β1 (10 ng/ml) 4h prior to stimulation with recombinant TNF-α (100 ng/ml) (R&D Systems). At the end of the experiment total RNA was isolated from the fibroblasts using the acid guanidinium thiocyanate-phenol-chloroform method. mRNA expression of the chemokines MIP-2, MCP-1, MIP-1α, MIP-β1 and IP-10 was assessed with a custom made RPA kit (Pharmingen).
7. Statistical analysis
Statistical analysis was performed using ANOVA followed by t-test corrected for multiple comparisons (Student-Newman-Keuls). Paired t-test was used to compare echocardiographic parameters prior to myocardial infarction and after 7 days of reperfusion. Data were expressed as mean ± SEM. Statistical significance was set at 0.05.
RESULTS
1. IL-10 null mice showed comparable mortality rates following reperfused infarction
Mortality during the initial instrumentation surgery was 10.9% in the IL-10 null group and 13.8% in WT animals. IL-10 -/- mice undergoing reperfused infarction protocols showed mortality rates comparable with WT animals (mortality rate: 8.1 % in -/- mice vs. 9.0 % in WT animals).
2. Il-10 gene disruption does not affect cardiac morphology and inflammatory gene expression in the absence of myocardial infarction
Sham IL-10 null hearts exhibited normal cardiac morphology and showed no evidence of spontaneous inflammation, or increased fibrosis. No infiltrating neutrophils were found in sham hearts from IL-10 -/- and WT animals. Mac-2 immunohistochemistry identified a small population of macrophages in both IL-10 null and WT sham hearts. Picrosirius red staining demonstrated that the interstitial matrix network was comparable in non-infarcted IL-10 -/- and WT hearts. Both IL-10 -/- and WT sham hearts demonstrated negligible mRNA expression of the chemokines MCP-1, MIP-1α, MIP-1β, IP-10 and MIP-2 and the pro-inflammatory cytokines IL-1β, TNF-β and IL-6. TGF-β isoform mRNA levels were comparable in sham hearts from IL-10 null and WT animals (not shown). Echocardiographic assessment prior to surgical instrumentation demonstrated that IL-10 null and WT hearts had similar left ventricular dimensions and normal systolic function (Table 1).
Table 1.
Assessment of echocardiographic parameters in the infarcted heart
| WT pre | WT 1h/7d | IL-10 -/- pre | IL-10 -/- 1h/7d | |
|---|---|---|---|---|
| LVEDD (mm) | 3.37±0.08 | 3.63±0.07** | 3.22±0.04 | 3.63±0.06** |
| LVESD (mm) | 1.96±0.08 | 2.32±0.02** | 1.84±0.05 | 2.44±0.11** |
| FS | 0.42±0.01 | 0.35±0.01** | 0.43±0.02 | 0.33±0.02** |
| WT | IL-10 -/- | |||
| Δ LVEDD (%) | 8.2±1.7 | 12.9±2.7 | ||
| Δ FS (%) | 13.63±3.4 | 22.5±5.9 | ||
| Δ LVESD (%) | 19.7±5.5 | 33.7±8.8 | ||
LVEDD, Left Ventricular End-diastolic Diameter; LVESD, Left ventricular End-Systolic Diameter; FS, Fractional Shortening;ΔLVEDD (%), (LVEDD post-LVEDD pre)X100/LVEDD pre; ΔFS, (FS pre-FS post)X100/FS pre
p<0.01 vs. corresponding pre-infarction values.
3. IL-10 null mice demonstrate timely resolution of the neutrophilic infiltrate
Reperfused myocardial infarction resulted in rapid and intense neutrophil infiltration in the ischemic myocardium (Fig. 1). After 24h of reperfusion, IL-10 null mice showed comparable peak neutrophil density with WT animals (neutrophil density after 24h of reperfusion: IL-10 -/-1169.5±67.9 cells/mm2 vs. WT 1008±30.6 cells/mm2, pNS, n=8). After 72h of reperfusion, both IL-10 null and WT mice exhibited a similar decrease in neutrophil density (72h reperfusion IL-10 -/- 803±70.9 vs. WT 756.2±53.4, pNS, n=8) indicating that resolution of the granulocytic infiltrate was not significantly affected by the absence of IL-10 (Fig. 1).
Figure 1.
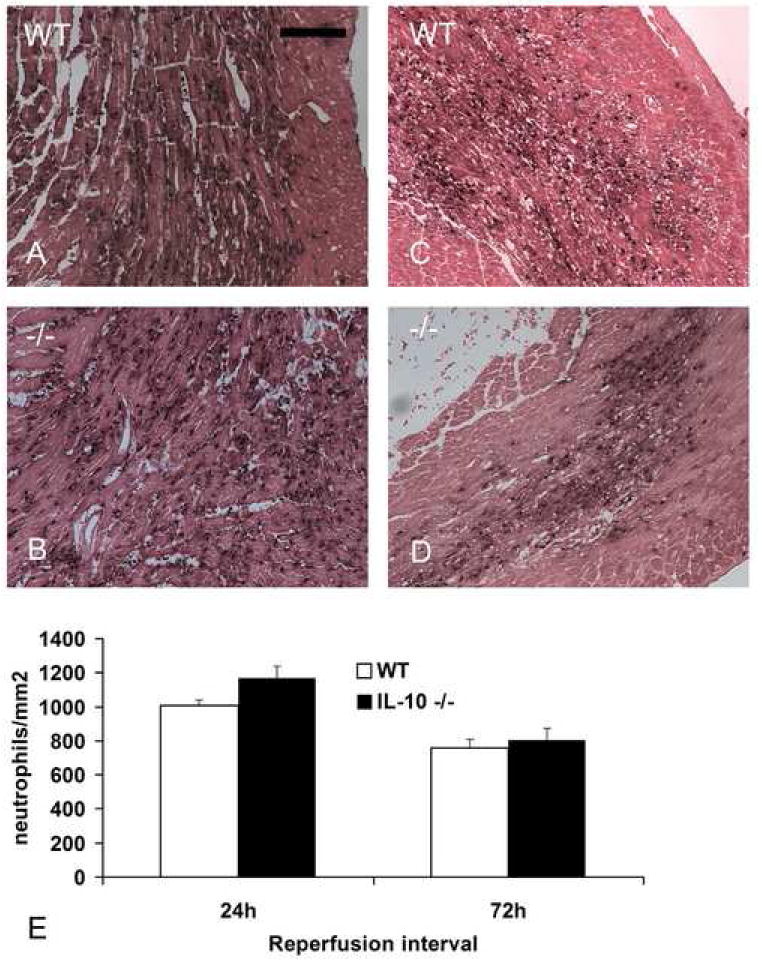
Neutrophil infiltration in the infarcted heart. Both WT (A) and IL-10 -/- mice (B) demonstrated intense neutrophil infiltration in the infarcted myocardium after 24h of reperfusion. Partial resolution of the neutrophilic infiltrate was noted after 72h of reperfusion in WT (C) and IL-10 null animals (D). E. Quantitative analysis shows no significant difference in neutrophil density between WT and -/- animals (scale bar=70 μm).
4. Time course of macrophage infiltration in IL-10 null infarcts
Mac-2 immunohistochemistry identified macrophages infiltrating the infarcted myocardium (Fig. 2). IL-10 -/- mice and their WT littermates showed no significant difference in the time course of macrophage infiltration and had comparable macrophage density in the infarcted area (Fig. 2E).
Figure 2.
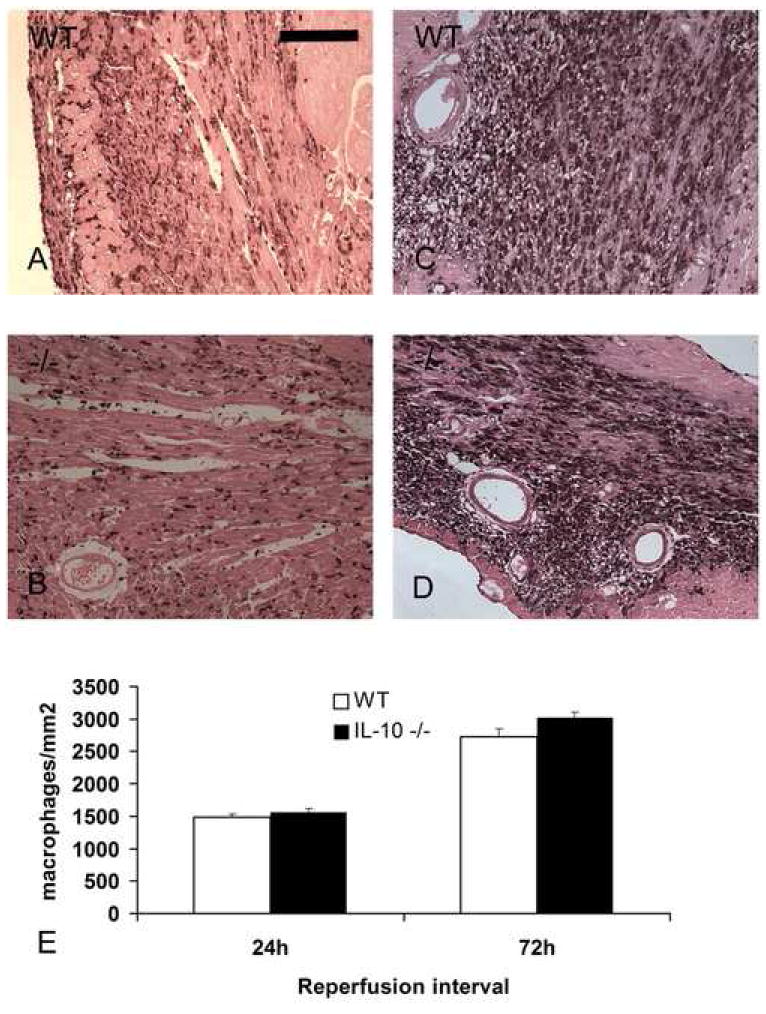
Mac-2 immunohistochemistry identifies macrophages in the infarcted mouse heart. Comparable macrophage numbers were found after 24h of reperfusion in WT (A) and IL-10 -/- (B) infarcts. The number of Mac-2 immunoreactive cells increased after 72h of reperfusion in both WT (C) and IL-10 null animals (D). E. Quantitative analysis shows comparable macrophage density in IL-10 -/- and WT infarcts (scale bar=70 μm).
5. IL-10 null animals exhibit increased peak TNF-α mRNA expression, but show timely resolution of the cytokine response
Reperfused mouse infarcts exhibit a marked but transient induction of the pro-inflammatory cytokines TNF-α, IL-1β and IL-6 that peaks after 6h of reperfusion. IL-10 null mice showed higher peak TNF-α mRNA levels than WT controls after 6h of reperfusion (fig. 3A); however, peak IL-1β and IL-6 mRNA expression was similar (Fig. 3B, 3C). Timely repression of pro-inflammatory cytokine mRNA expression was noted after 24-72h in both IL-10 null and WT animals (Fig. 3). The time course of TGF-β isoform expression was comparable in WT and IL-10 null infarcts (not shown).
Figure 3.
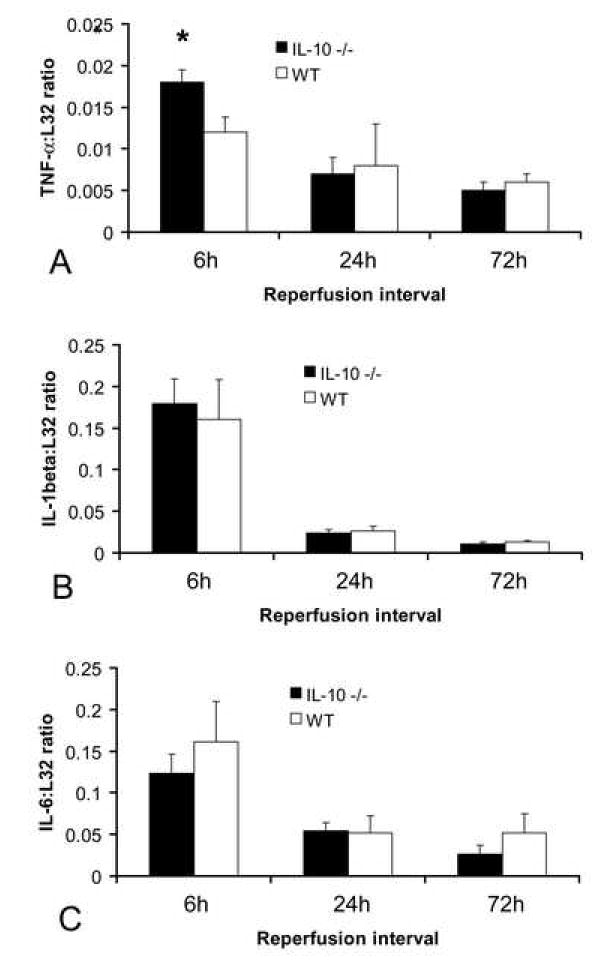
Cytokine mRNA expression in healing myocardial infarcts. A. RPA analysis showed that IL-10 null animals had slightly higher TNF-α mRNA levels in the infarcted heart than WT mice (*p<0.05). In contrast, IL-1β (B) and IL-6 (C) mRNA expression was comparable in both groups. Both WT and IL-10 null mice exhibited timely repression of pro-inflammatory cytokine synthesis after 24-72h of reperfusion (A-C).
6. IL-10 deficiency results in enhanced peak MCP-1 expression, but does not alter the time course of repression of the chemokine response
Reperfused mouse infarcts show rapid and transient induction of chemokine mRNA. IL-10 null animals exhibited enhanced and prolonged MCP-1 mRNA upregulation in the infarcted myocardium (Fig. 4A) (MCP-1: L32 ratio after 6h reperfusion: WT 0.586±0.05 vs. IL-10 -/- 0.89+ 0.055 **p≺0.01, and after 24h reperfusion: WT 0.21+0.017 vs. IL-10 -/- 0.334±0.039 *p<0.05). In contrast, MIP-2, MIP-1α (Fig. 4B), MIP-1β (Fig. 4C), and IP-10 mRNA expression was comparable in IL-10 null and WT infarcts. Timely repression of the chemokine response was noted in both IL-10 null and WT infarcts.
Figure 4.
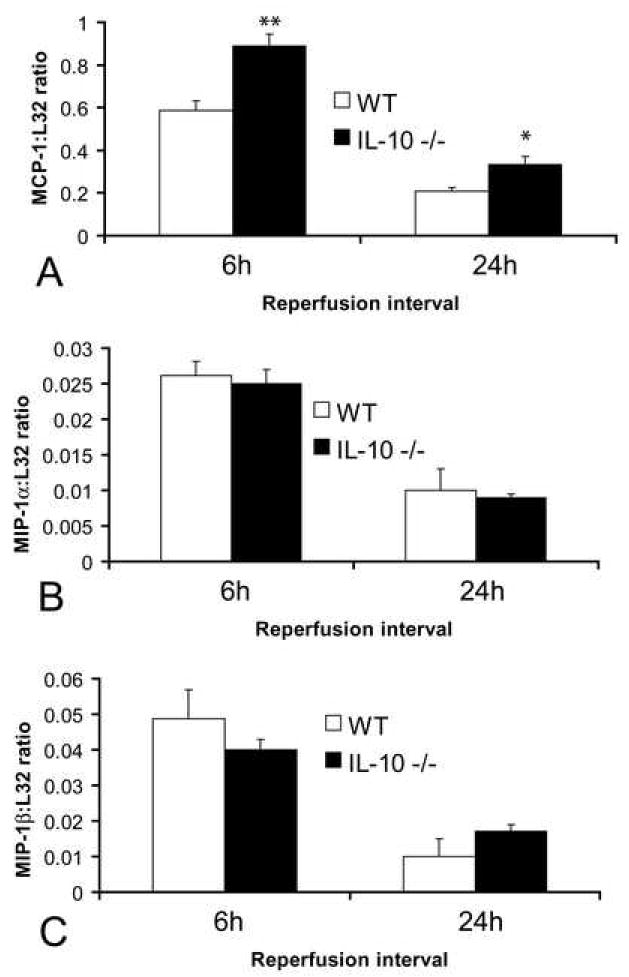
Chemokine mRNA expression in healing infarcts. A. IL-10 null mice had higher MCP-1 mRNA expression in the infarcted heart compared with WT animals. In contrast expression of the CC chemokines MIP-1α (B) and MIP-1β (C) was comparable between groups.
7. Myofibroblast infiltration in infarcted IL-10 -/- animals
In order to examine the effects of IL-10 absence on fibrous tissue deposition we quantitatively assessed myofibroblast density in IL-10 null and WT infarcts. In both WT and IL-10 -/- mice, dead cardiomyocytes are replaced with granulation tissue after 72h of reperfusion and abundant myofibroblasts infiltrate the infarcted myocardium, predominantly localized in the border zone (figure 5A). IL-10 null mice showed comparable myofibroblast density in the infarcted myocardium with their WT littermates (IL-10 -/- 856±88 cells/mm2 vs. WT 847.5±126.9 cells/mm2, pNS, n=8).
Figure 5.
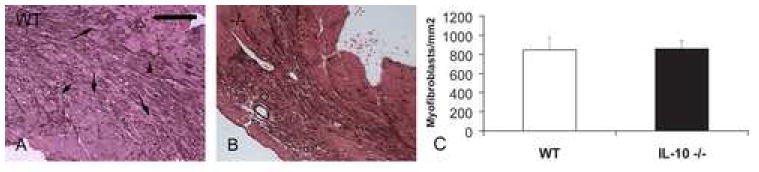
Myofibroblasts are identified in the healing infarct as extravascular α-SMA-expressing spindle-shaped cells. A. After 72 h of reperfusion dead cardiomyocytes are replaced with granulation tissue, and myofibroblasts infiltrate the infarcted area, predominantly localized in the border zone (arrows). B. IL-10 null mice show comparable myofibroblast accumulation in the infarct. C. Quantitative analysis of myofibroblast density in the infarcted myocardium (scale bar=70 μm).
8. IL-10 deficiency does not affect post-infarction cardiac remodeling
Independent assessment using echocardiography and quantitative morphometry of perfusion-fixed hearts showed that the absence of IL-10 had no significant effects on cardiac remodeling following reperfused myocardial infarction (Table 1, Fig. 6). Echocardiographic studies demonstrated that both WT and IL-10 null hearts developed systolic dysfunction and left ventricular dilation after 7 days of reperfusion (Table 1). LVEDD, LVESD and FS were comparable in infarcted IL-10 null and WT animals (Table 1, pNS). In addition, the percent change in the echocardiographic indices following infarction (∆LVEDD, ∆LVESD and ∆FS) was not significantly different between groups (Table 1). Quantitative morphometry demonstrated that after 7 days of reperfusion, IL-10 null and WT animals had similar LVEDV (WT: 55.75 mm3 ± 3.61, n=14 vs. IL-10 null: 51.4 mm3 ± 2.52 n=16; pNS) (fig. 6C) and comparable scar size (WT: 13.7±1.35%, n=14 vs. IL-10 -/-: 15.49±1.16%, n=16; pNS) (fig. 6D).
Figure 6.
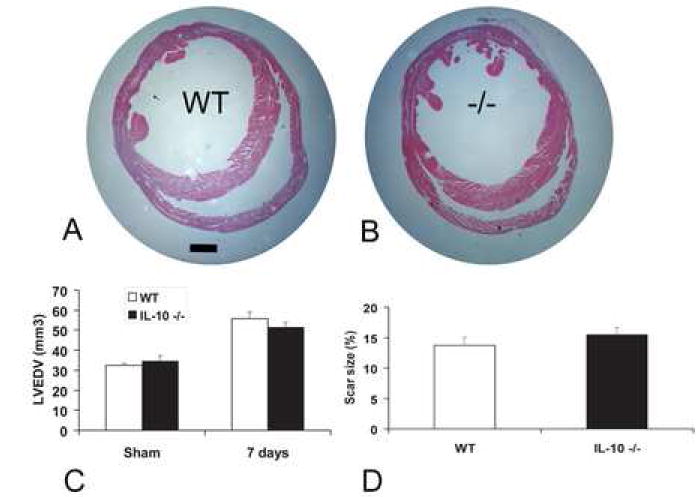
IL-10 gene disruption does not affect left ventricular remodeling following reperfused infarction. WT (A) and IL-10 null animals (B) showed comparable dilation of the left ventricle assessed by evaluation of perfusion-fixed hearts after 7 days of reperfusion. IL-10 null and WT mice showed similar LVEDV (pNS) (C), and comparable infarct size (D) (scale bar=1mm).
9. IL-10 does not alter pro-inflammatory chemokine synthesis by cardiac fibroblasts
IL-10 stimulation did not induce chemokine synthesis by isolated cardiac fibroblasts. In contrast, the pro-inflammatory cytokine TNF-α markedly induced fibroblast MCP-1 (fig. 7A), IP-10 (fig. 7B) and MIP-2 (fig. 7C) synthesis, but had no effect on MIP-1α and MIP-1β expression (not shown). In order to explore the potential ant-inflammatory actions of IL-10 on cardiac fibroblasts, we investigated the effects of IL-10 pre-treatment on chemokine synthesis by TNF-α stimulated fibroblasts. Pre-incubation with IL-10 did not affect TNF-α-induced chemokine upregulation (fig. 7A-C). In contrast, TGF-β incubation selectively suppressed TNF-α-induced IP-10 (but not MIP-2 and MCP-1) mRNA synthesis (fig. 7A-C).
Figure 7.
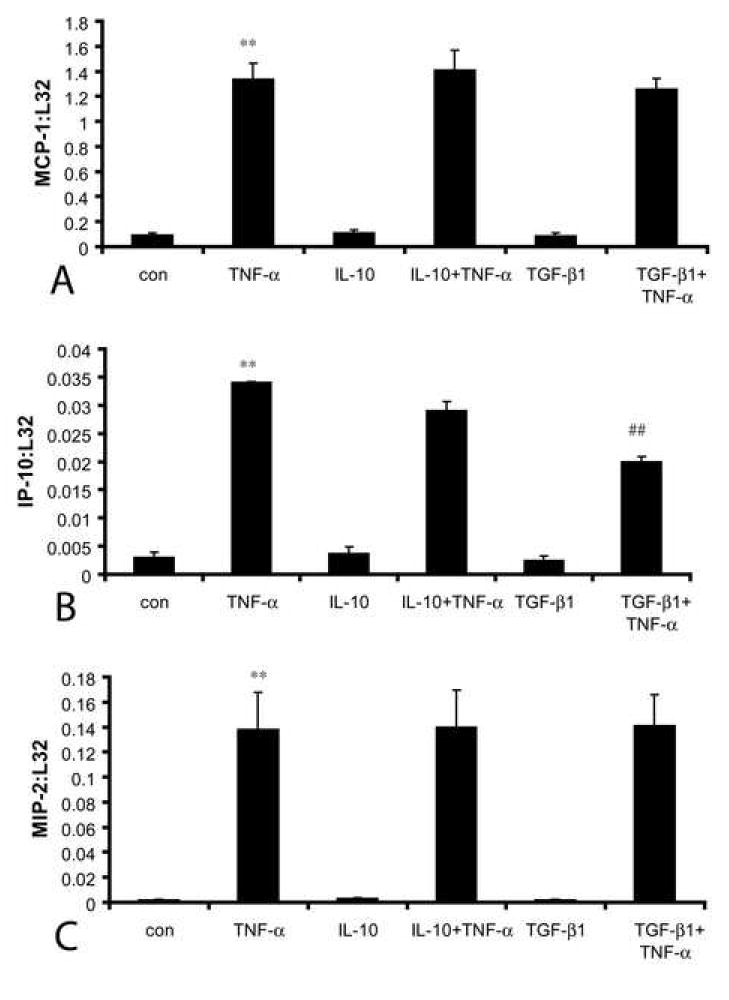
IL-10 does not alter inflammatory activation of cardiac fibroblasts exposed to TNF-α. IL-10 (100ng/ml) stimulation for 60 min did not induce mRNA expression of the chemokines MCP-1 (A), IP-10 (B), and MIP-2 (C). In contrast, TNF-α (100ng/ml) stimulation resulted in marked chemokine upregulation. Pre-incubation with IL-10 (100ng/ml) 4h prior to TNF-α stimulation did not inhibit TNF-α -mediated chemokine upregulation. In contrast, pre-incubation with TGF-β1 (10 ng/ml) significantly decreased TNF-α-induced IP-10 synthesis (B) (**p≺0.01), but did not affect MCP-1 and MIP-2 mRNA expression. TNF-α did not induce MIP-1α and MIP-1β synthesis by isolated cardiac fibroblasts (not shown).
DISCUSSION
Myocardial infarction sets into motion an inflammatory cascade that ultimately results in replacement of dead cardiomyocytes with a scar [21], [2], [22]. Cardiomyocyte death triggers Nuclear Factor (NF)-κB, complement and Toll-like receptor (TLR)-mediated pathways, resulting in marked induction of chemokines and cytokines in the infarcted myocardium [23], [1]. CXC chemokines mediate neutrophil recruitment in the infarct, whereas the CC chemokine MCP-1/CCL2 plays an important role in regulating monocyte infiltration [16]. Although induction of pro-inflammatory mediators is important for clearance of the wound from dead cardiomyocytes and matrix debris, activation of inflammatory pathways is transient and followed by repression of inflammatory gene synthesis and resolution of the leukocytic infiltrate [10]. Thus, optimal healing requires timely activation of inhibitory mechanisms suppressing expression of inflammatory mediators. Defective repression of the inflammatory reaction results in extension of granulation tissue formation and enhanced adverse remodeling [5].
We have previously identified IL-10 as a potentially important inhibitory mediator that may be involved in resolution of the post-infarction inflammatory reaction [9]. IL-10 is markedly induced in myocardial infarcts exhibiting a prolonged time course of expression, and possesses potent anti-inflammatory properties suppressing cytokine, chemokine and adhesion molecule synthesis by stimulated monocytes and macrophages [7], [24], [25]. IL-10 may also exert anti-inflammatory effects by generating functional decoy receptors on monocytes that act as molecular sinks and scavengers for inflammatory chemokines [26]. In addition, IL-10 signaling may regulate infarct healing and left ventricular remodeling through effects on extracellular matrix turnover and angiogenesis. In vitro studies suggested complex and cell-type specific actions of IL-10 on expression of MMPs and their inhibitors. IL-10 stimulation enhances TIMP-1 synthesis by human monocytes and macrophages promoting matrix deposition [11], but downregulates collagen synthesis and enhances MMP expression by human skin fibroblasts [27]. The in vivo role of IL-10 in fibrous tissue deposition is controversial and context-dependent: endogenous IL-10 increases collagen content in atherosclerotic plaques [28]; however, IL-10 gene therapy inhibits fibrosis in models of bleomycin-induced pulmonary injury, [29] and chronic renal disease [30]. Furthermore IL-10 exerts angiostatic effects in a model of angiogenesis induced by murine hindlimb ischemia [12].
In a previous study exploring the role of IL-10 in post-infarction inflammation, Yang and co-workers demonstrated that reperfused infarction in IL-10 null mice was associated with a 75% mortality rate, whereas no deaths occurred in WT animals [13]. Increased mortality was noted in the 24h-reperfusion group, but not in the 6h-reperfusion group; however, the mechanism of death in IL-10 null animals was not reported. Infarcted IL-10 null animals demonstrated enhanced inflammation evidenced by increased plasma levels of TNF-α and increased tissue expression of Intercellular Adhesion Molecule (ICAM)-1 [13]. In contrast, our findings do not support a critical role for IL-10 in resolution of post-infarction inflammation and regulation of infarct healing. Mortality in infarcted IL-10 -/- animals was low and comparable with the WT group. Although IL-10 null mice had higher peak TNF-α (Fig. 3) and MCP-1 (Fig. 4) mRNA expression in the infarcted heart than WT animals, suggesting a role for IL-10 in suppressing peak pro-inflammatory gene expression, both groups exhibited timely repression of inflammatory gene synthesis (Fig. 3-4). Furthermore, both IL-10 -/-and WT mice demonstrated partial clearance of the neutrophil infiltrate after 72 hours of reperfusion, exhibiting a comparable time course of neutrophil and macrophage infiltration (Fig. 1-2). The reason for the discrepancy between our findings and those of Yang and co-workers [13] is not clear. In both studies, IL-10 null and WT mice from the same source underwent reperfused infarction protocols. However, Yang et al. used an open-chest model where coronary ischemia and reperfusion was performed immediately after thoracotomy; this may have adversely affected survival in IL-10 null animals, which may be more susceptible to surgical trauma and the potential complications of surgical instrumentation. Our closed-chest model allows a 10-day recovery period after initial surgery, resulting in dissipation of the acute inflammation due to the surgical instrumentation and a more accurate evaluation of the consequences of myocardial infarction. In addition, our investigation assessed expression of a wide range of inflammatory mediators at several different timepoints providing a more detailed analysis of the time course of induction and suppression of post-infarction inflammation.
In contrast to the majority of experimental studies using permanent coronary artery ligation in order to induce myocardial infarction, our investigation used a model of reperfused infarction. Permanent coronary occlusion results in transmural necrosis, more extensive scar formation, and a delayed time course of infarct healing in comparison to reperfused infarction. Although evidence suggests that reperfusion accentuates the inflammatory reaction and increases IL-10 expression [9], the role of anti-inflammatory mediators (such as Il-10) may be more important in transmural infarcts induced by permanent occlusion. Thus, we cannot exclude the possibility that in the reperfused myocardium, the absence of transmural necrosis may limit the significance of mediators regulating the inflammatory and fibrotic response [31], [32].
Furthermore, the absence of IL-10 did not alter the cellular events associated with infarct healing. IL-10 null mice demonstrated timely replacement of dead cardiomyocytes with granulation tissue after 72h of reperfusion and had comparable myofibroblast density with WT animals (Fig. 5). IL-10 -/- and WT mice exhibited comparable LVEDD, LVESD and LVEDV 7 days after myocardial infarction suggesting similar extent of dilative post-infarction remodeling (Fig. 6, Table 1). In addition, IL-10 null and WT mice had comparable deterioration of systolic function following myocardial infarction (Table 1).
The non-critical role of IL-10 in resolution of post-infarction inflammation may be explained by the cell- and stimulus-specific nature of its anti-inflammatory properties. Although extensive evidence suggests that IL-10 suppresses cytokine and chemokine synthesis by mononuclear cells [6], [24], [7], its role in modulating pro-inflammatory activation of other cell types that actively participate in the post-infarction inflammatory response (such as endothelial cells and fibroblasts) is not established. IL-10 does not attenuate chemokine expression in stimulated canine endothelial cells [33] and does not inhibit pro-inflammatory activation of TNF-α and IL-1β-stimulated human umbilical vein endothelial cells [34]. Our findings demonstrated that pre-incubation with IL-10 did not modulate the pro-inflammatory phenotype of cardiac fibroblasts exposed to TNF-α, failing to inhibit chemokine mRNA synthesis (fig. 7). In contrast, pre-incubation with TGF-β1 suppressed TNF-α-induced IP-10 expression, supporting the potential role of TGF-β1 in repression of chemokine synthesis. In the infarcted myocardium, various cell types (such as endothelial cells, mononuclear cells and fibroblasts) are likely sources of inflammatory mediators. The limited role of IL-10 in suppressing the inflammatory response in healing infarcts may be due to the relative selectivity of its anti-inflammatory actions, with respect to cell type and stimulus.
Resolution of post-infarction inflammation is likely to involve multiple overlapping regulatory mechanisms controlling various pro-inflammatory pathways activated in the infarcted myocardium. Our study suggests that, although IL-10 is induced in the healing infarct, it plays a non-critical role in suppression of inflammatory mediators and in resolution of the inflammatory infiltrate. In addition, IL-10 signaling is of limited significance in regulation of the healing process and does not play a critical role in the pathogenesis of left ventricular remodeling. Other inhibitory mediators, such as TGF-β, may be important for repression of the cytokine and chemokine cascade and may play an important role in regulating fibrous tissue deposition.
Acknowledgments
This work was supported by NIH R01 HL-76246 (NGF) and a grant-in-Aid from the American Heart Association Texas affiliate.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errorsmaybe discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nian M, Lee P, Khaper N, Liu P. Inflammatory cytokines and postmyocardial infarction remodeling. Circ Res. 2004;94:1543–53. doi: 10.1161/01.RES.0000130526.20854.fa. [DOI] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. Targeting the inflammatory response in healing myocardial infarcts. Curr Med Chem. 2006;13:1877–93. doi: 10.2174/092986706777585086. [DOI] [PubMed] [Google Scholar]
- 3.Frangogiannis NG. The mechanistic basis of infarct healing. Antioxid Redox Signal. 2006;8:1907–39. doi: 10.1089/ars.2006.8.1907. [DOI] [PubMed] [Google Scholar]
- 4.Crawford SE, Stellmach V, Murphy-Ullrich JE, Ribeiro SM, Lawler J, Hynes RO, et al. Thrombospondin-1 is a major activator of TGF-beta1 in vivo. Cell. 1998;93:1159–70. doi: 10.1016/s0092-8674(00)81460-9. [DOI] [PubMed] [Google Scholar]
- 5.Frangogiannis NG, Ren G, Dewald O, Zymek P, Haudek S, Koerting A, et al. The critical role of endogenous Thrombospondin (TSP)-1 in preventing expansion of healing myocardial infarcts. Circulation. 2005;111:2935–2942. doi: 10.1161/CIRCULATIONAHA.104.510354. [DOI] [PubMed] [Google Scholar]
- 6.de Waal Malefyt R, Abrams J, Bennett B, Figdor CG, de Vries JE. Interleukin 10(IL-10) inhibits cytokine synthesis by human monocytes: an autoregulatory role of IL-10 produced by monocytes. J Exp Med. 1991;174:1209–20. doi: 10.1084/jem.174.5.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore KW, de Waal Malefyt R, Coffman RL, O’Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 8.Kopydlowski KM, Salkowski CA, Cody MJ, van Rooijen N, Major J, Hamilton TA, et al. Regulation of macrophage chemokine expression by lipopolysaccharide in vitro and in vivo. J Immunol. 1999;163:1537–44. [PubMed] [Google Scholar]
- 9.Frangogiannis NG, Mendoza LH, Lindsey ML, Ballantyne CM, Michael LH, Smith CW, et al. IL-10 is induced in the reperfused myocardium and may modulate the reaction to injury. J Immunol. 2000;165:2798–808. doi: 10.4049/jimmunol.165.5.2798. [DOI] [PubMed] [Google Scholar]
- 10.Dewald O, Ren G, Duerr GD, Zoerlein M, Klemm C, Gersch C, et al. Of mice and dogs: species-specific differences in the inflammatory response following myocardial infarction. Am J Pathol. 2004;164:665–77. doi: 10.1016/S0002-9440(10)63154-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lacraz S, Nicod LP, Chicheportiche R, Welgus HG, Dayer JM. IL-10 inhibits metalloproteinase and stimulates TIMP-1 production in human mononuclear phagocytes. J Clin Invest. 1995;96:2304–10. doi: 10.1172/JCI118286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Silvestre JS, Mallat Z, Duriez M, Tamarat R, Bureau MF, Scherman D, et al. Antiangiogenic effect of interleukin-10 in ischemia-induced angiogenesis in mice hindlimb. Circ Res. 2000;87:448–52. doi: 10.1161/01.res.87.6.448. [DOI] [PubMed] [Google Scholar]
- 13.Yang Z, Zingarelli B, Szabo C. Crucial role of endogenous interleukin-10 production in myocardial ischemia/reperfusion injury. Circulation. 2000;101:1019–26. doi: 10.1161/01.cir.101.9.1019. [DOI] [PubMed] [Google Scholar]
- 14.Jones SP, Trocha SD, Lefer DJ. Cardioprotective actions of endogenous IL-10 are independent of iNOS. Am J Physiol Heart Circ Physiol. 2001;281:H48–52. doi: 10.1152/ajpheart.2001.281.1.H48. [DOI] [PubMed] [Google Scholar]
- 15.Nossuli TO, Lakshminarayanan V, Baumgarten G, Taffet GE, Ballantyne CM, Michael LH, et al. A chronic mouse model of myocardial ischemia-reperfusion: essential in cytokine studies. Am J Physiol Heart Circ Physiol. 2000;278:H1049–55. doi: 10.1152/ajpheart.2000.278.4.H1049. [DOI] [PubMed] [Google Scholar]
- 16.Dewald O, Zymek P, Winkelmann K, Koerting A, Ren G, Abou-Khamis T, et al. CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ Res. 2005;96:881–9. doi: 10.1161/01.RES.0000163017.13772.3a. [DOI] [PubMed] [Google Scholar]
- 17.Michael LH, Ballantyne CM, Zachariah JP, Gould KE, Pocius JS, Taffet GE, et al. Myocardial infarction and remodeling in mice: effect of reperfusion. Am J Physiol. 1999;277:H660–8. doi: 10.1152/ajpheart.1999.277.2.H660. [DOI] [PubMed] [Google Scholar]
- 18.Eghbali M. Cardiac fibroblasts: function, regulation of gene expression, and phenotypic modulation. Basic Res Cardiol. 1992;87 (Suppl 2):183–9. doi: 10.1007/978-3-642-72477-0_16. [DOI] [PubMed] [Google Scholar]
- 19.Zeydel M, Puglia K, Eghbali M, Fant J, Seifter S, Blumenfeld OO. Properties of heart fibroblasts of adult rats in culture. Cell Tissue Res. 1991;265:353–9. doi: 10.1007/BF00398083. [DOI] [PubMed] [Google Scholar]
- 20.Lafontant PJ, Burns AR, Donnachie E, Haudek SB, Smith CW, Entman ML. Oncostatin M Differentially Regulates Cxc Chemokines In Mouse Cardiac Fibroblasts. Am J Physiol Cell Physiol. 2006 doi: 10.1152/ajpcell.00322.2005. [DOI] [PubMed] [Google Scholar]
- 21.Frangogiannis NG, Smith CW, Entman ML. The inflammatory response in myocardial infarction. Cardiovasc Res. 2002;53:31–47. doi: 10.1016/s0008-6363(01)00434-5. [DOI] [PubMed] [Google Scholar]
- 22.Ertl G, Frantz S. Healing after myocardial infarction. Cardiovasc Res. 2005;66:22–32. doi: 10.1016/j.cardiores.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 23.Frangogiannis NG. Chemokines in the ischemic myocardium: from inflammation to fibrosis. Inflamm Res. 2004;53:585–95. doi: 10.1007/s00011-004-1298-5. [DOI] [PubMed] [Google Scholar]
- 24.Fiorentino DF, Zlotnik A, Mosmann TR, Howard M, O’Garra A. IL-10 inhibits cytokine production by activated macrophages. J Immunol. 1991;147:3815–22. [PubMed] [Google Scholar]
- 25.Mostafa Mtairag E, Chollet-Martin S, Oudghiri M, Laquay N, Jacob MP, Michel JB, et al. Effects of interleukin-10 on monocyte/endothelial cell adhesion and MMP-9/TIMP-1 secretion. Cardiovasc Res. 2001;49:882–90. doi: 10.1016/s0008-6363(00)00287-x. [DOI] [PubMed] [Google Scholar]
- 26.D’Amico G, Frascaroli G, Bianchi G, Transidico P, Doni A, Vecchi A, et al. Uncoupling of inflammatory chemokine receptors by IL-10: generation of functional decoys. Nature Immunology. 2000;1:387–391. doi: 10.1038/80819. [DOI] [PubMed] [Google Scholar]
- 27.Reitamo S, Remitz A, Tamai K, Uitto J. Interleukin-10 modulates type I collagen and matrix metalloprotease gene expression in cultured human skin fibroblasts. J Clin Invest. 1994;94:2489–92. doi: 10.1172/JCI117618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mallat Z, Besnard S, Duriez M, Deleuze V, Emmanuel F, Bureau MF, et al. Protective role of interleukin-10 in atherosclerosis. Circ Res. 1999;85:e17–24. doi: 10.1161/01.res.85.8.e17. [DOI] [PubMed] [Google Scholar]
- 29.Arai T, Abe K, Matsuoka H, Yoshida M, Mori M, Goya S, et al. Introduction of the interleukin-10 gene into mice inhibited bleomycin-induced lung injury in vivo. Am J Physiol Lung Cell Mol Physiol. 2000;278:L914–22. doi: 10.1152/ajplung.2000.278.5.L914. [DOI] [PubMed] [Google Scholar]
- 30.Mu W, Ouyang X, Agarwal A, Zhang L, Long DA, Cruz PE, et al. IL-10 suppresses chemokines, inflammation, and fibrosis in a model of chronic renal disease. J Am Soc Nephrol. 2005;16:3651–60. doi: 10.1681/ASN.2005030297. [DOI] [PubMed] [Google Scholar]
- 31.Hudson MP, Armstrong PW, Ruzyllo W, Brum J, Cusmano L, Krzeski P, et al. Effects of selective matrix metalloproteinase inhibitor (PG-116800) to prevent ventricular remodeling after myocardial infarction: results of the PREMIER (Prevention of Myocardial Infarction Early Remodeling) trial. J Am Coll Cardiol. 2006;48:15–20. doi: 10.1016/j.jacc.2006.02.055. [DOI] [PubMed] [Google Scholar]
- 32.Carluccio E, Biagioli P, Alunni G, Murrone A, Giombolini C, Ragni T, et al. Patients with hibernating myocardium show altered left ventricular volumes and shape, which revert after revascularization: evidence that dyssynergy might directly induce cardiac remodeling. J Am Coll Cardiol. 2006;47:969–77. doi: 10.1016/j.jacc.2005.09.064. [DOI] [PubMed] [Google Scholar]
- 33.Frangogiannis NG, Mendoza LH, Lewallen M, Michael LH, Smith CW, Entman ML. Induction and suppression of interferon-inducible protein 10 in reperfused myocardial infarcts may regulate angiogenesis. FASEB J. 2001;15:1428–30. doi: 10.1096/fj.00-0745fje. [DOI] [PubMed] [Google Scholar]
- 34.Lisinski TJ, Furie MB. Interleukin-10 inhibits proinflammatory activation of endothelium in response to Borrelia burgdorferi or lipopolysaccharide but not interleukin-1beta or tumor necrosis factor alpha. J Leukoc Biol. 2002;72:503–11. [PubMed] [Google Scholar]