

The focus on the pathological changes underlying pulmonary hypertension (PH) have dominated the early investigations of this disease first described late in the 19th century. Pulmonary vascular pathology continues to play an important role in the present age of cell and molecular investigation of the pathogenesis of PH. This importance stems from the permanent quest to correlate pulmonary vascular remodeling with the altered pulmonary vascular hemodynamics, a critical advancement in the late 40’s and early 50’s with a wide impact on our present understanding of the disease. However, as it happens to most descriptive tools applied to medical sciences, the pathological insight into the extent and type of a particular form of pulmonary vascular remodeling has fallen short of establishing cause and effect relationships in the natural history of PH, and has had a limited impact on diagnosis and therapy. These limitations derive largely from the reliance of our current knowledge on studies of autopsies, as lung tissue is rarely available for histopathology during the course of the disease.
The pathological diagnosis of pulmonary vascular remodeling depends on the histological assessment of the cellular composition of pulmonary vascular walls, which if abnormal, is described as pulmonary vascular ‘lesions’. Although mostly reliant on examination of histological slides stained by hematoxylin and eosin stains, the pathological interpretation of PH has benefited from the progressive use of cell specific immunohistochemical markers to better define the structure and cellular composition of the pulmonary vascular lesions. Despite the advances in the understanding and treatments aimed at the disease, the pathology of PH clearly lags behind the comprehensive approach employed by pathologists in their assessment of other diseases such as cancer. This approach presently includes the screening for abnormal expression of the p53 tumor suppressor or adenomatous polyposis coli (APC) genes, abnormal expression of cytokeratins in breast adenocarcinomas, and markers of cell proliferation in sarcomas, among several tissue markers that help in the final diagnosis, staging prognosis, and treatment. By comparison, the histopathological diagnosis of PH infrequently fulfills some or all of these important tasks of the pathological work-up aimed at clinical management.
The functional status of the pulmonary circulation and the levels of pulmonary vascular resistance and pulmonary artery pressures ultimately determine the outcome and treatment of patients with PH. We have previously proposed that the functional status of the hypertensive pulmonary circulation could be broadly correlated with a specific type of pulmonary vascular remodeling present at the time of pathological evaluation of the pulmonary arteries [1]. We will frame this review on the pathology of PH within the functional categories proposed previously i.e., that PH can be broadly divided into mild/moderate vs. severe based on pulmonary artery pressures, their impact on right ventricular performance, and overall mortality [1] (Tables 1 and 2). Many non neoplastic lung diseases with intima thickening or medial hypertrophy, such as idiopathic interstitial pneumonias (IPF) [2] and chronic obstructive pulmonary diseases (COPD) [3], present with mild/moderate PH. On the other hand, conditions associated with endothelial cell proliferative lesions (including plexiform lesions), marked intima fibrosis (such as idiopathic pulmonary arterial hypertension (IPAH) and scleroderma), or medial and intimal smooth muscle cell growth (as observed in a fraction of IPF or COPD lungs) cause severe PH. As it will become apparent in this review, our ability to relate the mode of pulmonary vascular remodeling to the severity of the disease is rather limited. We will highlight the changes associated with IPAH as those paradigmatic of the pathology of severe PH.
Table 1.
Intima Remodeling.
| Cell | Smooth muscle cells | EC Matrix | Endothelial cells | |||
|---|---|---|---|---|---|---|
| Lesion | Eccentric | Concentric | Fibrotic | Plesiform | Concentric | Dilation/angiomatoid |
| Normal PAP | Yes | No | No | No | No | No |
| Mild/Moderate PH | Yes | No | Yes | No | No | No |
| Severe PH | Yes | Yes | Yes | Yes | Yes | Yes |
Table 2.
Medial Remodeling.
| Cell | Smooth muscle cells | EC Matrix |
|---|---|---|
| Normal PAP | Yes* | No |
| Mild/Moderate PH | Yes | Yes |
| Severe PH | Yes | Yes |
Medial hypertrophy is localized and restricted to some pulmonary arteries
In our goal to highlight the pathology of PH, we will follow the recommendations of the Evian meeting on Pulmonary Hypertension in 1998, which supported a more descriptive approach to the pulmonary vascular changes in PH [4]. We will also review the recent findings related to the pathogenesis of human PH (Table 3). We believe that their translation into the pathology of PH may eventually lead to a more refined and clinically useful approach towards the diagnosis and staging of the disease. The latest reorganization of the pathological nomenclature of PH by the international meeting in Venice in 2004 provides a useful framework of reference to the diagnosis of pulmonary vascular lesions in PH [5]. As a valuable background to this chapter, the readers are also referred to reviews on normal histology of pulmonary arteries [6;7], and a historical perspective of the research accomplishments in the last 100 years of studies of the pulmonary circulation [8].
Table 3.
| Molecules | Up or Down | Pathways Affected | Vascular Cells Affected |
|---|---|---|---|
| TGFβR2 | Down | b,e | EC, SMC |
| BMPR2 | Down | b,e | EC, SMC |
| Nitric oxide | Down | a,c,e | EC, SMC |
| Prostacyclin | Down | a,c | EC, SMC |
| Caveolin-1 | Down | a | EC |
| BAX | Down | b | EC |
| [K−] channels | Down | b,d | SMC |
| Thrombomodulin | Down | C | EC |
| Endothelin-1 | Up | a,bc,d,e | EC, SMC,AF |
| Tissue Factor | Up | a,bc | EC |
| von Willebrand Factor | Up | c,e | EC, SMC |
| 5-lipoxygenase | Up | a,de | EC |
| VEGF | Up | a,b,c,e | EC, SMC |
| VEGF reportors | Up | a,b,c,e | EC, SMC |
| HIF-1α | Up | a,b | EC, SMC,AF |
| Serotonin transporter | Up | a,d | SMC |
| Serotonin receptor | Up | a,d | SMC |
| RANTES | Up | e | Mφ |
| IL-6 | Up | e | EC |
| MIP-1α | Up | e | Mφ |
| Fractalkine | UP | e | T cells |
| PDGF | Up | a | EC, SMC, AF |
| TGF β | Up | a | SMC,AF |
| Tenascin | Up | a,b | SMC |
| MMP2 | Up | a | SMC |
| TIMP-1 | Up | a | SMC |
| MT-MMP1 | Up | A | SMC |
Pathobiological Processes
Increased cell proliferation
Decreased cell apoptosis
Increased clotting
Increased vasoconstriction
Inflammation
Abbreviations: EC, endothelial cells; SMC, smooth muscle cells; AF, adventitial fibroblast; Mφ, macrophages.
Intima Lesions
Pathology
Intimal lesions account for most of the reduction of luminal area of small pulmonary arteries and potentially largely influence the overall pulmonary vascular resistance. Intimal lesions consist of eccentric intima thickening, and fibrotic, plexiform, concentric, and dilation/angiomatoid lesions (Table 1, Figures 1–4). Focal eccentric lesions can be detected in normal lungs, but these lesions are more widespread and impinge to a larger extent on the vascular lumen in PH. It is conceivable that some of these lesions results from the organization (i.e., lysis of fibrin, recanalization by newly formed blood vessels, and/or ingrowth of myofibroblasts) of localized thrombi, which form the nidus for a localized growth of smooth muscle cells. More advanced lesions acquire a ‘fibrotic’ pattern, with interspersed myofibroblasts and marked accumulation of mucopolyssacharides (Figure 3). These ‘fibrotic’ lesions are widely present in explanted lungs of patients with severe PH, including lungs with IPAH or PAH associated with the CREST syndrome (acronym for calcinosis, Raynaud phenomenon, esophageal dysmotility, sclerodactily, and telengectasia) [9]. However, variable degrees of eccentric thickening can be seen in cigarette smoker’s lungs associated with pulmonary endothelial cell dysfunction, with or without evidence of PH [10].
Figure 1.
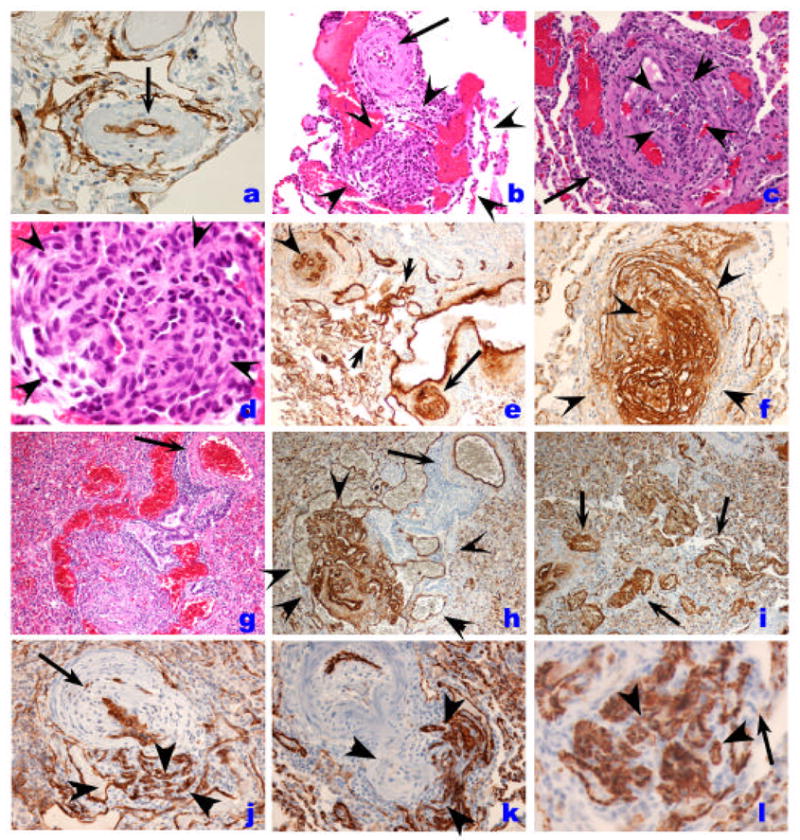
Histopathology of endothelial cell lesions in IPAH. A. Pulmonary artery showing medial hypertrophy and lined by a single layer of endothelial cells, as outlined by Factor VIII related antigen immunostaining (arrow). Plexiform lesion (outlined by the rim of arrowheads) with the proximal vascular arterial segment with marked intimal and medial thickening by smooth muscle cells (arrow). Note the proliferation of endothelial cells with the outer edge (3–5 o’clock) occupied by dilated blood vessel-like structures. C. Cross section of a plexiform lesion, outlined by arrowheads. Note perilesional inflammatory infiltrate (arrow). D. High magnification histology of plexiform lesions shown slit-like vascular channels lined by hyperchromatic and cuboidal endothelial cells. Cells in the core do not display distinct cytoplamic borders. E. Low magnification immunohistology with Factor VIII related antigen immunohistochemistry of different endothelial cell based vascular lesions. This area has re-vascularized lesions (possibly an organized thrombus), with well-formed and distinct small capillaries/vessels (arrowhead), a plexiform lesion (arrow), and dilated/angiomatoid lesions (between arrowheads). F. High magnification immunohistology of cellular plexiform lesion stained with Factor VIII related antigen (arrowheads). G and H. Histological identification of plexiform and dilation lesions (G) is markedly improved by Factor VIII related antigen immunohistochemistry (H) (arrowheads), while the parent vessel (arrow) shows mild medial remodeling. I. Highlight of vascular dilation/angiomatoid lesions with Factor VIII related antigen immunohistochemistry. J. Endothelial cells in plexiform lesion is highlighted by CD34 immunohisochemistry (arrowheads). Proximal pulmonary artery with marked intima and medial thickening is highlighted by the arrow. K and I. Endothelial cells are highlighted by CD31 immunohistochemistyr (arrowheads). Note that capillary endothelial cells express CD31 as well (arrow in I),
Figure 4.
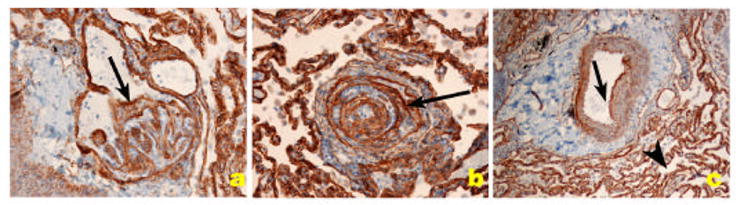
Distribution of collagen IV, a component of basement membrane of endothelial cells (arrows) in PH pulmonary arteries. A and B. Plexiform lesion. C. Pulmonary artery with medial thickening. The collagen IV basement membrane of alveolar capillaries shows the clear demarcation of capillary basement membrane.
Figure 3.
A. Fibrotic, relatively paucicellular intima thickening (outlined by arrowheads) in a pulmonary artery with the media highlighted with the arrow. B. Marked intima remodeling with almost complete obliteration by fibrous tissue with a marked intravascular and perivascular inflammatory infiltrate (arrows). C. Smooth muscle cell hypertrophy, with prominent thickening of medial layer (arrow). D. Highlight of medial hypertrophy with smooth muscle α actin immunohistochemistry. E. Markedly remodeled pulmonary artery with endothelial cell layer highlighted by Factor VIII related antigen immunohistochemistry. Note that the intima and medial smooth muscle cells are negative for Factor VIII related antigen reactivity. F. Ingrowth of smooth muscle cells in a plexiform lesions, highlighted by smooth muscle cell α actin immunohistochemistry (arrow).
The organization of myofibroblasts or endothelial cells in ‘onion-skin’ layers underlies the presentation of concentric lesions (Figures 1 and 2). These lesions are characteristic of in severe PH, as they significantly reduce significantly the luminal area. We documented the presence of concentric lesions in vascular segments proximal to plexiform lesions [11], suggesting that the concentric lesions arise from remodeled plexiform lesions. Concentric lesions composed of smooth muscle cells have also been described [12]. It is unclear whether endothelial- or smooth muscle-based lesions have a similar impact on the course of PH. However, it is conceivable that these lesions may arise from similar progenitors, as endothelial cells can transdifferentiate into smooth muscle cells when stimulated with platelet derived growth factor (PDGF)-BB [13] or transforming growth factor (TGF)-β [14]. Pulmonary arteries may also almost occluded by the accumulation of acellular matrix, suggestive of a terminal scarring process (Figure C). Approximately 20–25% of pulmonary arteries ranging between 25 and 200 um in diameter are compromised by intima occlusion or concentric lesions [15]. These two patterns of luminal obliteration might represent temporally related lesions, which start as an abnormal proliferation of endothelial and/or smooth muscle cells, progress into an ‘onion-skin’ lesion, and then become acellular with abundant extracellular matrix deposition (Figure 4).
Figure 2.
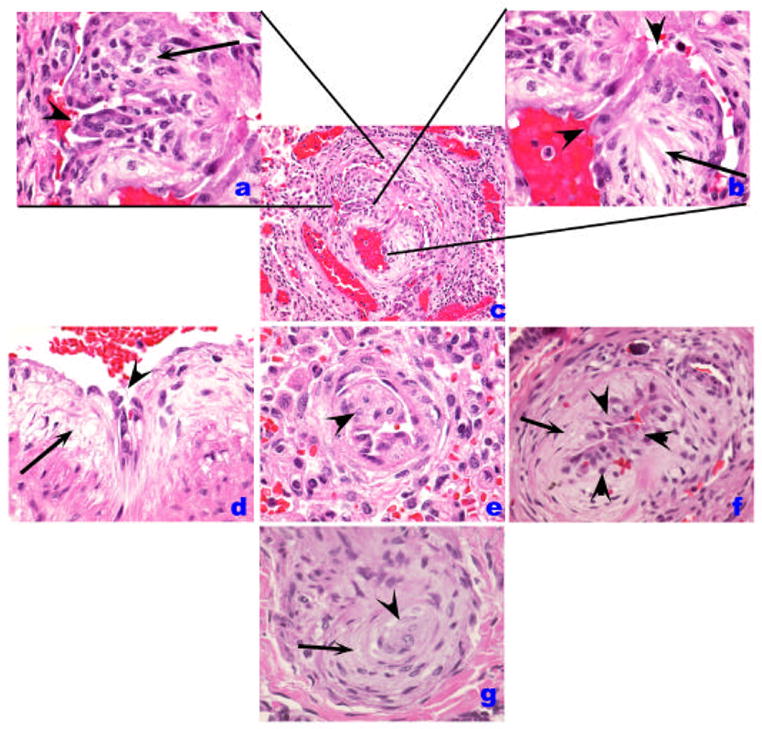
Histological patterns of endothelial cell injury and lesions in IPAH. (A–C). Plexiform lesion (B) with high magnification of proliferated endothelial cells in A. A luminal projection of endothelial cells is highlighted by an arrowhead while cells in cellular core are marked by an arrow. In B, the proximal segment is shown in high magnification, highlighting the atypical endothelial cell (arrowheads) facing the vascular lumen, while a myxoid component organized as concentric layers of smooth muscle cells is highlighted by the arrow. D. A similar pattern of abnormal endothelial cells (arrowhead) and the myxoid subendothelial layer (arrow) are highlighted. E. An intimal projection is highlighted (arrowhead). F. Increased number of endothelial cells (arrowheads) in the intima while the myxoid subintimal layer is highlighted by the arrow. These lesions (B, D–F) contain usually endothelial cells that can be highlighted by Factor VIII related antigen immunohistochemistry. The cluster of endothelial cells possibly represents early plexiform lesions. G. Shown is a concentric lesion (arrowhead) with adjacent smooth muscle-like cells (arrow).
Plexiform lesions, typically located in branching pointes of muscular arteries [11], consists of a network of vascular channels lined up by endothelial cells [16] (Figures 1 and 2) and a core of myofibroblastic or less well-differentiated cells (Figure 3) [17]. In our experience, these lesions are characteristically found in cases of severe PH, including IPAH, and PH associated with HIV infection, liver cirrhosis, CREST, congenital heart malformations, and schistosomiasis. As endothelial cells contribute significantly to both concentric and plexiform lesions, endothelial cell immunostaining with Factor VIII related antigen or CD31 may aid in the identification of the different stages of endothelial cell proliferation [6] (Figure 1 and 2). The detection of small clusters of endothelial cells can be aided by use of these stains [18] and cell proliferation markers such as MIB-1 may contribute to the identification of proliferating endothelial cells as well (normal pulmonary endothelial cells have a very low proliferation rate) [19]. Dilation lesions represent another potential endothelial cell lesion, with the formation of a rosary of dilated channels (also known as angiomatoid lesions), often distally to a plexiform lesion [6] (Figure 2). As with plexiform lesion, we have observed dilation lesions in cases of severe PH (Table 1). These lesions containing endothelial cells have abundant collagen IV expression, a component of basement membranes, and a useful marker for the outline of blood vessels (Figure 4).
Pathobiology
The prevailing concept is the one which dysfunctional endothelial cells plays the key pathobiological role in PH [20]. However, to the present day, the pathogenesis of intimal vascular lesions remains mostly undetermined. It is conceivable that these lesions result from injuries to the endothelium, followed by activation of smooth muscle cell migration, extracellular matrix deposition, and endothelial cell proliferation. As mentioned, it is also possible that these lesions occur due to organization of microthrombi. In animal models, only the association of the alkaloid monocrotaline (MCT) and high shear stress due to shunting between the systemic and pulmonary circulations leads to marked intima thickening by smooth muscle cells and severe PH in rats [21]. The intimal cells express smooth muscle cell markers, and fibronectin [19].
We have proposed that plexiform lesions represent a process of misguided angiogenesis based on the findings of expression of vascular endothelial growth factor (VEGF), its receptors 1 (flt) and 2(kdr), and hypoxia inducible factor (HIF)-1α and β [22]. The finding of monoclonality of endothelial cells in plexiform lesions in IPAH, but not in similar lesions in PH associated with congenital heart malformation, suggests that these lesions might arise from mutations in tumor suppressor genes [23]. Somatic loss of expression of transforming growth factor β receptor 2 ((TGFβ-R2) and the propapoptotic Bax, potentially due to microsatellite instability, is also documented in IPAH plexiform lesions [24]. Heterozygous germline mutations of bone morphogenetic protein receptor 2 (BMPR2) (a component of the TGF-β-related family) is found to underlie up to 60% of cases of familial IPAH [25;26]. BMPR2 expression is documented in plexiform lesions [12] and in remodeled pulmonary arteries in IPAH lungs, predominantly in endothelial cells. However, our studies reveal that the cells in the central core of plexiform lesions lack the expression of TGF-β receptor 2, TGF-β receptor 1 and their signaling smad(s) 2,1 (which shares common epitopes with smads 5 and 8), 3 and 4, including the phosphorylated smad 1/5/8 and 2. The absence of expression of these phosphorylated smads is the best indication that there is no TGF-β (via smad 2 or smads 1/5/8) or BMP (via smads 1/5/8) signaling in these cells [27]. Loss of cytostatic signaling from TGF-β would allow the plexiform cells to abnormally proliferate. Notwithstanding the evidence of preserved BMPR expression and signaling in IPAH endothelial cells, recent studies indicate that BMP signaling protects against endothelial cell apoptosis in vitro [28]; the loss of this protection by germline mutations in BMPR2 would thus favor enhanced susceptibility to apoptosis of lung endothelial cells.
The potential role for early endothelial cell apoptosis in the pathogenesis of uncontrolled proliferation of pulmonary endothelial cells was first documented in the rat model of severe pulmonary hypertension caused by the combination of VEGF receptor blockade with SU5416 and chronic hypoxia [29]. Moreover, the role of endothelial cell apoptosis in the pathogenesis of PH was also extended to the monocrotaline model [30]. Initial endothelial cell apoptosis might favor the emergence of apoptosis-resistant endothelial cells, with potential for uncontrolled proliferation [29;31].
VEGF [22], endothelin-1 [32], and survivin [33;34] are among the factors present in plexiform lesions that may enhance endothelial cell and smooth muscle cell proliferation or decrease vascular cell apoptosis. These lesions have decreased expression of anti-remodeling mediators such as nitric oxide synthase [35] and prostacyclin synthase [36], and tumor suppressors, such as caveolin-1[37]. These phenotypic characteristics lead us to propose that the plexiform lesions have characteristics in common with neoplasms [38]. As discussed below, there is emerging evidence that viral factors may play a role in endothelial cell proliferative lesions in severe PH.
A summary of the molecules that may contribute to lesion formation, their expression, and the signaling or metabolic pathways involved, as well as their potential target in the vasculature are shown in Table 3.
Medial Vascular Remodeling
Pathology
Medial smooth muscle cell hypertrophy is a characteristic pathological feature of PH that involves muscularized arteries (ranging between 70 and 500 μm in diameter), and precapillary vessels (below 70 μm in diameter) (Figure 3). The medial smooth muscle cell layer represents approximately 10–15% of the outside diameter of normal muscularized pulmonary arteries, while it approaches 30–60% of the outside diameter in vessels of IPAH lungs [15;39–41]. Although careful morphometric assessments of medial remodeling have not been carried out in non-IPAH PH, it is apparent that medial thickening occurs in mild/moderate or severe PH and in cases of normal individuals exposed to cigarette smoke with no evidence of PH [42] (Table 2). This diagnostic limitation of the finding of medial hypertrophy has led us to propose that the identification of pulmonary medial remodeling warrants additional clinical evidence of the presence of potential PH [6]. Several of these cases may have normal pulmonary artery pressure levels and thus do not have PH (Table 2). The limited number of smooth muscle cells markers, identified mostly in animal models, precludes the ‘functional’ dissection of pulmonary medial remodeling. Nevertheless, medial thickening probably plays an important role in the pathogenesis of PH, yet it is difficult to relate the morphological identification of medial remodeling to specific levels of pulmonary artery pressures, degrees of severity of PH [6], or potential for response to vasodilators.
Pathobiology
The expansion of the tunica media in pulmonary arteries in PH occurs by a combination of hypertrophy and hyperplasia. Smooth muscle cell hypertrophy together with accumulation of extracellular matrix, including thicker elastic laminae, accounts for most of medial thickening in PH [43]. Smooth muscle cell hyperplasia probably plays a minor role in medial thickening than cell hypertrophy or extracellular matrix deposition, since it is experimentally shown that the proliferation index of smooth muscle cells doubles in large pulmonary arteries in hypoxic rats, while those cells lining intraacinar arteries do not show evidence of [3H]-thymidine uptake [44]. A similar finding is observed in PH caused by monocrotaline in rats [45]. The apparent predominant role of smooth muscle cell hypertrophy over hyperplasia in medial thickening is also supported by human PH studies [19;46;47]. Based on the hypoxia model of PH, the appearance of smooth muscle cells in nonmuscularized precapillary arteries occurs by hypertrophy and metaplasia of potential precursor cells, i.e., pericytes or intermediate cells (pericytes-smooth muscle cells) [44]. A careful examination of the smooth muscle cell phenotypes in the tunica media of neonatal bovine pulmonary arteries reveals the existence of substantial cellular heterogeneity, with clusters of fully differentiated smooth muscle cells interspersed with cells negative for most smooth muscle cell markers [48]; such a cellular heterogeneity probably exists in the different forms of human PH as well, though a systematic dissection of these phenotypes has not been performed thus far. In spite of these data, most of the present day studies addressing the potential role of genetic mutations or alterations in ion channels have focused on smooth muscle cell proliferation, as noted below.
The mechanisms underlying the thickening of the pulmonary vascular medial layer have been linked mostly to cell proliferation and, more recently, to inhibition of cell apoptosis. The identification of mutations in BMPR2 led to several in vitro studies aimed at relating smooth muscle cell proliferation to abnormal BMPR2 signaling. IPAH smooth muscle cells isolated from proximal arteries (i.e., elastic vessels, >500 μm in diameter) exhibit decreased inhibitory effect on cell proliferation mediated by TGF-β1 or BMP-4 when compared with smooth muscle cells isolated from normal human pulmonary arteries [49]. Of note, these altered responses are not due to abnormal expression of smad signaling or ligand binding to its receptors in IPAH smooth muscle cells. Similar studies were extended to more peripheral smooth muscle cells (i.e., from arteries less than 2 mm in diameter-(that are still elastic arteries) obtained from normal pulmonary arteries, which undergo cell proliferation and protection against apoptosis when exposed to BMP-4, a ligand for BMPR2. Therefore, loss of function mutations in MPR2 would be predicted to cause smooth muscle cell arrest and increased cell death [50], i.e., paradoxically opposite to that predicted to occur in familial IPAH in which mutations would facilitate smooth muscle cell growth and remodeling. Given these somewhat discrepant results [50], the evidence of reduced expression of BMPR2 found in IPAH alveolar septa [12], and the finding of decreased levels of the activated form of smad 1 (the signaling smad for BMPR2) in smooth muscle cells of muscular pulmonary arteries in IPAH, it remains unclear how BMPR2 mutations either can cause or trigger the disease. Indeed, when compared with wild type mice, heterozygous mice lacking a single copy of BMPR2 have no pulmonary vascular phenotype at baseline or under chronic hypoxia [51], but show moderately increased pulmonary artery pressures and minimal remodeling when stressed with intratracheal delivery of a 5-lipoxygenase expressing vector [51].
The role of serotonin (5-hydroxytryptamine) in the growth stimulation of pulmonary artery smooth muscle cells has been intensively studied as a mechanism of medial remodeling in PH and a potential modifier gene to the familial IPAH. Serotonin is internalized in smooth muscle cells after binding to serotonin transporter (5-HTT) or to its receptors 5-HT1B-R, 5-HT-2A R, 5-HT7 -R, and 5-HT2B -R, when it promotes vasoconstriction, cell growth, and enhancement of hypoxia-induced remodeling and PH [52]. Vascular smooth muscle cells in IPAH lungs express higher levels of 5-HTT [53] or 5-HT2B R [52] by immunohistochemistry, and undergo enhanced cell proliferation when treated in vitro with serotonin compared with normal smooth muscle cells [53]. These findings are supported by the report that mice deficient in 5HTT or 5-HT-2B are protected against PH caused by hypoxia alone [52] or hypoxia combined with the anorexigen dexfenfluramine [54], respectively. Of interest is the observation that increased serotonin levels affect signaling pathways downstream of mutated BMPR2, as serotonin infusion enhances normoxic and hypoxic pulmonary pressures and vascular remodeling in BMPR2 heterozygous mice as compared with wild type mice [50].
Despite evidence supporting a loss of function for the TGF-β family signaling, particularly in endothelial cells of IPAH, TGF-β may produce gain-of-function alterations underlying medial smooth muscle cell growth and adventitial fibroblast activation. TGF-β isoforms 1, -2, and -3 are expressed in hypertensive pulmonary arteries [55], and could signal via activation of smad 2 or 3 by serine phosphorylation, whose expression was documented in pulmonary vascular smooth muscle cells [27]. Recent evidence implicates PDGF in the pathogenesis of both monocrotaline- and chronic hypoxia-induced PH [56]. Monocrotaline treatment or chronic hypoxia exposure leads to increased PDGF receptor (PDGFR)-β expression and phosphorylation and activation of ERK Map kinase in rats. The findings of positive response of hypertensive animals to Gleevac™, also an inhibitor of PDGFR signaling, and the evidence of increased expression of PDGFR-β and phosphorylated PDGFR-β in IPAH lungs, led the authors to translate their finding by treating an IPAH patient with Gleevac™, with promising early results [57].
The extracellular matrix is both a component of the thickened pulmonary vascular media and a regulator of smooth muscle cell growth. Tenascin, a glycoprotein involved in lung and vascular morphogenesis, is strongly expressed in remodeled intima and medial layers of human hypertensive pulmonary arteries [58]. Experimentally, products of enhanced elastolytic or proteolytic activity in pulmonary arteries can promote clustering of integrin receptors, which activate tenascin and epidermal growth factor (EGF) receptors, thus leading to smooth muscle cell proliferation [59]. These findings are pertinent to the monocrotaline model of PH [60;61], while pulmonary vascular remodeling in hypoxic pulmonary hypertension is improved by the action of metalloproteases [62]. Based on studies using cultured human smooth muscle cells isolated from elastic arteries from IPAH patients, it appears that Tissue Inhibitor of Metalloprotease-1 (TIMP-1) and metalloprotease (MMP)-2 are increased in expression; only the finding of MMP-2 enhanced expression is confirmed in situ in elastic pulmonary arteries in IPAH lungs [63]. It is unclear whether a similar pattern is present in distally remodeled pulmonary arteries; however, endothelial cells express moderate/intense immunohistochemical expression of MMP-2, while myofibroblasts display low levels of this extracellular protease [64]. Membrane type-1-MMP was also expressed in endothelial and myofibroblastic cells of concentric and plexiform lesions. These results and those in the monocrotaline model suggest that the inhibition of elastases or MMPs might be beneficial in IPAH as demonstrated in the monocrotaline model of PH [65], potentially leading to the apoptosis of smooth muscle cells.
It is becoming clear that the ultimate fate of vascular smooth muscle cells in PH is determined by their resistance to apoptosis. In fact, ‘apoptosis-resistance’ might play a central role in both the endothelial- and smooth muscle cell-based pulmonary vascular lesions [66], since IPAH lungs have lower number of apoptotic cells than normal or emphysematous lungs [67]. As growth signals originated by PDGF, TGF-β, EGF, serotonin, and extracellular matrix proteins are interrupted in animal models of PH, pulmonary arteries undergo de-remodeling associated with apoptosis of pulmonary artery smooth muscle cells [65]. Targeting apoptosis of the hypertrophic smooth muscle cells might represent a more viable approach towards treatment.
Recent studies of K+ channel activity have provided a novel insight of the lack of proapoptotic signals in IPAH smooth muscle cells. An exciting and complementary paradigm based on the interplay between K+ channels and apoptosis in pulmonary artery smooth muscle cells has emerged based on the demonstration of activation of K+ channels causes cytochrome C release from the mitochondria and water efflux from the dying cells [68]. Conversely, inhibition of K+ channels causes cell depolarization, enhances contractility, and decreases apoptosis of pulmonary artery smooth muscle cells. One potential mechanism linking this paradigm to BMPR2 mutations is the finding that BMPR2 activation upregulates K+ channels [69] and causes apoptosis of normal pulmonary artery smooth muscle cells, but not of cells from patients with IPAH [70]. McMurtry et al provide recent additional evidence that mechanisms akin to cancer operate in pulmonary vascular remodeling [34]. Survivin protects cancer cell against apoptosis by inhibiting caspase activation and apoptosis inducing factor [71]. Not only do pulmonary artery smooth muscle cells in IPAH lungs express higher levels of the antiapoptotic survivin, but monocrotaline-induced pulmonary vascular remodeling requires survivin expression. Indeed, transduction of a functionally deficient survivin in monocrotaline-treated lungs prevented pulmonary vascular remodeling and, when administered after monocrotaline treatment, it enhances apoptosis of pulmonary artery smooth muscle cells and reduces pulmonary artery pressures. This mutant survivin increases levels of K+ channel activity and leads to depolarization of mitochondria with enhanced cytochrome C release.
Adventitial Remodeling
Pathology
The adventitia is mostly composed of fibroblasts. There is growing evidence that, rather than just a structural support to pulmonary vessels, the adventitia may also play a role in the regulation of pulmonary vascular function from the ‘outside-in’ (as comprehensively reviewed in [72]). The normal adventitia represents approximately 15% of the external diameter of pulmonary arteries larger than 50 μm in diameter. In IPAH arteries, the adventitial thickness increases to 28% of artery diameter, predominantly due to collagen deposition [15]. We and others have not noted the presence of a vasa-vasorum in the adventitia of medium sized pulmonary arteries of PH. Whether the adventitia is thickened or presents with a heterogeneous stromal cell population in other forms of PH remains unclear. The diagnostic significance of the observation of the extent of adventitial thickening or its role in the differentiation of mild/moderate vs. severe PH is equally unknown. As discussed subsequently, the adventitia contains a perivascular cuff of inflammatory cells, which might modulate the growth of or transdifferentiate themselves into vascular structural cells in the pulmonary vascular wall.
Pathobiology
It is apparent that the adventitial fibroblasts react promptly to vascular stresses, notably those caused by hypoxia, high-perfusion flow models, and, to a lesser extent, the alkaloid monocrotaline treatment [72]. Fibroblasts undergo a marked increase in cell proliferation reaching 11-fold increase in labelling index in hilar vessels, at day 3 of chronic hypoxia exposure [44]. In the monocrotaline model, proliferation rate of adventitial fibroblasts reaches a 5-fold increase at day 21 [45]. These findings underscore the powerful abilities of adventitial fibroblasts to undergo proliferation, one of the most characteristic properties of these cells. Moreover, adventitial fibroblasts can: 1. either differentiate into smooth muscle cells and migrate into the remodeled media or; 2. trigger smooth muscle cell proliferation by secreting growth factors; 3. allow for the recruitment of inflammatory and bone marrow progenitor cells; 4. create a ‘vasculogenic and angiogenic’ niche for expansion of newly-formed vessels (vasa vasorum). It is apparent that adventitial fibroblasts might represent heterogeneous clusters of cells, like those found in the medial smooth muscle cells [72]; this biological heterogeneity is readily apparent in the neonatal bovine model of hypoxic PH. A potentially important finding is the contribution of circulating fibrocytes (precursor cells sharing the expression of the fibroblast marker α1-collagen and peripheral leukocyte markers CD45, CD34, CD11b, and CD14) to experimental hypoxic PH [72]. Adventitial fibroblasts sense alterations in their redox status, with the ensuing activation of cell growth, cytokine release, and generation of oxidants via activation of NADPH oxidase [72]. The redox regulation of adventitial cell function is supported by the identification of markers of oxidative stress in remodeled pulmonary arteries of IPAH lungs [73].
Venous pathology
Pathology
The pulmonary veins are primarily involved in the pathogenesis of postcapillary PH, such as that caused by venoocclusive disease, capillary hemangiomatosis, mitral valve and other forms of left heart dysfunction, and extrinsic main pulmonary vein obstructions. Venoocclusive disease and pulmonary capillary hemaniogiomatosis are rare causes of idiopathic PH. The first is characterized by variable luminal obstruction by intraluminal bands or eccentrically placed fibrous tissue, which is firmly attached to the intima, potentially representing organized thrombi. Interlobular septal veins become muscularized, and there is marked capillary distention in periseptal alveoli, leading to a pattern suggestive of interstitial lung disease (Figure 5). Pulmonary capillary hemangiomatosis is characterized by a neoplastic-like proliferation of endothelial cells, often forming capillaries, and infiltrating alveolar septa, venous, and arterial pulmonary arteries.
Figure 5.
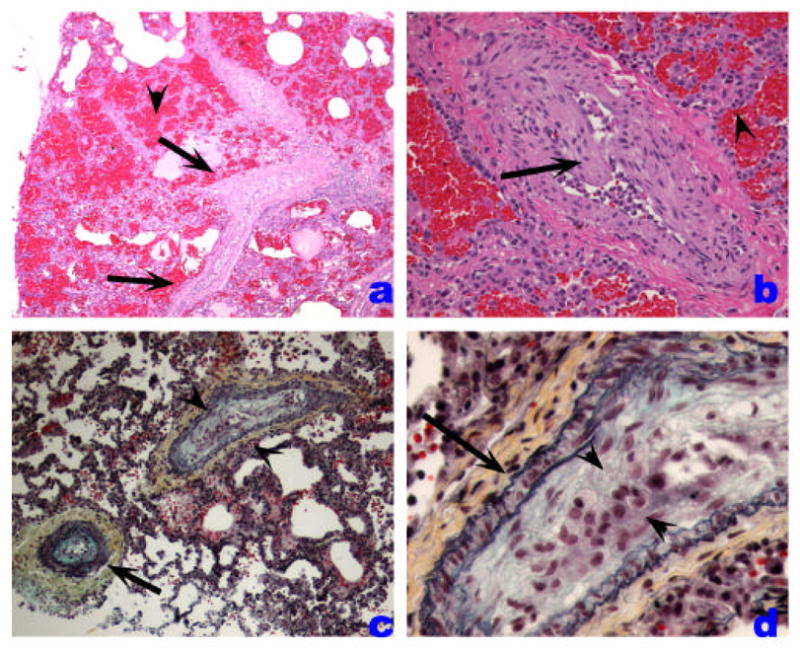
Veno-occlusive PH. A. Low-power histological view of thickened pulmonary veins running into the lung parenchyma from the pleural surface (left edge) (arrows). Note marked vein wall thickening and decreased lumen. Adjacent alveoli are filled with blood and show septal thickening with engorged capillaries (arrowhead). B. Marked vein thickening with intimal projection probably representing organized thrombus (arrow). Alveolar hemorrhage and septal thickening are highlighted with arrowhead. C and D. Movat stained pulmonary vein showing arterialization pattern with internal and external elastic layers (arrow). The vein shows marked intima thickening with organized thrombus (arrowheads).
PH caused by left heart failure or mitral valve dysfunction is often severe and one of the most frequent causes of PH. The lung vascular pathology is of limited help in its diagnosis and studies of its pathogenesis since most patients with left heart failure show chronic passive congestion with hemosiderin-laden macrophages. However, this finding is common to both patients with or without PH. Pulmonary arteries show mild medial thickening.
Pulmonary arteries also undergo remodeling in venous pulmonary hypertension, which might reflect the effect of the progressive buid-up of pressures in the pulmonary arterial side of the pulmonary circulation. It is important to consider that pulmonary arteries undergo a smooth muscle cell based remodeling with intima (eccentric lesions) and medial thickening. The extent and pattern of arterial remodeling is similar to that seen in arterial PH. Thus careful examination of pulmonary veins in any potential case of PH is warranted as pulmonary veins show intimal thickening in cases of precapillary PH [15].
Pathobiology
Little is known about the functional alterations of endothelial, smooth muscle, and adventitial fibroblasts in venous PH. Pulmonary veins are difficult to identify in lung sections, given the fact that they are located in the midst of the alveolated tissue. Only vein segments present in interlobular septa (i.e., invaginations of pleural connective tissue containing veins and lymphatics) can be more easily recognized. It is apparent that the development of endothelial cell markers unique to pulmonary veins would aid in the proper assessment of the contribution of endothelial cell dysfunction in venous pulmonary hypertension. The same limitations apply to venous smooth muscle cells and fibroblasts.
Inflammation, circulating progenitor cells, and viral agents
Pathology
Perivascular cuffing of remodeled pulmonary arteries is present in IPAH lungs and in severe PH associated with underlying conditions such as HIV infection and CREST [16;74]) (Figure 5). B- and T-cells, and macrophages infiltrate the vessel wall and are present within intimal lesions [16] (Figure 6). Both CD4- and CD8- cells are present and many of these express the memory T cell marker CD45RO, which might also indicate cell activation (Figure 6). Perivascular inflammation is more frequently seen in severe PH cases than in mild/moderate and normal lungs, but it is unclear whether this finding pertains to autopsied lung specimens or it occurs during the progression of the disease.
Figure 6.
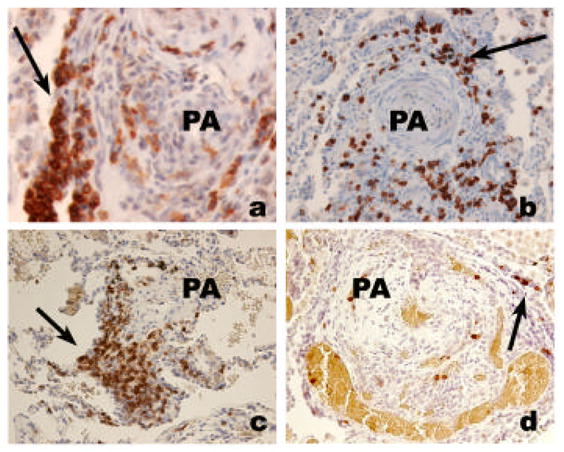
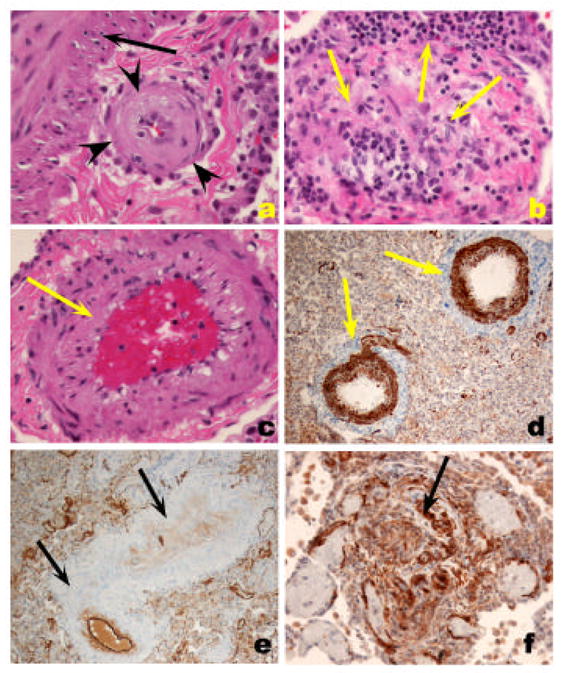
Inflammatory cell infiltrate in severe PH. A. Lymphomonuclear cells positive for CD45RO (arrow), surrounding a plexiform lesion (PA). B. Clustering of CD4 lymphocytes around a pulmonary artery (PA) with concentric thickening (arrow). C. Clustering of CD8 lymphocytes around a remodeled pulmonary artery (PA) (arrow). D. Few cells stain positively for CD45RA marker (indicative of naïve T cell cells).
As discussed in detail in the section below, it is important to consider that some of the so-called inflammatory cells might represent circulating blood progenitor cells. No detailed investigation for the lung homing of circulating blood progenitor cells has been carried out in PH. The lone evidence is provided by studies that reported AC133+ and CD45+ cells infiltration of remodeled intima of COPD patients [75].
Pathobiology
While perivascular inflammation has limited value in the pathological work up of cases of PH, it is presently one of the most promising areas of investigation in the disease. Inflammatory cells may interact with viral factors, which have emerged as potential etiological/pathogenetic agents in PH.
There is compelling evidence of global immunological alterations in IPAH patients [76;77] and PH occurs in the setting of profound immune deregulation underlying HIV infection and collagen vascular diseases. The recognition of an inflammatory component in PH [16;74] supports the investigation of expression of cytokines that might potentially drive perivascular inflammation and thus contribute to the disease. Remodeled pulmonary arteries express IL-1, IL-6, and PDGF in infiltrating inflammatory cells [78;79], the chemokine RANTES (acronym for regulated upon activation, normal T-cell expressed and secreted), an important chemoattractant for monocytes and T cells [80], and the macrophage inflammatory protein-1α (MIP-1 α) [76]. Lungs of IPAH patients have increased expression of fractalkine, a chemokine involved in T cell trafficking and monocyte recruitment, and their circulating CD4 and CD8 T-cells have higher levels of the fractalkine receptor CX3CR1 when compared with controls or samples of patients with thromboembolic PH [81].
Inflammatory cells infiltrating remodeled pulmonary arteries may include subpopulations of vascular precursor or early-progenitor cells, also potential contributors to pulmonary vascular remodeling in PH. Pulmonary arteries in PH caused by chronic hypoxia contain an infiltrating subpopulation of fibrocytes, identified by the expression mononuclear cell markers CD45, CD11b, CD14, and the fibroblast marker α1-procollagen. About 15% and 20% of these cells also undergo proliferation and express smooth muscle α-actin, respectively [82]. These studies also document that depletion of circulating monocytic cells alleviates pulmonary vascular remodeling caused by chronic hypoxia. Endothelial cell precursors may play a beneficial role in PH since their administration to monocrotaline-treated rats has dramatic healing effects in remodeled pulmonary arteries, notably when transfected with the endothelial nitric oxide synthase gene [83]. The potential for either deleterious or beneficial effects from manipulations that increase the progenitor cell pool underscores the need for careful and detailed studies in the human disease, prior to embarking on clinical trials.
Viral infection may disrupt normal immunoregulatory and homeostatic cellular pathways, which result in endothelial or smooth muscle cell injury and activate inflammation. Most of the pathways involved in virus pathogenesis converge on either pro-survival or pro-angiogenic signals, the same signals associated with severe PH.
The important role of inflammation is further highlighted in cases of PH where a viral etiology can be identified. For HIV-related pulmonary hypertension, the first clinical report of an association between infection and the development of lesions appeared in 1987 [84], followed by other reports [85–87] but with no evidence for the presence of the virus in PH vascular lesions. As PH is frequently diagnosed when it is advanced, the incidence of PH in HIV-infected patients is likely underestimated, although a recent report demonstrates a high prevalence of PH in HIV-infected children [88]. BMPR2 mutations are not required for severe PH to occur in HIV-infected patients, yet the vascular lesions in the lungs from HIV-infected [86] patients are identical to those with familial PH and sporadic IPAH with BMPR2 mutations.
Some studies showed no correlation between viral load and right heart changes [89;90]. However, a case report [91] and a recent unpublished observation showed that viral load control with HARRT therapy can be associated with an improved clinical outcome (M. Humbert, personal communication). Furthermore, bosentan, an endothelin receptor antagonist has been successfully used in some HIV-PH patients [92], suggesting that shear stress contributes to the disease independently of the viral load.
In the lung, HIV-1 primarily infects macrophages providing a potential reservoir not only for the transmission of the virus to circulating T-cells, but also a source for localized viral proteins such as Nef, Tat,and gp120, all of which may have direct effects on innocent by-stander cells. The chronic exposure to viral products in the lung, a deficiency in regulatory T cells, and an altered production of chemokines/cytokines, may all contribute to pulmonary vascular dysfunction, with endothelial cells being particularly sensitive target.
The HIV Nef (for negative factor) protein is found in plexiform lesions of macaques infected with a chimeric virus containing the simian immunodeficiency virus (SIV) backbone with the human immunodeficiency virus Nef (in place of SIV nef) [93]. Nef is also present in endothelial cells of HIV infected patients with PH (Figure 7) [93].
Figure 7.
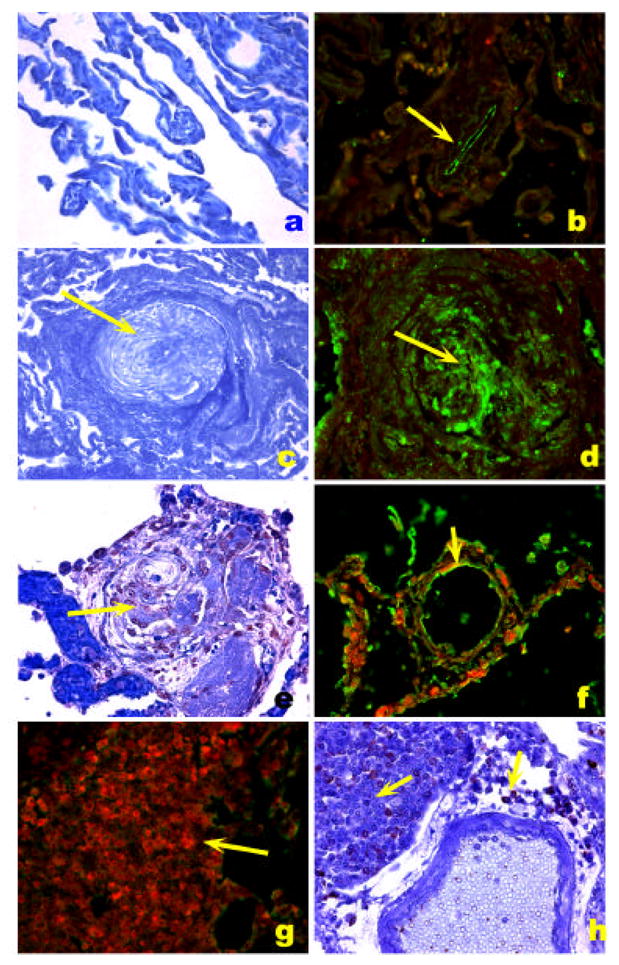
HIV-1 Nef localization in HIV-1 associated PH. A. Normal precapillary vessels (methyl green). B. Localization of Factor VIII related antigen stained with Alexa Fluor 488 (green) and Nef stained with Vector Red. Note lack of expression of Nef in normal pulmonary arteries, resulting in a green signal in the endothelial cells (arrow). C. Markedly remodeled pulmonary artery with concentric/plexiform lesion (arrow, methyl green). D. Co-localization of Factor VIII related antigen (green) with Nef (red), resulting in a yellow signal (arrow). E. Plexiform lesion in HIV-1 associated PH (arrow, methyl green). F. Co-localization of Factor VIII related antigen (green) with Nef (red), resulting in a yellow signal (arrow) in endothelial cells of pulmonary arteries with mild remodeling (arrow). G and H. Lymph node of an SIV infected monkey, showing extensive expression of Nef (red in G and brown in H, arrows) in a hilar lymph node. (Reproduced from Marecki, J. C., et al. “HIV-1 Nef is Associated with Complex Pulmonary Vascular Lesions in SHIV-nef-infected Macaques.” Am.J Respir Crit Care Med. 174.4 (2006): 437-45, with permission.)
Nef is also present in endothelial cells of HIV infected patients with PH. These recent studies suggest that the viral protein may exert direct effects on cells not necessarily permissive for viral replication. Foci of mononuclear cells and ectopic lymphoid tissues characteristically found in regions adjacent to the lesions may be sources of this viral protein (Figure 5).
The Nef protein appears to be dispensable for viral replication in vitro, but is a critical virulence factor for pathogenesis and maintenance of high viral loads in vivo [94;95]. Nef is an N-terminus-myristoylated protein with a relative molecular mass of 27 kDa is found associated with cellular membranes and the cytoskeleton [96]. Myristoylation is essential for almost all the functions ascribed to Nef, including membrane localization within lipid raft microdomains. The localization and adaptor functions recruit signaling proteins to discrete regions in the membrane and affect T cell signaling pathways [97]. Proteins associated with a survival and pro-angiogenic phenotype in severe PH such as PI-3 kinase, MAP kinases, and a p21 kinase-2 are all recruited to the rafts by Nef [98–101]. In human monocyte-derived macrophages (MDMs), Nef activates the STAT1 pathway and the secretion of MIP-1, IL-1-α, IL-6, and TNF-α [102]. Extracellular Nef found in HIV patients (approximately 10 ng/ml) enters the vascular endothelium in vivo via CXCR4 [103]. Finally, Nef can be proapoptotic or pro-survival, depending on the context of expression and the particular cell type [104]. Thus, localization of Nef to the lipid rafts may be sufficient to trigger the changes associated with the endothelial cell expansion characteristic of plexiform lesions. On the other hand, a second hit such as infection with other viruses (e.g. gammaherpesviruses such as HHV8) or a genetic susceptibility may be necessary as well.
Human herpesvirus 8 (HHV8) is a gammaherpesvirus, also known as Kaposi’s sarcoma-associated herpes virus (KSHV) [105;106]. Evidence of HHV8 is found in a large percentage of plexiform lesions of PH patients examined in Denver, USA. suggesting for the first time that this virus was a contributing factor [107]. There are several pro-angiogenic or oncogenic genes present in its genome, including a viral IL-8 and a viral IL-6 both shown to play a role in IPAH. In addition, the genome encodes a seven-transmembrane-spanning G protein-coupled receptor (GPCR) with extensive sequence similarity to cellular chemokine receptors [108]. When expressed in NIH 3T3 fibroblasts, this gene increases their ability to grow in soft agar and to induce tumor formation in nude mice [109]. GPCR increases secretion of vascular endothelial growth factor and activation of the ERK1/2 (p44/42) mitogen-activated protein kinase signaling pathway [110]. Endothelial cells that express this gene become immortalized with constitutive activation of the VEGF-receptor 2 (KDR) [111]. In addition, this gene can cause KS-like lesions in nude mice [109], and over-expression within hematopoietic cells results in angioproliferative lesions, resembling those found in KS [112]. These are yet more examples of viral factors with the potential of altering cellular phenotype in the absence of viral replication. Nevertheless, in spite of their recognized angioproliferative potential and the initial association with plexiform lesions, several groups do not reproduce these results with HHV8 [113]. Studies of patients from a San Francisco clinic along with Japanese and German cohorts find no evidence of latent virus in the lesions or serum antibodies against viral antigens [114–117]. The discrepancy between groups may be a reflection of the methodology used to detect the virus or of regional (genetic/environmental) differences in the study population. Of note, latency-associated transcripts may be undetectable if the virus is going through a lytic replication cycle. In addition, serological tests for viral antibodies are notoriously difficult and in many cases, hard to interpret. Thus, further studies are necessary to address these questions.
PH represents one of the extrahepatic complications of Hepatitis C virus infection, with a prevalence of 1–5% [118]. In the majority of patients, portal hypertension precedes pulmonary hypertension [118;119]. The pathogenesis is also poorly understood, but the histological hallmarks are similar to IPAH. Whether these lesions are secondary to increased inflammatory cytokine production or to direct viral replication or to presence of viral products in the lung remain to be determined. As in HIV-mediated pulmonary hypertension, an associated immune dysregulation may trigger uncontrolled intrapulmonary angiogenesis.
Some case report studies of pulmonary hypertension patients showed infection of lung endothelial cells with other viruses like herpes simplex type 2 or cytomegalovirus. The fact that both belong to the viral family Herpesviridae raises the intriguing possibility that they may share some of the angioproliferative potential of HHV8. Nevertheless, since both patients were HIV-positive, it is difficult to dissect the contribution of each virus to the angioproliferation.
Conclusions
The secondary role taken by the pathology in the present clinical management of PH reflects to some extent the limitations of our current understanding of the disease. As apparent by the preceding discussion, there is ample room for the diagnostic translation of the pathobiological studies, aimed at improving the diagnostic and prognostic power of the pathological assessment of pulmonary vascular remodeling. Indeed, this combined review of the pathology and pathobiology of PH seeks to show their complementarities. We forecast that the pathogenetic insights will allow one to further reclassify the disease as angioproliferative [120] or myofibroblastic (as in idiopathic pulmonary fibrosis), neoplastic-like, or a reaction to injury, with clear implications to biomarker discovery, disease-tailored therapies, genetic association studies, etc. Based solely on the advances in the past 10 years, there is the potential to refine the diagnostic tools using genomics of peripheral blood cells of PH patients [121] and also to breakdown the apparently unifying pathology of severe PH using genomics applied to lung tissue [122]. As illustrated in the color Figures, a lung with severe PH shows a range of ongoing vascular lesions, with distinct severities and cellular compositions. It is our hope that this chapter be rewritten in the future with a deeper insight into the role of specific pulmonary vascular lesions, ultimately aiding novel treatments aimed at PH.
Footnotes
This work was supported by the grant P01HL66254 to RMT and grant R01 1HL083491 to SCF, from the National Institutes of Health.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tuder RM, Cool CD, Yeager ME, Taraseviciene-Stewart L, Bull TM, Voelkel NF. The pathobiology of pulmonary hypertension: Endothelium. Chest Clin North Am. 2001;22(3):405–18. doi: 10.1016/s0272-5231(05)70280-x. [DOI] [PubMed] [Google Scholar]
- 2.Strange C, Highland KB. Pulmonary hypertension in interstitial lung disease. Curr Opin Pulm Med. 2005;11(5):452–5. doi: 10.1097/01.mcp.0000174250.38188.6d. [DOI] [PubMed] [Google Scholar]
- 3.Kessler R, Faller M, Weitzenblum E, Chaouat A, Aykut A, Ducolone A, et al. “Natural History” of pulmonary hypertension in a series of 131 patients with chronic obstructive lung disease. Am J Respir Crit Care Med. 2001;164(2):219–24. doi: 10.1164/ajrccm.164.2.2006129. [DOI] [PubMed] [Google Scholar]
- 4.Haworth SG, Rabinovitch M, Meyrick B, Michel R, Pietra GG, Polak JM, et al. In: Primary Pulmonary Hypertension: Executive Summary from the World Symposium-Primary Pulmonary Hypertension. Rich S, editor. World Health Organization; 1998. pp. 2–5. [Google Scholar]
- 5.Pietra GG, Capron F, Stewart S, Leone O, Humbert M, Robbins IM, et al. Pathologic assessment of vasculopathies in pulmonary hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):25S–32S. doi: 10.1016/j.jacc.2004.02.033. [DOI] [PubMed] [Google Scholar]
- 6.Tuder RM, Zaiman AL. Pathology of pulmonary vascular disease. In: Peacock A, Rubin LJ, editors. Pulmonary circulation. 2. London: Hodder Arnold, Health Sciences; 2003. [Google Scholar]
- 7.Wagenvoort CA, Wagenvoort N. Normal Circulation of the Lungs. In: Wagenvoort CA, Wagenvoort N, editors. Pathology of Pulmonary Hypertension. 1. New York: John Wiley & Sons; 1977. pp. 1–8. [Google Scholar]
- 8.Zaiman A, Fijalkowska I, Hassoun PM, Tuder RM. One hundred years of research in the pathogenesis of pulmonary hypertension. Am J Respir Cell Mol Biol. 2005;33(5):425–31. doi: 10.1165/rcmb.F307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cool CD, Kennedy D, Voelkel NF, Tuder RM. Pathogenesis and evolution of plexiform lesions in pulmonary hypertension associated with scleroderma and human immunodeficiency virus infection. Hum Pathol. 1997;28(4):434–42. doi: 10.1016/s0046-8177(97)90032-0. [DOI] [PubMed] [Google Scholar]
- 10.Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Respir J. 2002;19(4):632–8. doi: 10.1183/09031936.02.00245902. [DOI] [PubMed] [Google Scholar]
- 11.Cool CD, Stewart JS, Werahera P, Miller GJ, Williams RL, Voelkel NF, et al. Three-dimensional reconstruction of pulmonary arteries in plexiform pulmonary hypertension using cell specific markers: evidence for a dynamic and heterogeneous process of pulmonary endothelial cell growth. Am J Pathol. 1999;155(2):411–9. doi: 10.1016/S0002-9440(10)65137-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Atkinson C, Stewart S, Upton PD, Machado R, Thomson JR, Trembath RC, et al. Primary pulmonary hypertension is associated with reduced pulmonary vascular expression of type II bone morphogenetic protein receptor. Circulation. 2002;105(14):1672–8. doi: 10.1161/01.cir.0000012754.72951.3d. [DOI] [PubMed] [Google Scholar]
- 13.Yamashita J, Itoh H, Hirashima M, Ogawa M, Nishikawa S, Yurugi T, et al. Flk1-positive cells derived from embryonic stem cells serve as vascular progenitors. Nature. 2000;408(6808):92–6. doi: 10.1038/35040568. [DOI] [PubMed] [Google Scholar]
- 14.Frid MG, Kale VA, Stenmark KR. Mature vascular endothelium can give rise to smooth muscle cells via endothelial-mesenchymal transdifferentiation - In vitro analysis. Circ Res. 2002;90(11):1189–96. doi: 10.1161/01.res.0000021432.70309.28. [DOI] [PubMed] [Google Scholar]
- 15.Chazova I, Loyd JE, Newman JH, Belenkov Y, Meyrick B. Pulmonary artery adventitial changes and venous involvement in primary pulmonary hypertension. Am J Pathol. 199; 146(2):389–97. [PMC free article] [PubMed] [Google Scholar]
- 16.Tuder RM, Groves BM, Badesch DB, Voelkel NF. Exuberant endothelial cell growth and elements of inflammation are present in plexiform lesions of pulmonary hypertension. Am J Pathol. 1994;144(2):275–85. [PMC free article] [PubMed] [Google Scholar]
- 17.Heath D, Smith P, Gosney J. Ultrastructure of early plexogenic pulmonary arteriopathy. Histopathology. 1988;12(1):41–52. doi: 10.1111/j.1365-2559.1988.tb01915.x. [DOI] [PubMed] [Google Scholar]
- 18.Voelkel NF, Tuder RM, Cool CD, Geraci M, Quaife R, Bristow M. In: Severe chronic pulmonary hypertension and the pressure-overloaded right ventricle. 1. Polak J, editor. London: 2002. [Google Scholar]
- 19.Mitani Y, Ueda M, Komatsu R, Maruyama K, Nagai R, Matsumura M, et al. Vascular smooth muscle cell phenotypes in primary pulmonary hypertension. Eur Respir J. 2001;17(2):316–20. doi: 10.1183/09031936.01.17203160. [DOI] [PubMed] [Google Scholar]
- 20.Budhiraja R, Tuder RM, Hassoun PM. Endothelial dysfunction in pulmonary hypertension. Circulation. 2004;109(2):159–65. doi: 10.1161/01.CIR.0000102381.57477.50. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka Y, Schuster DP, Davis EC, Patterson GA, Botney MD. Role of vascular injury and hemodynamics in rat pulmonary artery remodeling. J Clin Invest. 1996;98(2):434–42. doi: 10.1172/JCI118809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tuder RM, Chacon M, Alger LA, Wang J, Taraseviciene-Stewart L, Kasahara Y, et al. Expression of angiogenesis-related molecules in plexiform lesions in severe pulmonary hypertension: evidence for a process of disordered angiogenesis. J Pathol. 2001;195(3):367–74. doi: 10.1002/path.953. [DOI] [PubMed] [Google Scholar]
- 23.Lee SD, Shroyer KR, Markham NE, Cool CD, Voelkel NF, Tuder RM. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest. 1998;101(5):927–34. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yeager ME, Halley GR, Golpon HA, Voelkel NF, Tuder RM. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88(1):e8–e11. doi: 10.1161/01.res.88.1.e2. [DOI] [PubMed] [Google Scholar]
- 25.The International PPH Consortium. Lane KB, Machado RD, Pauciulo MW, Thompson JR, Philips JA, III, et al. Heterozygous germline mutations in BMPR2 encoding a TGF-B receptor cause familiar pulmonary hypertension. Nat Genet. 2000;26(1):81–4. doi: 10.1038/79226. [DOI] [PubMed] [Google Scholar]
- 26.Deng Z, Morse JH, Slager SL, Cuervo N, Moore KJ, Venetos G, et al. Familial primary pulmonary hypertension (gene PPH1) Is caused by mutations in the bone morphogenetic protein receptor-II gene. Am J Hum Genet. 2000;67(3):737–44. doi: 10.1086/303059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Richter A, Yeager ME, Zaiman A, Cool CD, Voelkel NF, Tuder RM. Impaired transforming growth Factor β signaling in idiopathic pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170(12):1340–8. doi: 10.1164/rccm.200311-1602OC. [DOI] [PubMed] [Google Scholar]
- 28.Teichert-Kuliszewska K, Kutryk MJ, Kuliszewski MA, Karoubi G, Courtman DW, Zucco L, et al. Bone morphogenetic protein receptor-2 signaling promotes pulmonary arterial endothelial cell survival: implications for loss-of-function mutations in the pathogenesis of pulmonary hypertension. Circ Res. 2006;98(2):209–17. doi: 10.1161/01.RES.0000200180.01710.e6. [DOI] [PubMed] [Google Scholar]
- 29.Taraseviciene-Stewart L, Kasahara Y, Alger L, Hirth P, Mc Mahon GG, Waltenberger J, et al. Inhibition of the VEGF receptor 2 combined with chronic hypoxia causes cell death-dependent pulmonary endothelial cell proliferation and severe pulmonary hypertension. Faseb J. 2001;15(2):427–38. doi: 10.1096/fj.00-0343com. [DOI] [PubMed] [Google Scholar]
- 30.Campbell AIM, Zhao YD, Sandhu R, Stewart DJ. Cell-based gene transfer of vascular endothelial growth factor attenuates monocrotaline-induced pulmonary hypertension. Circulation. 2001;104(18):2242–8. doi: 10.1161/hc4201.097838. [DOI] [PubMed] [Google Scholar]
- 31.Sakao S, Taraseviciene-Stewart L, Lee JD, Wood K, Cool CD, Voelkel NF. Initial apoptosis is followed by increased proliferation of apoptosis-resistant endothelial cells. Faseb J. 2005;19(9):1178–80. doi: 10.1096/fj.04-3261fje. [DOI] [PubMed] [Google Scholar]
- 32.Giaid A, Yanagisawa M, Langleben D, Michel RP, Levy R, Shennib H, et al. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N Engl J Med. 1993;328(24):1732–9. doi: 10.1056/NEJM199306173282402. [DOI] [PubMed] [Google Scholar]
- 33.Bonnet S, Michelakis ED, Porter CJ, ndrade-Navarro MA, Thebaud B, Bonnet S, et al. An abnormal mitochondrial-hypoxia inducible factor-1 alpha-Kv channel pathway disrupts oxygen sensing and triggers pulmonary arterial hypertension in fawn hooded rats - Similarities to human pulmonary arterial hypertension. Circulation. 2006;113(22):2630–41. doi: 10.1161/CIRCULATIONAHA.105.609008. [DOI] [PubMed] [Google Scholar]
- 34.McMurtry MS, Archer SL, Altieri DC, Bonnet S, Haromy A, Harry G, et al. Gene therapy targeting survivin selectively induces pulmonary vascular apoptosis and reverses pulmonary arterial hypertension. J Clin Invest. 2005;115(6):1479–91. doi: 10.1172/JCI23203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giaid A, Saleh D. Reduced expression of endothelial nitric oxide synthase in the lungs of patients with pulmonary hypertension. N Engl J Med. 1995;333(4):214–21. doi: 10.1056/NEJM199507273330403. [DOI] [PubMed] [Google Scholar]
- 36.Tuder RM, Cool CD, Geraci MW, Wang J, Abman SH, Wright L, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med. 1999;159(6):1925–32. doi: 10.1164/ajrccm.159.6.9804054. [DOI] [PubMed] [Google Scholar]
- 37.Achcar RO, Demura Y, Rai PR, Taraseviciene-Stewart L, Kasper M, Voelkel NF, et al. Loss of caveolin and heme oxygenase expression in severe pulmonary hypertension. Chest. 2006;129(3):696–705. doi: 10.1378/chest.129.3.696. [DOI] [PubMed] [Google Scholar]
- 38.Voelkel NF, Cool CD, Lee SD, Wright L, Geraci MW, Tuder RM. Primary pulmonary hypertension between inflammation and cancer. Chest. 1999;114(Suppl 3):225S–30S. doi: 10.1378/chest.114.3_supplement.225s. [DOI] [PubMed] [Google Scholar]
- 39.Yamaki S, Wagenvoort CA. Comparison of primary plexogenic arteriopathy in adults and children. A morphometric study in 40 patients. Br Heart J. 1985;54(4):428–34. doi: 10.1136/hrt.54.4.428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yi ES, Kim H, Ahn H, Strother J, Morris T, Masliah E, et al. Distribution of obstructive intimal lesions and their cellular phenotypes in chronic pulmonary hypertension. A morphometric and immunohistochemical study. Am J Respir Crit Care Med. 2000;162(4):1577–86. doi: 10.1164/ajrccm.162.4.9912131. [DOI] [PubMed] [Google Scholar]
- 41.Palevsky HI, Schloo BL, Pietra GG, Weber KT, Janicki JS, Rubin E, et al. Primary pulmonary hypertension, vascular structure, morphometry, and responsiveness to vasodilator agents. Circulation. 1989;80(1207):1221. doi: 10.1161/01.cir.80.5.1207. [DOI] [PubMed] [Google Scholar]
- 42.Santos S, Peinado VI, Ramirez J, Melgosa T, Roca J, Rodriguez-Roisin R, et al. Characterization of pulmonary vascular remodelling in smokers and patients with mild COPD. Eur Resp J. 2002;19(4):632–8. doi: 10.1183/09031936.02.00245902. [DOI] [PubMed] [Google Scholar]
- 43.Balk AG, Dingemans KP, Wagenvoort CA. The ultrastructure of the various forms of pulmonary arterial intimal fibrosis. Virchows Arch A Pathol Anat Histol. 1979;382(2):139–50. doi: 10.1007/BF01102870. [DOI] [PubMed] [Google Scholar]
- 44.Meyrick B, Reid L. Hypoxia and incorporation of 3H-thymidine by cells of the rat pulmonary arteries and alveolar wall. Am J Pathol. 1979;96(1):51–70. [PMC free article] [PubMed] [Google Scholar]
- 45.Meyrick BO, Reid LM. Crotalaria-induced pulmonary hypertension. Uptake of 3H-thymidine by the cells of the pulmonary circulation and alveolar walls. Am J Pathol. 1982;106(1):84–94. [PMC free article] [PubMed] [Google Scholar]
- 46.Reid L, Anderson G, Simon G. Comparison of primary and thromboembolic pulmonary hypertension. Thorax. 1972;27(2):263–4. doi: 10.1136/thx.27.2.263-b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuder RM, Zaiman AL. Prostacyclin Analogs as the Brakes for Pulmonary Artery Smooth Muscle Cell Proliferation. Is it sufficient to treat severe pulmonary hypertension? Am J Respir Cell Mol Biol. 2002;26(2):171–4. doi: 10.1165/ajrcmb.26.2.f230. [DOI] [PubMed] [Google Scholar]
- 48.Frid MG, Aldashev AA, Dempsey EC, Stenmark KR. Smooth muscle cells isolated from discrete compartments of the mature vascular media exhibit unique phenotypes and distinct growth capabilities. Circ Res. 1997;81(6):940–52. doi: 10.1161/01.res.81.6.940. [DOI] [PubMed] [Google Scholar]
- 49.Morrell NW, Yang X, Upton PD, Jourdan KB, Morgan N, Sheares KK, et al. Altered growth responses of pulmonary artery smooth muscle cells from patients with primary pulmonary hypertension to transforming growth factor-beta(1) and bone morphogenetic proteins. Circulation. 2001;104(7):790–5. doi: 10.1161/hc3201.094152. [DOI] [PubMed] [Google Scholar]
- 50.Yang X, Long L, Southwood M, Rudarakanchana N, Upton PD, Jeffery TK, et al. Dysfunctional Smad signaling contributes to abnormal smooth muscle cell proliferation in familial pulmonary arterial hypertension. Circ Res. 2005;96(10):1053–63. doi: 10.1161/01.RES.0000166926.54293.68. [DOI] [PubMed] [Google Scholar]
- 51.Song YL, Jones JE, Beppu H, Keaney JF, Loscalzo J, Zhang YY. Increased susceptibility to pulmonary hypertension in heterozygous BMPR2-mutant mice. Circulation. 2005;112(4):553–62. doi: 10.1161/CIRCULATIONAHA.104.492488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Launay JM, Herve P, Peoc’h K, Tournois C, Callebert J, Nebigil CG, et al. Function of the serotonin 5-hydroxytryptamine 2B receptor in pulmonary hypertension. Nat Med. 2002;8(10):1129–35. doi: 10.1038/nm764. [DOI] [PubMed] [Google Scholar]
- 53.Eddahibi S, Humbert M, Fadel E, Raffestin B, Darmon M, Capron F, et al. Serotonin transporter overexpression is responsible for pulmonary artery smooth muscle hyperplasia in primary pulmonary hypertension. J Clin Invest. 2001;108(8):1141–50. doi: 10.1172/JCI12805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Eddahibi S, Hanoun N, Lanfumey L, Lesch KP, Raffestin B, Hamon M, et al. Attenuated hypoxic pulmonary hypertension in mice lacking the 5- hydroxytryptamine transporter gene. J Clin Invest. 2000;105(11):1555–62. doi: 10.1172/JCI8678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Botney MD, Bahadori L, Gold LI. Vascular remodeling in primary pulmonary hypertension. Potential role for transforming growth factor-beta. Am J Pathol. 1994;144(2):286–95. [PMC free article] [PubMed] [Google Scholar]
- 56.Schermuly RT, Dony E, Ghofrani HA, Pullamsetti S, Savai R, Roth M, et al. Reversal of experimental pulmonary hypertension by PDGF inhibition. J Clin Invest. 2005;115(10):2811–21. doi: 10.1172/JCI24838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ghofrani HA, Seeger W, Grimminger F. Imatinib for the treatment of pulmonary arterial hypertension. The New England Journal of Medicine. 2005;353(13):1412–3. doi: 10.1056/NEJMc051946. [DOI] [PubMed] [Google Scholar]
- 58.Jones PL, Cowan KN, Rabinovitch M. Tenascin-C, proliferation and subendothelial fibronectin in progressive pulmonary vascular disease. Am J Pathol. 1997;150(4):1349–60. [PMC free article] [PubMed] [Google Scholar]
- 59.Rabinovitch M. EVE and beyond, retro and prospective insights. Am J Physiol. 1999;277(1 Pt 1):L5–12. doi: 10.1152/ajplung.1999.277.1.L5. [DOI] [PubMed] [Google Scholar]
- 60.Cowan KN, Jones PL, Rabinovitch M. Elastase and matrix metalloproteinase inhibitors induce regression, and tenascin-C antisense prevents progression, of vascular disease. J Clin Invest. 2000;105(1):21–34. doi: 10.1172/JCI6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Merklinger SL, Jones PL, Martinez EC, Rabinovitch M. Epidermal growth factor receptor blockade mediates smooth muscle cell apoptosis and improves survival in rats with pulmonary hypertension. Circulation. 2005;112(3):423–31. doi: 10.1161/CIRCULATIONAHA.105.540542. [DOI] [PubMed] [Google Scholar]
- 62.Vieillard-Baron A, Frisdal E, Eddahibi S, Deprez I, Baker AH, Newby AC, et al. Inhibition of matrix metalloproteinases by lung TIMP-1 gene transfer or doxycycline aggravates pulmonary hypertension in rats. Circ Res. 2000;87(5):418–25. doi: 10.1161/01.res.87.5.418. [DOI] [PubMed] [Google Scholar]
- 63.Lepetit H, Eddahibi S, Fadel E, Frisdal E, Munaut C, Noel A, et al. Smooth muscle cell matrix metalloproteinases in idiopathic pulmonary arterial hypertension. Eur Resp J. 2005;25(5):834–42. doi: 10.1183/09031936.05.00072504. [DOI] [PubMed] [Google Scholar]
- 64.Matsui K, Takano Y, Yu ZX, Hi JE, Stetler-Stevenson WG, Travis WD, et al. Immunohistochemical study of endothelin-1 and matrix metalloproteinases in plexogenic pulmonary arteriopathy. Pathol Res Pract. 2002;198(6):403–12. doi: 10.1078/0344-0338-00273. [DOI] [PubMed] [Google Scholar]
- 65.Cowan KN, Heilbut A, Humpl T, Lam C, Ito S, Rabinovitch M. Complete reversal of fatal pulmonary hypertension in rats by a serine elastase inhibitor. Nat Med. 2000;6(6):698–702. doi: 10.1038/76282. [DOI] [PubMed] [Google Scholar]
- 66.Tuder RM, Yeager ME, Geraci M, Golpon HA, Voelkel NF. Severe pulmonary hypertension after the discovery of the familial primary pulmonary hypertension gene. Eur Respir J. 2001;17(6):1065–9. doi: 10.1183/09031936.01.00202701. [DOI] [PubMed] [Google Scholar]
- 67.Kasahara Y, Tuder RM, Cool CD, Lynch DA, Flores SC, Voelkel NF. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med. 2001;163(3):737–44. doi: 10.1164/ajrccm.163.3.2002117. [DOI] [PubMed] [Google Scholar]
- 68.Remillard CV, Yuan JXJ. Activation of K+ channels: an essential pathway in programmed cell death. Amer J Physiol -Lung Cell M PH. 2004;286(1):L49–L67. doi: 10.1152/ajplung.00041.2003. [DOI] [PubMed] [Google Scholar]
- 69.Fantozzi I, Platoshyn O, Wong AH, Zhang S, Remillard CV, Furtado MR, et al. Bone morphogenetic protein-2 upregulates expression and function of voltage-gated K+ channels in human pulmonary artery smooth muscle cells. AJP - Lung Cellular and Molecular Physiology. 2006:00191. doi: 10.1152/ajplung.00191.2005. [DOI] [PubMed] [Google Scholar]
- 70.Dawson CA. Hypoxic pulmonary vasocontriction: Heterogeneity. In: Yuan JX, editor. Hypoxic pulmonary vasoconstriction: Cellular and Molecule Mechanisms. Norwell; Massachussets: 2002. pp. 15–32. [Google Scholar]
- 71.Altieri DC. Validating survivin as a cancer therapeutic target. Nature Reviews Cancer. 2003;3(1):46–54. doi: 10.1038/nrc968. [DOI] [PubMed] [Google Scholar]
- 72.Stenmark KR, Davie N, Frid M, Gerasimovskaya E, Das M. Role of the adventitia in pulmonary vascular remodeling. Physiology (Bethesda) 2006;21(2):134–45. doi: 10.1152/physiol.00053.2005. [DOI] [PubMed] [Google Scholar]
- 73.Bowers R, Cool C, Murphy RC, Tuder RM, Hopken MW, Flores SC, et al. Oxidative stress in severe pulmonary hypertension. Am J Respir Crit Care Med. 2004;169(6):764–9. doi: 10.1164/rccm.200301-147OC. [DOI] [PubMed] [Google Scholar]
- 74.Caslin AW, Heath D, Madden B, Yacoub M, Gosney JR, Smith P. The histopathology of 36 cases of plexogenic pulmonary arteriopathy. Histopathology. 1990;16(1):9–19. doi: 10.1111/j.1365-2559.1990.tb01054.x. [DOI] [PubMed] [Google Scholar]
- 75.Peinado VI, Ramirez J, Roca J, Rodriguez-Roisin R, Barbera JA. Identification of vascular progenitor cells in pulmonary arteries of patients with chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol. 2006;34(3):257–63. doi: 10.1165/rcmb.2005-0255OC. [DOI] [PubMed] [Google Scholar]
- 76.Dorfmuller P, Perros F, Balabanian K, Humbert M. Inflammation in pulmonary arterial hypertension. Eur Resp J. 2003;22(2):358–63. doi: 10.1183/09031936.03.00038903. [DOI] [PubMed] [Google Scholar]
- 77.Nicolls MR, Taraseviciene-Stewart L, Rai PR, Badesch DB, Voelkel NF. Autoimmunity and pulmonary hypertension: a perspective. Eur Respir J. 2005;26(6):1110–8. doi: 10.1183/09031936.05.00045705. [DOI] [PubMed] [Google Scholar]
- 78.Humbert M, Monti G, Brenot F, Sitbon O, Portier A, Grangeot-Keros L, et al. Increased interleukin-1 and interleukin-6 serum concentrations in severe primary pulmonary hypertension. Am J Respir Crit Care Med. 1995;151(5):1628–31. doi: 10.1164/ajrccm.151.5.7735624. [DOI] [PubMed] [Google Scholar]
- 79.Humbert M, Monti G, Fartoukh M, Magnan A, Brenot F, Rain B, et al. Platelet-derived growth factor expression in primary pulmonary hypertension: comparison of HIV seropositive and HIV seronegative patients. Eur Resp J. 1998;11(3):554–9. [PubMed] [Google Scholar]
- 80.Dorfmuller P, Zarka V, Durand-Gasselin I, Monti G, Balabanian K, Garcia G, et al. Chemokine RANTES in severe pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165(4):534–9. doi: 10.1164/ajrccm.165.4.2012112. [DOI] [PubMed] [Google Scholar]
- 81.Balabanian K, Foussat A, Dorfmuller P, Durand-Gasselin I, Capel F, Bouchet-Delbos L, et al. CX3C chemokine fractalkine in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2002;165(10):1419–25. doi: 10.1164/rccm.2106007. [DOI] [PubMed] [Google Scholar]
- 82.Frid MG, Brunetti JA, Burke DL, Carpenter TC, Davie NJ, Reeves JT, et al. Hypoxia-induced pulmonary vascular remodeling requires recruitment of circulating mesenchymal precursors of a monocyte/macrophage lineage. Am J Pathol. 2006;168(2):659–69. doi: 10.2353/ajpath.2006.050599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhao YDD, Courtman DW, Deng YP, Kugathasan L, Zhang QW, Stewart DJ. Rescue of monocrotaline-induced pulmonary arterial hypertension using bone marrow-derived endothelial-like progenitor cells - Efficacy of combined cell and eNOS gene therapy in established disease. Circ Res. 2005;96(4):442–50. doi: 10.1161/01.RES.0000157672.70560.7b. [DOI] [PubMed] [Google Scholar]
- 84.Kim KK, Factor SM. Membranoproliferative glomerulonephritis and plexogenic pulmonary arteriopathy in a homosexual man with acquired immunodeficiency syndrome. Hum Pathol. 1987;18(12):1293–6. doi: 10.1016/s0046-8177(87)80417-3. [DOI] [PubMed] [Google Scholar]
- 85.Kanmogne GD, Kennedy RC, Grammas P. Is HIV involved in the pathogenesis of non-infectious pulmonary complications in infected patients? Curr HIV Res. 2003;1(4):385–93. doi: 10.2174/1570162033485177. [DOI] [PubMed] [Google Scholar]
- 86.Mette SA, Palevsky HI, Pietra GG, Williams TM, Bruder E, Prestipino AJ, et al. Primary pulmonary hypertension in association with human immunodeficiency virus infection. A possible viral etiology for some forms of hypertensive pulmonary arteriopathy. Am Rev Respir Dis. 1992;145(5):1196–200. doi: 10.1164/ajrccm/145.5.1196. [DOI] [PubMed] [Google Scholar]
- 87.Pellicelli AM, Barbaro G, Palmieri F, Girardi E, D’Ambrosio C, Rianda A, et al. Primary pulmonary hypertension in HIV patients: a systematic review. Angiology. 2001;52(1):31–41. doi: 10.1177/000331970105200105. [DOI] [PubMed] [Google Scholar]
- 88.Pongprot Y, Sittiwangkul R, Silvilairat S, Sirisanthana V. Cardiac manifestations in HIV-infected Thai children. Ann Trop Paediatr. 2004;24(2):153–9. doi: 10.1179/027249304225013439. [DOI] [PubMed] [Google Scholar]
- 89.Recusani F, Di M, Gambarin F, D’Armini A, Klersy C, Campana C. Clinical and therapeutical follow-up of HIV-associated pulmonary hypertension: prospective study of 10 patients. AIDS. 2003;17(Suppl 1):S88–S95. doi: 10.1097/00002030-200304001-00013. [DOI] [PubMed] [Google Scholar]
- 90.Barbaro G, Lucchini A, Pellicelli AM, Grisorio B, Giancaspro G, Barbarini G. Highly active antiretroviral therapy compared with HAART and bosentan in combination in patients with HIV-associated pulmonary hypertension. Heart. 2006;92(8):1164–6. doi: 10.1136/hrt.2005.076794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Speich R, Jenni R, Opravil M, Jaccard R. Regression of HIV-associated pulmonary arterial hypertension and long-term survival during antiretroviral therapy. Swiss Med Wkly. 2001;131(45–46):663–5. doi: 10.4414/smw.2001.09842. [DOI] [PubMed] [Google Scholar]
- 92.Sitbon O, Gressin V, Speich R, Macdonald PS, Opravil M, Cooper DA, et al. Bosentan for the treatment of human immunodeficiency virus-associated pulmonary arterial hypertension. Am J Respir Crit Care Med. 2001;170(11):1212–7. doi: 10.1164/rccm.200404-445OC. [DOI] [PubMed] [Google Scholar]
- 93.Marecki JC, Cool CD, Parr JE, Beckey VE, Luciw PA, Tarantal AF, et al. HIV-1 Nef is Associated with Complex Pulmonary Vascular Lesions in SHIV-nef-infected Macaques. Am J Respir Crit Care Med. 2006 doi: 10.1164/rccm.200601-005OC. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kirchhoff F, Greenough TC, Brettler DB, Sullivan JL, Desrosiers RC. Brief report: absence of intact nef sequences in a long-term survivor with nonprogressive HIV-1 infection. N Engl J Med. 1995;332(4):228–32. doi: 10.1056/NEJM199501263320405. [DOI] [PubMed] [Google Scholar]
- 95.Kestler HW, Ringler DJ, Mori K, Panicali DL, Sehgal PK, Daniel MD, et al. Importance of the nef gene for maintenance of high virus loads and for development of AIDS. Cell. 1991;65(4):651–62. doi: 10.1016/0092-8674(91)90097-i. [DOI] [PubMed] [Google Scholar]
- 96.Harris M. The role of myristoylation in the interactions between human immunodeficiency virus type I Nef and cellular proteins. Biochem Soc Trans. 1995;23(3):557–61. doi: 10.1042/bst0230557. [DOI] [PubMed] [Google Scholar]
- 97.Wang JK, Kiyokawa E, Verdin E, Trono D. The Nef protein of HIV-1 associates with rafts and primes T cells for activation. Proc Natl Acad Sci U S A. 2000;97(1):394–9. doi: 10.1073/pnas.97.1.394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Graziani A, Galimi F, Medico E, Cottone E, Gramaglia D, Boccaccio C, et al. The HIV-1 nef protein interferes with phosphatidylinositol 3-kinase activation 1. J Biol Chem. 1996;271(12):6590–3. doi: 10.1074/jbc.271.12.6590. [DOI] [PubMed] [Google Scholar]
- 99.Linnemann T, Zheng YH, Mandic R, Peterlin BM. Interaction between Nef and phosphatidylinositol-3-kinase leads to activation of p21-activated kinase and increased production of HIV. Virology. 1915;294(2):246–55. doi: 10.1006/viro.2002.1365. [DOI] [PubMed] [Google Scholar]
- 100.He JC, Husain M, Sunamoto M, D’Agati VD, Klotman ME, Iyengar R, et al. Nef stimulates proliferation of glomerular podocytes through activation of Src-dependent Stat3 and MAPK1,2 pathways. J Clin Invest. 2004;114(5):643–51. doi: 10.1172/JCI21004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Krautkramer E, Giese SI, Gasteier JE, Muranyi W, Fackler OT. Human immunodeficiency virus type 1 Nef activates p21-activated kinase via recruitment into lipid rafts. J Virol. 2004;78(8):4085–97. doi: 10.1128/JVI.78.8.4085-4097.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Olivetta E, Percario Z, Fiorucci G, Mattia G, Schiavoni I, Dennis C, et al. HIV-1 Nef induces the release of inflammatory factors from human monocyte/macrophages: involvement of Nef endocytotic signals and NF-kappa B activation. J Immunol. 1915;170(4):1716–27. doi: 10.4049/jimmunol.170.4.1716. [DOI] [PubMed] [Google Scholar]
- 103.James CO, Huang MB, Khan M, Garcia-Barrio M, Powell MD, Bond VC. Extracellular Nef protein targets CD4+ T cells for apoptosis by interacting with CXCR4 surface receptors. J Virol. 2004;78(6):3099–109. doi: 10.1128/JVI.78.6.3099-3109.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Choi HJ, Smithgall TE. HIV-1 Nef promotes survival of TF-1 macrophages by inducing Bcl-XL expression in an extracellular signal-regulated kinase-dependent manner. J Biol Chem. 2003;279(49):51688–96. doi: 10.1074/jbc.M410068200. [DOI] [PubMed] [Google Scholar]
- 105.Damania B, Desrosiers RC. Simian homologues of human herpesvirus 8. PhilosTrans R Soc Lond B Biol Sci. 1929;356(1408):535–43. doi: 10.1098/rstb.2000.0782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Desrosiers RC, Sasseville VG, Czajak SC, Zhang X, Mansfield KG, Kaur A, et al. A herpesvirus of rhesus monkeys related to the human Kaposi’s sarcoma-associated herpesvirus. J Virol. 1997;71(12):9764–9. doi: 10.1128/jvi.71.12.9764-9769.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Cool CD, Rai MD, Yeager ME, Hernandez-Saavedra D, Serls AE, Bull TM, et al. Expression of human herpesvirus 8 in primary pulmonary hypertension. N Engl J Med. 2003;349(12):1113–22. doi: 10.1056/NEJMoa035115. [DOI] [PubMed] [Google Scholar]
- 108.Boshoff C, Endo Y, Collins PD, Takeuchi Y, Reeves JD, Schweickart VL, et al. Angiogenic and HIV-inhibitory functions of KSHV-encoded chemokines. Science. 1997;278(5336):290–4. doi: 10.1126/science.278.5336.290. [DOI] [PubMed] [Google Scholar]
- 109.Guo HG, Sadowska M, Reid W, Tschachler E, Hayward G, Reitz M. Kaposi’s sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J Virol. 2003;77(4):2631–9. doi: 10.1128/JVI.77.4.2631-2639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Estep RD, Axthelm MK, Wong SW. A G protein-coupled receptor encoded by rhesus rhadinovirus is similar to ORF74 of Kaposi’s sarcoma-associated herpesvirus. J Virol. 2003;77(3):1738–46. doi: 10.1128/JVI.77.3.1738-1746.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bais C, Van G, Eroles P, Mutlu A, Chiozzini C, Dias S, et al. Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/KDR. Cancer Cell. 2003;3(2):131–43. doi: 10.1016/s1535-6108(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 112.Yang TY, Chen SC, Leach MW, Manfra D, Homey B, Wiekowski M, et al. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma. J Exp Med. 2000;191(3):445–54. doi: 10.1084/jem.191.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Galambos C, Montgomery J, Jenkins FJ. No role for kaposi sarcoma-associated herpesvirus in pediatric idiopathic pulmonary hypertension. Pediatr Pulmonol. 2006;41(2):122–5. doi: 10.1002/ppul.20349. [DOI] [PubMed] [Google Scholar]
- 114.Laney AS, De M, Peters JS, Malloy M, Teehankee C, Moore PS, et al. Kaposi sarcoma-associated herpesvirus and primary and secondary pulmonary hypertension. Chest. 2005;127(3):762–7. doi: 10.1378/chest.127.3.762. [DOI] [PubMed] [Google Scholar]
- 115.Katano H, Ito K, Shibuya K, Saji T, Sato Y, Sata T. Lack of human herpesvirus 8 infection in lungs of Japanese patients with primary pulmonary hypertension. J Infect Dis. 2001;191(5):743–5. doi: 10.1086/427824. [DOI] [PubMed] [Google Scholar]
- 116.Nicastri E, Vizza CD, Carletti F, Cicalini S, Badagliacca R, Poscia R, et al. Human herpesvirus 8 and pulmonary hypertension. Emerg Infect Dis. 2005;11(9):1480–2. doi: 10.3201/eid1109.040880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Henke-Gendo C, Mengel M, Hoeper MM, Alkharsah K, Schulz TF. Absence of Kaposi’s sarcoma-associated herpesvirus in patients with pulmonary arterial hypertension. Am J Respir Crit Care Med. 1915;172(12):1581–5. doi: 10.1164/rccm.200504-546OC. [DOI] [PubMed] [Google Scholar]
- 118.Moorman J, Saad M, Kosseifi S, Krishnaswamy G. Hepatitis C virus and the lung: implications for therapy. Chest. 2005;128(4):2882–92. doi: 10.1378/chest.128.4.2882. [DOI] [PubMed] [Google Scholar]
- 119.Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol. 1991;17(2):492–8. doi: 10.1016/s0735-1097(10)80121-4. [DOI] [PubMed] [Google Scholar]
- 120.Voelkel NF, Tuder RM. Severe pulmonary hypertensive diseases: a perspective. Eur Resp J. 1999;14(6):1246–50. doi: 10.1183/09031936.99.14612469. [DOI] [PubMed] [Google Scholar]
- 121.Bull TM, Coldren CD, Moore M, Sotto-Santiago SM, Pham DV, Nana-Sinkam SP, et al. Gene microarray analysis of peripheral blood cells in pulmonary arterial hypertension. Am J Respir Crit Care Med. 2004;170(8):911–9. doi: 10.1164/rccm.200312-1686OC. [DOI] [PubMed] [Google Scholar]
- 122.Geraci MW, Gao B, Hoshikawa Y, Yeager ME, Tuder RM, Voelkel NF. Genomic approaches to research in pulmonary hypertension. Respir Res. 2001;2(4):210–5. doi: 10.1186/rr59. [DOI] [PMC free article] [PubMed] [Google Scholar]