

Summary
Protein kinase D1 (PKD1) is involved in cellular processes including protein secretion, proliferation and apoptosis. Studies suggest PKD1 is activated by various stimulants including gastrointestinal (GI) hormones/neurotransmitters and growth factors in a protein kinase C (PKC)-dependent pathway. However, little is known about the mechanisms of PKD1 activation in physiologic GI tissues. We explored PKD1 activation by GI hormones/neurotransmitters and growth factors and the mediators involved in rat pancreatic acini. Only hormones/neurotransmitters activating phospholipase C caused PKD1 phosphorylation (S916, S744/748). CCK activated PKD1 and caused a time- and dose-dependant increase in serine phosphorylation by activation of high- and low-affinity CCKA receptor states. Inhibition of CCK-stimulated increases in phospholipase C, PKC activity or intracellular calcium decreased PKD1 S916 phosphorylation by 56%, 62% and 96%, respectively. PKC inhibitors GF109203X/Go6976/Go6983/PKC-ζ pseudosubstrate caused a 62/43/49/0% inhibition of PKD1 S916 phosphorylation and an 87/13/82/0% inhibition of PKD1 S744/748 phosphorylation. Expression of dominant negative PKC-δ, but not PKC-ε, or treatment with PKC-δ translocation inhibitor caused marked inhibition of PKD phosphorylation. Inhibition of Src/PI3K/MAPK/tyrosine phosphorylation had no effect. In unstimulated cells, PKD1 was mostly located in the cytoplasm. CCK stimulated translocation of total and phosphorylated PKD1 to the membrane. These results demonstrate that CCKA receptor activation leads to PKD activation by signaling through PKC-dependent and PKC-independent pathways.
Keywords: PKD1 activation, Pancreas, Cholecystokinin, Gastrointestinal hormones, Growth factors, PKC
1. Introduction
Protein kinase D1 (PKD1), PKD2 and PKD3 constitute the recently identified PKD family, a subclass of the AGC family of serine/threonine kinases, with structural and enzymological properties different from those of members of the PKC family [1,2]. PKD1 is composed of different domains: a N-terminal region, two cysteine-rich zinc-finger regions, a region rich in negatively charged amino acids, a pleckstrin-homology domain and a Ser/Thr kinase catalytic domain (for review, see [3]). PKD1 can be activated by growth factors, oxidative stress, thrombin, bioactive lipids, cross-linking of B- and T-cell receptors and some G-protein coupled receptors (GPCR) [4–7]. For most of these stimuli, direct phosphorylation by a member of the PKC family is reported to be the major mechanism of PKD1 activation in vivo [8]. This transphosphorylation occurs at the serine residues 744/748 in the activation loop of the kinase domain [8]. Mutational studies have shown that phosphorylation of these residues is required and sufficient for PKC-mediated PKD1 activation by a number of stimulants [9]. A second important phosphorylation event in PKD1 activation concerns serine 916 in the C-terminal region. This residue is autophosphorylated by activated PKD1, leading to a change in conformation that stabilizes PKD1 in an active state and influences duration of kinase activation [10]. Phosphorylation of S916 has been shown to tightly correlate with PKD1 activation status [11]. Whereas in resting cells, PKD1 is located mainly in the cytoplasm, upon stimulation, PKD1 migrates to the membrane where it is activated [12–14]. Activation of PKD1 leads to regulation of multiple cellular functions including Golgi organization and protein secretion [15], cell proliferation [16], invasion [17] and regulation of apoptosis [18].
Although PKD1 has been extensively studied in multiple cellular systems, several aspects of GPCR-mediated PKD1 activation remain controversial or not determined, including the role of changes in intracellular calcium levels in mediating PKD1 activation, the existence and relevance of PKC-independent pathways of PKD1 activation by GPCRs, the PKC isoforms or intracellular mediators other than PKC involved in PKD1 activation by CCK and the changes in the intracellular distribution of PKD1 upon activation. Furthermore, comparative data on kinetic and stoichiometric variables and intracellular distribution of the two phosphorylated PKD1 forms (S916 and S744/748) are lacking and only very limited information is available on PKD1 activation in native GI cells or tissues and the intracellular signaling cascades involved.
Intracellular signaling mechanisms for gastrointestinal (GI) growth factors as well as GI hormones/neurotransmitters have been extensively studied in dispersed pancreatic acini because these cells are highly responsive to various stimuli [19]. A number of GI hormones/neurotransmitters and growth factors that are known to alter pancreatic acinar cell function have been reported to activate PKD1 in other cells [20–22]. As PKD1 may play a central role in the regulation of protein trafficking, cell proliferation and induction of apoptosis in other cell systems [15–18], an understanding of the involvement of PKD1 in the signaling cascades of these GI hormones/neurotransmitters or growth factors in pancreatic tissue is important, because many of these stimuli play an important role in pancreatic disease. Therefore, to address the issues on PKD1 activation detailed above, we have analyzed the role of GI hormones/neurotransmitters and growth factors in PKD1 activation in pancreatic acinar cells and the intracellular signaling cascades involved.
2. Materials and Methods
2.1. Materials
Male Sprague-Dawley rats (150–250 g) were obtained from the Small Animals Section, Veterinary Resources Branch, National Institutes of Health (NIH), Bethesda, MD. Rabbit anti-phospho-PKD1-pS916, rabbit anti-phospho-PKD1-pS744/748, rabbit anti-PKD antibodies, rabbit anti-phospho-Src family (Y416), mouse anti-phospho p44/42 MAPK (T202/Y204) and mouse anti-phospho Akt (S473) were purchased from Cell Signaling Technology, Inc. (Beverly, MA). Rabbit anti-PKD (C20), rabbit anti-PKD1 (N20), bovine anti-goat horseradish peroxidase (HRP)-conjugate and anti-rabbit-HRP-conjugate antibodies were from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA). Anti-E-Cadherin, anti PKC-delta and anti PKC-epsilon were purchased from BD Biosciences (San Jose, CA). Anti-Calpain was from BioSource International, Inc. (Camarillo, CA). Rabbit anti-PKD2 was from Bethyl Laboratories (Montgomery, TX). Tris/HCl pH 8.0 and 7.5 were form Mediatech, Inc. (Herndon, VA). 2-mercaptoethanol, protein assay solution and SDS were from Bio-Rad Laboratories (Hercules, CA). CaCl2 and MgCl2 were from Quality Biological, Inc. (Gaithersburg, MD). Dulbecco’s phosphate buffered saline (DPBS), glutamine (200 mM), Tris/glycine/SDS buffer (10x), and Tris/glycine buffer (10x) were from Biosource. Minimal essential media (MEM) vitamin solution, basal medium Eagle (BME) amino acids 100x and Tris-Glycine gels were from Invitrogen (Carlsbad, CA). COOH-terminal octapeptide of cholecystokinin (CCK-8), hepatocyte growth factor (HGF), bombesin, insulin-like growth factor 1 (IGF-I), basic fibroblast growth factor (bFGF), vasoactive intestinal peptide (VIP), endothelin and secretin were from Bachem Bioscience Inc. (King of Prussia, PA). EGF, thapsigargin, PDGF, GF109203X, A23187, LY294002, Go6976, Go6983, PKCζ pseudosubstrate inhibitor, wortmannin, PD98059, U0126, AG527 (B44), PP2, PP3, syntide-2, U-74321, U-74343 and deoxycholic acid were from Calbiochem (La Jolla, CA). Carbachol, insulin, TGF-β1, dimethyl sulfoxide (DMSO), 12-O-tetradecanoylphorbol-13-acetate (TPA), L-glutamic acid, fumaric acid, pyruvic acid, trypsin inhibitor, HEPES, Triton-X, TWEEN®, phenylmethanesulfonylfluoride (PMSF), EGTA, sucrose, sodium-orthovanadate and sodium azide were from Sigma-Aldrich, Inc. (St. Louis, MO). Albumin standard, Protein G beads, Super Signal West (Pico, Dura) chemiluminescent substrate and stripping buffer were from Pierce (Rockford, IL). Protease inhibitor tablets were from Roche (Basel, Switzerland). Purified collagenase (type CLSPA) was from Worthington Biochemicals (Freehold, NJ). ATP and γ32P-ATP were from Amersham (Piscataway, NJ). P81 phosphocellulose paper was from Upstate (Charlottesville, VA). Nitrocellulose membranes were from Schleicher and Schuell BioSience, Inc. (Keene, NH). Alexa 594-conjugated anti-rabbit secondary antibody was from Molecular Probes (Eugene, OR). Poly-L-lysine coated slides and sample chambers were from Wescor (Logan, UT). Ad5-CMV-PKC-delta-DN and Ad5-CMV-PKC-epsilon-DN were from Seven Hill Bioreagents (Cincinnati, OH), Ad5-CMV-GFP and Ad5-CMV were from QBiogene (Montreal, Canada).
2.2. Methods
Tissue Preparation
Pancreatic acini were obtained by collagenase digestion as previously described [23]. Standard incubation solution contained 25.5 mM HEPES (pH 7.45), 98 mM NaCl, 6 mM KCl, 2.5 mM NaH2PO4, 5 mM sodium pyruvate, 5 mM sodium glutamate, 5 mM sodium fumarate, 11.5 mM glucose, 0.5 mM CaCl2, 1 mM MgCl2, 1 mM glutamine, 1% (w/v) albumin, 0.01% (w/v) trypsin inhibitor, 1% (v/v) vitamin mixture and 1% (v/v) amino acid mixture.
Acini Stimulation
After collagenase digestion, dispersed acini were pre-incubated in standard incubation solution for 2 hrs at 37 °C with or without inhibitors as described previously [23]. After pre-incubation 1 ml aliquots of dispersed acini were incubated at 37 °C with or without stimulants. Cells were lysed in lysis buffer (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 1% deoxycholate, 0.1% sodium azide, 1 mM EGTA, 0.4 mM EDTA, 0.2 mM sodium orthovanadate, 1 mM PMSF, and one protease inhibitor tablet per 10 ml). After sonication, lysates were centrifuged at 10,000x g for 15 min at 4 °C and protein concentration was measured using the Bio-Rad protein assay reagent. Equal amounts of samples were analyzed by SDS-PAGE and Western blotting.
Subcellular Fractionation
After stimulation acini were resuspended in membrane lysis buffer (50 mM Tris/HCl pH 7.5, 150 mM NaCl, 0.1% sodium azide, 1 mM EGTA, 0.4 mM EDTA, 0.2 mM sodium orthovanadate, 1 mM PMSF, and one protease inhibitor tablet per 10 ml). After homogenization for 30 seconds (Polytron homogenizer, Brinkmann Instruments) lysates were centrifuged at 500x g for 15 min to separate nuclei, debri and fat. The supernatant (cytosol and membrane fraction) was centrifuged for 30 min at 4°C and 60,000x g to separate cytosol and membrane fraction. The pellet containing the membrane fraction was washed twice in membrane lysis buffer, resuspended in lysis buffer (with detergents as described above), sonicated for 3 seconds and then centrifuged at 10,000x g for 15 min at 4°C. Protein concentration was measured using Bio-Rad protein assay reagents and equal amounts of protein were subjected to SDS-PAGE and analyzed by Western blotting.
Western Blotting
Whole cell lysates, immunoprecipitates or lysates of subcellular fractions were subjected to SDS-PAGE using 10% and 4–20% Tris-glycine gels. After electrophoresis, protein was transferred to nitrocellulose membranes. Membranes were blocked in blocking buffer (50 mM Tris/HCl pH 8.0, 2 mM CaCl2, 80 mM NaCl, 0.05% Tween® 20, 5% nonfat dry milk) at 4°C overnight or at room temperature for one hour. Membranes were then incubated with primary antibody. Membranes were incubated with HRP-conjugated secondary antibody (anti-mouse, anti-rabbit, anti-goat) for 45 minutes. Membranes were then washed twice in blocking buffer and twice in washing buffer (50 mM Tris/HCl pH 8.0, 2 mM CaCl2, 80 mM NaCl, 0.05% Tween® 20), incubated with chemiluminescense detection reagents and finally exposed to Kodak Biomax film (XAR, MR). The intensity of the protein bands was measured using Kodak ID Image Analysis. For re-probing, membranes were incubated in Stripping buffer for 30 minutes at room temperature, washed in washing buffer, blocked for 1 hour in blocking buffer and re-probed as described above.
Immunoprecipitation and PKD1 kinase assay
After treatment with or without 10 nM CCK for 5 minutes as indicated, pancreatic acini were lysed (50 mM Tris-HCL pH 7.5, 1% Triton-X, 2 mM EGTA, 2 mM EDTA, 2 mM DTT), lysates were sonicated, cleared by centrifugation at 10,000x g for 15 min at 4 °C and protein concentration was measured using the Bio-Rad protein assay reagent. 1800 μg of protein were incubated with a PKD antibody for 2 hours (C-20 from Santa-Cruz, diluted 1:100), conjugates were immunoprecipitated using protein G coupled to agarose beads and immunoprecipitates were washed twice in lysis buffer and twice in kinase buffer (30 mM Tris-HCl pH 7.5, 10 mM MgCl2, 2 mM DTT). For assessment of PKD1 autophosphorylation, 10 μl of kinase buffer containing 2.5 μCi 32P-ATP/sample (diluted with cold ATP to give a final concentration of 100 μM) was added to 20 μl of beads. Samples were incubated at 30°C for 10 minutes and the reaction was stopped by addition of 1 ml of ice-cold kinase buffer. Immunoprecipitates were recovered and an equal amount of 2x running buffer was added. Samples were submitted to SDS-PAGE and gels were dried prior to autoradiography. To determine phosphorylation of the synthetic substrate syntide-2, immunoprecipitates (20 μl) were incubated with 30 μl of kinase buffer containing 2 μCi/sample 32P-ATP (diluted with cold ATP to give a final concentration of 100 μM) and 5 μg/μl syntide-2 for 10 minutes at 30°C. Reactions were terminated by addition of 100 μl phosphoric acid (0.75%) and 75 μl of the supernatant was spotted onto P81 phosphocellulose squares. These were washed thoroughly 5 times 5 minutes in 0.75% phosphoric acid and then 5 minutes in acetone. Squares were allowed to dry and incorporation of 32P into syntide-2 was assessed by Cerenkov counting.
Immunocytochemistry and immunofluorescence imaging
After treating pancreatic acini with or without stimulants as indicated, cells were fixed, transferred to glass slides and blocked as previously reported [24]. Slides were incubated with an anti-PKD antibody (C-20, Santa Cruz) at a dilution of 1:500 and an anti-E-cadherin antibody (1:100) overnight at 4° C. Reactivity was demonstrated by incubation with Alexa Fluor555-conjugated goat anti-rabbit and Alexa Fluor468-conjugated goat anti mouse secondary antibodies at a dilution of 1:500 for 2 hours at room temperature. Negative controls consisted of replacement of primary antibody with an isotype-matched control. Slides were analyzed as previously reported [24].
Pancreatic acinar isolation, infection and culture
Pancreatic acini were isolated as described above, infected with adenovirus at various titers and cultured on dishes coated with rat tail collagen in Waymouth’s medium containing 0.5% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin and 0.2 mg/ml soybean trypsin inhibitor as described previously [25]. After 24 hours, stimulants were added and cells lysed as described above.
Statistical Analysis
All experiments were performed at least 4 times. Data are presented as mean ± SEM and were analyzed using the Student’s t-test for paired data using the software StatView (SAS Institute, Casy, NC). P values <0.05 were considered significant.
3. Results
3.1. PKD1 is expressed and phosphorylated by CCK in rat pancreatic acini
We first determined whether PKD1 was present in pancreatic acinar cells and whether it was phosphorylated in response to stimulation by CCK, a major physiological stimulant of these cells [26]. Using a specific PKD1 antibody for Western blotting on whole cell lysates from rat pancreatic acini treated with 10 nM CCK for 5 min, a single 115 kD protein was detected (Fig. 1, lane 1), whereas a single 105 kD protein was present when a specific PKD2 antibody was used (Fig. 1, lane 4), demonstrating the presence of both PKD1 and PKD2. When these lysates where submitted to Western blotting with specific antibodies directed against PKD1 phosphorylated at S916 and S744/748, proteins co-migrating with PKD1, but not PKD2, were visualized (Fig. 1, lanes 2 and 3), showing the presence of both forms of phosphorylated PKD1 in pancreatic acini treated with CCK. These findings demonstrate that PKD1 and PKD2 are present in rat pancreatic acini and that PKD1 is phosphorylated at S916 and S744/748 in response to CCK.
Fig. 1. PKDs and their phosphorylated forms occur in rat pancreatic acini.
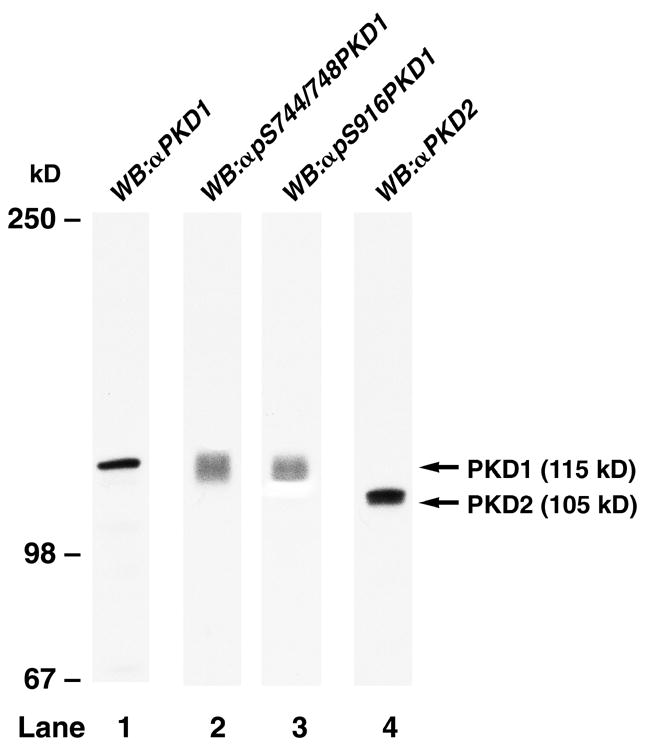
Rat pancreatic acini were treated for 5 min with 10 nM CCK and then lysed. Western blots were analyzed using either anti-PKD1 antibody (Ab) (lane 1), anti-pS744/748 PKD1 Ab (lane 2), anti-pS916 PKD1 Ab (lane 3) or anti PKD2 Ab (lane 4). Bands were visualized using chemiluminescence. Positions of molecular mass markers are shown on the left. The position of PKD1 and PKD2 is indicated with arrows. Results are representative of 2 other experiments.
3.2. PKD1 is phosphorylated on S916 and S744/748 by CCK, carbachol and bombesin, but not by growth factors in rat pancreatic acini
Numerous GI growth factors as well as GI hormones/neuropeptides activate pancreatic acinar cells [19]. To assess whether stimulation by GI hormones or growth factors can cause phosphorylation of PKD1, we analyzed the alterations in the phosphorylation of S916 (a PKD1 autophosphorylation site reflecting activation) and S744/748 (a site for transphosphorylation by PKC isoforms) in whole cell lysates from rat pancreatic acini treated with various stimuli (Fig. 2). Phosphorylation of PKD1 at both S916 and S744/748 was markedly increased after treatment with GI hormones causing mobilization of intracellular calcium and PKC activation (i.e. CCK, carbachol, bombesin) (Fig. 2, top panel, lanes 2–4) [19], but not after treatment with GI hormones that increase cellular cyclic AMP and cause PKA activation (i.e. secretin, VIP) (Fig. 2, top panel, lanes 5–8) [19] or treatment with various pancreatic growth factors (Fig. 2, bottom panel). To confirm that each stimulus tested can alter acinar cell function, we showed that each GI hormone stimulated an increase in phosphorylation of MAPK or Akt (Fig. 2) and that each growth factor stimulated an increase in Akt phosphorylation (Fig. 2), indicating that each stimulus had an effect on acinar cells in our assay.
Fig. 2. Ability of various pancreatic secretagogues and growth factors to stimulate PKD1 S916 and S744/748 phosphorylation in rat pancreatic acini.
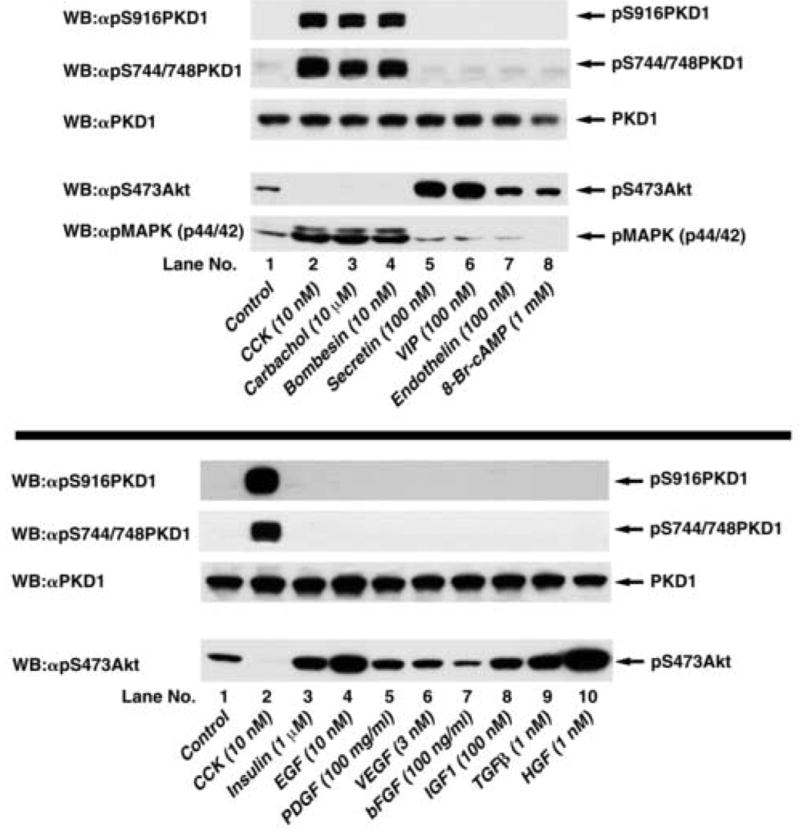
Top: Rat pancreatic acini were treated with no additions or with 10 nM CCK, 10 μM carbachol, 10 nM bombesin, 100 nM secretin or 100 nM VIP for 5 min, with 100 nM endothelin for 10 min, or with 1 mM 8-Br-cAMP for 30 min. Upper 3 panels: membranes were analyzed using either anti-pS916 PKD1 Ab (panel 1) or anti-pS744/748 PKD1 Ab (panel 2). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 3). Lower 2 panels: For positive controls for stimulation by pancreatic secretagogues, membranes were analyzed using anti-phospho Akt and anti-phospho MAPK Ab (panels 4, 5). The bands were visualized using chemiluminescence. Results from a representative experiment of 6 experiments are shown. Bottom: Rat pancreatic acini were treated with no additions, with 10 nM CCK for 5 min as positive control or with 10 nM EGF for 2.5 min, with 1 μM insulin or 100 mg/ml PDGF for 5 min, with 100 ng/ml bFGF or 1 nM HGF for 10 min, with 3 nM VEGF or 100 nM IGF1 for 15 min or with 1 nM TGFβ for 60 min. Upper 3 panels: membranes were analyzed using either anti-pS916 PKD1 Ab (panel 1) or anti-pS744/748 PKD Ab (panel 2). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 3). Lower panel: For positive controls for stimulation by growth factors, membranes were analyzed using anti-phospho Akt Ab (panel 4). The bands were visualized using chemiluminescence. Results from a representative experiment of 4 experiments are shown.
3.3. PKD1 is activated by CCK in rat pancreatic acini
We next wanted to determine if CCK not only causes PKD1 phosphorylation, but also activation. In-vitro kinase assays monitoring PKD autophosphorylation and phosphorylation of the synthetic substrate syntide-2 by PKD have been used in other experimental models to determine PKD activation by a given stimulus [5,27]. Incubation with 10 nM CCK for 5 minutes caused an 8.6±0.5-fold increase in PKD1 autophosphorylation (Fig. 3, upper panel) and a highly significant (p<0.001) increase in phosphorylation of the synthetic substrate syntide-2 (Fig. 3, lower panel), demonstrating activation of PKD kinase activity by CCK.
Fig. 3. CCK activates PKD1 in rat pancreatic acini.
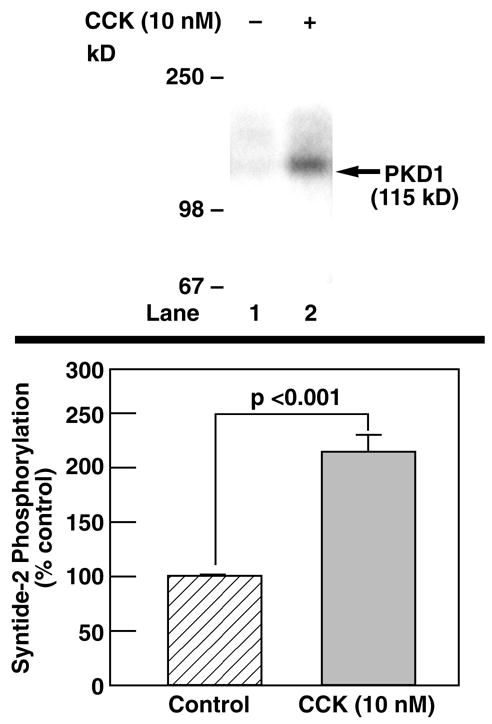
Rat pancreatic acini were treated for 5 min with 10 nM CCK or with no additions and then lysed. For determination of PKD1 autophosphorylation, immunoprecipitates were incubated with kinase buffer containing 32P-ATP and subjected to electrophoresis. Gels were dried and subjected to autoradiography (upper panel). Shown is a representative experiment of 3 others. For assessment of phosphorylation of the exogenous substrate syntide-2, immunoprecipitates were incubated with kinase buffer containing 32P-ATP and syntide-2. The reaction was stopped by adding 0.75% phosphoric acid and spotted onto P81 phosphocellulose squares. Incorporation of 32P was determined by scintillation counting. The values indicated are the means ± S.E. of 7 independent experiments and are expressed as percent of the pretreatment value.
3.4. Time-dependence and dose-dependence of PKD1 phosphorylation by CCK
Phosphorylation of both S916 and S744/748 of PKD1 rapidly increased after CCK stimulation with a detectable effect after 30 s and a maximal effect after 10–15 min of stimulation, lasting over 60 min (Fig. 4). The increase in S916 phosphorylation occurred significantly faster (p<0.05) than the increase in S744/748 phosphorylation (Table 1). The dose-response of PKD1 S916 and S744/748 phosphorylation by CCK was comparable for both phosphorylation sites (Fig. 5). Increase in phosphorylation was detectable at concentrations greater than 0.01 nM CCK and reached a maximum at 10 nM CCK (Fig. 5). Further increasing the CCK concentration to 100 or 1000 nM CCK did not increase the level of PKD1 S916 or S744/748 phosphorylation (Fig. 5). With maximal CCK stimulation, the fold increase over basal phosphorylation levels was significantly greater (p=0.013) for S916 than for S744/748PKD1 (Table 1).
Fig. 4. Time course of CCK stimulation of PKD1 S916 and S744/748 phosphorylation in rat pancreatic acini.
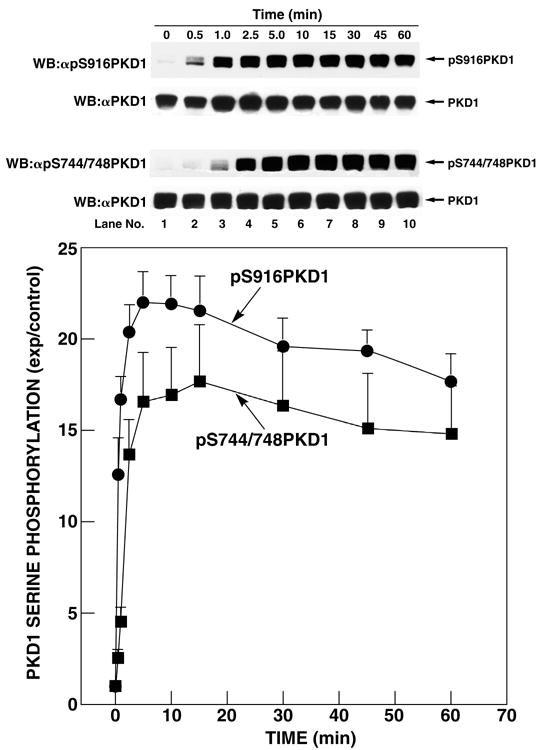
Rat pancreatic acini were treated with no additions or with 10 nM CCK for the indicated periods of time and then lysed. Upper 2 panels: membranes were analyzed using anti-pS916 PKD1 Ab (panel 1). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 2). Lower 2 panels: membranes were analyzed using anti-pS744/748 PKD Ab (panel 3). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 4). The bands were visualized using chemiluminescence and quantification of phosphorylation was assessed using scanning densitometry. The upper part shows a representative experiment. The values shown in the bottom part are the means ± S.E. of 4 independent experiments and are expressed as fold increases over the pretreatment level (exp/control).
Table 1.
Comparison of site-specific phosphorylation of PKD1 in rat pancreatic acinii.
| PKD1 serine phosphorylation | |||
|---|---|---|---|
| Variable | pS916 | pS744/748 | p |
| Stimulation by pancreatic secretagoguesii | |||
| Yes | CCK, carbachol, bombesin | CCK, carbachol, bombesin | |
| No | Secretin, VIP, endothelin | Secretin, VIP, endothelin | |
| Stimulation by pancreatic growth factorsiii | |||
| Yes | - | - | |
| No | Insulin, EGF, PDGF, VEGF, bFGF, IGF1, TGFβ, HGF | Insulin, EGF, PDGF, VEGF, bFGF, IGF1, TGFβ, HGF | |
| Stimulation by post-receptor activators | |||
| Yes | TPA | TPA | |
| No | Ionophore, 8-Br-cAMP | Ionophore, 8-Br-cAMP | |
| t1/2 of CCKiv (s) | 28.9 ± 3.1 | 103 ± 7.4 | 0.0031 |
| tmax of CCKiv (s) | 525 ± 75 | 900 ± 122 | NSv |
| EC50 of CCKvi (nM) | 0.14 ± 0.02 | 0.19 ± 0.05 | NS |
| EC50 of CCK-JMVv(nM)i | 1070 ± 725 | 119 ± 60 | NS |
| Fold increase with CCK (100nM)vi | 53.4 ± 7.2 | 11.2 ± 1.3 | 0.013 |
| Fold increase with CCK-JMV(10 μM)vi | 4.4 ± 0.9 | 2.5 ± 0.4 | NS |
| CCK stimulation with GF109203X(5 μM) (% CCK alone) | 38.0 ± 3.2 | 13.0 ± 0.8 | 0.026 |
| CCK stimulation with Thapsigargin (1 μM) (% CCK alone) | 4.2 ± 0.8 | 14.4 ± 1.4 | 0.032 |
| CCK stimulation with Go6976(10 μM) (% CCK alone) | 57.5 ± 20.0 | 86.9 ± 17.3 | NS |
| CCK stimulation with Go6983(10 μM) (% CCK alone) | 51.5 ± 9.1 | 17.6 ± 2.1 | 0.011 |
Results are calculated from the data shown in Figures 2–8. For CCK stimulation after pre-incubation with GF109203X, thapsigargin, Go6976 and Go6983, results are expressed as the percentages of the stimulation seen with CCK (10 nM) after pre-incubation in incubation buffer without any additions.
Concentrations and incubation times are reported in Figure 2.
Concentrations and incubation times are reported in Figure 2.
Stimulation with 10 nM CCK as described in Fig. 4.
NS: p value not significant (≥0.05).
Stimulation with CCK for 5 min as described in Fig. 5.
Fig. 5. Concentration-dependence of CCK and CCK-JMV stimulation of PKD1 serine 916 and serine 744/748 phosphorylation in rat pancreatic acini.
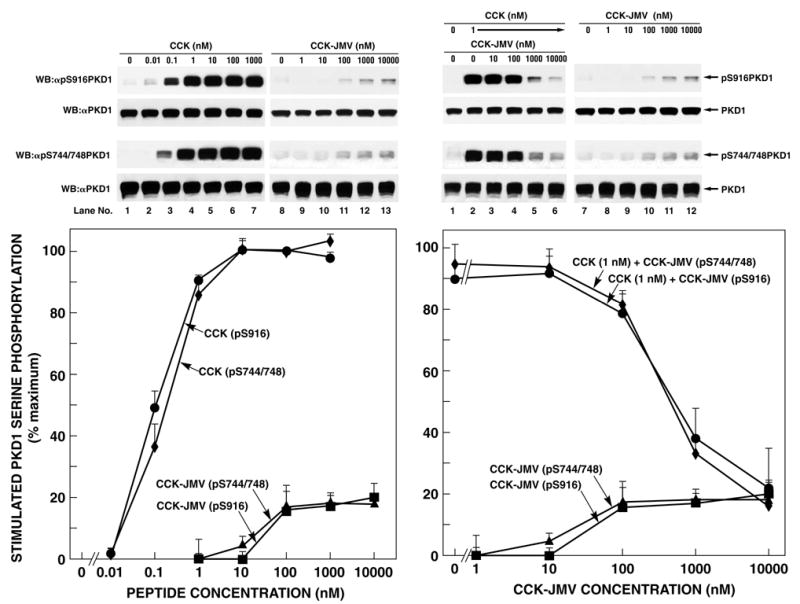
Left: Rat pancreatic acini were treated with no additions, with CCK or CCK-JMV at the indicated concentrations for 5 min and then lysed. Upper 2 panels: membranes were analyzed using anti-pS916 PKD1 Ab (panel 1). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 2). Lower 2 panels: membranes were analyzed using anti-pS744/748 PKD1 Ab (panel 3). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 4). Right: Rat pancreatic acini were treated with no additions or with CCK or/and CCK-JMV at the indicated concentrations for 5 min and then lysed. Whole cell lysates were submitted to SDS-PAGE and transferred to nitrocellulose membranes. Upper 2 panels: membranes were analyzed using anti-pS916 PKD1 Ab (panel 1). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 2). Lower 2 panels: membranes were analyzed using anti-pS744/748 PKD1 Ab (panel 3). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 4). The bands were visualized using chemiluminescence and quantification of phosphorylation was assessed using scanning densitometry. The upper part shows a representative experiment. The values shown in the bottom part are the means ± S.E. of 4 independent experiments and are expressed as percentages of maximal increase caused by 100 nM CCK.
3.5. CCK-induced PKD1 serine phosphorylation is mediated both by the high- and the low-affinity CCKA receptor
The CCKA receptor on pancreatic acini can exist in both a high and low affinity receptor state [19,26]. To analyze the possible role of each receptor state in causing CCK mediated PKD1 phosphorylation, we analyzed the effect of CCK-JMV, a CCK synthetic analog which in rat pancreatic acini functions as an agonist of the high-affinity CCKA state and as an antagonist at the low-affinity CCKA receptor state [28,29], on PKD1 serine phosphorylation. CCK-JMV caused an increase in PKD S916 and S744/748 phosphorylation detectable above 10 nM and reaching a maximum at 100 nM (Fig. 5). For both sites, the maximal stimulation by CCK-JMV was one-fifth of the maximal stimulation by CCK (Fig. 5, Table 1). Co-stimulation with CCK (1 nM) and CCK-JMV at variable concentrations led to a dose-dependent inhibition of CCK-mediated PKD1 S916 and S744/748 phosphorylation by CCK-JMV. After co-stimulation with CCK (1 nM) and maximal CCK-JMV concentrations (10 μM), phosphorylation of both S916 and S744/748PKD1 was comparable to phosphorylation stimulated by 10 μM CCK-JMV alone (Fig. 5). Together, these data demonstrate that 80% of the effect of CCK on PKD1 phosphorylation is mediated by activation of the low-affinity CCKA receptor and that 20% is mediated by activation of the high-affinity CCKA receptor state.
3.6. CCK-induced PKD1 phosphorylation is partly dependant on PKC and strongly inhibited by depletion of intracellular calcium
Binding of CCK to its receptor causes activation of PLC-β, leading to an increase of intracellular calcium levels and activation of PKC through diacylglycerol [26,30]. To assess the respective roles of PKC and intracellular calcium signaling on CCK-mediated PKD1 phosphorylation, we analyzed the effect of GF109203X, an inhibitor of classical and novel PKC isoforms [31], and thapsigargin, an agent that specifically inhibits the endoplasmic reticulum ATPase, which depletes Ca++ from intracellular compartments in a calcium-free medium after 1 h of incubation [23]. Furthermore, we analyzed the effect of TPA, an activator of classical and novel PKC isoforms, on PKD1 phosphorylation. TPA (1 μM) stimulated PKD1 S916 and S744/748 phosphorylation to a comparable degree to stimulation by 10 nM CCK (Fig. 6, top panel). TPA-induced PKD1 phosphorylation was completely inhibited by pre-incubation of pancreatic acini with GF109203X (Fig. 6, top panel), demonstrating effective inhibition of classical/novel PKC isoforms by GF109203X under our experimental conditions. In contrast, CCK-induced PKD1 phosphorylation was only partly inhibited by pre-incubation with GF109203X (Fig. 6, top panel). Specifically, CCK-stimulated pS916 PKD1 phosphorylation was inhibited by 62 ± 7% by GF109203X, which was significantly (p<0.026) less than the 87 ± 2% inhibition of pS744/748 PKD1 phosphorylation. Depletion of intracellular calcium by thapsigargin caused a 96 ± 2% and 87 ± 3% inhibition of CCK-induced PKD1 S916 and S744/748 phosphorylation, respectively (Fig. 6, top panel), but did not have a significant effect on TPA-mediated PKD1 phosphorylation (Fig. 6, top panel). Both CCK- and TPA-mediated PKD1 phosphorylation were completely inhibited after pre-incubation of pancreatic acini with GF109203X and thapsigargin in combination (Fig. 6, top panel). An acute increase in intracellular calcium by short-term incubation with thapsigargin in calcium-containing media did not cause PKD1 phosphorylation but potentiated TPA-mediated PKD1 phosphorylation (Fig. 6, bottom panel) as did the calcium ionophore A23187 (data not shown). These results demonstrate that CCK-stimulated PKD1 phosphorylation is mediated by activation of both arms of the phospholipase C-mediated cascade with participation of both PKC-dependent and -independent pathways, however, the contribution of these different pathways varies for the two serine phosphorylation sites. Furthermore, these data suggest an important role for intracellular calcium in CCK-mediated PKD1 activation.
Fig. 6.
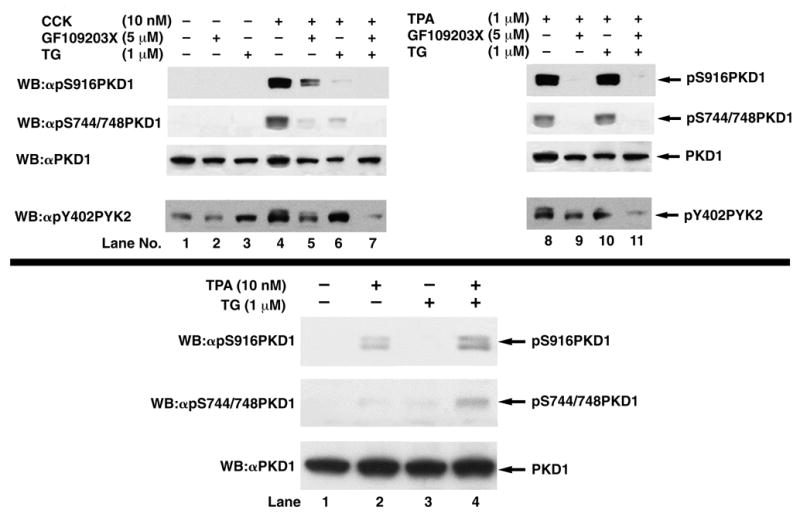
Top: Effect of the PKC inhibitor GF109203X and/or depletion of intracellular calcium with thapsigargin on CCK- (left panel) and TPA- (right panel) stimulated PKD1 S916 and S744/748 phosphorylation in rat pancreatic acini. Rat pancreatic acini were pretreated for with no additions, with GF109203X (5 μM) for 2 h or with thapsigargin (1 μM) in a calcium-free medium (with EGTA 5 μM) for 1 h. Acini were then incubated with no additions (control), with 10 nM CCK for 10 min or with 1 μM TPA for 5 min and then lysed. Upper 3 panels: membranes were analyzed using anti-pS916 PKD1 Ab (panel 1) or anti-pS744/748 PKD1 Ab (panel 2). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 3). Lower panel: For positive control for calcium-free conditions, membranes were analyzed using anti-phospho PYK2 (Y402) Ab (panel 4). The bands were visualized using chemiluminescence and quantification of phosphorylation was assessed using scanning densitometry. The upper part shows a representative experiment of 5 independent experiments. Bottom: Effect of an acute increase in intracellular calcium on PKD1 phosphorylation. Rat pancreatic acini were treated with TPA (10 nM) and/or Thapsigargin (1μM) for 10 min and then lysed. Membranes were analyzed using anti-pS916 PKD1 Ab (panel 1) or anti-pS744/748 PKD1 Ab (panel 2). To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab (panel 3). A representative blot of 3 independent experiments is shown.
3.7. PKD1 activation by CCK is partly dependent on PLC-mediated PKC activation
CCK-induced PKC activation is mediated by PLC beta in pancreatic acini [30]. To analyze the role of PLC in PKD1 activation by CCK, we studied the influence of the PLC inhibitor U-73122 on CCK-mediated PKD1 phosphorylation. U-73122 caused an almost complete inhibition (88.3±3.9%) of CCK-mediated PKD1 S744/748 and a partial inhibition (55.6±5.5%) of S916 phosphorylation (Fig. 7, top panel).
Fig. 7.
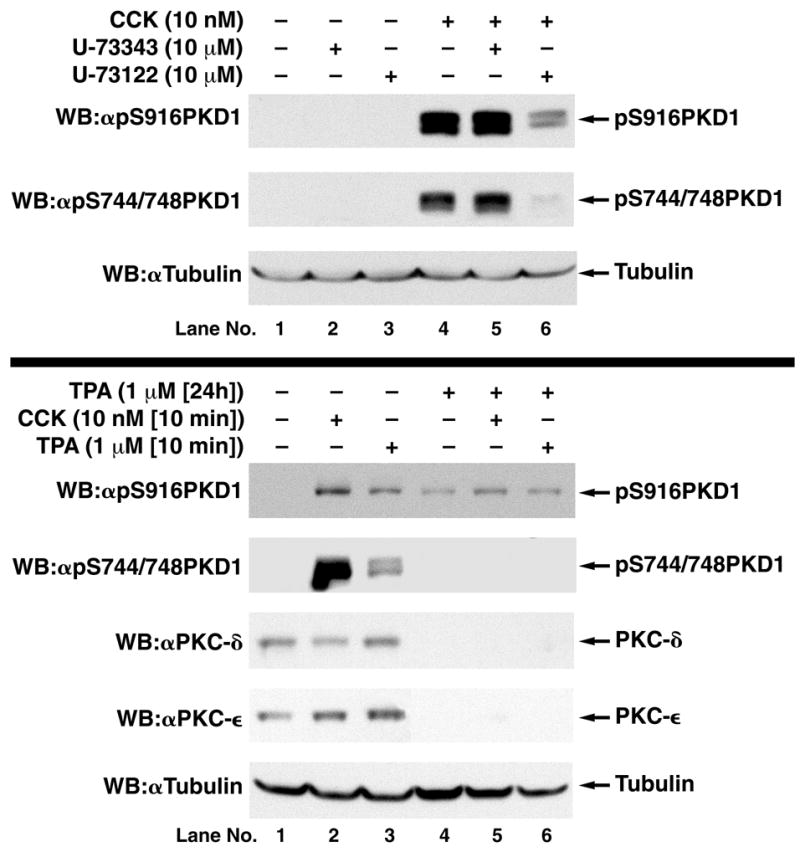
Top: Effect of the inhibition of phospholipase C on CCK stimulated PKD1 S916 and S744/748 phosphorylation in rat pancreatic acini. Rat pancreatic acini were pretreated for 3 hours with no addition, with the inactive compound U-73343 (10 μM) or the active compound U-73122 (10 μM). Subsequently, acini were incubated with no additions (control) or with 10 nM CCK for 10 min and then lysed. Upper panel: Membranes were analyzed using anti-pS916 PKD1 Ab. Middle panel: Membranes were analyzed using anti-pS744/748 PKD1 Ab. Lower panel: To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-Tubulin Ab. The bands were visualized using chemiluminescence and quantification of phosphorylation was assessed using scanning densitometry. A representative experiment of 3 independent experiments is shown. Bottom: Effect of PKC down-regulation by 24h TPA incubation on CCK and TPA stimulated PKD1 S916 and S744/748 phosphorylation in rat pancreatic acini. Rat pancreatic acini were cultured on collagen-coated dishes in Waymouth’s Medium with 5% FCS as described in Methods and pretreated with no addition or with 1 μM TPA for 24 h. Subsequently, acini were incubated with no additions (control), with 10 nM CCK for 10 min or with 1 μM TPA for 5 min and then lysed. Panel 1: Membranes were analyzed using anti-pS916 PKD1 Ab. Panel 2: Membranes were analyzed using anti-pS744/748 PKD1 Ab. Panel 3: Membranes were analyzed using anti-PKC-δ Ab. Panel 4: Membranes were analyzed using anti-PKC-ε Ab. Lower panel: To verify loading of equal amounts of protein, membranes were stripped and re-blotted with an anti-Tubulin Ab. The bands were visualized using chemiluminescence. A representative experiment of 3 independent experiments is shown.
Chronic treatment with phorbol ester for 24 hours has been shown to cause down-regulation of novel/classical PKC isoforms, but not PKD [32]. In pancreatic acini, TPA (1 μM/24h) caused complete down-regulation of PKC-δ and PKC-ε and completely inhibited TPA and CCK-stimulated PKD1 S744/748 phosphorylation (Fig. 7, bottom panel). However, after 24h TPA treatment, CCK could still trigger an increase in PKD1 S916 phosphorylation, which corresponds to 19% of the maximal increase without TPA pre-treatment. Together, these findings suggest that approximately 80% of PKD1 activation is mediated by a PLC-PKC dependent pathway but that 20% is mediated by a PKC-independent pathway.
3.8. Differential effect of inhibitors of classical, novel and atypical PKC isoforms on TPA- and CCK-induced PKD1 phosphorylation
Four PKC isoforms (α,δ, ε, ζ) have been reported to occur in pancreatic acini [33–36]. To elucidate which PKC isoforms are involved in CCK-mediated PKD1 phosphorylation, we used Go6976, a selective inhibitor of classical PKC isoforms and PKD1 [37], Go6983, a general PKC inhibitor [37] and PKC-ζ pseudosubstrate, a specific cell-permeable inhibitor of PKC-ζ [38]. TPA-mediated phosphorylation of the PKD1 auto-phosphorylation site S916 was significantly inhibited by classical PKC/PKD inhibitor Go6976 (Fig. 8, lane 8), demonstrating that this inhibitor was effective in our experimental system. However, TPA-mediated phosphorylation of the PKD1 trans-phosphorylation site S744/748, which does not require PKD1 catalytic activity, was not significantly altered by Go6976 (Fig. 8, lane 8), demonstrating that classical PKC isoforms are not involved in TPA-mediated PKD1 phosphorylation. Similarly, Go6976 had no significant effect on CCK-mediated PKD1 S744/748 phosphorylation, demonstrating classical PKC isoforms are not involved in CCK-mediated S744/748 phosphorylation. The general PKC inhibitor Go6983 completely inhibited TPA-mediated phosphorylation of both phospho-specific PKD1 sites studied (Fig. 8, lane 9). In contrast, Go6983 only partly inhibited CCK-induced PKD1 S744/748 phosphorylation and had a weak effect on CCK-mediated S916 phosphorylation (Fig. 8, right panel and lane 9). These results remained unchanged when different agonist concentrations and incubation times were used (i.e. CCK 100 nM for 5 min and TPA 1 μM for 10 min) (data not shown), suggesting that these results are not explained simply by different concentrations or incubation times of the agonists used. PKC-ζ pseudosubstrate, which has been shown to inhibit PKC-ζ kinase activity in rat pancreatic acini under experimental conditions identical to those used in the present study [33], had no significant effect on TPA- or CCK-induced PKD1 S916 or S744/748 phosphorylation (Fig. 8). These data support the conclusion that CCK-mediated PKD1 phosphorylation is partly dependant on novel PKC isoforms.
Fig. 8. Effect of the inhibition of classical/novel/atypical PKC isoforms on TPA (left panel) and CCK (right panel) stimulated PKD1 S916 and S744/748 phosphorylation in rat pancreatic acini.
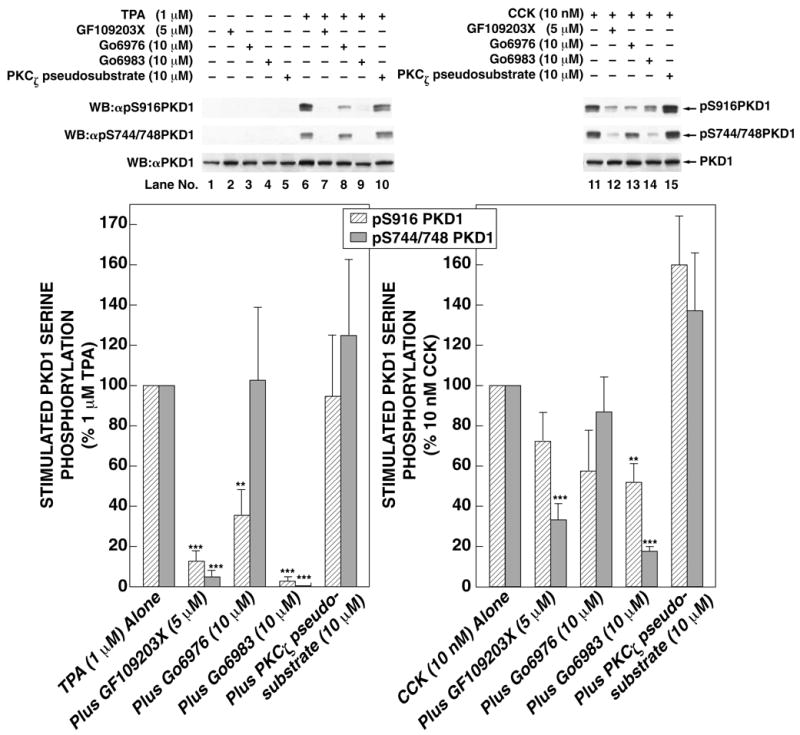
Rat pancreatic acini were pretreated for with no addition, with GF109203X (5 μM), Go6976 (10 μM), Go6983 (10 μM) for 2 h or with PKCζ pseudosubstrate inhibitor (10 μM) for 3 h. Subsequently, acini were incubated with no additions (control), with 10 nM CCK for 10 min or with 1 μM TPA for 5 min and then lysed. Upper panel: Membranes were analyzed using anti-pS916 PKD1 Ab. Middle panel: Membranes were analyzed using anti-pS744/748 PKD1 Ab. Lower panel: To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab. The bands were visualized using chemiluminescence and quantification of phosphorylation was assessed using scanning densitometry. The upper part shows a representative experiment. The values shown in the bottom part are the means ± S.E. of 6 independent experiments and are expressed as percentages of maximal increase caused by 1 μM TPA or 10 nM CCK. ** and *** indicate p<0.01 and p<0.001 compared to stimulation with CCK or TPA alone.
3.9. Effect of isoform-specific PKC translocation inhibitors and dominant-negative PKC isoforms on CCK-mediated PKD1 phosphorylation
To determine which novel PKC isoform(s) mediate(s) PKD1 phosphorylation, we used two independent approaches. First, we analyzed the effect of isoform-specific PKC translocation inhibitors [33] on CCK-mediated PKD1 phosphorylation of the S744/748 transphosphorylation site. While the PKC-δ translocation inhibitor δV1-1 caused a strong inhibition of PKD1 phosphorylation, the PKC-ε translocation inhibitor εV1-2 had no significant effect (Fig. 9). Second, using an adenoviral vector, we expressed dominant negative PKC-δ and PKC-ε isoforms as described previously [25]. These dominant negative (DN) mutants encode the full-length protein, but contain a single amino acid substitution in a critical ATP binding residue, rendering the kinase inactive [39]. As reported previously [25], infection with 109 pfu/mg of acinar protein leads to expression of the protein of interest in 100% of cells (Fig. 9, panel D). Infection with an empty adenoviral vector or with Ad-5-DN PKC-ε had no effect on PKD1 phosphorylation, but infection with Ad-5-DN PKC-δ strongly inhibited CCK-mediated PKD1 phosphorylation (Fig. 9). These data further demonstrate that PKC-δ is the main PKC isoform involved in PKD1 activation.
Fig. 9. CCK-induced PKD1 phosphorylation is mediated by PKC-δ.
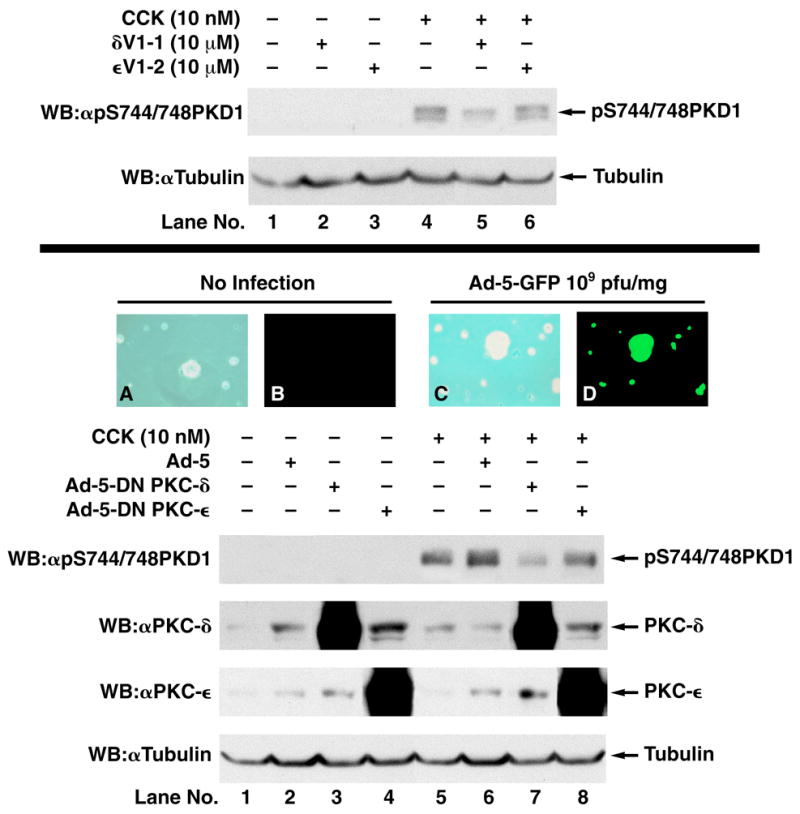
Top: Rat pancreatic acini were pretreated for with no addition, with the PKC-δ translocation inhibitor δV1–1 (10 μM) or the PKC-ε Translocation inhibitor εV1–2 (10 μM) for 4 h. Subsequently, acini were incubated with no additions (control) or with 10 nM CCK for 10 min. Upper panel: Membranes were analyzed using anti-pS916 PKD1 Ab. Lower panel: To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-tubulin Ab. These results show a representative experiment of 3 independent experiments. Bottom: Rat pancreatic acini were cultered as described in Methods and infected with no addition (Pictures A,C), with an empty adenoviral vector or with adenovirus encoding GFP (Pictures B,D), DN-PKC-δ or DN-PKC-ε. Pictures A/C show phase contrast microscopy and Pictures B/D show fluorescence microscopy after 24h. After 24h, acini for western blot experiments were incubated with no additions (control) or with 10 nM CCK for 10 min and then lysed. Whole cell lysates were submitted to SDS-PAGE and transferred to nitrocellulose membranes. Upper panel: Membranes were analyzed using anti-pS744/748 PKD1 Ab. Second panel: Membranes were analyzed using anti-PKC-δ Ab. Third panel: Membranes were analyzed using anti-PKC-ε Ab. Lower panel: To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-Tubulin Ab. The bands were visualized using chemiluminescence. These results show a representative experiment of 3 independent experiments.
3.10. CCK-stimulated PKD1 phosphorylation is not dependant on Src, PI3K, MAPK or tyrosine phosphorylation
To further characterize possible signaling cascades contributing to PKD1 phosphorylation in rat pancreatic acini, we analyzed the effect on CCK-stimulated PKD1 phosphorylation of the inhibition of several pathways reported to be important in mediating PKD1 activation in other cellular systems with other stimulants [4,5,40]. Inhibition of Src by PP2, PI3K by wortmannin or LY294002, MAPK by PD98059 or U0126 and inhibition of tyrosine phosphorylation by B44, a wide-spectrum tyrosine kinase inhibitor [41] had no effect on CCK-mediated S916 or S744/748 phosphorylation (Fig.10). These inhibitors have all been reported to inhibit CCK-mediated signaling cascades in pancreatic acini [42–45]. To ensure that this lack of effect was not due to a general non-responsiveness of our pancreatic acinar preparation to these inhibitors, controls to verify specific inhibition of a typical phosphorylation event were included for each inhibitor (Fig. 10).
Fig. 10. Inhibition of Src (A), PI3K (B), MAPK (C) or tyrosine phosphorylation (D) have no effect on CCK-stimulated PKD1 S916 and S744/748 phosphorylation in rat pancreatic acinar cells.

Rat pancreatic acini were pretreated for with no addition or with PP2 (10 μM) or PP3 (10 μM) (A), with LY294002 (100 μM) for 1 h or with wortmannin (10 μM) for 30 min (B), with U0126 (20 μM) for 1h or PD98059 for 30 min (C) and with B44 (300 μM) for 1 h. Subsequently, acini were incubated with no additions (control), with 10 nM CCK for 10 min or with 1 μM TPA for 5 min or with 1 nM HGF for 10 min and then lysed. Whole cell lysates were submitted to SDS-PAGE and transferred to nitrocellulose membranes. Upper panel: Membranes were analyzed using anti-pS916 PKD1 Ab. Second panel: Membranes were analyzed using anti-pS744/748 PKD1 Ab. Third panel: To verify loading of equal amounts of protein, membranes were stripped and re-blotted with anti-PKD1 Ab. Lower panel: to verify that the used inhibitors could influence cellular functions in our experimental model, membranes were analyzed with anti-pY416 Src (A,C), anti-pS473 Akt (B) or anti-pMAPK (C) as positive controls. The bands were visualized using chemiluminescence and quantification of phosphorylation was assessed using scanning densitometry. These results show a representative experiment of 3 others.
3.11. PKD1 translocates to the membrane after CCK stimulation in rat pancreatic acini
PKD membrane translocation is reported to be a crucial step in PKD activation in response to different stimuli in numerous cellular models [5,12,13,46]. To analyze intracellular distribution of total PKD in quiescent and stimulated intact pancreatic acini, cells were submitted to immunocytochemistry-immunofluorescence. In non-stimulated acini, PKD1 was distributed diffusely in the cytoplasm, with only minimal staining at the membrane (Fig. 11, panels B1-B2). After stimulation with CCK (Fig. 11, panels C1-2, D1-2) or TPA (Fig. 11, panels E1-2, F1-2), there was a rapid translocation to the membrane, obvious at 5 min (Fig. 12, panels C1-2, E1-2) and lasting at least 30 min (Fig. 11, panels D1-2, F1-2). The membrane translocation of PKD1 after stimulation is confirmed by a co-localization with the membrane marker E-cadherin (Fig. 11, panels C2-F2). To quantitate the PKD1 translocation response, subcellular fractions of rat pancreatic acini were analyzed by Western blotting (Fig. 12). In non-stimulated acini, 80 ± 7% of total PKD was found in the cytoplasmic fraction (Fig. 12, lanes 1, 6), confirming results of the immunocytochemistry. After stimulation with TPA, there was a marked increase of total PKD in the membrane fraction detectable at 1 min (data not shown), obvious at 5 min (Fig. 12, lane 7) and lasting at least 30 min (Fig. 12, lane 9) whereas PKD completely disappeared from the cytoplasmic fraction (Fig. 12, lanes 2, 4), indicating a pronounced and durable translocation of PKD to the membrane. Similarly, after CCK stimulation, total PKD increased by 209±57% at 1 min (data not shown), 357 ± 105% at 5 min and 305 ± 75% at 30 min in the membrane fraction (Fig. 12, lanes 8, 10). A marked increase in PKD1 phosphorylated at S916 and S744/748 was detectable in the membrane fraction after 1 (data not shown), 5 and 30 min CCK and TPA stimulation (Fig. 12, lanes 7–10) and in the cytoplasmic fraction after CCK stimulation only (Fig. 12, lanes 2–5). To verify separation of cytosolic and membrane fractions and equal loading of protein, lysates from subcellular fractions were analyzed for E-Cadherin (located in cell membranes) and Calpain (located in the cytosol). This analysis confirmed the purity of the subcellular fractions and showed equal loading.
Fig. 11. PKD immunofluorescense-cytochemistry in rat pancreatic acini with and without CCK treatment.
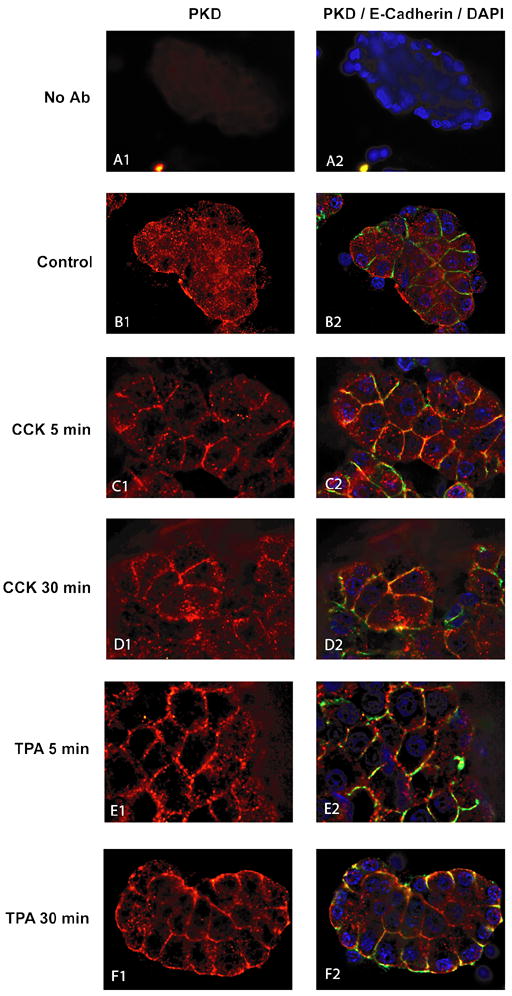
Pancreatic acini were incubated with no addition, CCK (10 nM) or TPA (1 μM) for time periods indicated below. After stimulation cells were washed, fixed, permeabilized and transferred onto poly-L-lysine coated glass slides by cytocentrifugation as described in Methods. Cells were then labeled using polyclonal rabbit anti-PKD (Santa Cruz, C-20) and mouse anti E-cadherin primary antibodies. Specific binding was detected using an Alexa Fluor555- and Fluor488-conjugated secondary antibodies so that red staining represents staining for total PKD and green staining represents E-caadherin. Nuclei were counterstained using DAPI (blue). Fluorescent images were collected using a Leica CTR5000 microscope. Panels A1–2 show acini without the addition of the primary antibodies but with normal isotype-matched serum as a negative control. Panels B1–2 shows acini treated with incubation buffer only. Panels C1-2 and E1-2 show cells treated for 5 min with 10 nM CCK (panels C1-2) or 1 μM TPA (panel E1-2). Panels D1-2 and F1-2 show cells treated for 30 min with 10 nM CCK (panel D1-2) or 1 μM TPA (panels F1-2). Shown are results of a typical experiment representative of 6 independent experiments. Cells shown are representative of >90% of total cells present.
Fig. 12. Ability of CCK and TPA to stimulate translocation of total PKD and phospho-PKD to the cell membranes in rat pancreatic acinar cells.

Acini were incubated with or without 1 μM TPA or 10 nM CCK for the time periods indicated. Samples were then processed as described in Methods to obtain subcellular fractions. Lysates from these fractions were submitted to SDS-PAGE and transferred to nitrocellulose membranes. Upper 3 panels: membranes were analyzed using anti-PKD Ab, anti pS916PKD1 Ab or anti pS744/748PKD1 Ab. Lower 2 panels: to assess the effectiveness of subcellular fractionation, the cytosol and membrane fractions were analyzed using anti-E-Cadherin Ab, a marker for the membrane fraction or anti-Calpain Ab, a marker for the cytosol, fraction. The bands were visualized using chemiluminescence and quantification of phosphorylation was assessed using scanning densitometry. The upper part shows a representative experiment. The lower part shows mean values ± S.E. of 8 independent experiments. * and ** indicate p<0.05 and p<0.001, respectively, compared to the control group.
4. Discussion
Our results demonstrate that PKD1 is present in rat pancreatic acini and is phosphorylated in response to GI hormones/neurotransmitters that activate phospholipase C, but not by GI hormones/neurotransmitters stimulating other transduction cascades or by pancreatic growth factors. We showed that CCK induced a 8.6-fold increase in PKD1 autophosphorylation and a 2.2-fold increase in phosphorylation of an exogenous substrate by PKD1, demonstrating PKD1 activation by CCK. These increases in PKD activity are comparable to the 4- to 8- fold increase in autophosphorylation and the 2.6–4 fold increase in syntide-2 phosphorylation reported in the literature [7,20,47]. The lower increase in activity of these kinase assays when compared to phosphorylation assays probably reflects different sensitivities of these assays. As rat pancreatic acini express exclusively CCKA and not CCKB receptors, our results for the first time show that CCKA receptors can lead to PKD1 activation. Moreover, because the extent of PKD1 S916 phosphorylation has been shown to closely reflect the activation status of PKD1 [11,32,48], the presence of phosphorylated S916 PKD1 after stimulation with CCK, carbachol or bombesin supports the conclusion that PKD1 is activated by these stimuli in pancreatic acini. Our results, showing lack of effect of endothelin or growth factors in pancreatic acini, differ from studies in other, mostly transformed and/or transfected cells which report activation of PKD1 can also be mediated by endothelin [20] or by various growth factors including PDGF [27] or by VEGF [4,49]. These data suggest that not only may the cellular basis of PKD1 activation by a given growth factor or GI hormone/neurotransmitter vary from one tissue to another, it may also vary from normal cells to transfected and/or neoplastic cells.
There are no studies that have compared in detail the kinetics and stoichiometry of PKD1 S916 (autophosphorylation site) and S744/748 (PKC transphosphorylation site) by a given stimulus. In general, for the individual serine phosphorylation sites, our kinetic and stoichiometric results agree with previous studies [21,50], which show a rapid stimulation of PKD1 activation with a sustained effect lasting over 60 min and an EC50 of most stimulants in the nanomolar range. However, we found that CCK stimulation of the serine phosphorylation sites S744/748 and S916 differed in kinetics and efficacy. Specifically, CCK caused a significantly faster onset of S916 phosphorylation by CCK (t1/2=28.9 vs. 103 s, p=0.0031, Table 1) with a comparable tmax. This result is somewhat unexpected because studies with PKD1 mutants have shown that S744/748 phosphorylation is required for PKC-mediated PKD1 activation [51] and in a recent model of PKD1 activation by GPCRs, S744/748 phosphorylation was proposed to be an essential phosphorylation step leading to PKD1 activation and autophosphorylation of the S916 site of PKD1 [52]. However, evidence exists from a study of BMP-2, which binds to a cell surface serine-threonine kinase receptor, leading to PKD1 activation by stimulating S916 serine phosphorylation without affecting S744/748 phosphorylation [53]. While multiple explanations for the more rapid serine phosphorylation of S744/748 by CCK could be proposed, including different levels of amplification, an important possibility is that PKC-dependent and -independent pathways of PKD1 phosphorylation with different kinetics could exist in rat pancreatic acini.
In pancreatic acini the CCKA-receptor has been shown to exist in both a low-affinity and a high-affinity receptor state, each of which can activate multiple, distinct downstream signaling cascades [19,54–56]. Our data support the conclusion that CCK-induced PKD1 activation and serine phosphorylation at both S744/748 and S916 is dependent on activation of both the low (80%) and the high affinity state (20%) of the CCKA-receptor. pThis is relevant because activation of the high-affinity CCKA receptor state by JMV does not lead to PLC or PKC activation in pancreatic acini [57], suggesting the participation of PLC/PKC-independent pathways of PKD1 activation in these cells. Nevertheless, 80% of PKD1 activation in pancreatic acini is mediated by the low affinity CCKA receptor, which is coupled to PLC activation, and the important role of PLC is illustrated by the pronounced inhibition of PKD1 S744/748 phosphorylation by the PLC inhibitor U-73122.
While the importance of PKC stimulation in mediating PKD1 activation has received considerable attention [52], the role of changes in intracellular calcium in mediating PKD1 activation is unclear because the few studies investigating the role of calcium in PKD1 activation and serine phosphorylation gave contradictory results. In pancreatic acini, activation of the CCKA-receptor leads to PLC activation, which in turn results in the formation of diacylglycerol (DAG) and inositol 1,4,5 triphosphate (IP3) [26,57,58]. DAG binds and activates pPKCs, while IP3 causes an increase in intracellular calcium levels [19,59]. Several lines of evidence in our study support the conclusion that CCK stimulation of both arms of the PLC-pathway, i.e. PKC activation and changes in cytosolic calcium, mediate PKD1 serine phosphorylation and that both PKC-dependent and PKC-independent signaling mechanisms are important. First, we found a >80% suppression of CCK-induced PKD1 serine phosphorylation at both the S744/748 and the 916 site after inhibiting CCK-mediated changes in cytosolic calcium using thapsigargin in a calcium-free medium. This result was not due to a toxic effect of thapsigargin on pancreatic acini because TPA-mediated PKD1 serine phosphorylation at neither S744/748 nor the S916 site was altered. Second, the general (classical, novel) PKC inhibitor GF109203X [31,38] caused only a partial inhibition of CCK-induced PKD1 S916 phosphorylation (Fig. 6). This lack of complete inhibition was not due to incomplete PKC inhibition because GF109203X was used at concentrations that completely inhibited S916 phosphorylation induced by the classical/novel PKC activator TPA. Third, CCK caused PKD1 activation through stimulation of both the high (20% of maximum) and low affinity (80% of maximum) CCKA receptor states. However, only activation of the low affinity CCKA receptor state is coupled to cascades activating PKC in pancreatic acini [56–58], supporting the conclusion that 20% of maximal PKD1 activation is mediated by a PKC-independent mechanism. Fourth, after 24h of TPA incubation, there was a complete down-regulation of novel PKC isoforms and, consequently, of PKD1 S744/748 transphosphorylation. However, CCK could still cause an increase in PKD1 S916 phosphorylation (20% of maximal increase), suggesting again that 20% of PKC activation is mediated independently of PKC. Our results, showing that both PKC-dependent and PKC-independent pathways are important in mediating serine phosphorylation of PKD1 at S916 and S744/748 sites, are in contrast to those of others showing complete or almost complete inhibition of PKD1 activation by different stimuli after pre-incubation with GF109203X in Swiss 3T3 cells or rat-1 cells transfected with the rat CCKB receptor [21,22] and in a number other cells [20,60–62], including rat parotid acinar cells stimulated with carbachol [63]. Our results therefore contrast with the conclusion of a number of other studies in other tissues reporting that stimulation of PKD1 by GPCR activation is exclusively PKC-mediated [21,22,60,63]. However, our finding of PKD1 activation mediated partly through PKC-independent signaling cascades in rat pancreatic acini is consistent with other studies of PKD1 activation in response to the growth hormone BMP-2 in osteoblasts and mesenchymal stem cells [53,64], the cytostatic agent Ara-C in myeloid leukemia cells [65] or oxidative stress in HeLa cells [40]. Different mechanisms of PKC-independent PKD activation have been described including direct interaction with Gβγ [66], cleavage by caspase-3 [65] or tyrosine phosphorylation of PKD [40]. The present study does not provide further insight into the exact mechanism(s) involved, but our results for the first time provide strong support for the conclusion that PKC-independent pathways can be important in GPCR agonist-mediated activation of PKD1. The marked differences in PKD1 activation pathways found in different cells in response to various stimuli suggest that not only the cell, but also the stimulus used can have an effect on the possible PKD1 activation pathways stimulated.
Previous studies have shown the presence of one classical PKC isoform (PKC-α), two novel PKCs (PKC-δ,ε) and one atypical PKC (PKC-ζ) in rat pancreatic acini [33–36]. A number of our results support the conclusion that PKC-δ is the important PKC isoform in mediating CCK-dependent PKD1 activation. First, GF109203X, which has been shown to be an inhibitor of classical and novel PKC isoforms in pancreatic acini and other cells [31,33,38] or the classical/novel PKC inhibitor Go6983 [37] significantly inhibited CCK stimulated PKD1 S744/748 phosphorylation, suggesting the involvement of either PKC-α, δ or ε in PKD1 phosphorylation in pancreatic acini. Second, Go6976, an inhibitor of classical PKC isoforms and PKD1 activity [37] had no significant effect on PKD1 S744/748 phosphorylation by CCK. This result supports the conclusion that the conventional isoform, PKC-α, is not involved in CCK-mediated PKD1 activation in pancreatic acini. This conclusion is consistent with most [33,35,36], but not all [34] recent studies which report CCKA receptor activation does not activate PKC-α in pancreatic acini. Third, PKC-ζ pseudosubstrate, a specific PKC-ζ inhibitor shown to inhibit PKC-ζ kinase activity in rat pancreatic acini under the same experimental conditions used in this study [33], had no effect on CCK mediated PKD1 S744/748 phosphorylation, illustrating that PKC-ζ is not involved in CCK-mediated PKD1 activation. Together, these data suggest that only the novel PKC isoforms δ and/or ε can mediate PKD activation. Our studies with isoform-specific PKC translocation inhibitors and dominant negative PKC isoforms clearly show that PKC-δ mediates CCK-induced PKD1 phosphorylation. This proposal is consistent with previous studies that demonstrate CCK is a potent activator of PKC-δ in pancreatic acini stimulating its tyrosine phosphorylation, translocation to plasma and nuclear membranes and its kinase activity [43,67]. This result is similar to recent studies showing PKD1 activation in rat aortic smooth muscle pcells, HeLa cells, BON neuroendocrine cells and intestinal epithelial cells is mediated by PKC-δ [48,68,69], but differs from other reports which show PKD1 activation can be mediated through PKC-α [49], PKC-ε [14,70], PKC-η [70,71] or PKC-θ [72] in other cells with other stimulants. These results suggest that PKD1 activation can not only vary in different cells with different stimulants in terms of the presence or absence of PKC-independent activation, but also in the PKC isoform mediating activation.
Various agents that activate PKD1 are reported to stimulate its translocation to membranes in transfected and tumor cells [5,12,13,46]. However, there are no studies in normal cells and there is no data on the distribution of the serine-phosphorylated PKD1 forms. In normal, unstimulated pancreatic acinar cells, we found that PKD1 was almost entirely located in the cytoplasm, which is similar to results reported in other cells. Furthermore, we found that both TPA and CCK stimulated total PKD translocation to membranes, however, there were several differences. With TPA, all detectable PKD translocates rapidly and durably to the membrane. This observation is in accordance with a previous study [12] showing a durable membrane translocation of a transfected GFP-PKD1 fusion protein after phorbol ester stimulation in Cos-7 cells. After TPA stimulation, PKD1 phosphorylated at S916 and S744/748 was detectable in the membrane fraction only. In contrast to our results with TPA, after CCK treatment, total PKD1 increased in membranes, however the total amount of PKD1 translocated was sufficiently small not to cause a change in the total PKD in the cytoplasm. PKD1 forms phosphorylated at S916 and S744/748 were absent in unstimulated cells and were found both in the membrane and in the cytoplasm after CCK stimulation. These results support the conclusion that after stimulation with TPA or CCK, PKD1 rapidly translocates to the plasma membrane, however, the magnitude and duration of translocation is much greater with TPA. While our results with CCK suggest that PKD1 serine phosphorylation could be occurring either in the cytoplasm or plasma membrane, our results with TPA suggest the phosphorylation is occurring in the membrane fraction, because no PKD1 was detected in the cytoplasm and the phosphorylated form of PKD1 was detected only in the membrane fraction. Similar to our study, transfected GFP-PKD1 translocated from the cytoplasm to the membrane after bombesin stimulation in fibroblasts and epithelial cells and then there was a rapid re-distribution from the membrane to the cytoplasm, but in contrast to our results, this re-distribution was complete within 10 min after stimulation [13]. In another study, transfected GFP-PKD1 translocated from the cytoplasm to the membrane in transfected ventricular fibroblasts and remained active in the membrane for over 90 min [46]. At present it is unknown whether the marked differences in the redistribution of PKD1 after stimulation with various agents in different cell systems are due to the fact that we are assessing a normal cell system, whereas others are analyzing transfected cells or whether there is a difference in the translocation of PKD1 with different stimulants in different cells.
With various stimulants in other cells, PKD1 activation is reported to be affected by Src kinases [14,68,73], PI3K [4] and tyrosine phosphorylation of various cellular proteins [5,6]. We found that CCK-mediated PKD1 activation is not dependent on Src, PI3K, MAPK, activation or stimulation of tyrosine phosphorylation. These results suggest that the importance of Src kinases, PI3K and tyrosine phosphorylation of various cellular proteins in PKD1 activation differs with different stimuli in various cells.
In conclusion, we report for the first time that PKD1 is expressed in rat pancreatic acini and activated by GI hormones/neurotransmitters leading to activation of phospholipase C, but not by GI hormones with other cellular mechanisms or by pancreatic growth factors. CCK-induced serine phosphorylation at both S744/748 and S916 sites was mediated by activation of both the high affinity (20%) and low affinity (80%) CCKA receptor state. In contrast to PKD1 activation in most other cells studied, CCK-induced PKD1 activation was only partially mediated by PLC/PKC in rat pancreatic acini and the principal PKC isoform involved is PKC-δ. Our results show that CCK-mediated PKD1 activation is highly dependent on changes in intracellular calcium but is not mediated by Src, PI3K, MAPK or tyrosine phosphorylation as reported for a number of other stimulants in other cell systems. Upon stimulation by CCK, PKD1 translocates to the membrane and marked increases of phospho-S916 and phospho-S744/748 occurred both in the membrane and, to a lesser degree, in the cytoplasm. However with TPA, total translocation to the membrane occurs and phosphorylated PKD1 forms are only present in the membrane, suggesting phosphorylation is occurring at the membrane. In agreement with some studies using transfected cell systems, but not others, our results show that the majority of activated PKD1 persists at the membrane after activation, whereas a fraction is redistributed to the cytoplasm. Our results demonstrate a number of novel aspects of PKD1 activation in normal pancreatic acinar cells by GI hormones/neurotransmitters not generally reported in studies of tumor cells or transfected cells. Particularly important are the demonstration of the importance of PKC-independent stimulation, the demonstration of the importance of calcium-mediated cellular processes for activation of PKD1 and the lack of involvement of a number of other cellular signaling cascades reported important with other stimuli. These results emphasize the importance of studying the effects of stimulants of PKD1 in normal cell systems.
Acknowledgments
This work is partly supported by the Intramural Research Program of the NIDDK and NEI, NIH. Jose A. Tapia is supported by Junta de Extremadura (grant 2PR04C015) and Michelle Thill is supported by a Bourse Formation Recherche (BFR) grant from the Ministry of Culture, Higher Education and Research of the Grand-Duchy of Luxembourg. The authors thank Dr. S. Pandol and Dr. J. Reeve for providing the PKC translocation inhibitors.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Johannes FJ, Prestle J, Eis S, Oberhagemann P, Pfizenmaier K. PKCu is a novel, atypical member of the protein kinase C family. J Biol Chem. 1994;269:6140–6148. [PubMed] [Google Scholar]
- 2.Valverde AM, Sinnett-Smith J, Van Lint J, Rozengurt E. Molecular cloning and characterization of protein kinase D: a target for diacylglycerol and phorbol esters with a distinctive catalytic domain. Proc Natl Acad Sci U S A. 1994;91:8572–8576. doi: 10.1073/pnas.91.18.8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Lint J, Rykx A, Maeda Y, Vantus T, Sturany S, Malhotra V, Vandenheede JR, Seufferlein T. Protein kinase D: an intracellular traffic regulator on the move. Trends Cell Biol. 2002;12:193–200. doi: 10.1016/s0962-8924(02)02262-6. [DOI] [PubMed] [Google Scholar]
- 4.Qiang YW, Yao L, Tosato G, Rudikoff S. Insulin-like growth factor I induces migration and invasion of human multiple myeloma cells. Blood. 2004;103:301–308. doi: 10.1182/blood-2003-06-2066. [DOI] [PubMed] [Google Scholar]
- 5.Waldron RT, Rozengurt E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J Biol Chem. 2000;275:17114–17121. doi: 10.1074/jbc.M908959199. [DOI] [PubMed] [Google Scholar]
- 6.Storz P, Toker A. Protein kinase D mediates a stress-induced NF-kappaB activation and survival pathway. EMBO J. 2003;22:109–120. doi: 10.1093/emboj/cdg009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zugaza JL, Sinnett-Smith J, Van Lint J, Rozengurt E. Protein kinase D (PKD) activation in intact cells through a protein kinase C-dependent signal transduction pathway. EMBO J. 1996;15:6220–6230. [PMC free article] [PubMed] [Google Scholar]
- 8.Waldron RT, Rey O, Iglesias T, Tugal T, Cantrell D, Rozengurt E. Activation loop Ser744 and Ser748 in protein kinase D are transphosphorylated in vivo. J Biol Chem. 2001;276:32606–32615. doi: 10.1074/jbc.M101648200. [DOI] [PubMed] [Google Scholar]
- 9.Iglesias T, Rozengurt E. Protein kinase D activation by mutations within its pleckstrin homology domain. J Biol Chem. 1998;273:410–416. doi: 10.1074/jbc.273.1.410. [DOI] [PubMed] [Google Scholar]
- 10.Vertommen D, Rider M, Ni Y, Waelkens E, Merlevede W, Vandenheede JR, Van Lint J. Regulation of protein kinase D by multisite phosphorylation. Identification of phosphorylation sites by mass spectrometry and characterization by site-directed mutagenesis. J Biol Chem. 2000;275:19567–19576. doi: 10.1074/jbc.M001357200. [DOI] [PubMed] [Google Scholar]
- 11.Matthews SA, Rozengurt E, Cantrell D. Characterization of serine 916 as an in vivo autophosphorylation site for protein kinase D/Protein kinase Cmu. J Biol Chem. 1999;274:26543–26549. doi: 10.1074/jbc.274.37.26543. [DOI] [PubMed] [Google Scholar]
- 12.Matthews S, Iglesias T, Cantrell D, Rozengurt E. Dynamic re-distribution of protein kinase D (PKD) as revealed by a GFP-PKD fusion protein: dissociation from PKD activation. FEBS Lett. 1999;457:515–521. doi: 10.1016/s0014-5793(99)01090-x. [DOI] [PubMed] [Google Scholar]
- 13.Rey O, Young SH, Cantrell D, Rozengurt E. Rapid protein kinase D translocation in response to G protein-coupled receptor activation. Dependence on protein kinase C. J Biol Chem. 2001;276:32616–32626. doi: 10.1074/jbc.M101649200. [DOI] [PubMed] [Google Scholar]
- 14.Waldron RT, Rey O, Zhukova E, Rozengurt E. Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J Biol Chem. 2004;279:27482–27493. doi: 10.1074/jbc.M402875200. [DOI] [PubMed] [Google Scholar]
- 15.Liljedahl M, Maeda Y, Colanzi A, Ayala I, Van Lint J, Malhotra V. Protein kinase D regulates the fission of cell surface destined transport carriers from the trans-Golgi network. Cell. 2001;104:409–420. doi: 10.1016/s0092-8674(01)00228-8. [DOI] [PubMed] [Google Scholar]
- 16.Zhukova E, Sinnett-Smith J, Rozengurt E. Protein kinase D potentiates DNA synthesis and cell proliferation induced by bombesin, vasopressin, or phorbol esters in Swiss 3T3 cells. J Biol Chem. 2001;276:40298–40305. doi: 10.1074/jbc.M106512200. [DOI] [PubMed] [Google Scholar]
- 17.Bowden ET, Barth M, Thomas D, Glazer RI, Mueller SC. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation. Oncogene. 1999;18:4440–4449. doi: 10.1038/sj.onc.1202827. [DOI] [PubMed] [Google Scholar]
- 18.Trauzold A, Schmiedel S, Sipos B, Wermann H, Westphal S, Roder C, Klapper W, Arlt A, Lehnert L, Ungefroren H, Johannes FJ, Kalthoff H. PKCmu prevents CD95-mediated apoptosis and enhances proliferation in pancreatic tumour cells. Oncogene. 2003;22:8939–8947. doi: 10.1038/sj.onc.1207001. [DOI] [PubMed] [Google Scholar]
- 19.Jensen RT. Receptors on pancreatic acinar cells. In: Johnson LR, Jacobson ED, Christensen J, Alpers DH, Walsh JH, editors. Physiology of the Gastrointestinal Tract. 3. Raven Press; New York: 1994. pp. 1377–1446. [Google Scholar]
- 20.Zugaza JL, Waldron RT, Sinnett-Smith J, Rozengurt E. Bombesin, vasopressin, endothelin, bradykinin, and platelet-derived growth factor rapidly activate protein kinase D through a protein kinase C-dependent signal transduction pathway. J Biol Chem. 1997;272:23952–23960. doi: 10.1074/jbc.272.38.23952. [DOI] [PubMed] [Google Scholar]
- 21.Zhukova E, Sinnett-Smith J, Wong H, Chiu T, Rozengurt E. CCK(B)/gastrin receptor mediates synergistic stimulation of DNA synthesis and cyclin D1, D3, and E expression in Swiss 3T3 cells. J Cell Physiol. 2001;189:291–305. doi: 10.1002/jcp.10018. [DOI] [PubMed] [Google Scholar]
- 22.Chiu T, Rozengurt E. CCK2 (CCK(B)/gastrin) receptor mediates rapid protein kinase D (PKD) activation through a protein kinase C-dependent pathway. FEBS Lett. 2001;489:101–106. doi: 10.1016/s0014-5793(01)02076-2. [DOI] [PubMed] [Google Scholar]
- 23.Tapia JA, Ferris HA, Jensen RT, Marin LJ. Cholecystokinin activates PYK2/CAKβ, by a phospholipase C-dependent mechanism, and its association with the mitogen-activated protein kinase signaling pathway in pancreatic acinar cells. J Biol Chem. 1999;274:31261–31271. doi: 10.1074/jbc.274.44.31261. [DOI] [PubMed] [Google Scholar]
- 24.Hoffmann KM, Tapia JA, Berna MJ, Thill M, Braunschweig T, Mantey S, Moody T, Jensen RT. Gastrointestinal hormones cause rapid c-Met receptor down-regulation by a novel mechanism involving clathrin-mediated endocytosis and a lysosome dependent mechanism. J Biol Chem. 2006 doi: 10.1074/jbc.M602583200. [DOI] [PubMed] [Google Scholar]
- 25.Nicke B, Tseng MJ, Fenrich M, Logsdon CD. Adenovirus-mediated gene transfer of RasN17 inhibits specific CCK actions on pancreatic acinar cells. Am J Physiol. 1999;276:G499–G506. doi: 10.1152/ajpgi.1999.276.2.G499. [DOI] [PubMed] [Google Scholar]
- 26.Jensen RT, Wank SA, Rowley WH, Sato S, Gardner JD. Interaction of CCK with pancreatic acinar cells. Trends Pharmacol Sci. 1989;10:418–423. doi: 10.1016/0165-6147(89)90192-2. [DOI] [PubMed] [Google Scholar]
- 27.Van Lint J, Ni Y, Valius M, Merlevede W, Vandenheede JR. Platelet-derived growth factor stimulates protein kinase D through the activation of phospholipase Cgamma and protein kinase C. J Biol Chem. 1998;273:7038–7043. doi: 10.1074/jbc.273.12.7038. [DOI] [PubMed] [Google Scholar]
- 28.Stark HA, Sharp CM, Sutliff VE, Martinez J, Jensen RT, Gardner JD. CCK-JMV 180: a peptide that distinguishes high affinity cholecystokinin receptors from low affinity cholecystokinin receptors. Biochim Biophys Acta. 1989;1010:145–150. doi: 10.1016/0167-4889(89)90154-7. [DOI] [PubMed] [Google Scholar]
- 29.Sato S, Stark HA, Martinez J, Beaven MA, Jensen RT, Gardner JD. Receptor occupation, calcium mobilization and amylase release in pancreatic acini: effect of CCK-JMV-180. Am J Physiol. 1989;257:G202–G209. doi: 10.1152/ajpgi.1989.257.2.G202. [DOI] [PubMed] [Google Scholar]
- 30.Williams JA, Yule DI. Stimulus-secretion coupling in pancreatic acinar cells. In: Go VLW, DiMagno EP, Gardner JD, Lebenthal E, Reber HA, Scheele GA, editors. PANCREAS: Biology, Pathobiology, and Disease. Raven Press; New York: 1993. pp. 167–189. [Google Scholar]
- 31.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–15781. [PubMed] [Google Scholar]
- 32.Chiu T, Rozengurt E. PKD in intestinal epithelial cells: rapid activation by phorbol esters, LPA, and angiotensin through PKC. Am J Physiol Cell Physiol. 2001;280:C929–C942. doi: 10.1152/ajpcell.2001.280.4.C929. [DOI] [PubMed] [Google Scholar]
- 33.Satoh A, Gukovskaya AS, Nieto JM, Cheng JH, Gukovsky I, Reeve JR, Jr, Shimosegawa T, Pandol SJ. PKC delta and epsilon regulate NF-κB activation induced by cholecystokinin and TNF-α in pancreatic acinar cells. Am J Physiol (Gastrointest Liver Physiol) 2004;287:G582–G591. doi: 10.1152/ajpgi.00087.2004. [DOI] [PubMed] [Google Scholar]
- 34.Li C, Chen X, Williams JA. Regulation of CCK-induced amylase release by PKC-delta in rat pancreatic acinar cells. Am J Physiol Gastrointest Liver Physiol. 2004;287:G764–G771. doi: 10.1152/ajpgi.00111.2004. [DOI] [PubMed] [Google Scholar]
- 35.Bastani B, Yang L, Baldassare JJ, Pollo DA, Gardner JD. Cellular distribution of isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1995;1269:307–315. doi: 10.1016/0167-4889(95)00120-0. [DOI] [PubMed] [Google Scholar]
- 36.Pollo DA, Baldassare JJ, Honda T, Henderson PA, Talkad VD, Gardner JD. Effects of cholecystokinin (CCK) and other secretagogues on isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta. 1994;1224:127–138. doi: 10.1016/0167-4889(94)90120-1. [DOI] [PubMed] [Google Scholar]
- 37.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C mu by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 38.Satoh A, Gukovskaya AS, Daghighian MS, Reeve JR, Jr, Pandol SJ. Novel protein kinase C isoforms δand ε mediate cholecystokinin-induced activation of PKY2 and NF-κB in pancreatic acinar cells by diverging pathways. Gastroenterology. 2004;126(4):A-530. Ref Type: Abstract ] [Google Scholar]
- 39.Braz JC, Gregory K, Pathak A, Zhao W, Sahin B, Klevitsky R, Kimball TF, Lorenz JN, Nairn AC, Liggett SB, Bodi I, Wang S, Schwartz A, Lakatta EG, Paoli-Roach AA, Robbins J, Hewett TE, Bibb JA, Westfall MV, Kranias EG, Molkentin JD. PKC-alpha regulates cardiac contractility and propensity toward heart failure. Nat Med. 2004;10:248–254. doi: 10.1038/nm1000. [DOI] [PubMed] [Google Scholar]
- 40.Storz P, Doppler H, Johannes FJ, Toker A. Tyrosine phosphorylation of protein kinase D in the pleckstrin homology domain leads to activation. J Biol Chem. 2003;278:17969–17976. doi: 10.1074/jbc.M213224200. [DOI] [PubMed] [Google Scholar]
- 41.Levitzki A. Tyrophostins-tyrosine kinase blockers as novel antiproliferative agents and dissectors of signal transduction. FASEB J. 1992;6:3275–3282. doi: 10.1096/fasebj.6.14.1426765. [DOI] [PubMed] [Google Scholar]
- 42.Ferris HA, Tapia JA, Garcia LJ, Jensen RT. CCKA receptor activation stimulates p130cas tyrosine phosphorylation, translocation, and association with Crk in rat pancreatic acinar cells. Biochemistry. 1999;38:1497–1508. doi: 10.1021/bi981903w. [DOI] [PubMed] [Google Scholar]
- 43.Tapia JA, Garcia-Marin LJ, Jensen RT. Cholecystokinin-stimulated protein kinase C-delta activation, tyrosine phosphorylation and translocation is mediated by Src tyrosine kinases in pancreatic acinar cells. J Biol Chem. 2003;278:35220–35230. doi: 10.1074/jbc.M303119200. [DOI] [PubMed] [Google Scholar]
- 44.Tapia JA, Camello C, Jensen RT, Garcia LJ. EGF stimulates tyrosine-phosphorylation of focal adhesion kinase (p125FAK) and paxillin in rat pancreatic acinar cells by a phospholipase C-independent process that depends on P13-K, the small GTP-binding protein, Rho and the integrity of the actin cytoskeleton. Biochim Biophys Acta. 1999;1448:486–499. doi: 10.1016/s0167-4889(98)00157-8. [DOI] [PubMed] [Google Scholar]
- 45.Bragado MJ, Groblewski GE, Williams JA. p70s6k is activated by CCK in rat pancreatic acini. Am J Physiol. 1997;273:C101–C109. doi: 10.1152/ajpcell.1997.273.1.C101. [DOI] [PubMed] [Google Scholar]
- 46.Oancea E, Bezzerides VJ, Greka A, Clapham DE. Mechanism of persistent protein kinase D1 translocation and activation. Dev Cell. 2003;4:561–574. doi: 10.1016/s1534-5807(03)00087-x. [DOI] [PubMed] [Google Scholar]
- 47.Paolucci L, Rozengurt E. Protein kinase D in small cell lung cancer cells: rapid activation through protein kinase C. Cancer Res. 1999;59:572–577. [PubMed] [Google Scholar]
- 48.Tan M, Xu X, Ohba M, Cui MZ. Angiotensin II-induced protein kinase D activation is regulated by protein kinase Cdelta and mediated via the angiotensin II type 1 receptor in vascular smooth muscle cells. Arterioscler Thromb Vasc Biol. 2004;24:2271–2276. doi: 10.1161/01.ATV.0000148449.92035.3a. [DOI] [PubMed] [Google Scholar]
- 49.Wong C, Jin ZG. Protein kinase C-dependent protein kinase D activation modulates ERK signal pathway and endothelial cell proliferation by vascular endothelial growth factor. J Biol Chem. 2005;280:33262–33269. doi: 10.1074/jbc.M503198200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sinnett-Smith J, Zhukova E, Hsieh N, Jiang X, Rozengurt E. Protein kinase D potentiates DNA synthesis induced by Gq-coupled receptors by increasing the duration of ERK signaling in swiss 3T3 cells. J Biol Chem. 2004;279:16883–16893. doi: 10.1074/jbc.M313225200. [DOI] [PubMed] [Google Scholar]
- 51.Iglesias T, Waldron RT, Rozengurt E. Identification of in vivo phosphorylation sites required for protein kinase D activation. J Biol Chem. 1998;273:27662–27667. doi: 10.1074/jbc.273.42.27662. [DOI] [PubMed] [Google Scholar]
- 52.Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280:13205–13208. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 53.Lemonnier J, Ghayor C, Guicheux J, Caverzasio J. Protein kinase C-independent activation of protein kinase D is involved in BMP-2-induced activation of stress mitogen-activated protein kinases JNK and p38 and osteoblastic cell differentiation. J Biol Chem. 2004;279:259–264. doi: 10.1074/jbc.M308665200. [DOI] [PubMed] [Google Scholar]
- 54.Matozaki T, Martinez J, Williams JA. A new CCK analogue differentiates two functionally distinct CCK receptors in rat and mouse pancreatic acini. Am J Physiol. 1989;257:G594–600. doi: 10.1152/ajpgi.1989.257.4.G594. [DOI] [PubMed] [Google Scholar]
- 55.Matozaki T, Goke B, Tsunoda Y, Rodriguez M, Martinez J, Williams JA. Two functionally distinct cholecystokinin receptors show different modes of action on Ca2+ mobilization and phospholipid hydrolysis in isolated rat pancreatic acini. Studies using a new cholecystokinin analog, JMV-180. J Biol Chem. 1990;265:6247–6254. [PubMed] [Google Scholar]
- 56.Lankisch TO, Nozu F, Owyang C, Tsunoda Y. High-affinity cholecystokinin type A receptor/cytosolic phospholipase A2 pathways mediate Ca2+ oscillations via a positive feedback regulation by calmodulin kinase in pancreatic acini. Eur J Cell Biol. 1999;78:632–641. doi: 10.1016/S0171-9335(99)80048-X. [DOI] [PubMed] [Google Scholar]
- 57.Rowley WH, Sato S, Huang SC, Collado-Escobar DM, Beaven MA, Wang LH, Martinez J, Gardner JD, Jensen RT. Cholecystokinin-induced formation of inositol phosphates in pancreatic acini. Am J Physiol. 1990;259:G655–G665. doi: 10.1152/ajpgi.1990.259.4.G655. [DOI] [PubMed] [Google Scholar]
- 58.Tsunoda Y, Yoshida H, Owyang C. Structural requirements of CCK analogues to differentiate second messengers and pancreatic secretion. Am J Physiol. 1996;271:G8–19. doi: 10.1152/ajpgi.1996.271.1.G8. [DOI] [PubMed] [Google Scholar]
- 59.Noguchi M, Adachi H, Gardner JD, Jensen RT. Calcium-activated, phospholipid-dependent protein kinase in pancreatic acinar cells. Am J Physiol. 1985;248:G692–G701. doi: 10.1152/ajpgi.1985.248.6.G692. [DOI] [PubMed] [Google Scholar]
- 60.Paolucci L, Sinnett-Smith J, Rozengurt E. Lysophosphatidic acid rapidly induces protein kinase D activation through a pertussis toxin-sensitive pathway. Am J Physiol Cell Physiol. 2000;278:C33–C39. doi: 10.1152/ajpcell.2000.278.1.C33. [DOI] [PubMed] [Google Scholar]
- 61.Chiu T, Wu SS, Santiskulvong C, Tangkijvanich P, Yee HF, Jr, Rozengurt E. Vasopressin-mediated mitogenic signaling in intestinal epithelial cells. Am J Physiol Cell Physiol. 2002;282:C434–C450. doi: 10.1152/ajpcell.00240.2001. [DOI] [PubMed] [Google Scholar]
- 62.Guha S, Rey O, Rozengurt E. Neurotensin induces protein kinase C-dependent protein kinase D activation and DNA synthesis in human pancreatic carcinoma cell line PANC-1. Cancer Res. 2002;62:1632–1640. [PubMed] [Google Scholar]
- 63.Bradford MD, Soltoff SP. P2X7 receptors activate protein kinase D and p42/p44 mitogen-activated protein kinase (MAPK) downstream of protein kinase C. Biochem J. 2002;366:745–755. doi: 10.1042/BJ20020358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Celil AB, Campbell PG. BMP-2 and insulin-like growth factor-I mediate Osterix (Osx) expression in human mesenchymal stem cells via the MAPK and protein kinase D signaling pathways. J Biol Chem. 2005;280:31353–31359. doi: 10.1074/jbc.M503845200. [DOI] [PubMed] [Google Scholar]
- 65.Endo K, Oki E, Biedermann V, Kojima H, Yoshida K, Johannes FJ, Kufe D, Datta R. Proteolytic cleavage and activation of protein kinase C [micro] by caspase-3 in the apoptotic response of cells to 1-beta -D-arabinofuranosylcytosine and other genotoxic agents. J Biol Chem. 2000;275:18476–18481. doi: 10.1074/jbc.M002266200. [DOI] [PubMed] [Google Scholar]
- 66.Jamora C, Yamanouye N, Van Lint J, Laudenslager J, Vandenheede JR, Faulkner DJ, Malhotra V. Gbetagamma-mediated regulation of Golgi organization is through the direct activation of protein kinase D. Cell. 1999;98:59–68. doi: 10.1016/S0092-8674(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 67.Tapia JA, Bragado MJ, Garcia-Marin LJ, Jensen RT. Cholecystokinin stimulated tyrosine phosphorylation of PKC-delta in pancreatic acinar cells is regulated bidirectionally by PKC activation. Biochim Biophys Acta (Mol Cell Res) 2002;1593:99–113. doi: 10.1016/s0167-4889(02)00346-4. [DOI] [PubMed] [Google Scholar]
- 68.Storz P, Doppler H, Toker A. Protein kinase Cdelta selectively regulates protein kinase D-dependent activation of NF-kappaB in oxidative stress signaling. Mol Cell Biol. 2004;24:2614–2626. doi: 10.1128/MCB.24.7.2614-2626.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Li J, O’Connor KL, Hellmich MR, Greeley GH, Jr, Townsend CM, Jr, Evers BM. The role of protein kinase D in neurotensin secretion mediated by protein kinase C-alpha/-delta and Rho/Rho kinase. J Biol Chem. 2004;279:28466–28474. doi: 10.1074/jbc.M314307200. [DOI] [PubMed] [Google Scholar]
- 70.Brandlin I, Hubner S, Eiseler T, Martinez-Moya M, Horschinek A, Hausser A, Link G, Rupp S, Storz P, Pfizenmaier K, Johannes FJ. Protein kinase C (PKC)eta-mediated PKC mu activation modulates ERK and JNK signal pathways. J Biol Chem. 2002;277:6490–6496. doi: 10.1074/jbc.M106083200. [DOI] [PubMed] [Google Scholar]
- 71.Waldron RT, Iglesias T, Rozengurt E. The pleckstrin homology domain of protein kinase D interacts preferentially with the eta isoform of protein kinase C. J Biol Chem. 1999;274:9224–9230. doi: 10.1074/jbc.274.14.9224. [DOI] [PubMed] [Google Scholar]
- 72.Yuan J, Bae D, Cantrell D, Nel AE, Rozengurt E. Protein kinase D is a downstream target of protein kinase Ctheta. Biochem Biophys Res Commun. 2002;291:444–452. doi: 10.1006/bbrc.2002.6469. [DOI] [PubMed] [Google Scholar]
- 73.Cabrera-Poch N, Sanchez-Ruiloba L, Rodriguez-Martinez M, Iglesias T. Lipid raft disruption triggers protein kinase C and Src-dependent protein kinase D activation and Kidins220 phosphorylation in neuronal cells. J Biol Chem. 2004;279:28592–28602. doi: 10.1074/jbc.M312242200. [DOI] [PubMed] [Google Scholar]