

Abstract
Several approaches are used to survey genomic DNA methylation patterns, including Southern blot, PCR, and microarray strategies. All of these methods are based on the use of methylation-sensitive isoschizomer restriction enzyme pairs and/or sodium bisulfite treatment of genomic DNA. They have many limitations, including PCR bias, lack of comprehensive assessment of methylated sites, labor-intensive protocols, and/or the need for expensive equipment. Since the presence of 5-methylcytosine alters the melting properties of DNA molecules, denaturing gradient gel blots (DGG blots), a gene scanning technique which detects differences in DNA fragments based on differential melting behavior, were used to examine genomic modification patterns in normal tissues. Variations in melting behavior, observed as restriction fragment melting polymorphisms (RFMPs), were detected in various tissues from single individuals in all human and mouse genes tested, suggesting the presence of widespread differential cell type-specific DNA modification. Additional DGG blot experiments comparing genomic DNA to unmethylated cloned DNA suggested that the melting variants were most likely caused by DNA methylation differences. The results suggest that the use of DGG blots can provide a comprehensive and rapid method for comparing complex in vivo DNA modification patterns in normal adult somatic cells.
Keywords: DGGE, methylation, DGG blots
1. Introduction
Genetic effects on the phenotype of an organism are the result of sequence changes in genes. Similarly, chemical modifications to DNA and histones that result in altered gene expression, but not changes in DNA sequence, can cause phenotypic differences among individuals; these modifications are referred to as epigenetic effects. In vertebrates, widespread epigenetic modification of genomic DNA has been detected most commonly as methylation at the 5′ position of cytosine (5-methylcytosine) within symmetric 5′-CpG-3′ (CpG) dinucleotide sequences. In addition, 5-methylcytosines have been found within asymmetric, non-CpG sequences in mammals, including humans, as well as in plants (Clark et al., 1995; Wada et al., 2003; Inoue and Oishi, 2005; Vanyushin, 2005). In the genomes of bacteria, protists, fungi, and plants, N6-methyladenine is found at the internal adenine within 5′-TGATCA-3′ sequences (Vanyushin, 2005). Although detection of N6-methyladenine in animal cells has not been reported, open reading frame DNA sequences homologous to bacterial adenine DNA-methyltransferases have been found in the genomes of insects, nematodes, and vertebrates, including humans, raising the possibility of the presence of adenine methylation in higher eukaryotes (Baniushin, 2005; Vanyushin, 2005). In addition, post-translational modifications of histone proteins, including acetylation, phosphorylation, methylation, and ubiquitination, together with DNA methylation, modify chromatin structure and cause changes in patterns of gene expression (Cheung and Lau, 2005).
A major barrier to the study of the role of DNA modification and epigenetic effects is the need for sensitive and practical techniques for identifying methylated sites in genomic DNA. Sites and patterns of DNA methylation have been studied by Southern blot- and PCR-based methods that rely on methylation-sensitive isoschizomer restriction enzyme pairs, in which digestion by one enzyme is prevented by methylation within the restriction enzyme recognition site. Because of the paucity of CpG dinucleotides in mammalian genomes (present at only 10–20% of the predicted frequency if they occurred at random), any method for the detection of methylated cytosines that relies on testing restriction enzyme recognition sequences can only assess 5% or less of methylated sites (Fazzari and Greally, 2004; Tollefsbol, 2004). This limitation exists for all restriction enzyme-based methods, making a comprehensive analysis of all methylated sites impossible.
Denaturing gradient gel electrophoresis (DGGE), a gene scanning technique that utilizes separation of DNA fragments on the basis of their melting behavior, provides an alternative opportunity for studying covalent modification patterns to genomic DNA. Both single nucleotide sequence differences and modification of DNA fragments by addition of 5-methylcytosines or N6-methyladenines affect the melting behavior of genomic and cloned DNA fragments (Collins and Myers, 1987; Abrams and V.P. Stanton, 1992). Such subtle variations in otherwise identical DNA fragments are detectable by DGGE as abnormal upward or downward band shifts. Consequently, both DNA sequence polymorphisms and methylation differences in purified DNA fragments have been revealed by DGGE. Since detection by DGGE does not rely on the modified base occurring within a restriction enzyme recognition site, methylated nucleotides can be detected indiscriminately if the modifications occur within the DNA sequence of the least stable melting domain of the DNA fragment (Fischer and Lerman, 1983; Abrams and V.P. Stanton, 1992).
Denaturing gradient gel blots (DGG blots) were used in previous experiments to identify single base change mutations and polymorphisms in genomic fragments from several genes as sequence restriction fragment melting polymorphisms (RMFPs) (Gray et al., 1991; Gray, 1992; Laprise et al., 1998). In an earlier study, genomic DNA fragments from paired human sperm and leukocyte samples were compared using DGG blots (Reindollar et al., 2000). In these experiments, cell type- and allele-specific melting patterns were detected between sperm and leukocyte DNA from the same individuals in several genes (Reindollar et al., 2000). The absence of melting differences on DGG blots in PCR-amplified DNA fragments strongly suggested that these melting variants were caused by differential cell type-specific DNA methylation (Reindollar et al., 2000).
The present study investigates whether or not cell type-specific in vivo DNA methylation patterns exist in other human and mouse tissues besides sperm and leukocytes. The results demonstrate that cell type-specific DNA fragment melting differences are common, and are most likely caused by differential methylation at sites not assayable by methylation-sensitive restriction enzyme analysis.
2. Materials and methods
2.1 Blood and tissue samples
Controls
Normal control blood samples were obtained from outpatient volunteers at the clinics of the Department of Obstetrics and Gynecology at New England Medical Center, Boston, Massachusetts. All blood samples were obtained following standard venipuncture and consent, according to applicable IRB guidelines. Tissue and semen samples were also obtained from volunteers at the clinics of the Department of Obstetrics and Gynecology at New England Medical Center following consent, according to applicable IRB guidelines.
Animals
Female CF-1 mice (Charles River Laboratories, Wilmington, MA) were injected with 5–7.5 IU of pregnant mare's serum gonadotropin and human chorionic gonadotropin and ovaries removed following CO2 anesthesia and cervical dislocation for an unrelated experiment (Ducibella and Buetow, 1994). Nine different tissues (brain, heart, kidney, liver, lung, skeletal muscle, small intestine, spleen and uterus) were then removed for the experiments described here. Tissues were also removed from male CF-1 mice, including testis.
2.2 Preparation of genomic DNA
Preparation of genomic DNA from human peripheral blood leukocytes was performed as described elsewhere (Gray, 1992). Genomic DNA was extracted from sperm as described elsewhere (Reindollar et al., 2000). Genomic DNA from human and mouse tissue was prepared by the same procedure, with some modifications. Briefly, tissue was placed in 3 ml of tissue extraction buffer (0.1 M EDTA, 0.05 M Tris-HCl, pH 8.0) in a large glass test tube and ground with a tissue homogenizer until no large clumps were visible. The suspension was transferred to a 15 ml polypropylene tube. The glass test tubes were rinsed with 2 ml tissue extraction buffer using the tissue homogenizer, and the suspension added to the 15 ml tube. 250 μl of 20% sodium dodecyl sulfate (SDS) and 50 μl proteinase K (10 μg/μl) was added to each tube and incubated at 55°C overnight, followed by one phenol/chloroform (1:1) extraction. DNA was precipitated at room temperature using 1/10 volume of 3 M sodium acetate and 3 volumes of isopropanol. DNA was pelleted by centrifugation at 14,000 rpm for 10 minutes, washed with 70% ethanol, and resuspended in 200 μl sterile 1X TE (10mM Tris-HCl, 1 mM EDTA).
2.3 Preparation and hybridization of DGG and Southern blots
DGG blots and agarose Southern blots were prepared as previously described (Laprise et al., 1998). Southern blots were also made from 4.5% non-denaturing polyacrylamide gels in TBE buffer (0.089 M Tris-borate, 0.089 M boric acid, 2 mM EDTA, pH 8.0) by electrotransfer to nylon membrane as previously described for DGG blots (Laprise et al., 1998).
Blots were hybridized as described previously (Laprise et al., 1998; Reindollar et al., 2000) with several genomic and cDNA clones: plasmid p625.8, containing sequences 30 kb downstream from the human F8 gene (Gitschier et al., 1984; Wood et al., 1984); mouse F8 cDNA (Elder et al., 1993); human ADRB2 cDNA (Kobilka et al., 1987); human GCCR genomic fragments (Reindollar et al., 2000); human MYB genomic and cDNA fragments (Franchini et al., 1983); human COL2AI genomic fragments (Cheah et al., 1985); human TP53 cDNA (Nogueira, 1997); human AMH cDNA (Cate et al., 1986); and mouse Th genomic fragments (Iwata et al., 1992).
2.4 Transformation and cloning
For preparation of cloned DNA fragments for DGGE analysis, plasmids containing cDNA or genomic DNA fragments were used to transform competent dam−, dcm− GM2163 bacteria cells (New England Biolabs, Beverly, MA). Preparation of competent cells and transformation procedures were adapted from previously published protocols (Sambrook et al., 1989; Inoue et al., 1990).
3. Results
3.1 Cell type-specific RFMPs in human and mouse genes
To investigate the in vivo DNA modification patterns of various genes, DGG blots were prepared using genomic DNA from human and mouse tissues digested with frequent cutting restriction enzymes. These enzymes (mostly 4-bp cutters) usually produce small (200–700 bp) DNA fragments that contain at least two melting domains. Since DGGE can reveal differences in all but the highest temperature (most stable) melting domain of a DNA fragment, the use of frequent cutting restriction enzymes is best for DGGE analysis (Fischer and Lerman, 1983; Abrams and V.P. Stanton, 1992). In comparisons of various tissue DNA samples from both human and mouse specimens, cell type-specific RFMPs were observed in all genes tested (Figs. 1–3).
Figure 1.
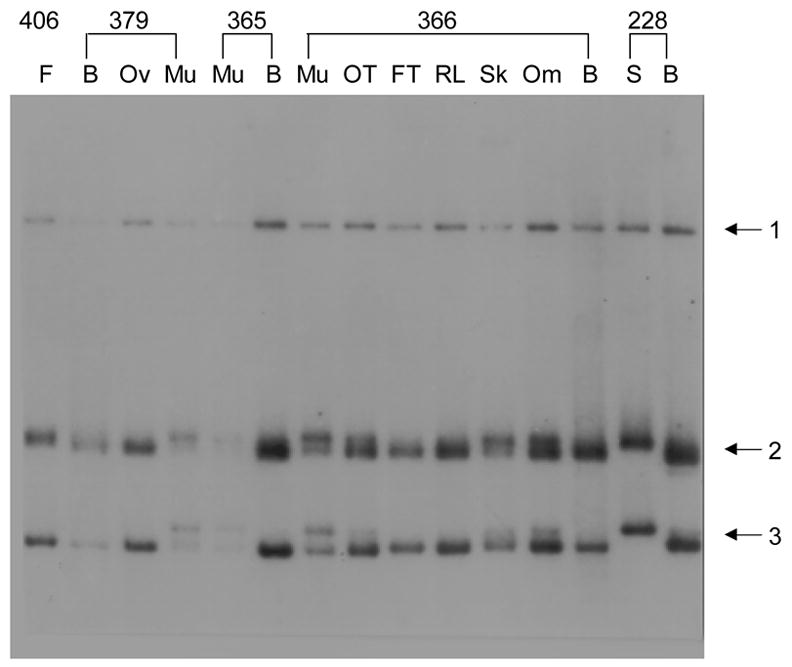
Tissue-specific modifications in the human ADRB2 gene. DGG blot made using RsaI-digested DNA isolated from various tissues from single individuals, electrophoresed in a 20–80% DGG for 1500 volt-hours (VH), and hybridized with a 2.0 kb EcoRI ADRB2 cDNA fragment. B = blood leukocyte, S = sperm, Om = omentum, Sk = skin, RL = round ligament, FT = fallopian tube, OT = malignant ovarian tumor, F = fibroid, Ov = ovary, Mu = muscle, Myo = myometrium. Numbers indicate individual patients from whom tissues were collected.
Figure 3.
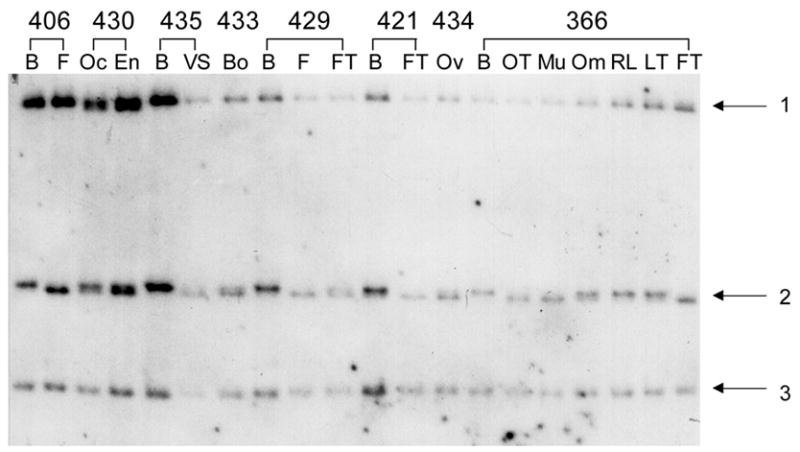
Tissue-specific modification of the human F8 gene. DGG blot containing HinfI-digested DNA isolated from various tissues from single individulas, prepared from a 20–80% DGG, electrophoresed for 1500 VH, and hybridized with a 2 kb exon 26 F8 gene probe. Fragment 2 varies, depending upon the tissue source of DNA. B = blood leukocyte, Om = omentum, RL = round ligament, FT = fallopian tube, LT = left tube, Ov = ovary, Mu = muscle, OT = malignant ovarian tumor, F = fibroid, OC = ovarian capsule, En = endometrium, VS = vaginal septum, Bo = bowel. Numbers indicate individual patients from whom tissues were collected.
3.1.1. Human genes
A DGG blot revealed three RsaI fragments when hybridized with a beta2 adrenergic receptor (ADRB2) gene probe. Two fragments, the second (diffuse band) and third (sharper band), exhibit cell type- and tissue-specific melting patterns (Fig. 1). Four melting variants were found for each of these fragments that differ according to the tissue origin of the DNA. In each tissue, either one or two variants were present. In cases of two variants of a single fragment, the ratio of band intensity of the doublet bands differed among tissues (for example, in individual 366, muscle, ovarian tumor, and skin; Fig. 1). These doublets do not represent allelic variants, because these would present as two bands of equal intensity. Multiple bands are probably caused by differential modification of the ADRB2 gene from the mixture of cell types within each tissue. Similar observations were visualized in the ADRB2 gene when DNA was digested with other restriction enzymes, including MvaI and DdeI (data not shown). Since the melting variants were found in DNA from several tissues from all single individuals tested, the band differences cannot be the result of somatic mutations. The RFMPs observed in these RsaI fragments are probably caused by cell type-specific differential DNA modification of the ADRB2 gene.
To compare the DGG blot results to patterns revealed using methylation-sensitive enzymes, Southern blot assays of genomic DNA digested with the methylation-sensitive isoschizomer restriction enzymes MspI and HpaII were performed. MspI and HpaII recognize the sequence 5′-CCGG-3′; however, HpaII activity is inhibited by methylation at the internal cytosine. Three MspI sites within the human intronless ADRB2 gene were detected on non-denaturing polyacrylamide Southern blots using the same DNA probe as that used for DGG blots. All tissues digested with HpaII showed the same pattern of restriction fragments as that produced by MspI digestion (data not shown). This shows that all three of these CpG sites are unmethylated, and no tissue- or cell type-specific differences were found, in contrast to DGG blot results.
An mRFMP was also detected on a DGG blot prepared with RsaI-digested DNA and hybridized with a probe made from the human glucocorticoid receptor (GCCR) gene (Figure 2). In contrast to the diffuse ADRB2 gene fragment 2, the GCCR gene bands were sharp and well focused. In GCCR fragment 2, only single bands were observed in leukocyte and sperm DNA, while in DNA derived from other tissues, doublets were seen; often, both single and doublet bands occurred in different tissues from the same individuals (Figure 2, individuals 406 and 379). This cell type-specific DNA modification was not revealed on MspI/HpaII Southern blots (data not shown).
Figure 2.
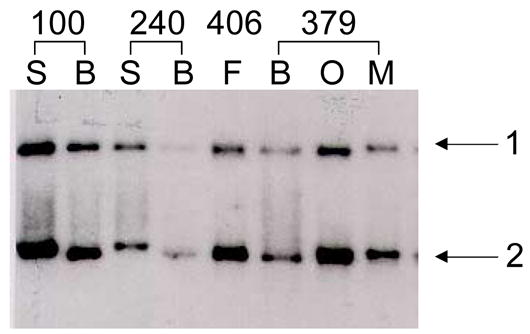
Tissue-specific modification of the human GCCR gene. DGG blot containing RsaI-digested DNA isolated from various tissues from single individuals, prepared from a 20–80% DGG, electrophoresed for 1500 VH, and hybridized with a 3.5 kb EcoRI GCCR gene fragment from the plasmid p24RI. Fragment 2 varies, depending upon the tissue source of DNA (doublets are seen in individual 406, fibroid, and individual 379 ovary). B = blood leukocyte, S = sperm, F = fibroid, Ov = ovary, Mu = muscle. Numbers indicate individual patients from whom tissues were collected.
A third example of cell type-specific mRFMPs was revealed in a human X-linked gene. Three HinfI fragments were observed on a DGG blot hybridized with a probe including exon 26 of the human factor VIII gene (F8) gene (Figure 3). In fragment 2, two different melting variants were present, representing differential modification of the F8 gene in the many cell types within these tissues. In individual 8, only blood leukocytes contained the high variant, while some tissues contained the low variant (for example, ovarian tumor and muscle); other tissues contained both variants (for example, omentum). Similar observations were made in other individuals, in that either the high, low, or both variants were present, depending upon the tissue source of DNA. Similar to the situation present in the ADRB2 gene, the differences seen in these variants between tissues could represent differential allelic modification. In addition, melting variation is detected between two different tissue DNA samples from the fallopian tube from the same individual (366; FT and LT). This is probably due to the functional and physiological differences of various regions of the fallopian tube. No MspI sites exist within this region of the F8 gene (Gitschier et al., 1984; Toole et al., 1984; Wood et al., 1984); therefore, MspI/HpaII Southern blot methods were not informative for examining methylation patterns at this locus (data not shown).
3.1.2. Mouse genes
To test additional tissue types and other mammals, we analyzed tissue-specific genes for differential DNA modification patterns in mouse tissues (Fig. 4). In the mouse FVIII (mouse F8) gene, cell type-specific RFMPs were observed with a cDNA probe including exons 1–8. Two DdeI fragments displayed melting variants depending upon the tissue source of DNA (Fig. 4A). The upper band exhibited subtle shifts among various murine tissues (for example, brain vs. heart). The lower band also displayed subtle migration shifts, with larger variations detected in some tissues (for example, brain, heart, skeletal muscle, and small intestine; Fig. 4A). An additional RFMP was detected with the same probe in analysis of HaeIII-digested DNA. In this case, the difference is qualitative, in that here, two bands were present in every tissue, and the intensity of the upper band varied relative to the lower band (Fig. 4B). These melting and intensity variations are probably caused by differential DNA modification in this region of the mouse F8 gene; in other words, this loss of intensity seen in the second band is most likely caused by one or more alleles of this band appearing as a diffuse RFMP due to DNA modification. If one compressed the diffuse staining back into a thin line, it is likely that all of the lanes would then have a 1:1 ratio. No differences were observed in this gene on non-denaturing polyacrylamide and agarose Southern blots containing mouse genomic DNA digested with MspI/HpaII (data not shown).
Figure 4.
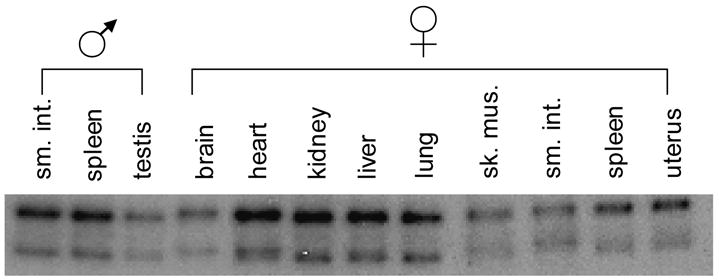
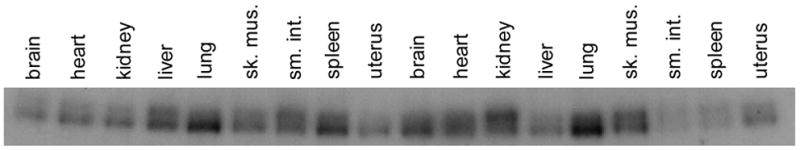
Tissue-specific modifications in the mouse F8 gene. (A) DGG blot containing DdeI-digested DNA isolated from various tissues from a single mouse, electrophoresed on a 20–80% DGG for 1500 VH, and hybridized with a 1.5 kb NcoI mouse F8 cDNA fragment that included exons 1–8. The position of each band varies depending on the tissue source of DNA. (B) DGG blot containing HaeIII-digested DNA isolated from tissues from a single mouse, electrophoresed and hybridized as in (A). Each tissue displays two bands. The relative intensities of the bands vary depending upon the tissue.
In the interpretation of the mouse F8 data, it is important to note that many bands on DGGE appear as diffuse bands. These bands are not gel artifacts; rather, they are just as informative as sharp band shifts. All differences seen on DGGE, whether shifts of sharply focused bands or qualitative transitions to diffuse bands, can be attributed to either sequence changes or DNA modification differences (Fischer and Lerman, 1983; Gray et al., 1991; Abrams and V.P. Stanton, 1992; Gray, 1992; Laprise et al., 1998).
Cell type-specific RFMPs were also observed in DNA fragments of the mouse tyrosine hydroxylase gene (mouse Th) in the same tissues analyzed in the previous mouse F8 gene experiment (data not shown). MspI/HpaII Southern blots did not reveal any differential tissue-specific methylation patterns in the mouse Th gene (data not shown).
3.2. Comparison of genomic DNA fragments to unmethylated cloned DNA fragments
As shown above and in numerous other experiments (data not shown), DGG blots revealed in vivo cell type-specific melting patterns in all human and mouse genes tested. To demonstrate that the differential melting behavior is associated with chemical DNA modification, tissue-derived and unmethylated DNA fragments were compared by DGGE. Paired human leukocyte and sperm DNA samples from single individuals were digested with restriction enzymes and electrophoresed alongside similarly digested DNA fragments made from cloned DNA produced in bacteria. The Escherichia coli bacteria strain used as the host to propagate the plasmids lacked the ability to methylate cytosines or adenines (dam−, dcm−). Such side-by-side analysis allowed the direct comparison of tissue-derived genomic fragments to the same fragments with no methylated cytosines or adenines.
Paired leukocyte and sperm genomic DNA samples from single individuals were chosen for these experiments for two reasons: (1) in previous experiments, the most dramatic DNA melting differences were observed between these two cell types (Reindollar et al., 2000), and (2) experimental results would not be complicated by the presence of several melting variants produced in cell populations more heterogeneous than sperm. Since whole plasmid clones were used in these experiments, the extra "end" fragments produced by enzyme digestion were usually present in lanes containing cloned DNA. These end fragments probably included small portions of the cloned insert adjacent to vector sequences. Such fragments were not mistaken for those corresponding to leukocyte and sperm derived fragments because they appeared as one or two fragments at positions in the gradient that did not correspond to any sperm or leukocyte fragment.
Eight different human and mouse loci were screened using this strategy; all loci tested showed extensive methylation in DNA from both leukocytes and sperm. In general, bacteria-derived fragments stopped higher in the denaturing gradient compared to genomic fragments. Because the addition of a methyl group on a cytosine has a stabilizing effect on DNA fragment melting in a dose- and position-dependent manner, the downward shift of the genomic fragments might indicate a stabilizing chemical modification to the DNA (Collins and Myers, 1987). In addition, the homogeneous unmethylated cloned fragments often produced sharper band patterns when compared to those produced by their tissue-derived counterparts.
In each gene, differences were usually present in DNA fragments from more than one restriction enzyme digest and usually in more than one fragment. Among seven human loci tested (ADRB2, GCCR, MYB, COL2A1 [type II collagen gene], TP53 [tumor protein p53], AMH [anti-Mullerian hormone], and one extragenic locus nearby the F8 gene), fifty different leukocyte- and sperm-derived DNA fragments had altered mobility compared to bacteria-derived cloned DNA fragments (Fig. 5). Four patterns were observed in the comparison of human genomic fragments to cloned DNA fragments: (a) both leukocyte and sperm DNA fragments co-migrated with cloned DNA; (b) both leukocyte and sperm DNA had altered mobility, but to similar extents; (c) both leukocyte and sperm DNA displayed altered mobility, but to different extents; (d) leukocyte DNA fragments displayed altered mobility and sperm DNA did not. None of these common patterns were unique to any gene; in addition, more than one pattern was often present in the same gene and often in the same enzyme digest. In general, fragments derived from leukocyte genomic DNA migrated further into the denaturing gradient than sperm DNA fragments, with occasional exceptions.
Figure 5.
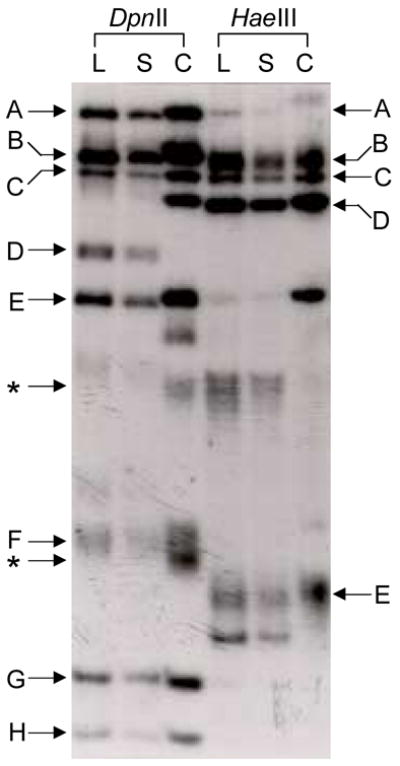
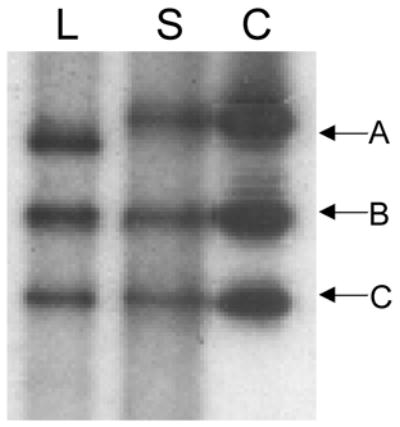
DGG blots showing differential methylation of human genomic DNA versus unmethylated bacterially-grown DNA fragments. L = leukocyte DNA, S = sperm DNA, C = cloned DNA. (A) DpnII- and HaeIII-digested genomic and cloned DNA, electrophoresed on a 20–80% DGG for 1500 VH, and hybridized with a 7.3 kb EcoRI COL2AI gene fragment. Letters indicate fragments that either are not modified or show similar modification in blood and sperm cells (see text for details). The fragments that are from the ends of the plasmid insert and do not have the same lengths as the intact genomic fragments are indicated by an asterisk. (B) HinfI-digested genomic and cloned DNA. This blot was prepared as in (A) and was hybridized with a probe that includes a region 30 kb downstream from the human F8 gene. Fragment A includes a leukocyte/sperm mRFMP; the leukocyte-derived fragment is modified compared with the sperm-derived fragment and the unmethylated cloned fragment.
For some fragments, there was no difference in DGGE mobility between those from leukocytes, sperm, or grown in bacteria. It is likely that there are no chemical modifications in the analyzed regions of these fragments. This was observed in a region from the human COL2AI gene in DpnII- and HaeIII-digested DNA (Fig. 5A). Several fragments in these lanes did not show any mobility differences, since fragments from all three sources migrated to the same position in the denaturing gradient (Fig. 5A; DpnII fragments C,G,H; HaeIII fragments B,C,D).
For other fragments, such as several DpnII fragments in the COL2AI gene, both the sperm- and leukocyte-derived fragments traveled to the same position in the gel while the corresponding unmethylated fragment stopped at a position higher in the gradient (Fig. 5A, DpnII fragments A,B,D,E,F). In these DpnII fragments, the genomic fragments were shifted either subtly (fragments A,B,E) or by several millimeters (fragments D,F,G). Other examples were seen in the same gene in HaeIII-digested DNA (fragments A,E). In all of these examples, both genomic fragments were shifted downward by the same distance compared to the corresponding cloned fragments, suggesting that both leukocyte and sperm derived DNA fragments were modified to a similar extent.
When differential modification of leukocyte- and sperm-derived fragments was observed, the leukocyte DNA fragments typically migrated further into the gradient, most likely due to stabilization of genomic fragments by the presence of 5-methylcytosines (Collins and Myers, 1987). HinfI-digested leukocyte and sperm genomic DNA were electrophoresed in a denaturing gradient gel alongside a HinfI-digested 3.2 kb genomic fragment from a site 30kb downstream from the human F8 gene (Fig. 5B). The leukocyte fragment A was shifted downward by 2 mm compared to the position of the corresponding sperm and cloned fragments; this downward shift of the leukocyte-derived fragment suggests that the fragments from leukocytes had more extensive modifications than sperm-derived fragment DNA fragments. Also, since the sperm-derived fragment co-migrated with the unmethylated fragment, it is likely that it is unmodified. No mobility differences were observed in fragments B and C between leukocyte, sperm, and cloned DNA (Fig. 5B).
In some cases, there were downward shifts of both sperm- and leukocyte-derived fragments compared to corresponding cloned fragments, usually with the leukocyte fragment migrating further into the gradient (data not shown). The opposite pattern of sperm-derived fragments traveling further into the gradient compared to those from leukocytes was less common; only two examples were detected (data not shown).
4. Discussion
The hypothesis for the present experiments was that DGG blots can be used for rapid and comprehensive in vivo detection of cell type-specific DNA modifications, including cytosine methylation. The limitations of widely-used techniques for detecting DNA methylation include CpG island bias, assessment of 5-methylcytosines only within CpG dinucleotides and/or within restriction enzyme recognition sites, PCR bias, and/or the need for specialized equipment (Yan et al., 2000; Fraga and Esteller, 2002; Liu and Maekawa, 2003; Cottrell, 2004; Tollefsbol, 2004). In addition, these methods cannot detect other forms of chemical DNA modification, such as N6-methyladenine. Other chemically-induced covalent DNA modifications occur, as well. For example, chemotherapeutic alkylating agents can cause the formation of O6-methylguanine, which is mutagenic because it can base pair with thymine (Soejima et al., 2005). Since the gene encoding O6-methylguanine-DNA methyltransferase (MGMT), a DNA repair enzyme that removes the methyl group from O6-methylguanine, is not expressed in many tumors, detection of epigenetic silencing of MGMT can be useful during chemotherapy (Soejima et al., 2005). Similarly, detection of O6-methylguanine in tumor genomic DNA may offer another useful approach for monitoring the alkylating activity of chemotherapeutics. Thus, methylation detection methods that can reveal any type of chemical modification to DNA may be clinically beneficial.
4.1. Cell type-specific RFMPs
In our earlier studies, cell type-specific melting differences were investigated in sperm, a homogeneous cell type population, and leukocytes (Reindollar et al., 2000). Leukocyte- and sperm-specific melting variants were observed in DNA fragments from several human genes (Reindollar et al., 2000). In the present experiments, genomic DNA from additional tissues from both humans and mice were analyzed to see if cell type-specific DNA melting variants can be found among the various cell types among different single tissues.
To compare DNA modification patterns in several human and mouse genes, DGG blots were made using restriction enzyme-digested genomic DNA isolated from several tissues from a single individual or animal. Since small restriction fragments with at least two melting domains were analyzed, DGGE analysis of each fragment cannot detect chemical modifications in the most stable melting domain. To address this, several DGG blots were made, each with genomic DNA digested with a different restriction enzyme, maximizing the likelihood that most of the DNA sequence in a region will be included in a lowest melting domain (Gray et al., 1991).
Results from the experiments presented here suggest that human and mouse genes undergo widespread DNA modification in a cell type- and tissue-specific manner. The presence of a variety of melting variants of many fragments in almost all genes analyzed suggest that normal somatic cells have extensive and complex patterns of in vivo DNA modification. In some tissue DNA samples, several different melting variants within the same fragment were detected. This may be a consequence of all of the different cell types within each tissue, each with its own pattern of genomic DNA modification. DNA sequence differences, such as somatic mutations, cannot be the cause of the observed cell-type specific RFMPs, because they were present in all subjects tested, have the same patterns in various tissues from many individuals, and disappear after PCR amplification (Reindollar et al., 2000).
4.2. Comparison of methylated and unmethylated DNA fragments
Results from methylation-sensitive restriction enzyme Southern blot analyses suggest that cell type-specific methylation patterns are neither abundant nor easily found in the genes tested. The Southern blot strategy can detect only a few potential methylation sites and only at restriction enzyme recognition sites, leaving the majority of CpGs untested; this is true of any method that relies on methylation-sensitive isoschizomer enzymes. In contrast, the DGG blot data demonstrate widespread and complex patterns of cell type-specific DNA modification, in all genes tested. It is most likely that the DNA modifications that cause cell-type specific RFMPs are 5-methylcytosines not within restriction enzyme recognition sites.
To provide evidence that the cell type-specific RFMPs are caused by differences in DNA methylation, we compared the patterns of tissue-derived fragments to those of the same fragments in the unmethylated state (propagated in bacteria). There is no methyltransferase activity in either the host bacteria used or in the PCR reactions; therefore, since the unmethylated fragments migrated differently from the tissue-derived fragments in all subjects and tissues tested, we believe that the tissue-derived fragments are methylated to various extents in vivo.
Differential migration of DNA fragments in DGGE does not reveal the precise sites of modification or if the modifications occur within CpG dinucleotides or non-CpG sequences. Methylation at cytosines within non-CpG sequences has been observed in human sequences (Woodcock et al., 1997; Vu et al., 2000; Inoue and Oishi, 2005). In contrast, non-CpG cytosines were rarely methylated in human LI promoter regions (Burden et al., 2005). Nonetheless, DGG blots can detect methylation outside of CpG dinucleotides, unlike other common methods. The loci and sequence motifs associated with modified residues detected by DGG blots can be revealed by genomic sequencing of sodium bisulfite treated DNA (Frommer et al., 1992).
In our previous comparisons of leukocyte and sperm genomic DNA, most of the leukocyte/sperm cell type-specific RFMPs revealed prominent downward shifts of the leukocyte derived fragments (Reindollar et al., 2000). This suggests that genomic DNA isolated from leukocytes might be modified to a greater extent compared to sperm genomic DNA, if the RFMPs are entirely due to 5′-cytosine methylation.
Other methylation detection strategies that rely on DNA melting behavior have been developed, such as PCR and fluorescence melting curve analysis and two-dimensional DNA fingerprinting (2-D fingerprinting) (Liu and Maekawa, 2003). The former method requires the use of a fluorometer to analyze the melting behavior of sodium-bisulfite treated DNA during PCR amplification. This method depends on the observation that unconverted methylated cytosines will stabilize double-stranded fragments more than fragments with unmethylated cytosines that are converted to uracils (Liu and Maekawa, 2003). This technique is sensitive and can resolve allele-specific methylation patterns, but it is susceptible to PCR bias and is not quantitative (Liu and Maekawa, 2003). Two-dimensional fingerprinting has been used to reveal differential methylation patterns between glioma tumor and leukocyte DNA samples (Uhlmann et al., 1999). In this study, two to eleven methylation differences were revealed in twenty-eight blood/tumor pairs (Uhlmann et al., 1999). We found fifty tissue-specific modified DNA fragments among seven loci using the very simple and rapid DGG blot method described above (Reindollar et al., 2000).
4.3. Summary
In summary, DGG blots can be used to detect cell type-specific melting behavior differences in several human and mouse genes in tissues with heterogeneous cell populations. The presence of melting variants within more than one fragment and in more than one enzyme digest indicates the presence of complex DNA modification patterns that cannot be characterized by analysis of recognition sites for methylation-sensitive isoschizomer restriction enzyme pairs. A DGG blot-based strategy for screening in vivo chemical modification sites (not limited to detecting cytosine methylation only) has several advantages over commonly used techniques to identify sites of DNA methylation, including: (1) detection of cytosine methylation is not limited to those within restriction enzyme recognition sequences; (2) detection of cytosine methylation is not limited to those within CpG dinucleotides; (3) detection of other forms of chemical DNA modification that would alter DNA melting behavior, including the presence of N6-methyladenine or O6-methylguanine; (4) no requirement for complex primer design or concerns about PCR bias; and (5) DGG blots are simple and do not require expensive automated equipment. The use of DGG blots is a potentially powerful method for rapid analysis of chemical DNA modification patterns and study of the role of cell type-specific epigenetic modification in the mammalian genome.
Acknowledgments
This work was supported by NIH; Grant number HL43202
Gene abbreviations
- ADRB2,
human beta-2-adrenergic receptor
- AMH
human anti-mullerian hormone
- COL2AI
human type II collagen
- F8
human factor VIII
- GCCR
human glucocorticoid receptor
- MGMT
O6-methylguanine-DNA methyltransferase
- MYB
mouse F8, mouse factor VIII
- mouse Th
mouse tyrosine hydroxylase
- TP53
tumor protein p53
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abrams ESVP, Stanton J. Use of denaturing gradient gel electrophoresis to study conformational transitions in nucleic acids. Methods in Enzymology. 1992;212:71–104. doi: 10.1016/0076-6879(92)12006-c. [DOI] [PubMed] [Google Scholar]
- Baniushin BF. Methylation of adenine residues in DNA of eukaryotes. Molecular Biology (Moscow) 2005;39:557–566. [PubMed] [Google Scholar]
- Burden AF, Manley NC, Clark AD, Gartler SM, Laird CD, Hansen RS. Hemimethylation and non-CpG methylation levels in a promoter region of human LINE-1 (L1) repeated elements. Journal of Biological Chemistry. 2005;280:14413–14419. doi: 10.1074/jbc.M413836200. [DOI] [PubMed] [Google Scholar]
- Cate RL, Mattaliano RJ, Hession C, Tizard R, Farber NM, Cheung A, Ninfa EG, Frey AZ, Gash DJ, Chow EP, et al. Isolation of the bovine and human genes for Mullerian inhibiting substance and expression of the human gene in animal cells. Cell. 1986;45:685–698. doi: 10.1016/0092-8674(86)90783-x. [DOI] [PubMed] [Google Scholar]
- Cheah KSE, Stoker NG, Griffin JR, Grosveld FG, Solomon E. Identification and characterization of the human type II collagen gene (COL2AI) Proceedings of the National Academy of Sciences, USA. 1985;82:2555–2559. doi: 10.1073/pnas.82.9.2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung P, Lau P. Epigenetic regulation by histone methylation and histone variants. Molecular Endocrinology. 2005;19:563–573. doi: 10.1210/me.2004-0496. [DOI] [PubMed] [Google Scholar]
- Clark SJ, Harrison J, Frommer M. CpNpG methylation in mammalian cells. Nature Genetics. 1995;10:20–27. doi: 10.1038/ng0595-20. [DOI] [PubMed] [Google Scholar]
- Collins M, Myers RM. Alterations in DNA helix stability due to base modifications can be evaluated using denaturing gradient gel electrophoresis. Journal of Molecular Biology. 1987;198:737–744. doi: 10.1016/0022-2836(87)90214-2. [DOI] [PubMed] [Google Scholar]
- Cottrell SE. Molecular diagnostic applications of DNA methylation technology. Clinical Biochemistry. 2004;37:595–604. doi: 10.1016/j.clinbiochem.2004.05.010. [DOI] [PubMed] [Google Scholar]
- Ducibella T, Buetow J. Competence to undergo normal, fertilization-induced cortical activation develops after metaphase I of meiosis in mouse oocytes. Developmental Biology. 1994;165:95–104. doi: 10.1006/dbio.1994.1237. [DOI] [PubMed] [Google Scholar]
- Elder B, Lakich D, Gitschier J. Sequence of the murine factor VIII cDNA. Genomics. 1993;16:374–379. doi: 10.1006/geno.1993.1200. [DOI] [PubMed] [Google Scholar]
- Fazzari MJ, Greally JM. Epigenomics: beyond CpG islands. Nature Reviews. 2004;5:446–455. doi: 10.1038/nrg1349. [DOI] [PubMed] [Google Scholar]
- Fischer SG, Lerman LS. DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: Correspondance with melting theory. Proceedings of the National Academy of Sciences, USA. 1983;80:1579–1583. doi: 10.1073/pnas.80.6.1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraga MF, Esteller M. DNA methylation: A profile of methods and applications. BioTechniques. 2002;33:632–649. doi: 10.2144/02333rv01. [DOI] [PubMed] [Google Scholar]
- Franchini R, Wong-Staal F, Baluda MA, Lengel C, Tronick SR. Structural organization and expression of human DNA sequences related to the transforming gene of avian myeloblastosis virus. Proceedings of the National Academy of Sciences, USA. 1983;80:7385–7390. doi: 10.1073/pnas.80.24.7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Gregg GW, Malloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proceedings of the National Academy of Sciences, USA. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gitschier J, Wood WI, Goralka TM, Wion KL, Chen EY, Eaton DH, Vehar GA, Capon DJ, Lawn RM. Characterization of the human factor VIII gene. Nature. 1984;312:326–330. doi: 10.1038/312326a0. [DOI] [PubMed] [Google Scholar]
- Gray MR. Detection of DNA sequence polymorphisms in human genomic DNA by using denaturing gradient gel blots. American Journal of Human Genetics. 1992;50:331–346. [PMC free article] [PubMed] [Google Scholar]
- Gray MR, Charpentier A, Walsh K, Wu P, Bender W. Mapping point mutations in the Drosophila rosy locus using denaturing gradient gel blots. Genetics. 1991;127:139–149. doi: 10.1093/genetics/127.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue H, Nojima H, Okayama H. High efficiency transformation of Escherichia coli with plasmids. Gene. 1990;96:23–28. doi: 10.1016/0378-1119(90)90336-p. [DOI] [PubMed] [Google Scholar]
- Inoue S, Oishi M. Effects of methylation of non-CpG sequence in the promoter region on the expression of human synaptotagmin XI (syt11) Gene. 2005;348:123–134. doi: 10.1016/j.gene.2004.12.044. [DOI] [PubMed] [Google Scholar]
- Iwata N, Kobayashi K, Sasaoka T, Hidaka H, Nagatsu T. Structure of the mouse tyrosine hydroxylase gene. Biochemistry Biophysics Research Communication. 1992;182:348–354. doi: 10.1016/s0006-291x(05)80151-2. [DOI] [PubMed] [Google Scholar]
- Kobilka BK, Dixon RAF, Frielle T, Dohlman HG, Bolanowski MA, Sigal IS, Yang-Feng TL, Francke U, Caron MG. cDNA for the human beta2-adrenergic receptor: A protein with multiple membrane-spanning domains and encoded by a gene whose chromosomal location is shared with that of the receptor for platelet-derived growth factor. Proceedings of the National Academy of Sciences, USA. 1987;84:46–50. doi: 10.1073/pnas.84.1.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laprise SL, Mak EK, Killoran KA, Layman LC, Gray MR. Use of denaturing gradient gel blots to screen for point mutations in the factor VIII gene. Human Mutation. 1998;12:393–402. doi: 10.1002/(SICI)1098-1004(1998)12:6<393::AID-HUMU5>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- Liu ZJ, Maekawa M. Polymerase chain reaction-based methods of DNA methylation analysis. Analytical Biochemistry. 2003;317:259–265. doi: 10.1016/s0003-2697(03)00169-6. [DOI] [PubMed] [Google Scholar]
- Nogueira C. Identification of polymorphisms in the p53 gene by denaturing gradient gel blots. Human Heredity. 1997;47:52–57. doi: 10.1159/000154390. [DOI] [PubMed] [Google Scholar]
- Reindollar RH, Fusaris KW, Gray MR. Methylation-dependent melting polymorphisms in genomic fragments of deoxyribonucleic acid. American Journal of Obstetrics and Gynecology. 2000;182:785–793. doi: 10.1016/s0002-9378(00)70327-9. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning: A laboratory manual. Cold Spring Harbor Press; 1989. [Google Scholar]
- Soejima H, Zhao W, Mukai T. Epigenetic silencing of the MGMT gene in cancer. Biochemistry and Cell Biology. 2005;83:429–437. doi: 10.1139/o05-140. [DOI] [PubMed] [Google Scholar]
- Tollefsbol TO. Methods of epigenetic analysis. Methods in Molecular Biology: Epigenetics Protocols. 2004;287:1–8. doi: 10.1385/1-59259-828-5:001. [DOI] [PubMed] [Google Scholar]
- Toole JJ, Knopf JL, Wozney JM, Sultzman LA, Buecker JL, Pittman DD, Kaufman RJ, Brown E, Shoemaker C, Orr EC, Amphlett GW, Foster WB, Coe ML, Knutson GJ, Fass DN, Hewick RM. Molecular cloning of a cDNA encoding human anti-haemophilic factor. Nature. 1984;312:342–347. doi: 10.1038/312342a0. [DOI] [PubMed] [Google Scholar]
- Uhlmann K, Marczinek K, Hampe J, Thiel G, Nurnberg P. Changes in methylation patterns identified by two-dimensional DNA fingerprinting. Electrophoresis. 1999;20:1748–1755. doi: 10.1002/(SICI)1522-2683(19990101)20:8<1748::AID-ELPS1748>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- Vanyushin BF. Enzymatic DNA methylation is an epigenetic control for genetic functions of the cell. Biochemistry (Moscow) 2005;70:597–603. doi: 10.1007/s10541-005-0143-y. [DOI] [PubMed] [Google Scholar]
- Vu TH, Li T, Nguyen D, Nguyen BT, Yao XM, Hu JF, Hoffman AR. Symmetric and asymmetric DNA methylation in the human IGF2-H19 imprinted region. Genomics. 2000;64:132–143. doi: 10.1006/geno.1999.6094. [DOI] [PubMed] [Google Scholar]
- Wada Y, Ohya H, Yamaguchi Y, Koizumi N, Sano H. Preferential de novo methylation of cytosine residues in non-CpG sequences by a domains rearranged DNA methyltransferase from tobacco plants. Journal of Biological Chemistry. 2003;278:42386–42393. doi: 10.1074/jbc.M303892200. [DOI] [PubMed] [Google Scholar]
- Wood WI, Capon DJ, Simonsen CC, Eaton DL, Gitschier J, Keyt B, Seeburg PH, Smith DH, Hollingshead P, Wion KL, Delwart E, Tuddenham EGD, Vehar GA, Lawn RM. Expression of active human factor VIII from recombinant DNA clones. Nature. 1984;312:330–337. doi: 10.1038/312330a0. [DOI] [PubMed] [Google Scholar]
- Woodcock DM, Lawler CB, Linsenmeyer ME, Doherty JP, Warren WD. Asymmetric methylation in the hypermethylated CpG promoter region of the human LI retrotransposon. Journal of Biological Chemistry. 1997;272:7810–7816. doi: 10.1074/jbc.272.12.7810. [DOI] [PubMed] [Google Scholar]
- Yan PS, Perry MR, Laux DE, Asare AL, Caldwell CW, Huang THM. CpG island arrays: An application toward deciphering epigenetic signatures of breast cancer. Clinical Cancer Research. 2000;6:1432–1438. [PubMed] [Google Scholar]