

Abstract
Many studies have supported a genetic aetiology for autism. Here we report mutations in two X-linked genes, neuroligins NLGN3 and NLGN4, in siblings with autism spectrum disorders. These mutations affect cell adhesion molecules localised at the synapse and suggest that a defect of synaptogenesis may predispose to autism.
Keywords: Amino Acid Sequence; Autistic Disorder; genetics; metabolism; Base Sequence; Brain; metabolism; Carrier Proteins; genetics; Chromosomes, Human, X; genetics; DNA, Complementary; genetics; Female; Gene Expression Profiling; Humans; Linkage (Genetics); Male; Membrane Proteins; genetics; Molecular Sequence Data; Mutation; Nerve Tissue Proteins; genetics; Pedigree; RNA, Messenger; genetics; metabolism; Sequence Homology, Amino Acid
Autism is characterised by impairments in reciprocal social interaction and communication as well as restricted and stereotyped patterns of interests and activities1. Asperger syndrome (AS) refers to subjects with higher cognitive abilities and more normal language function2. The recurrence risk of autism in sib-ships is approximately 45 times greater than in the general population and twin studies have documented a higher concordance rate in monozygotic (60%–91%) than in dizygotic twins (0%–6%)3. The male-to-female ratio is 4:1 in autism and 8:1 in AS. Male predisposition t o autistic disorder remains unexplained, although abnormalities of the sex chromosomes are frequently associated with autistic spectrum disorders4. At least two loci for a predisposition t o autism have been suggested on the X chromosome. At Xp22.3, de novo chromosomal deletions have been observed in three autistic females5 and a second locus at Xq13-21 is supported by two independent genome scans, showing increased allele sharing around markers DXS7132 (52 cM) and DXS6789 (62 cM) in affected sib-pair (ASP) analyses6,7.
Within the Xp22.3 deleted interval, we identified the transcript KIAA1260, which corresponds to NLGN4, a member of the neuroligin family8. Interestingly, NLGN39, a homologue of NLGN4, is located at Xq13 (55–56 cM), within the second chromosome X locus for autism. These genes encode cell adhesion molecules present at the postsynaptic side of the synapse10–12 and may be crucial factors for the formation of functional synapses13. Five NLGN genes have been identified in the human genome, which are localised at 3q26 (NLGN1), 17p13 (NLGN2), Xq13 (NLGN3), Xp22.3 (NLGN4), and Yq11.2 (NLGN4Y). Neuroligin phylogeny suggests that NL3 is the common ancestor of NLGN4 and NLGN4Y (not shown). Sequence comparison showed that all critical amino acids known to be essential for neuroligins are conserved in NLGN4/Y, including the cysteines, the transmembrane domain, and the postsynaptic density 95/discs large/zona occludens-1 (PDZ) domain.
The expression profile of NLGNs was determined by specific RT-PCR in individual male and female adult brain tissues (Fig. 1). All RT-PCR products were sequenced and correspond t o alternative splicing isoforms. NLGN1-3 transcripts were detected in all brain regions. NLGN4 and NLGN4Y transcripts were detected at similar levels in male brains, without significant differences in regional distribution. There is an alternative promoter, 693 bp downstream of NLGN4 exon E1a, substituting exon E1a by exon E1b. Exon E1b is specific to NLGN4 and contains an alternative donor splice site.
Fig. 1.
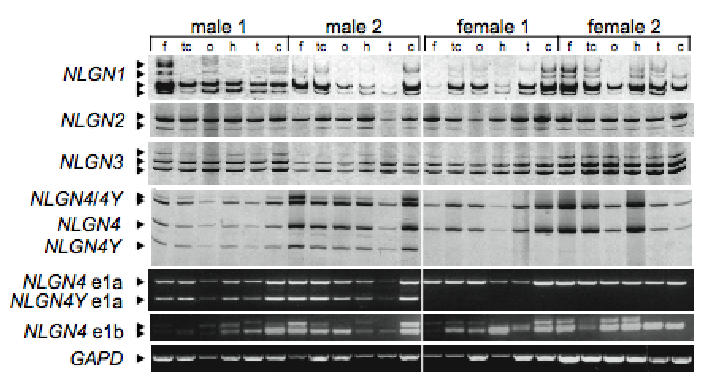
Expression of NLGNs in the human brain. Specific RT-PCRs were performed on total RNA from different brain regions using primers in exon 2 and 5 in order to analyse the different alternative spliced transcripts. To distinguish between NLGN4 and NLGN4Y mRNA, RT–PCRs were digested by NcoI. Using specific forward primers in exon 1a or exon 1b and reverse primers in exon 2, two alternative NLGN4 promoters were identified substituting the first exon 1a for 1b. Size of NLGN4 and NLGN4Y PCR differs by 193 bp. Exon 1b is specific to NLGN4 and contains an internal alternative donor splice site. The ages of the two males and the two females studied were 74, 42, 55, and 36 years old with a post-mortem delay of 10, 21, 24, and 2 h, respectively. f: frontal cortex, tc: temporal cortex, o: occipital cortex, h: hippocampus, t: thalamus; c: cerebellum. Normal control human brains were obtained at autopsy under guidelines approved by the ethics committee. DNA amplification and RT-PCRs were performed as described17. Alignment and sequences of all NLGN proteins and isoforms are available from the authors.
We screened for NLGN3 and NLGN4/Y mutations in 36 ASP and 122 trios with autism or AS (140 males and 18 females). In one Swedish family with two affected brothers, one with typical autism and the other with AS, a frameshift mutation (1186insT) was identified in NLGN4 (Fig. 2a). This mutation creates a stop codon at position 396, leading to premature termination of the protein before the transmembrane domain. The mutation is present in the mother but is absent in the maternal grandmother and in two maternal aunts. The unaffected maternal grandfather was deceased and could not be studied. A false paternity of the maternal grandfather was excluded by studying eight microsatellites markers from the X chromosome (not shown). Together, these results indicate that 1186insT is a de novo mutation in the mother. The mutation was not found in the unaffected brother and in 350 unrelated controls (250 females and 100 males). In a second Swedish family with two affected brothers, one with typical autism and the other with AS, we identified a C to T transition in NLGN3 inherited from the mother and changing a highly conserved arginine residue into cysteine (R451C) within the esterase domain (Fig. 2b). R451 is located in a predicted EF-hand domain conserved in all known neuroligins (including D. melanogaster NL and gliotactin) and in all sequenced esterases from mammals, fish and birds14. EF-hand domains are known to confer structural integrity and Ca2+ dependent functional properties. R451C may therefore modify the binding of neuroligins to their presynaptic partners neurexins since binding is only observed in the presence of Ca2+ 15. This mutation was absent in 200 controls (100 females and 100 males). Detailed clinical information for these families is available as Supplementary Note.
Fig. 2.
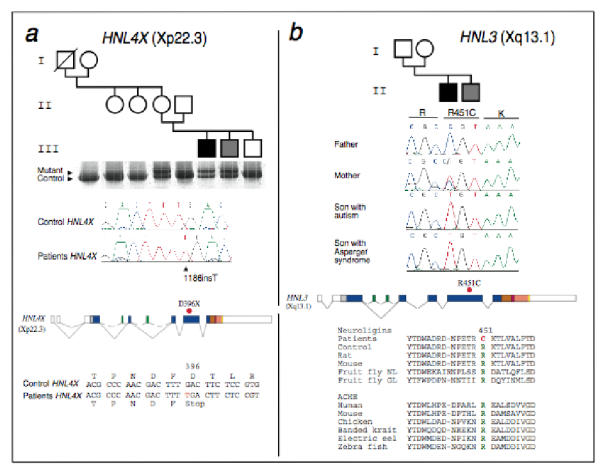
Mutation screening of NLGN3 and NLGN4 in autistic subjects. a, Identification of the frameshift NLGN4 mutation in two affected individuals with autism and Asperger syndrome. Pedigree structure and the abnormal SSCP conformer are shown. Using NLGN4 specific primers, sequence of the PCR product revealed a one-base-pair (T) insertion in the mother and her two affected sons, absent in the father and the unaffected son. This insertion occurs in exon 5 of NLGN4 and causes a frameshift that leads to the premature termination of NLGN4 which thus lacks 421 amino acids (51% of the protein) including the transmembrane domain. b, Identification of the NLGN3 mutation. The family comprises a son with autism and his younger brother with AS. The R451C mutation is localised in the esterase domain of the protein and modifies a highly conserved arginine residue present in neuroligins and acetylcholine esterase (ACHE). Affected individuals are indicated by filled symbols (black for autism and grey for AS). Squares are males, circles are females, and a line through a symbol indicates that the person is deceased. SSCP analysis was performed with a GenePhor apparatus (Pharmacia-Biotech). We carried out direct sequencing with BigDye terminator cycle Sequencing Kit (Applied Biosystems) on an ABI 3100 Automated Sequencer.
Three independent lines of evidence strongly suggest that NLGN3 and NLGN4 mutations are involved in autistic spectrum disorders. First, deletions at Xp22.3 that include NLGN4 have been reported in several autistic subjects5. Second, the NLGN3 and NLGN4 point mutations cause severe alterations of the predicted protein structure. Third, there is the de novo appearance of the NLGN4 mutation in the patients’ mother. Among the various proteins involved in the establishment of the neural networks, cell adhesion molecules are crucial factors for the identification of the appropriate partner cell and the formation of a functional synapse16. Therefore, we hypothesise that a defect in NLGN3 or NLGN4 may abolish formation, stabilisation and/or recognition of specific synapses essential for the communication processes that are deficient in subjects with autistic spectrum disorder.
Supplementary Note
The Swedish family presenting the NLGN4 mutation comprised a 32 year-old man with autistic disorder, his younger brother (28 years old) with AS, and an unaffected male sibling, born t o healthy, unrelated parents. The first child was diagnosed with autism when he was 3 years old. Physical examination showed no dysmorphic features or other associated malformations, except for mild thoracic kyphosis. On evaluation at 22 years of age, he met DSM-III-R criteria for autistic disorder. On the Childhood Autism Rating Scale (CARS) he scored 44 points, reflecting severe autism. Neuropsychological evaluation showed normal intelligence. He met all criteria for autistic disorder on the Autism Diagnostic Interview-Revised (ADI-R) (scores of 29, 14, and 8 for social, communication, and repetitive behaviour domains, respectively). At present, he communicates with words only when prompted, using a limited number of single words and sentences; most communication is by writing. The second brother was diagnosed with Asperger syndrome and dysarthria at the age of 17 years. Physical examination revealed no dysmorphic features; he has a minor thoracic kyphosis, as his brother. Neuropsychological evaluation showed normal intelligence. When evaluated at the age of 19 years, he did not quite meet DSM-III-R criteria for autistic disorder (total symptom score of 7, cut-off score 8). He scored 22.5 points on the CARS (cut-off score 30). According to the AS Diagnostic Interview (ASDI), he fulfilled criteria for AS: social impairment, narrow interests, repetitive routines, speech and language peculiarities, non-verbal communication problems, and motor clumsiness. On the ADI-R, he did not quite meet the algorithm criteria for autistic disorder (scores of 10, 6, and 1 for social, communication, and repetitive behaviour domains, respectively; the cut-off scores for verbal individuals are 10, 8, and 3, respectively).
The family with the NLGN3 mutation (R451C) comprised a 24-year-old man with classic autism and severe mental retardation, and his younger (22-year-old) brother with AS and a non-verbal learning disability according to WISC-testing at age 10 years (verbal IQ 116, performance IQ <73). The man with classic autism developed epilepsy (clonic-tonic generalised seizures) at age 19 years. His autism diagnosis was established before age 3 years on the basis of extensive clinical examination by CG, whereas the AS diagnosis of his brother was established only at age 10 years. On evaluation at ages 20 and 22 years respectively, both men met full criteria for autistic disorder according to the DSM-III-R and the DSM-IV. The man with classic autism had scored 44 on the CARS at age 15 years (equalling severe autism), whereas his brother with an original clinical diagnosis of AS scored 30.5 at age 16 years (indicating mild autism).
Acknowledgments
We thank the patients and their families for agreeing to participate in this study. We also thank the Centre d’Investigations Cliniques de l’Hôpital Robert Debré for obtaining the blood samples from the French families, the DNA and cell bank from the INSERM U289 (IFR des Neurosciences, Hôpital Pitié-Salpêtrière, Paris), V. Sazdovitch for providing brain samples, and C. Bouchier and S. Duthoy for the sequencing facilities at the Génopole Pasteur. This work was funded by the French Research Ministry (Actions Concertées Incitatives), the INSERM (Institut National de la Santé et la Recherche Médicale), France Télécom, and the Swedish Medical Research Council.
Footnotes
URLs. All primers used in this study are available upon request or at http://www.im3.inserm.fr/autism/Recherche.htm.
GenBank accession number. The cDNA sequences of NLGN2, NLGN4, and NLGN4Y are deposited in GenBank under accession numbers AF376802, AF376803, and AF376804, respectively.
Competing interests statement
The authors declare that they have no competing financial interests.
References
- 1.American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 4. American Psychiatric Press; Washington D.C.: 1994. [Google Scholar]
- 2.Gillberg C. Br J Psychiatry. 1998;172:200–209. doi: 10.1192/bjp.172.3.200. [DOI] [PubMed] [Google Scholar]
- 3.Folstein SE, Rosen-Sheidley B. Nat Rev Genet. 2001;2:943–955. doi: 10.1038/35103559. [DOI] [PubMed] [Google Scholar]
- 4.Gillberg C. J Autism Dev Disord. 1998;28:415–425. doi: 10.1023/a:1026004505764. [DOI] [PubMed] [Google Scholar]
- 5.Thomas NS, et al. Hum Genet. 1999;104:43–48. doi: 10.1007/s004390050908. [DOI] [PubMed] [Google Scholar]
- 6.Shao Y, et al. Am J Med Genet. 2002;114:99–105. doi: 10.1002/ajmg.10153. [DOI] [PubMed] [Google Scholar]
- 7.Auranen M, et al. Am J Hum Genet. 2002;71:777–390. doi: 10.1086/342720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bolliger MF, Frei K, Winterhalter KH, Gloor SM. Biochem J. 2001;356:581–588. doi: 10.1042/0264-6021:3560581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Philibert RA, Winfield SL, Sandhu HK, Martin BM, Ginns EI. Gene. 2000;246:303–310. doi: 10.1016/s0378-1119(00)00049-4. [DOI] [PubMed] [Google Scholar]
- 10.Ichtchenko K, et al. Cell. 1995;81:435–43. doi: 10.1016/0092-8674(95)90396-8. [DOI] [PubMed] [Google Scholar]
- 11.Ichtchenko K, Nguyen T, Sudhof TC. J Biol Chem. 1996;271:2676–2682. doi: 10.1074/jbc.271.5.2676. [DOI] [PubMed] [Google Scholar]
- 12.Song JY, Ichtchenko K, Sudhof TC, Brose N. Proc Natl Acad Sci USA. 1999;96:1100–1105. doi: 10.1073/pnas.96.3.1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Scheiffele P, Fan JH, Choih J, Fetter R, Serafini T. Cell. 2000;101:657–669. doi: 10.1016/s0092-8674(00)80877-6. [DOI] [PubMed] [Google Scholar]
- 14.Tsigelny I, Shindyalov IN, Bourne PE, Sudhof TC, Taylor P. Prot Sci. 2000;9:180–185. doi: 10.1110/ps.9.1.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nguyen T, Sudhof TC. J Biol Chem. 1997;272:26032–26039. doi: 10.1074/jbc.272.41.26032. [DOI] [PubMed] [Google Scholar]
- 16.Brose N. Naturwissenschaften. 1999;86:516–524. doi: 10.1007/s001140050666. [DOI] [PubMed] [Google Scholar]
- 17.Jamain S, et al. Mol Psychiatry. 2002;7:302–310. doi: 10.1038/sj.mp.4000979. [DOI] [PMC free article] [PubMed] [Google Scholar]