

Abstract
RyR1 is an intracellular calcium channel with a central role in muscle contraction. We obtained a three‐dimensional reconstruction of the RyR1 in the closed state at a nominal resolution of ∼10 Å using cryo‐EM. The cytoplasmic assembly consists of a series of interconnected tubular structures that merge into four columns that extend into the transmembrane assembly. The transmembrane assembly, which has at least six transmembrane α‐helices per monomer, has four tilted rods that can be fitted with the inner helices of a closed K+ channel atomic structure. The rods splay out at the lumenal side and converge into a dense ring at the cytoplasmic side. Another set of four rods emerges from this ring and shapes the inner part of the four columns. The resulting constricted axial structure provides direct continuity between cytoplasmic and transmembrane assemblies, and a possible mechanism for control of channel gating through conformational changes in the cytoplasmic assembly.
The ryanodine receptor isoform 1 (RyR1) is a large homotetrameric intracellular calcium channel of skeletal muscle. Its opening releases Ca2+ stored in the sarcoplasmic reticulum (SR) into the myoplasm, and this increase of cytoplasmic Ca2+ triggers the interaction of actin and myosin that causes contraction of the muscle fibers. Ca2+ release by RyR1 is a tightly controlled process. RyR1 opening is directly triggered by activation of the dihydropyridine receptor (DHPR), which acts as the voltage sensor in the cell membrane (sarcolemma), and initiates the process known as excitation‐contraction coupling. Other cytoplasmic environmental variables (Ca2+, Mg2+ and ATP concentrations, and redox conditions), as well as some ligands (calmodulin (CaM) and FK‐506 binding protein 12 kDa (FKBP12)) modulate key RyR1 gating parameters (channel‐open probability and mean open time). These variables act through the high‐ and low‐affinity cation‐binding sites, nucleotide‐binding sites, redox sensors and CaM‐ and FKBP12‐binding sites in RyR1, respectively1,2. Most of the mass of the RyR1 tetramer is exposed to the cytoplasm, carrying out the task of receiving and coordinating all of the cytoplasmic inputs, and translating them into a signal that is transmitted to the gate in the transmembrane domain. Considerable progress in the elucidation of RyR1’s structure in the last decade3 has allowed investigators to begin delineating the area of DHPR interaction on the peripheral domains of RyR1 (refs. 4,5), to find the areas of apo‐ and Ca2+‐CaM interaction6,7 and to begin to define the structural changes associated with gating8,9. However, the resolution of these three‐dimensional reconstructions (between 33 and 14 Å; refs. 7,10,11) has been insufficient both to provide information about the signaling pathway between cytoplasmic and transmembrane assemblies and to provide a basic architecture of the transmembrane assembly.
The transmembrane assembly of RyR seems to have a complex structure. Based on hydropathy plots it has been estimated that there are between 4 and 12 transmembrane helices per subunit12,13. The TM9 segment in the 10‐transmembrane model was later identified as a lumenal segment using site‐specific antibodies14. More recently, sequence alignments of the RyR, IP3R and K+ channels including KcsA have identified the selectivity filter signature motif in RyR and IP3R15,16. Together with mutational studies, the TM10 segment of RyR has been proposed as the inner helix of the ion pore16–20, and a theoretical model for the pore‐forming region of RyR was built17. The structure of the bacterial K+ channel KcsA, determined at atomic resolution21, consists of four pairs of membrane‐spanning α‐helices arranged around a four‐fold symmetry axis. In the closed state the inner helices form a right‐handed bundle, giving a characteristic inverted tepee appearance that constricts the pore diameter near the intracellular membrane surface. On the extracellular side of the channel, short pore helices and lumenal loops connect the inner and outer helices and form the selectivity filter.
We carried out cryo‐EM of RyR1 at increased resolution to better understand RyR’s intraprotein signaling and to resolve substructure within the transmembrane assembly. Our three‐dimensional map at a nominal resolution of ∼10 Å allows a better understanding of the cytoplasmic assembly organization, and reveals four dense rod‐like structures organized as a tepee and other secondary structure elements as constituents of the transmembrane assembly. This allowed us to directly compare RyR with the K+ channel atomic model and to propose a general model on RyR signal integration and gating.
RESULTS
The improved resolution of our present three‐dimensional reconstruction, 10.3 Å (Fourier shell correlation (FSC) cutoff criterion at 0.15; ref. 22) and 13.6 Å (FSC cutoff criterion at 0.5; ref. 23) (Fig. 1a), was achieved using several improvements over our previous single‐particle cryo‐EM preparation and analysis7,24: (i) the purified RyR1 was applied to electron microscope grids and imaged in ice suspended over holes in the carbon support, which increased the randomness of RyR1 views, and hence the isotropy of the resolution ( Fig. 1b); (ii) the contrast transfer function (CTF) correction was applied; and (iii) a larger data set (36,000 particles) was used. The ability to distinguish secondary structure in parts of the macromolecule indicates that it is more appropriate to use the higher‐resolution estimate25,26. The overall structure agrees with previous three‐dimensional reconstructions of RyR1 (ref. 3), exhibiting a large square prism (corresponding to the cytoplasmic assembly27,28) connected to a smaller, square tapering prism (transmembrane assembly). The substantially higher resolution reveals a more scaffold‐like structure for the cytoplasmic assembly (Fig. 2). The overall dimensions are 275 × 275 × 100 ų for the cytoplasmic assembly, 115 × 115 × 60 ų maximum dimensions for the tapering transmembrane assembly, with a space of 14 Å between these two structures that is spanned by intra‐assembly columns (see below). The two square prism structures have a rotation of ∼38° with respect to each other (Fig. 2b). This three‐dimensional structure, in which the RyR1s were prepared in buffer containing submicromolar Ca2+, should correspond to RyR1 in the closed state29.
Figure 1.
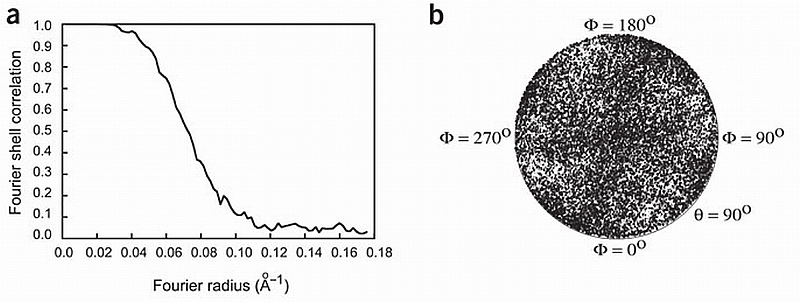
Image processing. (a) Fourier shell correlation curve indicating a resolution of 10.3 Å according to the 0.15 cutoff criterion. (b) Plot of the angular distribution of the particles used in the three‐dimensional reconstruction showing a uniform distribution of orientations of the vitrified RyR.
Figure 2.
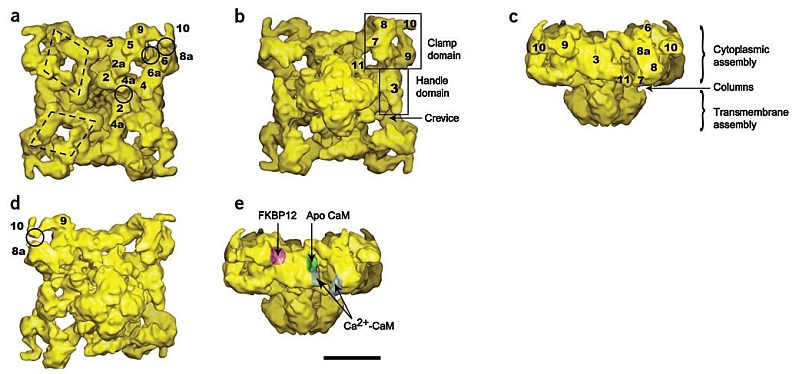
Isosurface representation of the RyR1 in different views. (a–e) T‐tubule view (a), SR view (b,d), side view (c,e). The SR view in d is slightly tilted with respect to b and contoured at a slightly higher threshold in order to expose the self‐standing structure formed by domains 9‐10 and the separation between domains 10‐8a. In e the approximate positions of the binding sites for FKBP12 (ref. 6), apo CaM7 and Ca2+‐CaM6 are indicated with surface shadowing. The clamp, handle, crevice, column (cl) and numeral domain assignations are indicated throughout. The two polygons highlight the rhomboid disposition of the tubular structures defined by domains 2‐4‐5‐6 at their corners. The midpoints of three of the sides of the rhomboids are labeled as domains 2a, 4a and 6a. The full circles indicate areas of close apposition between neighboring domains 2‐4a, 5‐6 and 10‐8a. Scale bar, 10 nm.
Cytoplasmic assembly interconnectivity
The ‘clamp’ domains, which define the corners of the cytoplasmic assembly, can now be seen to be formed by two separate structures: a flat tubular structure that loops around the outer corners of the square prism (domains 8a‐8‐7), and a smaller elongated and tapering structure that extends by the side of the square toward its corner (domains 9‐10) (Fig. 2). A slight threshold increase reveals that domains 9‐10 constitute a self‐standing elment that protrudes toward domain 8a (Fig. 2d). Domain 10, at the tip, is thinner and characterized by a pronounced kink (Fig. 2b, Fig. 2d). The T‐tubule facing surface of this reconstruction is also formed of tubular structures, with the rhomboid structure defined by domains 2‐4‐5‐6 at its corners as the main feature (Fig. 2a). The rhomboid structure appears to be connected to most of RyR1’s domains (Fig. 3a–Fig. 3d). Further inter‐relation among domains seems to occur in regions where separate bulks of mass appear in close proximity. There are at least three such regions per subunit. The pairs of domains involved are: 2‐4a′ (4a from the adjacent rhomboid structure), 5‐6 and 10‐8a (indicated by circles in Fig. 2a,Fig. 2d).
Figure 3.
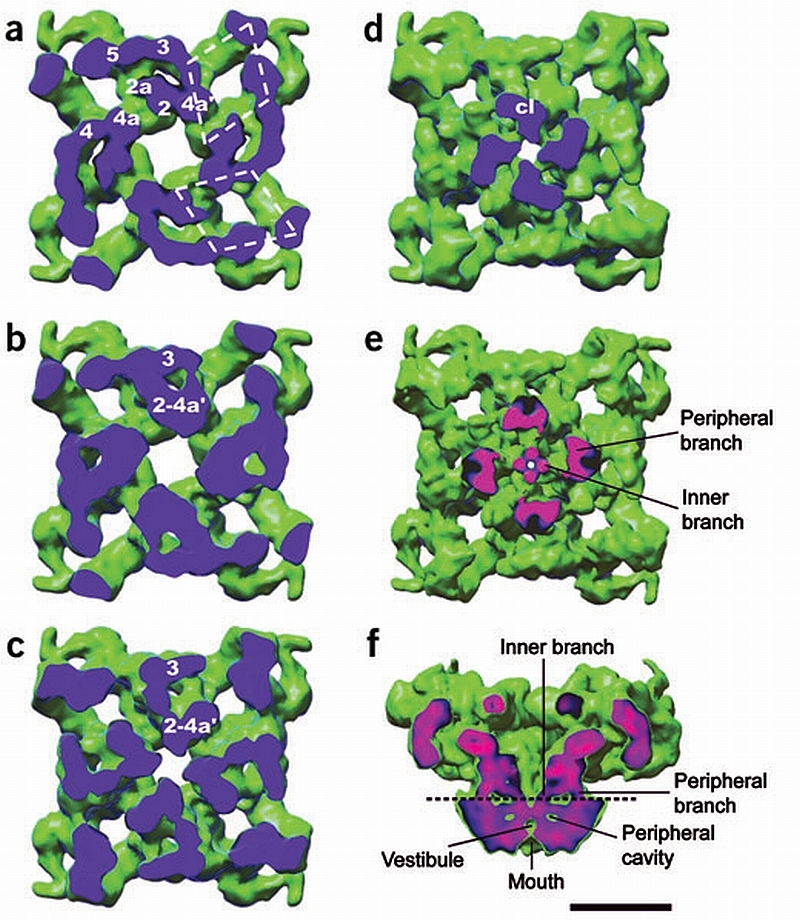
Internal structure of the cytoplasmic assembly and the columns. (a–d) Successive sections of the cytoplasmic assembly seen from the SR face show internal mass arrangements and interdomain connections. The sectioning plane is blue. The cytoplasmic domains are interconnected as follows. Below the T‐tubule‐facing surface domains 2 and 4a′ (from adjacent rhomboids) fuse (a). At a lower level, domains 2a and 3 interconnect through a second bridge of density and merge with domains 2‐4a′ (b,c). Therefore each structure formed by the fusion of domains 2‐2a‐3‐4a′ becomes a column of density (d). The four columns of density (cl) in (d) merge into the transmembrane assembly. (e) Section of RyR1 cut along the plane indicated by the dashed line in f seen from the SR lumen, displayed at a slightly higher threshold to show the internal and peripheral regions of the columns. (f) Slice across the side view showing the two branches of each column. The plane of the cut in e,f is color‐coded, with pink corresponding to denser structures and blue to less dense structures. Scale bar, 10 nm.
Domain 3, the flat, slab‐shaped ‘handle’ domain, defines each side of the square cytoplasmic assembly. The crevice, which is bounded by domains 3, 4, 8a and 8, seems to define a tilted canyon that extends from the SR side to the T‐tubule side (Fig. 2c). This reconstruction has a new distinct domain (labeled as domain 11) that protrudes from the central part of the cytoplasmic assembly, on the SR‐proximal side.
With our increased resolution, a region of separation between the cytoplasmic and transmembrane assemblies can now be distinguished. Four discrete, thick columns ∼14 Å in height span the gap (Fig. 2c). These columns are formed from the SR‐proximal part of the cyto plasmic assembly (Fig. 3a–Fig. 3d). The columns are not of uniform density, but rather a peripheral branch and an internal branch of higher density are apparent (see Fig. 3e,Fig. 3f).
Transmembrane assembly substructure
The four peripheral branches of the columns merge, defining the external boundaries of the transmembrane assembly prism structure (Fig. 3e and Fig. 4a). The four inner branches, shaped as thin bent rods, merge into a ring of high density inside the transmembrane assembly, on the cytoplasmic side (Fig. 4a). Directly underneath the ring, another set of four high‐density rods emerges toward the luminal side forming a tepee structure (Fig. 4a). These, and other dense structures visible on the side view slices of the transmembrane assembly, define a central cavity in the lumenal part of the transmembrane assembly (vestibule) and four peripheral cavities (Fig. 3f and Fig. 4c). The mouth of the presumed ion channel is separated from the vestibule by a constricted region (Fig. 3f).
Figure 4.
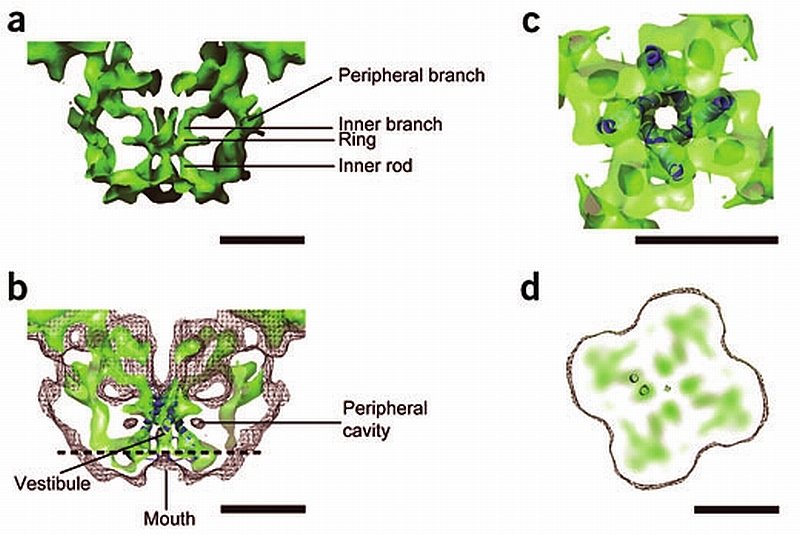
Substructure of the transmembrane assembly. (a) Central slice of the side view of the transmembrane assembly shown at high threshold to display the rod‐like structures of high density. The two sets of four rods of high density are connected to the ring of high density. (b) Fitting of the inner helices of the K+ channel atomic structure into the central four rods of high density of RyR1. The surface representation is in semitransparent color. A mesh with the surface of RyR1 at a threshold corresponding to RyR1’s molecular mass shows the fitting of the vestibule with the central cavity of the K+ channel. (c) A section of the transmembrane assembly after a 90° rotation shown at the same threshold as in b shows the fitting of the high‐density ring of RyR1 with KcsA’s gate. (d) Slice of the transmembrane assembly of RyR1 perpendicular to the four‐fold symmetry axis with discrete high‐density regions. The position of the slice is indicated by the horizontal dashed line in c. The separation between adjacent high‐density regions is compatible with neighboring α‐helices as shown here with the inner and outer helices of a subunit of the K+ channel in dark blue. Scale bars, 5 nm.
An analysis of a RyR1 three‐dimensional reconstruction from the same data set in which the volume was limited to a region comprising the transmembrane assembly, excluding most of the cytoplasmic periphery, yielded a resolution of 9 Å (FSC cutoff at 0.15) and 11.7 Å (FSC cutoff at 0.5). The improved resolution of structural details in this region is probably due to a higher degree of structural rigidity. Although in general it is estimated that a resolution of 7 Å is required before it is possible to distinguish α‐helices26, visualization of α‐helices has been reported at a resolution of 9 Å, for example in the case of the inner helices of the acetylcholine receptor30. This suggests that, considering the structural details seen here, the resolution measured at the lower FSC cutoff is more appropriate.
Structural homology with the K+ channel
We have docked the atomic structure of a closed K+ channel, KcsA21, into our three‐dimensional map of the transmembrane assembly of RyR1 (see Methods). There is notable architectural compatibility between part of the KcsA channel and part of the transmembrane assembly of RyR1 (Fig. 4b). The central rod‐shaped densities of RyR1’s transmembrane assembly, with their tepee‐like arrangement, overlap with the inner helices of KcsA such that the selectivity filter of the K+ channel is oriented toward the lumen of the sarcoplasmic reticulum and the gate oriented toward the cytoplasm (Fig. 4b). The K+ channel inner helices span from the lumenal mouth to the other face of RyR1’s transmembrane assembly. The central cavity of the K+ channel overlaps with RyR1’s vestibule, whereas the narrow ion selectivity filter in the K+ channel corresponds to the higher density region between RyR1’s vestibule and lumenal mouth. Closer to the cytoplasmic face, the inner rods of RyR1 merge into the ring‐shaped density, parallel to the plane of the membrane (Fig. 4a) in a position equivalent to the gate in the K+ channel. The dimensions of the high‐density ring of RyR1 are also similar to those of the KcsA gate (Fig. 4c). The set of four outer helices of the K+ channel fitted poorly with the corresponding region of our structure. Thus, for the docking shown in Figure 4b, Figure 4c, residues 23–71 corresponding to the outer helix of KcsA were deleted.
Other discrete regions of high density with diameter similar to that of the central rods are distinguishable in the slices parallel to the membrane (z slices) of the transmembrane assembly (Fig 4d). Although they too could correspond to cross‐sections of other α‐helices, they show less continuity than the central rod structures. We superposed a section of the inner and outer helices for one of the subunits of the KcsA atomic structure to two neighboring densities in our density map (Fig. 4d). The fact that the separation between centers is practically equivalent in both cases supports the possibility that the other discrete densities in the z slices of the transmembrane assembly three‐dimensional map actually correspond to neighboring individual α‐helices. The slightly elongated shape in some of these dense features could represent α‐helices in a tilted orientation. Although at our current level of resolution we cannot assert with certainty the number of transmembrane‐spanning domains in RyR1’s transmembrane assembly, from the examination of our density map we can predict that in the region of the transmembrane assembly between the mouth and the dense ring, the number is not less than six per subunit.
DISCUSSION
Comparison with previous reconstructions
The clamp domain, which forms each of the corners of the cytoplasmic assembly, can now be seen to be a distinct curved ribbon structure (domains 8a‐8‐7) that contacts an elongated self‐standing structure formed by domains 9‐10. The separation between domains 10 and 8a had not been observed before. In previous three‐dimensional reconstructions domain 10, at the corners of the structure, appeared connected on either side to both domains 8 and 9 (refs. 3,6–11), or connected to domain 8 and disconnected from domain 9 (ref. 8). Although this part of RyR1's structure is farthest away from the channel gate, this region shows conformational changes associated with channel opening8,9.
Domain 3 contains the binding site for FKBP12 (ref. 6). The higher resolution of this model indicates a more concave surface for this binding site, resembling the shape of a nest, and suggesting a more intricate interaction between RyR1 and FKBP12 (Fig. 2e). The hollow in domain 3, which is the binding site for apoCaM7, can be readily identified (Fig. 2e) and appears to be deeper than in the earlier three‐dimensional reconstruction. Ca2+‐CaM was found in the cleft between domains 3 and 7 on the SR‐facing side of the receptor6. This region shows a much more complex substructure (Fig. 2e) than was observed in the three‐dimensional reconstruction where this ligand was mapped. With the ability to identify the binding sites for CaM at higher resolution, the change of CaM position under different Ca2+ concentrations seems more likely to be due to a physical change of location than to a movement of the binding site.
Domain 11 and the four columns are features that were seen in a previous three‐dimensional reconstruction11, but not described in detail. The high‐density rods forming the inner branches of the columns and in the central region of the transmembrane assembly have never been described before. These structures have much higher density than the surrounding mass, which makes them apparent by a simple increase in threshold. This characteristic, together with the good agreement with the inner helices of the K+ channel structure, strongly suggests that these features correspond to true secondary structure.
Intraprotein signaling
The several points of intra‐ and/or intermonomer interconnection are a notable feature of RyR. On the outer surface, the rhomboid structures are continuous with the clamp and the handle domains (Fig. 2). In the interior, there is a fusion of domains 2 and 4a′ from adjacent ‘rhomboids’ and this mass is subsequently merged with domain 3. Each of these merged structures becomes a column of density (Fig. 3). This complex domain arrangement may facilitate the transmission of signals so that a signal received by one domain is, by design, transmitted to the other subunits, and facilitate allosteric cooperative responses31–34.
In addition to this domain interconnection, there are three regions where separate bulks of mass are in very close proximity. The pair of domains 10‐8a seems to be separated in our three‐dimensional reconstruction. Another region where separate domains in close proximity change their relative positioning upon gating is the pair of domains 5‐6. These domains have appeared to be either connected through a bridge of density, or separate, with domain 6 more stretched toward the T‐tubule8,9. There is a third region with similar structural characteristics, involving the pair of domains 2‐4a′. It is conceivable that regions with protein surfaces in very close proximity can support a scenario for RyR1's intraprotein regulation whereby changes in the cytoplasmic environment could promote, or inhibit, interdomain contacts, and in this manner endorse signaling‐related conformational changes such as have been suggested for the domain zipping and unzipping hypothesis35,36.
Altogether, it seems that RyR’s cytoplasmic structure, with its characteristic domain network, may facilitate the coordination of incoming signals from the surface sensors and provide a basis for the hierarchy of the different inputs. In addition, this scaffold‐like structure might provide the elasticity needed to allow a rapid return to a ‘basal’ state of energy.
For ligands or modulators that bind to the cytoplasmic portions of the receptor to cause the receptor to gate, all the information gathered by the cytoplasmic assembly must somehow be codified and transmitted to the gate. From our new data we would suggest that this transmission is through the four columns of density that merge into the transmembrane assembly. Thus, it seems that these columns constitute an important component in the orthograde RyR1 signaling between cytoplasmic and transmembrane assemblies (for example, depolarization‐induced Ca2+‐release and modulation by cytoplasmic factors). The angle between cytoplasmic and transmembrane assemblies has been shown to change conformation when the channel gates8,9. It seems likely that upon activation, the four columns undergo a twist that results in the observed rotation between cytoplasmic and transmembrane assemblies.
Gating of the ion pore
The good fitting of the central rod‐like densities of RyR1’s transmembrane assembly with the K+ channel inner helices presented here suggests that the RyR ion pore has a common architecture with that of the K+ channels. According to the model of the main functional aspects of RyR that are supported by the new features observed here (Fig. 5), the central high‐density rods of the transmembrane assembly represent the inner helices of the RyR ion channel, the dense region under the lumenal mouth represents the selectivity filter, and the high‐density ring represents the ion gate. Although the possibility of an overall organization of RyR’s pore similar to the K+ channel has been proposed before17,20, this is the first time that rod‐like densities suggestive of α‐helices have been directly seen within RyR1 allowing overlay of the atomic model of the K+ channel with RyR1.
Figure 5.
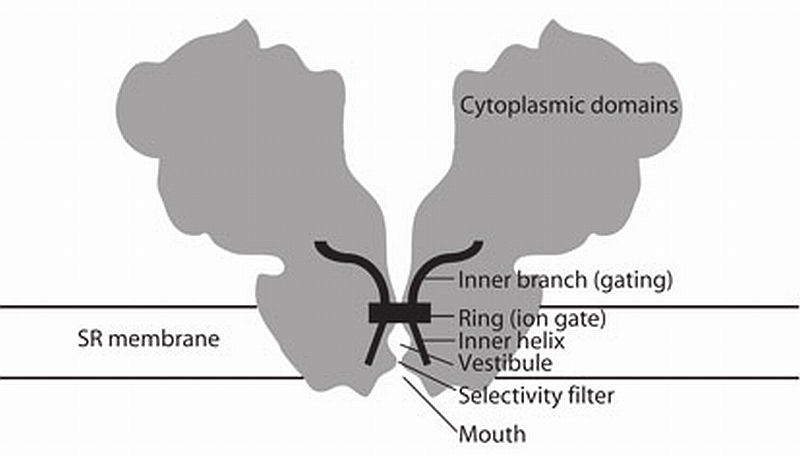
Schematic of the architecture of the RyR. A section across the RyR (gray) is overlaid with the higher‐density, secondary structure—like elements (black). The distinguishable secondary structure elements, and their hypothetical function, are indicated. According to the model proposed here, input from the cytoplasmic assembly would result in a conformational change of the inner branches of the columns. This conformational change would be transmitted directly to the pore inner helices and effectively change the diameter of the gate. An outward pulling of the inner helices in the ring region would open a central path for Ca2+ ions to flow through.
The emerging model has the four transmembrane inner helices, one contributed by each subunit, forming a tepee structure that frames the ion pore. The inner helices are splayed out surrounding a central cavity (vestibule) that is separated from the mouth by a constricted tunnel, presumably formed by one short pore helix from each subunit, and lined by the Ca2+ selectivity filter. On the cytoplasmic side of the transmembrane assembly the inner helices converge into a ring that makes up the ion gate. In RyR, the putative gate is facing the cytoplasm, and the selectivity filter is facing the sarcoplasmic reticulum lumen.
The inner branches are also directly connected to the ion gate. The resulting continuity between these outwardly bending inner branches and the inner helices also has a parallel in the MthK K+ channel. Studies on C‐terminal fragments downstream from the inner helices of the MthK calcium‐gated potassium channel have suggested a model for ligand‐gated channel opening. In this model, conformational changes in the RCK domains outside the transmembrane domain caused by Ca2+ binding are directly coupled to movement of the inner helices that in turn controls the pore opening37. In the RyR, the axial continuity of the inner helices and the inner branches found here (diagram shown to scale in Fig. 5) allows for a simple means to change the diameter of the central constriction that shapes the ion gate. This would provide a mechanism to control channel conductivity as the result of a change in conformation of the large remaining cytoplasmic domain.
METHODS
Purification of RyR1
RyR1 was isolated from rabbit skeletal muscle as described38 with some modifications. Briefly, the solubilized sarcoplasmic reticulum vesicles were applied to a Sephacryl S‐300 HR chromatography column (Amersham Biosciences) and the RyR1 peak was further purified on a 5–20% (w/v) sucrose gradient. The RyR1 peak from the sucrose gradients were concentrated on a HiTrap Heparin HP column (Amersham Biosciences), and eluted with 20 mM Na‐MOPS pH 7.4, 0.9 M NaCl, 0.5% (w/v) CHAPS, 2 mM DTT, 2 mM EGTA, 5 µg ml−1 aprotinin, 5 µg ml−1 leupeptin, and 2.5 µg ml−1 pefabloc. Purity of the RyR1 fraction was assessed by SDS‐PAGE electrophoresis and specific activity was determined by [³H]ryanodine binding. DTT was purchased from Calbiochem. All other chemicals with the highest available purity grade were purchased from either Fisher or Sigma‐Aldrich.
Cryo‐EM and image processing
To prepare grids for cryo‐EM, a 5 µl aliquot of a 2–4 mg ml−1 solution of RyR1 was adsorbed to a glow‐discharged quantifoil holey grid, and the excess of buffer blotted off with Whatman 540 filter paper. The sample was vitrified by plunging the grid into liquid ethane at −180°C using a custom‐made cryo‐plunging apparatus. Cryo‐EM was carried out on a FEI Tecnai F20 field emission gun microscope operated at an accelerating voltage of 200 kV. The grid was transferred to the electron microscope using an Oxford cryotransfer holder. Images were recorded onto Kodak SO‐163 film under standard low‐dose procedures at a nominal magnification of either 50,000X or 62,000X. A range of 2.5–4 µm underfocus was used. Micrographs (818) were digitized on a Zeiss SCAI scanner at a step size of 7 µm, and subsequently images were binned down, with a final pixel size of 2.8 Å. A total of 36,454 RyR1 particles were selected interactively using WEB39. Defocus parameters for every particle were calculated using CTFTILT40, and images were submitted to a projection‐matching refinement scheme starting from an initial three‐dimensional model of RyR1 (ref. 7) that was filtered to a resolution of 40 Å to avoid bias. Rotation and shift parameters were refined until they stabilized. At that point, and to minimize the possibility that the alignment process fell into a local minimum, the volume was low‐pass filtered to 20 Å and several loops of search at a large angular step were carried out, followed by several cycles of refinement until the shifts and rotations stabilized again. To obtain the final reconstruction, this series of iterations was repeated once more. FREALIGN41 and SPIDER39 were used. A threshold of cross‐correlation was set to include only the 70% best‐correlating particles into the final three‐dimensional reconstruction. Resolution values of 10.3 and 13.6 Å were estimated from the FSC at a 0.15 threshold22 and at a 0.5 threshold23, respectively. The three‐dimensional structure was filtered to 10.3 Å using a B‐factor of −500 Ų. Except where indicated otherwise, the three‐dimensional map was contoured according to the molecular mass of RyR assuming a protein density of 1.22 g cm−3 (ref. 42). Image rendering and manual docking with the atomic coordinates of the K+ channel was carried out with Chimera43 ( http://www.cgl.ucsf.edu/chimera). Manual docking was carried out taking the distinguishable features into account: the tepee arrangement of the high‐density rods, the central cavity, the high‐density ring and the mouth. To fit the two models along the four‐fold axis, the selectivity filter of the KcsA was fitted between the central cavity and the depression of the mouth. In this position, the diameter of the gate of KcsA accounts for the diameter of the RyR1 high‐density ring, and the center of the cavities of RyR1 and KcsA are in equivalent position. The tepee‐like organization of the high‐density rods of RyR1 and the inner helices of KcsA facilitated the fitting around the four‐fold axis. The optimal fitting between both structures as a whole is also the best fitting for each feature (high density rods, cavity, ring and mouth) considered independently.
ACKNOWLEDGMENTS
We thank T. Walz for his generous sharing of the microscopy and image processing facilities at the Department of Cell Biology at Harvard Medical School, and for helpful discussions. We also acknowledge N. Grigorieff for allowing the use of CTFTILT and FREALIGN software, and the Resource for Visualization of Biological Complexity (US National Institutes of Health (NIH) RR01219) of Wadsworth Center for the use of the cryo‐plunger apparatus. We thank Y. Cheng for his helpful discussions, advice with data analysis and technical support with the electron microscope, J. Frank for helpful discussions and X. Shen and J. Berkowitz for help with the RyR1 purification. This work was supported by RO1 AR43140 and PO1 AR17605 (to P.D.A.), AHA 0530147N (to M.S.), and AR40615 (to T.W.). The molecular EM facility at Harvard Medical School was established by a generous donation from the Giovanni Armenise Harvard Center for Structural Biology and is maintained by funds from NIH‐GM62580 (to T. Walz).
Footnotes
COMPETING INTERESTS STATEMENT The authors declare that they have no competing financial interests.
References
- 1.Fill M, Copello JA. Ryanodine receptor calcium release channels. Physiol. Rev. 2002;82:893–922. doi: 10.1152/physrev.00013.2002. [DOI] [PubMed] [Google Scholar]
- 2.Dulhunty AF, Pouliquin P. What we don’t know about the structure of ryanodine receptor calcium release channels. Clin. Exp. Pharmacol. Physiol. 2003;30:713–723. doi: 10.1046/j.1440-1681.2003.03904.x. [DOI] [PubMed] [Google Scholar]
- 3.Wagenknecht T, Samsoó M. Three‐dimensional reconstruction of ryanodine receptors. Front. Biosci. 2002;7:d1464–d1474. doi: 10.2741/A853. [DOI] [PubMed] [Google Scholar]
- 4.Wolf M, Eberhart A, Glossmann H, Striessnig J, Grigorieff N. Visualization of the domain structure of an L‐type Ca2+ channel using electron cryo‐microscopy. J. Mol. Biol. 2003;332:171–182. doi: 10.1016/s0022-2836(03)00899-4. [DOI] [PubMed] [Google Scholar]
- 5.Paolini C, Protasi F, Franzini‐Armstrong C. The relative position of RyR feet and DHPR tetrads in skeletal muscle. J. Mol. Biol. 2004;342:145–153. doi: 10.1016/j.jmb.2004.07.035. [DOI] [PubMed] [Google Scholar]
- 6.Wagenknecht T, et al. Locations of calmodulin and FK506‐binding protein on the three‐dimensional architecture of the skeletal muscle ryanodine receptor. J. Biol. Chem. 1997;272:32463–32471. doi: 10.1074/jbc.272.51.32463. [DOI] [PubMed] [Google Scholar]
- 7.Samsó M, Wagenknecht T. Apocalmodulin and Ca2+‐calmodulin bind to neighboring locations on the ryanodine receptor. J. Biol. Chem. 2002;277:1349–1353. doi: 10.1074/jbc.M109196200. [DOI] [PubMed] [Google Scholar]
- 8.Orlova EV, Serysheva II, van Heel M, Hamilton SL, Chiu W. Two structural configurations of the skeletal muscle calcium release channel. Nat. Struct. Biol. 1996;3:547–552. doi: 10.1038/nsb0696-547. [DOI] [PubMed] [Google Scholar]
- 9.Sharma MR, Jeyakumar LH, Fleischer S, Wagenknecht T. Three‐dimensional structure of ryanodine receptor isoform three in two conformational states as visualized by cryo‐electron microscopy. J. Biol. Chem. 2000;275:9485–9491. doi: 10.1074/jbc.275.13.9485. [DOI] [PubMed] [Google Scholar]
- 10.Radermacher M, et al. Cryo‐electron microscopy and three‐dimensional reconstruction of the calcium release channel/ryanodine receptor from skeletal muscle. J. Cell Biol. 1994;127:411–423. doi: 10.1083/jcb.127.2.411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Serysheva II, Hamilton SL, Chiu W, Ludtke SJ. Structure of Ca2+ release channel at 14 Å resolution. J. Mol. Biol. 2005;345:427–431. doi: 10.1016/j.jmb.2004.10.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Takeshima H, et al. Primary structure and expression from complementary DNA of skeletal muscle ryanodine receptor. Nature. 1989;339:439–445. doi: 10.1038/339439a0. [DOI] [PubMed] [Google Scholar]
- 13.Zorzato F, et al. Molecular cloning of cDNA encoding human and rabbit forms of the Ca2+ release channel (ryanodine receptor) of skeletal muscle sarcoplasmic reticulum. J. Biol. Chem. 1990;265:2244–2256. [PubMed] [Google Scholar]
- 14.Grunwald R, Meissner G. Lumenal sites and C terminus accessibility of the skeletal muscle calcium release channel (ryanodine receptor) J. Biol. Chem. 1995;270:11338–11347. doi: 10.1074/jbc.270.19.11338. [DOI] [PubMed] [Google Scholar]
- 15.Balshaw D, Gao L, Meissner G. Luminal loop of the ryanodine receptor: a poreforming segment? Proc. Natl. Acad. Sci. 1999;96:3345–3347. doi: 10.1073/pnas.96.7.3345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhao M, et al. Molecular identification of the ryanodine receptor pore‐forming segment. J. Biol. Chem. 1999;274:25971–25974. doi: 10.1074/jbc.274.37.25971. [DOI] [PubMed] [Google Scholar]
- 17.Welch W, Rheault S, West DJ, Williams AJ. A model of the putative pore region of the cardiac ryanodine receptor channel. Biophys. J. 2004;87:2335–2351. doi: 10.1529/biophysj.104.044180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Du GG, Sandhu B, Khanna VK, Guo XH, MacLennan DH. Topology of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum (RyR1) Proc. Natl. Acad. Sci. USA. 2002;99:16725–16730. doi: 10.1073/pnas.012688999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang R, et al. The predicted TM10 transmembrane sequence of the cardiac Ca2+ release channel (ryanodine receptor) is crucial for channel activation and gating. J. Biol. Chem. 2004;279:3635–3642. doi: 10.1074/jbc.M311367200. [DOI] [PubMed] [Google Scholar]
- 20.Williams AJ, West DJ, Sitsapesan R. Light at the end of the Ca2+‐release channel tunnel: structures and mechanisms involved in ion translocation in ryanodine receptor channels. Q. Rev. Biophys. 2001;34:61–104. doi: 10.1017/s0033583501003675. [DOI] [PubMed] [Google Scholar]
- 21.Doyle DA, et al. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 22.Rosenthal PB, Henderson R. Optimal determination of particle orientation, absolute hand, and contrast loss in single‐particle electron cryomicroscopy. J. Mol. Biol. 2003;333:721–745. doi: 10.1016/j.jmb.2003.07.013. [DOI] [PubMed] [Google Scholar]
- 23.Malhotra A, et al. Escherichia coli 70 S ribosome at 15 Å resolution by cryo‐electron microscopy: localization of fMet‐tRNAfMet and fitting of L1 protein. J. Mol. Biol. 1998;280:103–116. doi: 10.1006/jmbi.1998.1859. [DOI] [PubMed] [Google Scholar]
- 24.Samsó M, Trujillo R, Gurrola GB, Valdivia HH, Wagenknecht T. Three‐dimensional location of the imperatoxin A binding site on the ryanodine receptor. J. Cell Biol. 1999;146:493–499. doi: 10.1083/jcb.146.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jiang W, Baker ML, Ludtke SJ, Chiu W. Bridging the information gap: computational tools for intermediate resolution structure interpretation. J. Mol. Biol. 2001;308:1033–1044. doi: 10.1006/jmbi.2001.4633. [DOI] [PubMed] [Google Scholar]
- 26.Fujiyoshi Y. The structural study of membrane proteins by electron crystallography. Adv. Biophys. 1998;35:25–80. doi: 10.1016/s0065-227x(98)90004-1. [DOI] [PubMed] [Google Scholar]
- 27.Block BA, Imagawa T, Campbell KP, Franzini‐Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. J. Cell Biol. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagenknecht T, Hsieh CE, Rath BK, Fleischer S, Marko M. Electron tomography of frozen‐hydrated isolated triad junctions. Biophys. J. 2002;83:2491–2501. doi: 10.1016/S0006-3495(02)75260-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Meissner G. Ryanodine activation and inhibition of the Ca2+ release channel of sarcoplasmic reticulum. J. Biol. Chem. 1986;261:6300–6306. [PubMed] [Google Scholar]
- 30.Unwin N. Nicotinic acetylcholine receptor at 9 Å resolution. J. Mol. Biol. 1993;229:1101–1124. doi: 10.1006/jmbi.1993.1107. [DOI] [PubMed] [Google Scholar]
- 31.Pessah IN, Zimanyi I. Characterization of multiple [³H]ryanodine binding sites on the Ca2+ release channel of sarcoplasmic reticulum from skeletal and cardiac muscle: evidence for a sequential mechanism in ryanodine action. Mol. Pharmacol. 1991;39:679–689. [PubMed] [Google Scholar]
- 32.Welch W, et al. Structural components of ryanodine responsible for modulation of sarcoplasmic reticulum calcium channel function. Biochemistry. 1997;36:2939–2950. doi: 10.1021/bi9623901. [DOI] [PubMed] [Google Scholar]
- 33.Carroll S, Skarmeta JG, Yu X, Collins KD, Inesi G. Interdependence of ryanodine binding, oligomeric receptor interactions, and Ca2+ release regulation in junctional sarcoplasmic reticulum. Arch. Biochem. Biophys. 1991;290:239–247. doi: 10.1016/0003-9861(91)90615-p. [DOI] [PubMed] [Google Scholar]
- 34.Fessenden JD, et al. Ryanodine receptor point mutant E4032A reveals an allosteric interaction with ryanodine. Proc. Natl. Acad. Sci. USA. 2001;98:2865–2870. doi: 10.1073/pnas.041608898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kobayashi S, Yamamoto T, Parness J, Ikemoto N. Antibody probe study of Ca2+ channel regulation by interdomain interaction within the ryanodine receptor. Biochem. J. 2004;380:561–569. doi: 10.1042/BJ20040112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shtifman A, et al. Interdomain interactions within ryanodine receptors regulate Ca2+ spark frequency in skeletal muscle. J. Gen. Physiol. 2002;119:15–32. doi: 10.1085/jgp.119.1.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jiang Y, et al. Crystal structure and mechanism of a calcium‐gated potassium channel. Nature. 2002;417:515–522. doi: 10.1038/417515a. [DOI] [PubMed] [Google Scholar]
- 38.Wagenknecht T, et al. Cryoelectron microscopy resolves FK506‐binding protein sites on the skeletal muscle ryanodine receptor. Biophys. J. 1996;70:1709–1715. doi: 10.1016/S0006-3495(96)79733-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank J, et al. SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fields. J. Struct. Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- 40.Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J. Struct. Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- 41.Grigorieff N. Three‐dimensional structure of bovine NADH:ubiquinone oxidoreductase (complex I) at 22 Å in ice. J. Mol. Biol. 1998;277:1033–1046. doi: 10.1006/jmbi.1998.1668. [DOI] [PubMed] [Google Scholar]
- 42.Quillin ML, Matthews BW. Accurate calculation of the density of proteins. Acta Crystallogr. D. 2000;56:791–794. doi: 10.1107/s090744490000679x. [DOI] [PubMed] [Google Scholar]
- 43.Huang CC, Couch GS, Pettersen EF, Ferrin TE. Chimera: an extensive molecular modeling application constructed using standard components. Pac. Symp. Biocomput. 1996;1:724. [Google Scholar]