

Abstract
The folding of some proteins appears to be a two-state kinetic process. A two-state kinetic model is justified if protein molecules rapidly equilibrate between different unfolded conformations prior to complete folding. Here I show that this rapid equilibration is a natural consequence of reasonable assumptions about reaction rate constants and folding thermodynamics.
Folding Kinetics
The folding of some proteins appears to be a two-state kinetic process (1). The rate equation is
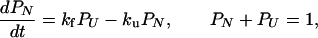 |
1 |
where PN is the fraction of protein in its native state N and PU is the fraction of protein in the unfolded state U. The folding rate is kf and the unfolding rate is ku.
How can a two-state kinetic model of protein folding ever be valid? We must be careful about what the word “state” means. It denotes a region of configuration space, usually the neighborhood of a potential minimum. The native state is associated with the deepest minimum. The “unfolded state” is the rest of configuration space. It is made up of a large number of distinct regions, each one associated with a local minimum or conformation of the polypeptide chain. (In a commonly used illustration, a protein of 100 amino acids may have of the order of 3100 conformations.) If the regions of configuration space are properly chosen, the protein remains in one state long enough to reach local equilibrium, and then jumps to another state. Thus folding appears to be intrinsically a many-state kinetic process, described by the more general rate equation
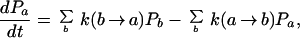 |
2 |
where Pa is the fraction of protein in a particular region or conformational state a. The native state is a = N, and the “unfolded state” is all a ≠ N. The first sum is the total gain in state a due to transitions from other states b, and the second sum is the total loss from state a due to transitions to other states b. The long time limit Pa(t → ∞) of the solution of these equations is the equilibrium fraction Pa(eq).
In justifying a kinetic two-state model, Creighton (2, 3) observed that “the experimental evidence … is largely consistent with the following general scheme: under folding conditions, unfolded protein molecules rapidly equilibrate between different conformations prior to complete refolding.” In this view, the “unfolded state” is actually an equilibrium distribution of many unfolded or partially folded conformational states. I show here that this rapid equilibration is a consequence of reasonable assumptions about rate constants and the thermodynamics of folding.
Rate Constants
Where do the rate constants come from? One source is transition state theory; this may be more appropriate for gas phase reactions than for protein folding in solution. Another more likely source is Kramers’ theory of Brownian motion over potential barriers; this is probably more appropriate for folding in solution.
In both transition state theory and Kramers’ theory, the rate constant k(a → b) has a special structure which leads to rapid equilibration of the unfolded states. It is determined by a quantity Ba,b that depends only on the boundary dividing the initial state a and the final state b, and by the partition function Qa of the initial state,
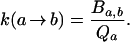 |
3 |
In quantum transition state theory, the boundary factor in Eq. 3 is
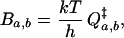 |
4 |
where 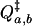 is the partition function of the “activated complex” or transition state. It is determined solely by the boundary between the regions of configuration space associated with the two states a and b. In the limit of small Planck constant, all factors of h cancel, and the classical transition state theory rate constant has a similar form, but with classical partition functions instead.
is the partition function of the “activated complex” or transition state. It is determined solely by the boundary between the regions of configuration space associated with the two states a and b. In the limit of small Planck constant, all factors of h cancel, and the classical transition state theory rate constant has a similar form, but with classical partition functions instead.
The transition rate in Kramers’ theory has the same general form. The standard example is Brownian motion over a one-dimensional potential barrier; however, the theory has been extended to more general multidimensional processes. Ref. 4 provides an excellent review of Kramers’ theory and the surprisingly deep relation to transition state theory that was discovered by Pollak (5). As in transition state theory, the boundary factor in Kramers’ theory is determined by the potential barrier or saddle point separating the regions of configuration space corresponding to the states a and b. In Kramers’ theory, the time scale is set by a friction coefficient, which is usually attributed to the viscosity of the surrounding medium. In a study of the dynamics of conformational changes in myoglobin, Ansari et al. (6) found that Kramers’ theory worked quite well, but both solvent viscosity and internal friction are needed.
The transition rates that are widely used in Metropolis Monte Carlo simulations of lattice models of protein folding have a structure that is different from Eq. 3. The partition functions of both the initial and final states are involved. If the free energy of the final state b is lower than that of the initial state a, or Qb > Qa, the rate from a to b is k1 and is the same for all initial states. Otherwise the rate from a to b is k1Qb/Qa. The Metropolis Monte Carlo rules were designed for convenience and for a computationally fast approach to equilibrium, but they have no theoretical basis, and they may not be an accurate representation of actual kinetics.
In both transition state theory and Kramers’ theory, the boundary factor is the same whether the protein moves from a to b or from b to a,
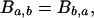 |
5 |
The reaction rate constants must satisfy the principle of detailed balance; at equilibrium, forward and backward rates must be equal,
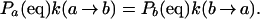 |
6 |
This condition is automatically satisfied, because Pa(eq) is proportional to the partition function Qa that appears in the denominator of k(a → b).
The rate constants in transition state theory and Kramers’ theory depend on boundary factors and on partition functions for the various conformational states. In a folding transition, the partition functions change substantially as the folding conditions are varied slightly. I assume that the boundary factors, associated with barriers, are much less sensitive to folding conditions. Further, I assume that there are no dramatic differences between barriers which might lead to trapping in special unfolded states. (An example of trapping is the slow cis-trans isomerization of proline peptide bonds.) Then the variation of the rate constants is determined mainly by folding thermodynamics.
Folding Thermodynamics
Each individual state a has its own partition function Qa. The native state has the partition function QN. The purpose of the present section is to show that under folding conditions, Qa for any single unfolded state is much smaller than QN.
The total partition function Qtotal is the sum over all states,
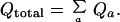 |
7 |
The equilibrium fraction of protein in the single state a is
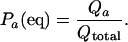 |
8 |
In particular, the fraction of protein in the native state a = N is
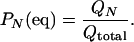 |
9 |
The folding transition is produced by a change in folding conditions, for example in temperature or in the concentration of a denaturant. If the folding conditions favor a completely unfolded protein, all of the partition functions Qa are roughly the same size and all of the rate constants are comparable.
Suppose that experimental conditions are changed to favor folding, so that the equilibrium fraction of native protein varies from small, say PN(eq) ≈ 0.01, to large, PN(eq) ≈ 1. Then in this range of folding conditions, the partition function of the native state is a substantial fraction of the total partition function, QN/Qtotal > 0.01. The remaining partition function of all the unfolded states,
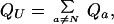 |
10 |
is always smaller than Qtotal, and the ratio QU/QN is always smaller than 100. The ratio of the partition function Qa of a single unfolded state to the partition function QN of the native state is limited by
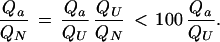 |
11 |
If there are a very large number of unfolded states, each one can make only a small contribution to QU, and the ratio Qa/QU is expected to be very much smaller than 1/100. Under folding conditions, any individual Qa is much smaller than QN.
Rapid Equilibration
The rate constants, from either transition state theory or Kramers’ theory, are inversely proportional to partition functions. Then under folding conditions, the rate constant for escape from any single unfolded state is much larger than the rate constant for escape from the native state. However, the connectivity of the transition rate matrix must also be taken into account. Each state corresponds to a region of configuration space. In transition state theory or Kramers’ theory, transitions can occur only if the two regions have a common boundary; then they are “connected.” If conformational transitions are due to local rearrangements, then any single unfolded state in a protein with n amino acids can probably connect to the order of n other states. The actual number of connections may be larger because of the possibility of more global motions—for example, the diffusion of two segments of the chain relative to each other. The native state connects to a number of “gateway” unfolded states. If the protein is in a gateway state, it can make transitions to the single native state or to many other unfolded states, all with comparable rates. Then transitions between unfolded states are statistically more likely than transitions into the native state. This observation, along with the earlier estimate of the relative order of magnitude of rate constants, leads to the conclusion that folding kinetics involves two distinctly different time scales.
Another way to make transitions to other unfolded states more likely than transitions to the native state is to impose a free energy barrier between unfolded and native states. However, if one wants to use a free energy barrier, then it must be a function of some coordinate, a measure of the distance from an unfolded state to the native state. For a barrier to be useful in kinetics, the coordinate must vary slowly with time, and local equilibrium with respect to that coordinate must be reached rapidly. It is hard to verify that any coordinate except the occupancy of the native state satisfies these requirements.
According to experiment (7), the fast time scale extends from microseconds to milliseconds, and the slow time scale may require seconds or minutes. In the fast time scale, the unfolded protein moves rapidly between unfolded or partially folded conformational states. After a short time these states come to local thermodynamic equilibrium, and all details about the initial state and the sequence of transitions (the “folding mechanism”) are forgotten. The fraction of folded protein varies on the slow time scale. This is precisely the scheme proposed by Creighton.
Two-State Kinetics
Now it is easy to show explicitly how the many-state rate equation reduces to the two-state equation. Transitions between unfolded states are fast, and the ensemble of unfolded states relaxes rapidly to local thermodynamic equilibrium. However, after this fast relaxation the total fraction of unfolded states will still change, as the fraction in the native state changes. This can be handled by a time-dependent normalization coefficient c(t), so that after the fast relaxation, the time-dependent Pa(t) is proportional to the equilibrium Pa(eq),
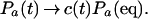 |
12 |
The coefficient is determined by a normalization condition,
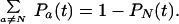 |
13 |
Then, after the fast relaxation, the fraction in any unfolded state is approximately
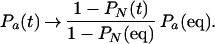 |
14 |
This is the approximation that leads to two-state kinetics. When it is inserted into the rate equation for the native state,
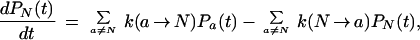 |
15 |
the result is
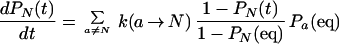 |
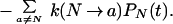 |
16 |
The first term can be simplified by using the detailed balance condition
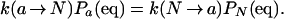 |
17 |
Then Eq. 15 becomes
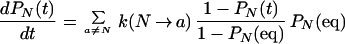 |
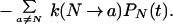 |
18 |
This has exactly the structure of the two-state kinetic model,
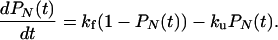 |
19 |
The unfolding rate is the sum of all transition rates from the native state to all gateway states,
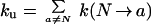 |
20 |
and the folding rate is
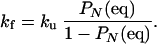 |
21 |
Summary
This qualitative justification of a two-state kinetic model was based on a number of assumptions. (i) The statistical thermodynamics of the folding transition is well described by a single folded state and a large ensemble of unfolded states. (ii) Any individual unfolded state makes a very small contribution to the total partition function of unfolded states. (iii) Transition rate constants are inversely proportional to the partition functions of single conformational states, and unfolded states make transitions to many other unfolded states. (iv) The boundary factors in rate constants are insensitive to changes in folding conditions. (v) There are no exceptional barriers between particular unfolded states that might lead to trapping for long periods of time. If a particular protein meets these requirements, then one should expect that its folding kinetics is well described by a two-state model.
Acknowledgments
I thank T. E. Creighton, W. A. Eaton, A. Szabo, and P. G. Wolynes for helpful comments.
References
- 1.Jackson S E, Fersht A R. Biochemistry. 1991;30:10428–10435. doi: 10.1021/bi00107a010. [DOI] [PubMed] [Google Scholar]
- 2.Creighton T E. Proc Natl Acad Sci USA. 1988;85:5082–5086. doi: 10.1073/pnas.85.14.5082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Creighton T E. In: Mechanisms of Protein Folding. Pain R H, editor. Oxford: Oxford Univ. Press; 1994. pp. 1–25. [Google Scholar]
- 4.Hängii P, Talkner P, Borkovec M. Rev Mod Phys. 1990;62:251–341. [Google Scholar]
- 5.Pollak E. J Chem Phys. 1986;85:865–867. [Google Scholar]
- 6.Ansari A, Jones C M, Henry E R, Hofrichter J, Eaton W A. Science. 1992;256:1796–1798. doi: 10.1126/science.1615323. [DOI] [PubMed] [Google Scholar]
- 7.Eaton W A, Thompson P A, Chan C-K, Hagen S J, Hofrichter J. Structure. 1996;4:1133–1139. doi: 10.1016/s0969-2126(96)00121-9. [DOI] [PubMed] [Google Scholar]