

Abstract
A method is described that allows the sequence-specific ligation of DNA. The method is based on the ability of RecA protein from Escherichia coli to selectively pair oligonucleotides to their homologous sequences at the ends of fragments of duplex DNA. These three-stranded complexes were protected from the action of DNA polymerase. When treated with DNA polymerase, unprotected duplex fragments were converted to fragments with blunt ends, whereas protected fragments retained their cohesive ends. By using conditions that greatly favored ligation of cohesive ends, a second DNA fragment could be selectively ligated to a previously protected fragment of DNA. When this second DNA was a vector, selected fragments were preferentially cloned. The method had sufficient power to be used for the isolation of single-copy genes directly from yeast or human genomic DNA, and potentially could allow the isolation of much longer fragments with greater fidelity than obtainable by using PCR.
In 1991, we described RecA-assisted restriction endonuclease (RARE) cleavage (1). The method was a general and efficient way to target restriction enzyme cleavage to unique, predetermined sites. The method utilized the ability of RecA protein to pair oligonucleotides to homologous sequences in duplex DNA to form three-stranded complexes. These complexes protected the selected sites from enzymatic methylation, and after dissociation of the complexes, restriction enzyme cleavage was limited to the selected unmethylated sites. We later improved the method to cleave fragments greater than a megabase in size (2), and it has been used by numerous investigators to map and manipulate large segments of DNA (3–18). We have now developed a complementary method that is functionally the reverse of RARE cleavage, in that the method uses RecA protein and oligonucleotides to direct the sequence-specific ligation of DNA. It represents the first report of targeting of the action of DNA ligase. When one of the DNA segments is a vector, it is possible to perform sequence-specific cloning of a selected genomic DNA segment. When used in this manner, we refer to the technique as RecA-assisted cloning (RAC).
Several methods are available to amplify genomic DNA and to isolate selected fragments in a pure form. The most widely used method is PCR. A major limitation of PCR is the small size that may be reliably amplified, although improvements have allowed amplification of up to 20–30 kb by using human genomic DNA as the template (19–21). The other widely used general method to isolate genomic fragments involves constructing and screening DNA libraries. Libraries based on λ phage or cosmid vectors have long been in use, and several other vectors are now available for cloning large (>100 kb) segments of DNA in the form of yeast artificial chromosomes, bacterial artificial chromosomes, and P1 phage-derived artificial chromosomes. Such libraries, however, are tedious to construct and screen.
Several groups have described strategies to use RecA protein from E. coli to screen libraries or enrich a selected DNA fragment (22–27). These strategies utilized the ability of RecA protein to pair single-stranded DNA of any sequence to homologous target duplex DNA to create a three-stranded complex (28), or to pair two complementary single-strands to the target duplex DNA to create a four-stranded complex (25, 26). The use of a biotinylated single-strand and avidin or streptavidin allowed the physical purification of the target duplex. These strategies have never been applied to practical problems in molecular biology. In this report, we describe a general method by using RecA protein that allows one to clone almost any selected fragment directly from human genomic DNA.
MATERIALS AND METHODS
Preparation of DNA.
λ DNA was purchased from New England Biolabs. Saccharomyces cerevisiae DNA used as the source of the RAD51 gene was purified as described (29). Human DNA used as the source of the int-2 gene was purchased from Sigma (D 7011). The DNA had been isolated from multiple placentas, and before use it was extracted twice with phenol/chloroform and then three times with chloroform or diethyl ether alone, precipitated with ethanol in the presence of 0.3 M sodium acetate, and washed with 70% ethanol. The DNA was digested to completion with EcoRI and BamHI, and the extraction, precipitation, and washing steps were repeated. Yields of this step were typically about 60%. Digested DNA was loaded on a 0.8% SeaPlaque GTG agarose gels (FMC) in Tris/acetate/EDTA buffer (30). Multiple wells, each with an area of 9 × 7 mm, were loaded with 150–200 μg of DNA per well. The gels were run until the 1.4-kb fragment to be cloned had migrated 4–6 cm. The marker lane was then removed, stained with ethidium bromide, and used as a guide to excise 0.5 cm above and below the expected position of the 1.4-kb fragment. DNA was extracted from the excised gel by using GELase (Epicentre) according to the manufacturer’s directions; complete digestion was required for good yields. The final yield of DNA from the excised region was typically 2–4% of the starting material. Comparable yields were obtained with a silicagel extraction kit (Qiagen) or by electroelution (31). The size of the extracted DNA from the heavily overloaded gels was checked on analytical gels. Depending on the amount available, DNA was quantified by absorbance, fluorescence (32), or spotting in an ethidium bromide solution (30).
Oligonucleotides.
Oligonucleotides >30 bases in length were purified on acrylamide gels, and concentrations were determined by assuming that a 33 μg/ml solution has an absorbance at 260 nm of 1. For the experiment in Fig. 2, the sequence of the L oligonucleotide was 5′-gattatAGCTTTTCTAAT-TTAACCTTTGTCAGGTTTACCA-3′, and the R oligonucleotide was 5′-gattatAG-CTTTGTGTGCCACCCACTACGACCTGCATAA-3′. The lowercase letters were the sequences of nonhomologous tails, and the uppercase letters were the sequences of the portions homologous to the proximal (L) and distal (R) ends of the 2.3-kb λ DNA fragment. The short radioactive duplex ligated to the fragment had the structure:
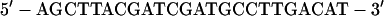 |
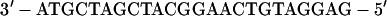 |
The HindIII cohesive end is at the left, and the bottom strand was labeled with [γ-32P]ATP by using polynucleotide kinase. The kinase was inactivated at 65°C for 10 min, and unreacted [γ-32P]ATP was removed by gel filtration (Chroma Spin+TE-10 columns, CLONTECH) before adding the top strand.
Figure 2.

Sequence-specific labeling of a fragment of λ DNA. (A) Schematic showing the position of the 2.3-kb λ DNA HindIII fragment labeled. The arrow points to the 2.3-kb fragment, and L and R show the positions of the two oligonucleotides used to direct sequence-specific ligation to a short radioactive duplex with a HindIII cohesive end. (B) Agarose gel stained with ethidium bromide showing the HindIII fragments of λ DNA in lanes 1–5 and a ScaI digest in lane 6. The two shortest HindIII fragments have run off the bottom of the gel. (C) Autoradiogram of the dried gel. Lane 1 shows λ DNA where KF was omitted and every fragment was labeled. The band at 4.4 kb is less intense because of ligation to the 23.1-kb band via the terminal λ cos sites. Lane 2 shows labeling of both ends of the 2.3-kb fragment. Lanes 3, 4, and 5 show the effect of omitting either one or both of the oligonucleotides. Lane 6 shows the results by using λ DNA fragments with blunt ends.
The 1.4-kb human int-2 gene fragment was cloned by using one oligonucleotide with a sequence identical to the genomic sequence from position 2290–2347, and a second oligonucleotide complementary to position 3621–3677. EcoRI and BamHI cleave 3′ of residues 2304 and 3660, respectively (33, 34). The 1.2-kb yeast RAD51 gene fragment was cloned by using oligonucleotides complementary to positions 1–48 and 1164–1204 (35).
Streptavidin beads were saturated with the following duplex that contained an EcoRI cohesive end:
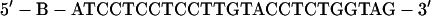 |
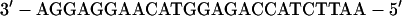 |
The upper oligonucleotide was synthesized with a biotin (B) group at the 5′ end by using the LC Biotin-ON phosphoramidite (CLONTECH).
Enzymes.
RecA protein was prepared as described (1) by using an overproducing strain provided by Barbara McGrath of the Brookhaven National Laboratory (Upton, NY). RecA protein purchased from Boehringer Mannheim gave equivalent results. Unless otherwise stated, all other enzymes were from New England Biolabs.
RAC Reaction Conditions.
Except where indicated, conditions and concentrations of the step using RecA protein to protect against the action of the polymerase were identical in all experiments. The components of the reaction are identical to those used in RARE cleavage (2), with the exception that the exonuclease-free Klenow fragment (KF) of E. coli DNA polymerase I (36) and the four deoxynucleoside triphosphates were used instead of a methylase and S-adenosylmethionine. Volumes and ligation conditions were varied depending on the goal of the experiment. In the experiment shown in Fig. 2, the RecA protein protection reaction volume was 100 μl and contained 25 mM Tris acetate, pH 7.85, 4 mM magnesium acetate, 0.4 mM DTT, 0.5 mM spermidine, 1.1 mM ADP, 0.3 mM ATP-γ-S (Fluka), 13 μg of RecA protein, 0.32 μg of oligonucleotide (lane 2 had 0.16 μg each of two oligonucleotides), 2.5 μg of a HindIII digest of λ DNA, and 40 μg of BSA (Sigma, A 7638). After 10 min at 37°C, 12.5 units of KF (United States Biochemical, 70057) and 38 μM each of dATP, dCTP, dGTP, and TTP were added and the reaction was allowed to proceed for 30 min at 37°C. The RecA protein and KF were then inactivated by extraction with phenol/chloroform (1:1), followed by extraction three times with diethyl ether, addition of sodium acetate to 0.3 M, and precipitation with ethanol. The DNA pellets were washed with 70% ethanol, dried, and used in the subsequent ligation reactions.
When cloning from human DNA, the reaction volume was 1.44 ml and contained 3.2 μg each of the two int-2 oligonucleotides, 360 μg of RecA protein, 26 μg of size-fractionated human DNA, 570 μg of BSA, and 450 units of KF. When cloning from yeast DNA, the two RAD51 oligonucleotides were used and the reaction was scaled down by a factor of 14.
Ligation Conditions.
Several commercially available ligases were tested for efficiency and specificity in the RAC procedure. We confirmed the observation that E. coli DNA ligase joins cohesive ends much more rapidly than blunt ends whereas T4 DNA ligase has reduced specificity (37, 38). In the Fig. 2 labeling experiment, the short radioactive duplex was present at a relatively high concentration so that E. coli ligase was able to function efficiently. Fragments with blunt ends were nonspecifically labeled when T4 ligase was used. For the cloning experiments such as in Fig. 3, only T4 ligase was commercially available at the concentration necessary for an acceptable ligation yield.
Figure 3.

Sequence-specific cloning of a portion of the human int-2 gene. (A) Agarose gel stained with ethidium bromide. Lane 1 shows λ BstEII size standards. Lane 2 shows 20 μg of human genomic DNA that had been digested with EcoRI and BamHI. Lanes 3 and 4 show 0.25 μg of pooled plasmid DNA prepared by using the RAC procedure. The DNA in lane 3 has been digested with EcoRI and BamHI. (B) Southern blot of the gel in A. A 0.6-kb portion of the int-2 gene was used as the probe. Arrows show the positions of both of the alleles of the int-2 fragment. The amount of radioactivity in each band was quantitated by using a BAS 2000 Fujix Bio-Imaging Analyzer. Specifically, using the software of the instrument, each band was centered in a rectangle and the total amount of signal within the rectangle was determined. A rectangle was also placed just above each band to measure the background signal. All rectangles were the same size and shape. In lane 2, the lower allele band gave a reading of 1,420 with a background of 721. In lane 4, the two bands from the plasmid had a combined reading of 15,711 with a background of 1,076. After subtracting the background signal, the ratio of the plasmid signal in lane 4 to the lower allele signal in lane 2 was 20. Because 80 times more DNA was loaded in lane 2 than in lane 4, the total enrichment was 1,600.
In the Fig. 2 experiment, the reaction (40 μl) contained 1.0 μg of the λ DNA fragments, 0.8 μg of the short radioactive duplex, 8 units of E. coli DNA ligase, and the buffer recommended by New England Biolabs without BSA. After ligation for 1 hr at room temperature, unligated radioactive duplex was removed by gel filtration (Chroma Spin+TE-400 columns, CLONTECH). Samples were heated to 65°C for 3 min and run on an agarose gel. Yields were calculated by comparison with the 2.3-kb band in lane 1 after a small correction for the decrease in intensity (3%) of the band because of ligation to other fragments. Quantitation of radioactivity in all experiments was done on a BAS 2000 Fujix Bio-Imaging Analyzer.
Ligation of Vector to the Human int-2 Gene Fragment.
Human DNA treated as above was used in the ligation scheme outlined in Fig. 4. Two milligrams of streptavidin beads (Dynabeads M-280, Dynal) were used according to the manufacturer’s directions. They were saturated with the short biotinylated duplex described above. After binding, excess duplex was removed by washing the beads with 1 M NaCl, 50 mM Tris chloride, pH 7.5, and then with T4 DNA ligase buffer from New England Biolabs. Vector was prepared by treating pBluescript SK(+) (Stratagene) with EcoRI, BamHI, and calf intestinal phosphatase. The small polylinker fragment liberated from the vector was removed by gel filtration with Chroma Spin+TE-400 columns. The ligation reaction contained the washed beads and 80 μl of T4 DNA ligase buffer with 2.6 μg of the human DNA, 3.4 μg of vector, and 3,200 units of T4 DNA ligase. After 16 hr at 16°C, unligated DNA and vector were removed by washing the beads. The lower oligonucleotide of the biotinylated duplex (0.3 μg) was added to replace any removed by washing. Immobilized fragment-vector DNA was removed by treatment with 80 units of EcoRI in 100 μl of its buffer. The solution containing the fragment-vector DNA was removed from the beads and extracted and precipitated in the presence of 20 μg of glycogen. To circularize the fragment-vector molecules, the DNA was treated with 1,600 units of T4 DNA ligase in 100 μl of its buffer for 16 hr at 16°C. The DNA was concentrated by ethanol precipitation and used to transform 50 μl of XL1-Blue MRF′ bacteria (Stratagene).
Figure 4.

Diagram of the construct made when selected fragment-vector molecules were separated from unligated vector. An explanation is given in the text. EcoRI and BamHI cohesive ends are shown. S, streptavidin; B, biotin; P, 5′ phosphate.
Generation and Analysis of Clones.
Preparation of bacteria and electroporation in 0.1-cm cuvettes a Gene Pulser apparatus (Bio-Rad) was done according the manufacturer’s directions. The efficiency of transformation was 8 × 108 colonies per μg of intact plasmid DNA by using standard 0.2-cm cuvettes. Cells were plated on Luria–Bertani (LB) agar containing ampicillin, tetracycline (to maintain the bacterial episome), isopropyl β-d-thiogalactoside, and X-gal (30). In the human DNA experiment described above, plasmid DNA was obtained by scraping and washing the resulting 7,000 white and 900 blue colonies from the plates with LB. Plasmid DNA was prepared by using a plasmid isolation kit (Qiagen) and extracted with cetyltrimethylammonium bromide to remove enzyme inhibitors (29). When this DNA was used as the substrate for a second round of RAC, the reaction volume was scaled down by a factor of 14, pBC SK(+) (Stratagene) was used as the vector, and clones were selected on chloramphenicol plates. This eliminated any background from the previous plasmid vector that contained an ampicillin resistance gene.
The plasmid DNA enriched for the int-2 gene was digested with EcoRI and BamHI and analyzed by the Southern blot shown in Fig. 3. The SS6 fragment containing 0.6 kb of the selected int-2 fragment was used as the probe (33, 34). The fraction of colonies containing selected fragments was calculated by determining the band intensity from such blots or from other blots prepared with known quantities of colony and pure probe DNA. In experiments where the fraction was high, individual colonies were analyzed by lysing the bacteria on membranes, spotting purified plasmids, or by blotting restriction digests of purified plasmids after agarose gel electrophoresis. Sequencing was performed on an Applied Biosystems model 373 sequencer with their PRISM DyeDeoxy Terminator Cycle Sequencing kit and the M13 −20 and reverse primers.
RESULTS
Outline of the Method.
The cloning strategy is outlined in Fig. 1. The first step was to select a particular restriction enzyme fragment of DNA to clone and to digest the DNA with that enzyme. The enzyme had to be one of the many available that produce 3′ recessed ends. Alternatively, two such enzymes could be used to clone a fragment created by a double digest. Two oligonucleotides, generally 30–60 bases in length, were synthesized such that each had complete homology to one of the ends of the selected fragment. The oligonucleotides and RecA protein were incubated with the duplex DNA and complexes formed at the ends of the selected fragment. KF and the four deoxynucleoside triphosphates were then added and allowed to fill in all available sites, sparing the ends protected by the oligonucleotide and RecA protein complexes. The complexes were dissociated and the KF was inactivated. Plasmid vector with the appropriate cohesive ends and DNA ligase were added, and the product was used to transform bacteria. Because DNA fragments with complementary cohesive ends were ligated to the vector much more readily than DNA fragments with blunt ends, the resulting clones were highly enriched for the selected fragment.
Figure 1.

Schematic of the strategy used for sequence-specific cloning of DNA. An explanation is given in the text.
In principle, any polymerase or exonuclease that acted on cohesive ends could have been used in this strategy. Exonuclease-free KF was used because it efficiently converted 3′ recessed ends to blunt ends, could be added in excess without degrading DNA, was blocked by RecA protein–oligonucleotide complexes, and was easily inactivated once the reaction was complete.
Demonstration of Sequence-Specific Ligation.
A simple demonstration of the method is shown in Fig. 2. This figure shows sequence-specific ligation of a short radioactive duplex with a cohesive end to a selected fragment of λ DNA. For reasons described in the Materials and Methods, E. coli DNA ligase was used in labeling experiments and T4 DNA ligase was used in cloning experiments. λ DNA was cleaved by the restriction enzyme HindIII into eight fragments, and we chose to ligate the short radioactive duplex to the 2.3-kb fragment shown in Fig. 2A. The agarose gel stained with ethidium bromide is shown in Fig. 2B, and the autoradiogram of the dried gel is shown in Fig. 2C. All of the fragments were labeled when the KF was omitted (Fig. 2C, lane 1). Efficient labeling of only the 2.3-kb band occurred when a complete experiment was performed (lane 2). In this lane, two oligonucleotides were used to protect both the left and right ends of the fragment with subsequent ligation of the short radioactive duplex to both ends. When only the left oligonucleotide (lane 3) or the right oligonucleotide (lane 4) was used to protect, each band was about half the intensity of the band in lane 2. No specific labeling was seen when no ends were protected (lane 5) or when the restriction enzyme used to fragment the starting DNA produced blunt ends (lane 6).
Efficiency, Specificity, and Oligonucleotide Parameters.
In the experiment shown in Fig. 2, the protection efficiency at each end of the 2.3-kb fragment was 90%. Nonspecific protection of other ends was detectable but less than 0.5%, and labeling of the DNA with blunt ends in lane 6 was not detected. Only 29 bases of sequence information at each end of the duplex were used in designing the oligonucleotides (33 bases if the 4-base, single-stranded tail produced by HindIII is counted). A series of nine oligonucleotides was synthesized to investigate the parameters that determine protection efficiency. Efficiency was the same when 41 bases of homology was used, but dropped to 76% with 19 bases, and to less than 1% with 10 bases. The oligonucleotides could have the same sequence as either strand at the end of the duplex without changing the efficiency. Addition of a tail that extended the oligonucleotide past the end of the fragment did not change the efficiency. These results were slightly more favorable than protection efficiencies with RARE cleavage (1) and probably reflected the increased stability of complexes formed at the end of duplexes (39) rather than in the middle as in RARE cleavage.
Cloning of a Single-Copy Human Gene.
To demonstrate RAC with genomic DNA, we chose to clone a 1.4-kb EcoRI-BamHI fragment of the human int-2 protooncogene. This gene has been mapped and sequenced, and one EcoRI site lies just upstream of exon 2, and in about half of the alleles, a BamHI site lies 1.4 kb downstream of the EcoRI site (33, 34). In the other half of the alleles, this BamHI site is not present, and the next BamHI site is 6.9 kb downstream of the EcoRI site. Fig. 3 shows the results of a cloning experiment. A gel stained with ethidium bromide (Fig. 3A) shows the starting genomic DNA after digestion with EcoRI and BamHI (lane 2), the similarly digested DNA from a pool of 7,000 colonies obtained by RAC using size-fractionated DNA as the starting material (lane 3), and the same cloned DNA without digestion (lane 4). In lane 3, only vector DNA is visible, although a faint smear of insert DNA centered around 1.4 kb was seen on another gel with twice as much DNA.
Yield and Analysis of the Cloned Products.
Quantification of the Southern DNA blot (Fig. 3B) revealed that the amount of the human int-2 fragment was 20 times greater in the cloned DNA lanes even though the genomic DNA lane contained 80 times more DNA than the cloned DNA lanes. Thus, a 1,600-fold enrichment of the fragment was obtained. Lane 2 also shows the genomic DNA 6.9-kb allele that was almost as intense as the 1.4-kb allele (44% versus 56% of the total int-2 signal, respectively). The other faint bands were not consistently seen and probably arose from binding of the int-2 probe to cross-hybridizing sequences. Overall, the result demonstrated cloning of a fragment present at only about one copy per diploid human genome. As with any amplification technique, contamination with a previously amplified fragment may lead to a misleading estimation of the amount of enrichment. Before the first successful RAC trial, the int-2 and yeast RAD51 fragment described later had never been in the lab in either a cloned or amplified form.
Multiple cloning trials were carried out on yeast and human DNA. With human DNA, the typical enrichment was 1,000- to 2,000-fold, and one int-2 clone was present for every 2,000–4,000 colonies. At least one int-2 clone was obtained for every 70 μg of starting genomic DNA. When the pooled DNA after one round of RAC (Fig. 3, lanes 3 and 4) was run through the procedure a second time, 24% of the colonies contained the int-2 fragment. This demonstrated an additional 500-fold enrichment and showed that incorrect clones arose mainly through a stochastic process, and not through a biased selection based on partial homology to the int-2 sequence. We also chose to clone a 1.2-kb EcoRI-BamHI yeast genomic DNA fragment containing the proximal portion of the RAD51 gene (35). Three percent of the clones contained the RAD51 fragment, and one RAD51 clone was obtained for each 3 μg of starting genomic DNA.
Plasmids from 10 int-2 clones and 10 rad51 gene clones were analyzed by restriction enzyme mapping. No rearrangements were detected. Single-pass sequencing of the two vector-insert junctions and about 400 bases of insert DNA of each clone revealed no deviations from the published sequences. Although not proven, it is likely that the error rate of the method is closer to the in vivo error rate in E. coli of 10−10 mutations/bp per chromosome duplication (40, 41) rather than the best-reported PCR error rate of about 10−5–10−6 (19, 42).
Ancillary Steps: Fractionating Input DNA and Removing Unligated Vector.
When cloning fragments directly from genomic DNA, the application of RAC was aided by two steps not shown in Fig. 1. First, the digested genomic DNA used was coarsely size-fractionated on a short agarose gel and the DNA from the relevant region was used in the RecA protein/KF reaction. This was done for three reasons: a modest (about 10-fold) enrichment was obtained, reagent requirements were thus reduced 10-fold, and small fragments that would preferentially be represented in the final clones were eliminated (30). The second additional step was constructing a “sandwich” as shown in Fig. 4 to reduce the number of background colonies containing only vector DNA. Magnetic streptavidin beads were coated with a short biotinylated duplex that terminated with an EcoRI-cohesive end that lacked a 5′ phosphate. The beads were included when ligating genomic DNA to a vector that had been treated with EcoRI, BamHI, and phosphatase. The selected fragment was able to be immobilized by ligation to the short biotinylated duplex. The BamHI site on the other side of the fragment was available to ligate to the vector. Unligated vector and DNA with blunt ends were removed by washing the beads, and the immobilized DNA was released by treatment with EcoRI. The linear fragment-vector molecules were then circularized by performing a second ligation reaction that joined the EcoRI sites present on both ends of the fragment-vector molecules.
The streptavidin bead step was necessary because when plasmid or λ DNA vectors were simply ligated to DNA from the RecA protein/KF reaction, the overwhelming majority of clones contained no insert. Efforts were made to reduce this background problem by lowering the concentration of the vector, but this also lowered the efficiency of the cloning procedure. Given the low amount of the selected fragment in genomic DNA, a large concentration of the vector aided in driving the intermolecular ligation of the vector to the fragment. Removal of unligated vector from fragment-vector molecules on a short agarose gel did not reduce the background to an acceptable level, but this convenient procedure would likely be effective when cloning larger fragments.
DISCUSSION
As now developed, RAC has sufficient specificity to allow cloning directly from genomic DNA and is an alternative to constructing or screening genomic DNA libraries. In essence, RecA protein biochemically carries out the steps now done by manually constructing and screening libraries. Although the specificity and fidelity of the technique are excellent, the major drawback of the current technique, especially in comparison to PCR, is the large amount of genomic DNA required. Model experiments analyzing each step of the method indicated that the intermolecular ligation of the selected fragment to the vector was the least efficient step. The literature of commercially available ligases was reviewed, and promising conditions were tested (such as in refs. 36 and 37). Only the straightforward strategies of increasing reagent concentrations and ligation times were found to be helpful. If the quantity of DNA is limiting, there are now methods capable of producing competent bacteria that transform more than an order of magnitude more efficiently than those used in this study (43–45).
Many modifications of the method can be imagined. The RecA protein and KF reactions were found to work well on DNA embedded in agarose; this will be useful when cloning large molecules that would tend to shear in solution. Fragments several hundred kilobases in length should be able to be cloned in currently available yeast artificial chromosome, bacterial artificial chromosome, and P1 phage-derived artificial chromosome vectors. For applications where increased specificity is desired, RAC can be used after RARE cleavage, or with type II restriction enzymes that create varied and asymmetric staggered ends unrelated to their recognition sites (46). Increases in specificity would be useful for labeling specific genomic DNA fragments in a manner similar to Fig. 2 and may be an alternative to detection methods such as Southern DNA blotting. RAC should also work well on cDNA.
Although the method is not intended for routine cloning of short segments of DNA, it holds promise for the cloning of long or highly repetitive fragments of DNA, especially if absolute fidelity is required. An especially timely application of the method is to close gaps in DNA contigs, because closing such gaps by using conventional methods is now the rate-limiting step when sequencing large genomes (47). Another useful application, currently being pursued in this laboratory, is to apply RARE cleavage and RAC to rapidly map and clone large, rearranged regions from multiple individual tumor specimens. In addition, RAC represents an expansion of the repertoire of reactions that can be driven by RecA protein and, coupled with the recent discovery of homologous DNA pairing promoted by a 20-aa peptide derived from RecA protein (48), suggests the possibility of directing DNA rearrangements inside cells.
Acknowledgments
We thank P. Hsieh, D. Kastner, R. Proia, and S. Zimmerman of the National Institutes of Health for invaluable comments on the manuscript, and G. Poy and L. Robinson for their assistance.
ABBREVIATIONS
- RARE cleavage
RecA-assisted restriction endonuclease cleavage
- RAC
RecA-assisted cloning
- KF
Klenow fragment of E. coli DNA polymerase I
References
- 1.Ferrin L J, Camerini-Otero R D. Science. 1991;254:1494–1497. doi: 10.1126/science.1962209. [DOI] [PubMed] [Google Scholar]
- 2.Ferrin L J, Camerini-Otero R D. Nat Genet. 1994;6:379–383. doi: 10.1038/ng0494-379. [DOI] [PubMed] [Google Scholar]
- 3.Ferrin L J. In: Genetic Engineering: Principles and Methods. Setlow J, editor. Vol. 17. New York: Plenum; 1995. pp. 21–30. and references therein. [PubMed] [Google Scholar]
- 4.Barton M C, Emerson B M. Genes Dev. 1994;8:2453–2465. doi: 10.1101/gad.8.20.2453. [DOI] [PubMed] [Google Scholar]
- 5.Boren J, Lee I, Callow M J, Rubin E M, Innerarity T L. Genome Res. 1996;6:1123–1130. doi: 10.1101/gr.6.11.1123. [DOI] [PubMed] [Google Scholar]
- 6.Callow M J, Ferrin L J, Rubin E M. Nucleic Acids Res. 1994;22:4348–4349. doi: 10.1093/nar/22.20.4348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farman M L, Leong S A. Genetics. 1995;140:479–492. doi: 10.1093/genetics/140.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Feder J N, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy D A, Basava A, Dormishian F, Domingo R, Jr, Ellis M C, Fullen A, et al. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 9.Gnirke A, Iadonato S P, Kwok P, Olson M V. Genomics. 1994;24:199–210. doi: 10.1006/geno.1994.1607. [DOI] [PubMed] [Google Scholar]
- 10.Gourdon G, Sharpe J A, Wells D, Wood W G, Higgs D R. Nucleic Acids Res. 1994;22:4139–4147. doi: 10.1093/nar/22.20.4139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heineman T C, Cohen J I. J Virol. 1994;68:3317–3323. doi: 10.1128/jvi.68.5.3317-3323.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Macina R A, Barr F G, Galili N, Riethman H C. Genomics. 1995;26:1–8. doi: 10.1016/0888-7543(95)80076-x. [DOI] [PubMed] [Google Scholar]
- 13.Macina R A, Morii K, Hu X, Negorev D G, Spais C, Ruthig L A, Riethman H C. Genome Res. 1995;5:225–232. doi: 10.1101/gr.5.3.225. [DOI] [PubMed] [Google Scholar]
- 14.Martin-Gallardo A, Lamerdin J, Sopapan P, Friedman C, Fertitta A L, Garcia E, Carrano A, Negorev D, Macina R A, Trask B J, Riethman H C. Cytogenet Cell Genet. 1995;71:289–295. doi: 10.1159/000134129. [DOI] [PubMed] [Google Scholar]
- 15.Mason M M, Lee E, Westphal H, Reitman M. Mol Cell Biol. 1995;15:407–414. doi: 10.1128/mcb.15.1.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ning Y, Roschke A, Smith A C M, Macha M, Precht K, Riethman H, Ledbetter D H, Flint J, Horsley S, Regan R, et al. Nat Genet. 1996;14:86–89. [Google Scholar]
- 17.Reston J T, Hu X, Macina R A, Spais C, Riethman H C. Genomics. 1995;26:31–38. doi: 10.1016/0888-7543(95)80079-2. [DOI] [PubMed] [Google Scholar]
- 18.Wang Y, Huff E J, Schwartz D C. Proc Natl Acad Sci USA. 1995;92:165–169. doi: 10.1073/pnas.92.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Barnes W M. Proc Natl Acad Sci USA. 1994;91:2216–2220. doi: 10.1073/pnas.91.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheng S, Fockler C, Barnes W M, Higuchi R. Proc Natl Acad Sci USA. 1994;91:5695–5699. doi: 10.1073/pnas.91.12.5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Foord O S, Rose E A. PCR Methods Appl. 1994;3:S149–S161. doi: 10.1101/gr.3.6.s149. [DOI] [PubMed] [Google Scholar]
- 22.Rigas B, Welcher A A, Ward D C, Weissman S M. Proc Natl Acad Sci USA. 1986;83:9591–9595. doi: 10.1073/pnas.83.24.9591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Honigberg S M, Rao B J, Radding C M. Proc Natl Acad Sci USA. 1986;83:9586–9590. doi: 10.1073/pnas.83.24.9586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Taidi-Laskowski B, Tyan D, Honigberg S M, Radding C R, Grumet F C. Nucleic Acids Res. 1988;16:8157–8169. doi: 10.1093/nar/16.16.8157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sena E P, Zarling D A. Nat Genet. 1993;3:365–371. doi: 10.1038/ng0493-365. [DOI] [PubMed] [Google Scholar]
- 26.Jayasena V K, Johnston B H. J Mol Biol. 1993;230:1015–1024. doi: 10.1006/jmbi.1993.1216. [DOI] [PubMed] [Google Scholar]
- 27.Hakvoort T B M, Spijkers J A A, Vermeulen J L M, Lamers W H. Nucleic Acids Res. 1996;24:3478–3480. doi: 10.1093/nar/24.17.3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Camerini-Otero R D, Hsieh P. Cell. 1993;73:217–223. doi: 10.1016/0092-8674(93)90224-e. [DOI] [PubMed] [Google Scholar]
- 29.Ausubel F, editor. Current Protocols in Molecular Biology. New York: Wiley; 1995. [Google Scholar]
- 30.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 31.Pun K K, Kam W. Prep Biochem. 1990;20:123–135. doi: 10.1080/00327489008050184. [DOI] [PubMed] [Google Scholar]
- 32.Labarca C, Paigen K. Anal Biochem. 1980;102:344–352. doi: 10.1016/0003-2697(80)90165-7. [DOI] [PubMed] [Google Scholar]
- 33.Brookes S, Smith R, Casey G, Dickson C, Peters G. Oncogene. 1989;4:429–436. [PubMed] [Google Scholar]
- 34.Casey G, Smith R, McGillivray D, Peters G, Dickson C. Mol Cell Biol. 1986;6:502–510. doi: 10.1128/mcb.6.2.502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shinohara A, Ogawa H, Ogawa T. Cell. 1992;69:457–470. doi: 10.1016/0092-8674(92)90447-k. [DOI] [PubMed] [Google Scholar]
- 36.Derbyshire V, Freemont P S, Sanderson M R, Beese L, Friedman J M, Joyce C M, Steitz T A. Science. 1988;240:199–201. doi: 10.1126/science.2832946. [DOI] [PubMed] [Google Scholar]
- 37.Hayashi K, Nakazawa M, Ishizaki Y, Obayashi A. Nucleic Acids Res. 1985;13:3261–3271. doi: 10.1093/nar/13.9.3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hayashi K, Nakazawa M, Ishizaki Y, Hiraoka N, Obayashi A. Nucleic Acids Res. 1985;13:7979–7992. doi: 10.1093/nar/13.22.7979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim M G, Zhurkin V B, Jernigan R L, Camerini-Otero R D. J Mol Biol. 1995;247:874–889. doi: 10.1006/jmbi.1994.0187. [DOI] [PubMed] [Google Scholar]
- 40.Drake J W. Proc Natl Acad Sci USA. 1991;88:7160–7164. doi: 10.1073/pnas.88.16.7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schaaper R M. J Biol Chem. 1993;268:23762–23765. [PubMed] [Google Scholar]
- 42.Cline J, Braman J C, Hogrefe H H. Nucleic Acids Res. 1996;24:3546–3551. doi: 10.1093/nar/24.18.3546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chuang S, Chen A, Chao C. Nucleic Acids Res. 1995;23:1641. doi: 10.1093/nar/23.9.1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Michelsen B K. Anal Biochem. 1995;225:172–174. doi: 10.1006/abio.1995.1130. [DOI] [PubMed] [Google Scholar]
- 45.Sheng Y, Mancino V, Birren B. Nucleic Acids Res. 1995;23:1990–1996. doi: 10.1093/nar/23.11.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Berger S L. Anal Biochem. 1994;222:1–8. doi: 10.1006/abio.1994.1445. [DOI] [PubMed] [Google Scholar]
- 47.Goffeau A, Barrell B G, Bussey H, Davis R W, Dujon B, Feldmann H, Galibert F, Hoheisel J D, Jacq C, Johnston M, et al. Science. 1996;274:546–567. doi: 10.1126/science.274.5287.546. [DOI] [PubMed] [Google Scholar]
- 48.Voloshin O N, Wang L, Camerini-Otero R D. Science. 1996;272:868–872. doi: 10.1126/science.272.5263.868. [DOI] [PubMed] [Google Scholar]