

Abstract
We demonstrate herein dramatic acceleration of aqueous nitric oxide (NO) reaction with O2 within the hydrophobic region of either phospholipid or biological membranes or detergent micelles and demonstrate that the presence of a distinct hydrophobic phase is required. Per unit volume, at low amounts of hydrophobic phase, the reaction of NO with O2 within the membranes is approximately 300 times more rapid than in the surrounding aqueous medium. In tissue, even though the membrane represents only 3% of the total volume, we calculate that 90% of NO reaction with O2 will occur there. We conclude that biological membranes and other tissue hydrophobic compartments are important sites for disappearance of NO and for formation of NO-derived reactive species and that attenuation of these potentially damaging reactions is an important protective action of lipid-soluble antioxidants such as vitamin E.
Nitric oxide (NO) is an important mediator and messenger in mammalian systems and subserves an astonishing variety of roles in physiology and pathophysiology (1). One of its distinctive properties is its relatively short half-life (reported to be on the order of several seconds) in biological systems, which determines its spatial range and temporal extent of actions (2). One generally recognized mechanism for the disappearance of NO is reaction with O2, which is responsible for the formation of nitrite as a product of NO oxidation. Intermediates in this reaction are responsible for nitrosative reactions that result in the formation of biologically important species such as nitrosamines and nitrosothiols (2).
The aqueous reaction of NO with dioxygen occurs with the following overall stoichiometry:
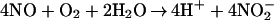 |
1 |
and the rate of disappearance of NO is given by
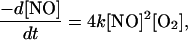 |
2 |
with k = 2 × 106 M−2⋅s−1 at 25°C (3, 4). Because NO is approximately nine times more soluble in a hydrophobic solvent such as hexane than in water (5, 6), we [and others (7, 8)] have suspected that the presence of a hydrophobic phase (such as the interior of a lipid bilayer membrane) might accelerate the autooxidation of NO because of the concentration of reactants within the hydrophobic phase. Thus, biological membranes may act as a “lens” that can focus and magnify the autooxidation of NO. That is, even if the intrinsic rate constant of the reaction within the membrane hydrophobic phase is the same as in the aqueous cytosol, the reaction is accelerated overall because of the increased reactant concentrations within the membrane.
To test this possibility, we used an electrochemical method to measure the rate of disappearance of NO in an aerobic buffered solution upon addition of various hydrophobic phases.
METHODS
Hepatocyte Isolation and Cell Membrane Preparation.
Rat hepatocytes were isolated as described (9). For membrane isolation, cells were suspended in 50 mM potassium phosphate (pH 7.4) containing 0.5 mM EDTA and sonicated (two 10-s bursts) while cooled on ice. The sonicated preparations were centrifuged at 5,000 × g for 5 min at 4°C. The supernatant was subjected to ultracentrifugation at 78,000 × g for 60 min at 4°C in a Beckman L8–55M ultracentrifuge. The pellet was washed once with buffer.
Determination of Total Lipids in Hepatocyte Membranes.
The total lipids in the membranes were extracted and estimated by using modifications of the methods described by Bragdon (10). Ten milliliters of a 2:1 mixture of chloroform and methanol was added to 0.5 ml of membrane sample, mixed, and incubated at room temperature for 15 min. Two milliliters of dilute H2SO4 was then added, and the mixture was incubated for another 10 min to allow the phases to equilibrate and centrifuged at 3,000 × g for 15 min. The lower chloroform phase containing lipids was collected and used for lipid estimation. For measurement of lipids, 1 ml of palmitic acid (0–2 mg) as the standard or a 1-ml sample was dried with N2 gas in a water bath at 57°C. Two milliliters of K2Cr2O7 solution (1 g/10 ml of concentrated H2SO4) was added, and the samples were boiled 30 min with the tubes covered with marbles. After cooling, 5 ml of water was added and the samples were incubated 30 min at room temperature. Absorbance was measured at 580 nm. From this measurement, the amount of lipid (in g/ml; Ch) was determined for each experiment.
Measurement of NO Reaction with O2 In the Presence of Various Hydrophobic Phases.
NO was scrubbed of higher nitrogen oxides by passage through 1 M deaerated (bubbled with argon) KOH solution. A sample of buffer (0.1 M potassium phosphate, pH 7.4/0.1 M KCl) was deaerated and then saturated with scrubbed NO. All electrochemical measurements of NO were carried out at 25°C by using a BAS 100B Electrochemical Analyzer with a PA-1 preamplifier and C2 cell stand from Bioanalytical Systems (West Lafayette, IN). The potential on the working electrode was held at 0.55 V vs. Ag/AgCl reference. The detecting electrode was a Nafion-coated electrode of our own design (11), a platinum disc electrode (200 μm in diameter) was used as the working electrode, and a platinum wire and an Ag/AgCl electrode were used as the auxiliary and reference electrodes, respectively. The response time for the electrode was approximately 1 s and current was linear with NO concentration from 0.05 to 60 μM. The reaction vessel (6 ml) was capped, contained small holes for inserting probes, and was filled with aerated buffer and with or without indicated hydrophobic phases. Small surface area compared with volume with this apparatus assured that under these conditions the volatilization of NO was negligible compared with reaction in solution. NO autooxidation was initiated by injection of a measured volume of saturated NO solution with a gas-tight syringe with rapid stirring. Unilamellar soy phospholipid vesicles were prepared as described (12). Measurement of nitrite and nitrate was performed as described (13).
RESULTS
As shown in Fig. 1A, immediately subsequent to injection of 30 μM NO to a well-stirred, aerated solution the NO disappears, and as shown in Fig. 1B, the disappearance closely follows a course predicted by a second-order dependence on NO concentration with a rate constant of 6.6 × 106 M−2⋅s−1, similar to the rate constant reported previously (3, 4). However, when a suspension of soy phospholipid vesicles at 40 mg/ml is present (slightly higher than the amount of phospholipid in tissue, see below), NO disappears much more rapidly and the rate is still second order with rate constant 8.8 × 107 M−2⋅s−1, approximately 13 times more rapid than in the absence of the liposomes.
Figure 1.

(A) Accelerated NO autooxidation in the presence of phospholipid vesicles (liposomes; 40 mg/ml). NO (30 μM) was added in the well-stirred aerated vessel at t = 0 and detected electrochemically. (B) Second-order linear transformation of the data.
The fact that the reaction in the presence of phospholipid membrane is still second order with respect to NO concentration indicates that the disappearance of NO is still occurring via reaction with O2. We tested the possibility that this increase is caused by increased reactivity within the hydrophobic phase. If the partition coefficient for NO within the hydrophobic membrane interior is similar to that for organic phases [approximately 9 (5)], then this means that in a heterogeneous system (containing both aqueous and hydrophobic compartments) the reaction within the hydrophobic phase will be accelerated by a factor of 92 = 81 compared with the aqueous phase (because the rate is proportional to the square of the NO concentration, Eq. 2). In addition, the rate will also be accelerated because of the increased partitioning of O2 within the membrane interior (14). We therefore developed a model to predict and thus test this phenomenon (Fig. 2). The rate constants for the autooxidation of NO in the aqueous and hydrophobic phases, respectively, are given by k2 and k2′. Thus, k2 is equal to 4k in Eq. 2. The partitioning of NO and O2 between the aqueous (aq) and hydrophobic (h) phases are described by k1 and k−1 (for NO) and k3 and k−3 (for O2). In a rapidly stirred vessel, such as was used in this study, transport of NO and O2 between the two phases is rapid (compared with the reaction rates) because of convection. When the aqueous volume is greater than the hydrophobic volume (in these experiments we use hydrophobic phase of less than 10 mg/ml) and if the standard steady-state assumption is made, the following expression can be derived to describe the concentration of NO as a function of time after addition of NO:
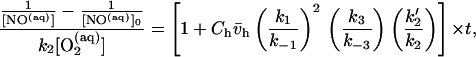 |
3 |
where [NO(aq)] is the aqueous concentration of NO at time t, [NO(aq)]0 is the initial aqueous concentration of NO (at t = 0), Ch is the concentration of hydrophobic phase present (in g/ml), and v̄h is the partial specific volume of the hydrophobic phase (in ml/g). The product Ch × v̄h is thus the fraction of the total volume that is in the hydrophobic phase.
Figure 2.

Model to predict the effects of a membrane on NO autooxidation.
It is clear from this equation that when no hydrophobic phase is present (Ch = 0), the disappearance of NO will simply follow that for a homogeneous aqueous second-order reaction. It is thus the second term in the brackets [Chv̄h(k1/k−1)2(k3/k−3) (k2′/k2)], which determines the disappearance of NO within the hydrophobic phase, and we define a new term λ, which we designate the acceleration factor: λ = (k1/k−1)2(k3/k−3)(k2′/k2). This term is a product of the three factors that will accelerate the disappearance of NO via oxidation in the hydrophobic phase: the square (second order) of the increased concentration of NO within the hydrophobic membrane (k1/k−1)2, the first-order increase caused by concentration of O2 within the membrane (k3/k−3), and the increase (or decrease) in the intrinsic reaction rate in the hydrophobic phase compared with the aqueous reaction (k2′/k2). The extent of acceleration is also dependent on the amount of the total volume which is in the hydrophobic phase. This parameter is reflected by the quantity Chv̄h, as described above.
For any given concentration of hydrophobic phase (Ch), a plot of the terms on the left side of Eq. 3 vs. time will yield a straight line with slope equal to 1 + Chv̄hλ. Thus, this is a two-step process: (i) Plots of the quantity on the left side of Eq. 3 vs. time are made for each of several experiments with increasing amounts of hydrophobic phase (Ch) and the slopes of the resultant straight line for each is obtained. (ii) A plot of the values of these slopes as a function of the amount of hydrophobic phase for each (Ch) will also yield a straight line, with an intercept of one and a slope equal to v̄hλ.
Fig. 3A is a second-order plot of the time course of NO disappearance in the presence of three different hydrophobic phases at 0.003 g/ml, soy phospholipid vesicles, the nonionic detergent Triton X-100, and isolated membranes from rat hepatocytes. Two important points are evident: (i) the disappearance of NO in the presence of these three phases is indistinguishable (meaning that the accelerated reaction is independent of the chemical nature of the hydrophobic phase) and (ii) the reaction is second-order for all three. Fig. 3B shows a plot of the slopes of these time courses for different concentrations of these three phases. This plot shows that the rate of disappearance is significantly accelerated by quite small amounts of hydrophobic phase (0.5–3 mg/ml). In addition, these different phases accelerate the rate of NO reaction with O2 to approximately identical extents, meaning the quantity v̄hλ is similar for each.
Figure 3.

(A) Second-order plot of NO disappearance in the presence of soy phospholipid vesicles (0.003 g/ml), Triton X-100 (0.003 g/ml), and isolated hepatocyte membranes (0.003 g/ml). (B and C) Determination of v̄hλ for soy vesicles, Triton X-100, and hepatocyte membranes (B) and three detergents with different CMCs, as denoted by the arrows (C). Z-12, Zwittergent-12 [CMC = 1.21 mg/ml (15)]; OTG, octyl thioglucoside [CMC = 2.77 mg/ml (16)]; OG, octyl glucoside [CMC = 7.3 mg/ml (17)].
For detergents, the critical micelle concentration (CMC) is the concentration of detergent at which micelles form; below this concentration the detergent is present as monomeric species (no hydrophobic phase). Because of the cooperative nature of micellar formation, above the CMC, addition of more detergent results in increased micelle number with no change in free monomer, in other words, increased hydrophobic phase volume (18). If the effect of these amphipathic molecules on NO autooxidation is simply to provide a hydrophobic phase within which both NO and O2 accumulate, then there should be little acceleration of NO autooxidation below the CMC and linear increase above it. As shown in Fig. 3B, for a series of three detergents with relatively high CMCs, for each detergent there is indeed no increase in NO disappearance until the CMC is reached (denoted by the arrows), above which the reaction is greatly accelerated, with the slope of the lines above the CMCs (v̄hλ) being similar for each.
Table 1 lists the values for v̄hλ for eight hydrophobic phases. Other detergents accelerate NO disappearance with magnitudes similar to Triton X-100. This is true for other nonionic (octylthioglucoside and octyl glucoside), anionic (SDS), cationic (cetyltrimethylammonium bromide), and zwitterionic (Z-12) detergents. In addition, acceleration of autooxidation by phospholipid membranes and a biological membrane (isolated from rat hepatocytes) is similar to each other and also to the detergents.
Table 1.
Rate of NO autooxidation within various hydrophobic phases compared to the aqueous phase
| Hydrophobic phase | v̄hλ |
|---|---|
| Triton X-100 | 307 ± 2 |
| SDS | 327 ± 10 |
| CTAB | 344 ± 1 |
| Z-12 | 313 ± 2 |
| OTG | 311 ± 2 |
| OG | 301 ± 9 |
| Liposomes | 322 ± 2 |
| Hepatocyte membranes | 291 ± 3 |
CTAB, cetyltrimethylammonium bromide; OTG, octyl thioglucoside; OG, octyl glucoside.
Measurement of the final products of NO autooxidation by the Griess reagent plus enzymatic nitrate conversion to nitrite (13) showed that the exclusive reaction product was nitrite: the percentage of product that is nitrite (compared with total nitrite plus nitrate) for overnight incubation of 50 μM NO donor DEA [sodium (Z)-1-(N,N-diethylamino)diazen-1-ium-1,2-diolate]/NO in a sealed stirred vessel with no headspace gas was 96.8 ± 3.1% for buffer alone and 92.6 ± 2.2% for buffer plus 4% Triton X-100.
DISCUSSION
By using an electrochemical detection method, we find that disappearance of NO in aqueous solution is dramatically accelerated by the presence of hydrophobic phases. The facts that (i) the accelerated reaction is still second order with respect to NO concentration, (ii) the acceleration is independent of the chemical nature of the hydrophobic phase (Table 1), and (iii) the exclusive product is nitrite demonstrates that the disappearance is caused by NO autooxidation. In addition, the acceleration in the presence of biological membranes (isolated from rat hepatocytes) follows closely the rate that is predicted based solely on the volume of the phospholipid phase in these membranes (i.e., predicted on the basis of phospholipid in g/ml, Ch; see Fig. 3A). In other words, the presence of proteins and other components in biological membranes does not appreciably alter the rate of disappearance away from that predicted only on the basis of the hydrophobic partitioning phenomenon we describe herein.
On the basis of the values for the acceleration factor (λ) that we determined at low amounts of hydrophobic phase (Table 1) and the model shown in Fig. 2, it is possible to predict the effects of any concentration of hydrophobic phase on NO autooxidation. From these calculations, we can obtain the fold increase in total NO disappearance that occurs when hydrophobic phase is added to purely aqueous solution and also obtain the proportion of total NO autooxidation that occurs within the hydrophobic phase (compared with the total rate). For these calculations, we choose the data obtained by using phospholipid bilayers, where v̄hλ = 322 (Table 1). We also use the experimentally determined value for the partition coefficient of oxygen between the aqueous phase and the bilayer of dimyristoylphosphatidylcholine vesicles (k3/k−3) of 3 (14), and the value for v̄ (the total partial specific volume) for phosphatidylcholine vesicles of 0.9885 ml/g (12). Note that this value is the total partial specific volume, which includes both hydrophilic and hydrophobic volumes, the latter being v̄h. When the organization of phospholipid bilayers is considered, we assume that the hydrophilic volume is small compared with the hydrophobic volume and so we use the value 0.9 as an approximation for v̄.
From this modeling, Fig. 4 shows that relatively low amounts of lipid phase dramatically increases the total rate of NO disappearance, as reflected by the fold increase in total NO reaction when compared with the rate in the absence of hydrophobic phase (Ch = 0). Perhaps more importantly, even at very low amounts of lipid (as low as 5 mg/ml), the majority of NO autooxidation occurs within the membrane. For NO-producing tissue such as brain and liver, which have a phospholipid content of approximately 0.03 g/ml (19) [slightly below the amount (40 mg/ml) of phospholipid used for the data in Fig. 1], this means that the rate of NO reaction with O2 is greater than 8 times more rapid than if phospholipid were not present and that greater than 90% of the reaction takes place within the membrane as opposed to the cytosol.
Figure 4.

Predicted effect of increasing amounts of membrane phase on the total and the membrane reaction of NO with O2. The upper curve is the percent of the total reaction of NO with O2 that occurs within the membrane interior as the amount of membrane increases. The lower curve is the fold increase in total reaction of NO with O2 that results from increasing amounts of membrane.
The contribution of membrane-associated NO reactivity may provide alternative explanations to previously published results. Borutaité and Brown (20) found that addition of mitochondria results in accelerated NO disappearance, and the rate of this disappearance is only modestly affected by inhibition of mitochondrial respiration. Our results suggest that the increase in NO disappearance may largely be caused by autooxidation within the mitochondrial membrane rather than metabolism via electron transfer. Also, in an elegant and thorough analysis of the kinetics of NO oxidation by macrophages, Lewis et al. (21) found an excess of nitrite above that predicted from previously determined kinetic constants. Increased autooxidation within the macrophage membrane could explain this excess since the rate constant used in the calculations was the reaction solely in the aqueous phase.
We point out that the NO/O2 reaction in Fig. 2 does not refer to molecularity or mechanism but is a representation of the observed order of the overall reaction (rate ∝ [NO]2[O2]). In the aqueous phase, NO disappears by reaction with both O2 and NO2 (3, 4). Because it is reasonable to assume that NO2, like NO, will partition into the hydrophobic milieu, this could also contribute to the overall acceleration in NO disappearance. These effects (as well as partitioning of other potential nitrogen oxide intermediates) will be incorporated into the observed rate constants k2 and k2′ in Eq. 3.
Unless its half-life is exceptionally short (which will not likely be solely because of the phenomenon described herein), the diffusional spread of NO will still be substantial (22, 23) and NO will diffuse large distances. The findings herein show that the autooxidation of NO is not homogeneously distributed throughout these relatively large tissue volumes as it spreads out from its source but is instead confined (>90%) to the hydrophobic compartments.
Biological membranes and perhaps other hydrophobic compartments are thus important locations for the nitrosative chemistry, which occurs upon NO autooxidation, such as formation of nitrosothiol (24), DNA damage (25), and genotoxicity (26), as well as formation of nitrosamine (27) and lipid peroxide (28). Especially important environments include cells and structures that possess relatively high ratios of surrounding membrane surface area to internal aqueous volume (such as neurons and mitochondria) and also hydrophobic structures such as the lining of the lung (within the volume containing pulmonary surfactant), lipoproteins, atherosclerotic plaque, adipose tissue, and myelin sheath. The exceptionally rapid diffusibility of NO ensures that all hydrophobic compartments within a large radius of a point source of NO [approximately 100–200 μm (2)] will be important sites in tissue for this chemistry. We also suggest that reaction of NO with hydroperoxyl radical (HO2), the conjugate acid of superoxide anion (pK = 4.8), may occur within lipid bilayers, resulting in the formation of the highly reactive peroxynitrous acid (ONOOH), which (also within the membrane hydrophobic phase) could react further with CO2 (29). Accumulation of HO2 within lipid bilayers and reaction with NO may be especially important in acidic environments such as the phagolysosome (30). Finally, we speculate that scavenging of reactive nitrogen oxides that are formed within the membrane may be an important protective function of the lipid-soluble antioxidant vitamin E (31, 32).
Acknowledgments
This work was supported in part by National Institutes of Health Grants DK46935 (J.R.L.) and HD31885 (M.J.S.M.).
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviation: CMC, critical micelle concentration.
References
- 1.Kerwin J F J, Lancaster J R, Jr, Feldman P L. J Med Chem. 1995;38:4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 2.Lancaster J R., Jr Nitric Oxide. 1997;1:18–30. doi: 10.1006/niox.1996.0112. [DOI] [PubMed] [Google Scholar]
- 3.Ford P C, Wink D A, Stanbury D M. FEBS Lett. 1993;326:1–3. doi: 10.1016/0014-5793(93)81748-o. [DOI] [PubMed] [Google Scholar]
- 4.Awad H H, Stanbury D M. Int J Chem Kinet. 1993;25:375–381. [Google Scholar]
- 5.Shaw A W, Vosper A J. J Chem Soc Faraday Trans 1. 1977;8:1239–1244. [Google Scholar]
- 6.Malinski T, Taha Z, Grunfeld S, Patton S, Kapturczak M, Tomboulian P. Biochem Biophys Res Commun. 1993;193:1076–1082. doi: 10.1006/bbrc.1993.1735. [DOI] [PubMed] [Google Scholar]
- 7.Singh R J, Hogg N, Mchaourab H S, Kalyanaraman B. Biochim Biophys Acta. 1994;1201:437–441. doi: 10.1016/0304-4165(94)90073-6. [DOI] [PubMed] [Google Scholar]
- 8.Stamler J S. In: Biochemical, Pharmacological, and Clinical Aspects of Nitric Oxide. Weissman B A, Allon N, Shapira S, editors. New York: Plenum; 1995. pp. 67–78. [Google Scholar]
- 9.Kim Y M, Bergonia H A, Muller C, Pitt B R, Watkins W D, Lancaster J R., Jr J Biol Chem. 1995;270:5710–5713. doi: 10.1074/jbc.270.11.5710. [DOI] [PubMed] [Google Scholar]
- 10.Bragdon J H. In: Lipids and the Steroid Hormones in Clinical Medicine. Sunderman F W, Sunderman F W Jr, editors. Philadelphia: Lippincott; 1960. pp. 6–14. [Google Scholar]
- 11.Sandoval M, Liu X, Mannick E E, Clark D A, Miller M J S. Gastroenterology. 1997;113:1480–1488. doi: 10.1053/gast.1997.v113.pm9352850. [DOI] [PubMed] [Google Scholar]
- 12.Huang C. Biochemistry. 1969;8:344–351. doi: 10.1021/bi00829a048. [DOI] [PubMed] [Google Scholar]
- 13.Grisham M B, Johnson G G, Lancaster J R., Jr Methods Enzymol. 1996;268:237–246. doi: 10.1016/s0076-6879(96)68026-4. [DOI] [PubMed] [Google Scholar]
- 14.Subczynski W K, Hyde J S. Biophys J. 1983;41:283–286. doi: 10.1016/S0006-3495(83)84439-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Navarette R, Serrano R. Biochim Biophys Acta. 1983;728:403–408. doi: 10.1016/0005-2736(83)90512-6. [DOI] [PubMed] [Google Scholar]
- 16.Tsuchiya T, Saito S. J Biochem. 1984;96:1593–1597. doi: 10.1093/oxfordjournals.jbchem.a134989. [DOI] [PubMed] [Google Scholar]
- 17.Helenius A, McCaslin D R, Fries E, Tanford C. Methods Enzymol. 1979;56:734–749. doi: 10.1016/0076-6879(79)56066-2. [DOI] [PubMed] [Google Scholar]
- 18.Helenius A, Simons K. Biochim Biophys Acta. 1975;415:29–79. doi: 10.1016/0304-4157(75)90016-7. [DOI] [PubMed] [Google Scholar]
- 19.Long C, editor. Anonymous. Biochemists’ Handbook. Princeton, NJ: Van Nostrand; 1961. p. 677. [Google Scholar]
- 20.Borutaité V, Brown G C. Biochem J. 1996;315:295–299. doi: 10.1042/bj3150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis R S, Tamir S, Tannenbaum S R, Deen W M. J Biol Chem. 1995;270:29350–29355. doi: 10.1074/jbc.270.49.29350. [DOI] [PubMed] [Google Scholar]
- 22.Lancaster J R., Jr Proc Natl Acad Sci USA. 1994;91:8137–8141. doi: 10.1073/pnas.91.17.8137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wood J, Garthwaite J. Neuropharmacology. 1994;33:1235–1244. doi: 10.1016/0028-3908(94)90022-1. [DOI] [PubMed] [Google Scholar]
- 24.Stamler J S. Cell. 1994;78:931–936. doi: 10.1016/0092-8674(94)90269-0. [DOI] [PubMed] [Google Scholar]
- 25.Wink D A, Kasprzak K S, Maragos C M, Elespuru R K, Misra M, et al. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 26.Tamir S, deRojas-Walker T, Wishnok J S, Tannenbaum S R. Methods Enzymol. 1996;269:230–243. doi: 10.1016/s0076-6879(96)69025-9. [DOI] [PubMed] [Google Scholar]
- 27.Bartsch H, Ohshima H, Pignatelli B, Calmels S. Pharmacogenetics. 1992;2:272–277. doi: 10.1097/00008571-199212000-00005. [DOI] [PubMed] [Google Scholar]
- 28.Pryor W A, Lightsey J W. Science. 1981;214:435–437. doi: 10.1126/science.214.4519.435. [DOI] [PubMed] [Google Scholar]
- 29.Uppu R M, Squadrito G L, Pryor W A. Arch Biochem Biophys. 1996;327:335–343. doi: 10.1006/abbi.1996.0131. [DOI] [PubMed] [Google Scholar]
- 30.Aikens J, Dix T A. J Biol Chem. 1991;266:15091–15098. [PubMed] [Google Scholar]
- 31.Hogg N, Singh R J, Goss S P, Kalyanaraman B. Biochem Biophys Res Commun. 1996;224:696–702. doi: 10.1006/bbrc.1996.1086. [DOI] [PubMed] [Google Scholar]
- 32.Kim Y K, Tannenbaum S R, Wishnok J S. IARC Sci Publ. 1980;31:207–214. [PubMed] [Google Scholar]