

Abstract
Restriction endonucleases such as EcoRI bind and cleave DNA with great specificity and represent a paradigm for protein–DNA interactions and molecular recognition. Using osmotic pressure to induce water release, we demonstrate the participation of bound waters in the sequence discrimination of substrate DNA by EcoRI. Changes in solvation can play a critical role in directing sequence-specific DNA binding by EcoRI and are also crucial in assisting site discrimination during catalysis. By measuring the volume change for complex formation, we show that at the cognate sequence (GAATTC) EcoRI binding releases about 70 fewer water molecules than binding at an alternate DNA sequence (TAATTC), which differs by a single base pair. EcoRI complexation with nonspecific DNA releases substantially less water than either of these specific complexes. In cognate substrates (GAATTC) kcat decreases as osmotic pressure is increased, indicating the binding of about 30 water molecules accompanies the cleavage reaction. For the alternate substrate (TAATTC), release of about 40 water molecules accompanies the reaction, indicated by a dramatic acceleration of the rate when osmotic pressure is raised. These large differences in solvation effects demonstrate that water molecules can be key players in the molecular recognition process during both association and catalytic phases of the EcoRI reaction, acting to change the specificity of the enzyme. For both the protein–DNA complex and the transition state, there may be substantial conformational differences between cognate and alternate sites, accompanied by significant alterations in hydration and solvent accessibility.
The EcoRI endonuclease cleaves double-stranded GAATTC sequences on both strands between G and A, at a rate at least 105 faster than the next best nucleic acid sequence as quantitated under standard conditions (1). EcoRI utilizes two α-helices to contact this cognate recognition sequence DNA in the major groove via an intricate network of hydrogen bonding interactions, which is thought to give rise to its high specificity (2, 3). The cleavage mechanism of this endonuclease is believed to involve activation of a water molecule by a basic group from the enzyme, followed by nucleophilic attack at the phosphodiester bond. The reaction is thought to involve only one step and is assisted by Mg2+ as cofactor. The rate-limiting step may be release of doubly cleaved product, release of nicked product followed by rebinding and second strand cleavage, or conformational changes in the enzyme–substrate complex (4, 5). Binding constants and catalytic rate constants have been established for all single base substitutions of the canonical sequence and are in good agreement with the structural model for recognition and specificity (1, 6).
Under certain buffer conditions, EcoRI cleavage is enhanced at noncognate DNA sequences, which differ from GAATTC by a single base pair (7–9). This change in EcoRI specificity has been termed EcoRI∗ (“star”) activity. Analogous effects have been observed in many other restriction enzyme systems and represent a challenge to our understanding of protein–DNA and enzyme–substrate recognition (10). Furthermore, knowledge of the origin of this phenomenon is important for effective use of EcoRI and other restriction enzymes in research, diagnostic, and forensic applications.
Of particular interest is the influence of bound water on site-specific recognition and cleavage of DNA by EcoRI and other enzymes. The importance of interstitial water molecules in mediating specificity of protein–DNA interactions has been clearly demonstrated by structural studies of the binding of the trp repressor to its target operator sequence (11–13). Additional examples of structural evidence for water-mediated contacts with DNA include the lac repressor complex (14), the Arc repressor (15), glucocorticoid receptor (16, 17), and the Hin recombinase (18).
In addition to observing structural roles for water by its presence in x-ray crystal or NMR structures, a number of investigators have recently examined the roles of water in macromolecular interaction, stability, and function. Release of bound water has been shown to accompany oxygen binding to hemoglobin (19), electron transfer in cytochrome c oxidase (20), glucose binding to hexokinase (21), DNA binding by the gal repressor (22) and EcoRI endonuclease (23, 36), stabilization of lysozyme, ribonuclease, and other proteins (24, 25), binding of cytochrome c to cytochrome c oxidase (26), turnover in the catalytic cycle of cytochrome P450cam (27), and activation of adenosine deaminase (28). It has also been demonstrated that binding of about 10 water molecules accompanies formation of the Hin recombinase–DNA complex (29).
In many cases these proposals that discretely bound water molecules play an integral part in the recognition process utilize application of osmotic stress to perturb protein conformation or binding equilibria. Because osmotic pressure (π) controls the activity of water in an aqueous compartment inaccessible to neutral solutes (osmolytes), osmotic stress induces the release of bound water from macromolecules into bulk solvent. Macromolecular conformations are thus shifted toward the state with the smallest volume, which is the state with the least amount of bound water. Therefore, in the simplest case, a correlation between osmotic pressure and a biomolecular process, such as ligand binding, is commonly interpreted as evidence for a role of bound water in the process (30).
These phenomena are not limited to those cases in which solutes are excluded from water in a cavity or pore. Macromolecular surfaces also appear to be “preferentially hydrated” (31) and to exclude neutral solutes or cosolvents (32). Thus solvent release from protein and DNA surfaces or clefts can be probed by changing water activity through application of osmotic stress.
Application of osmotic pressure is clearly a useful and versatile technique for studying the role of water in molecular recognition and is readily applied to the study of protein–DNA interactions (22, 23, 29, 33). Typically application of osmotic stress favors formation of the protein–DNA complex, because a sizable dehydration of both molecules accompanies binding (22, 23, 33). In the rare situations where water binding accompanies formation of the complex, osmotic stress weakens the interaction (29). In either case, correlating the change in equilibrium constant with osmotic pressure allows the change in volume and number of waters bound to be derived. It is important to note that not all waters that are affected by osmotic stress are likely to be well enough resolved to be detected either by x-ray crystallography (which depends on a high fractional occupancy) or NMR (which relies on a long residence time) (34). Osmotic stress is believed to be more sensitive than either of these methods for detecting functionally significant waters (35).
Previously we have shown that for EcoRI (36), BamHI, and PvuII (37) increases in osmotic pressure (by addition of neutral solutes and cosolvents) are uniformly and uniquely correlated with increases in cleavage at “star” sites. Changes in EcoRI specificity were measured in the presence of a wide range of cosolvents and neutral solutes, including glycerol, ethylene glycol, isopropyl alcohol, ethanol, dimethyl sulfoxide, N-methyl formamide, sucrose, and dextrose. We demonstrated that the effects of these compounds on EcoRI specificity are uniquely correlated to osmotic pressure; other solvent properties such as dielectric constant, viscosity, and water activity do not display uniform correlations with star activity for all compounds tested (36, 38). The effects of osmotic pressure can be reversed by application of hydrostatic pressure (37, 38). It was recently shown that binding of EcoRI to cognate DNA releases substantially fewer waters than binding to a poly(dI⋅dC) DNA (23). These findings indicate that release of bound water is associated with changes in the specificity of the enzymes and strongly suggest that a population of water molecules is involved in substrate recognition at the canonical sites. We examine here the mechanism of water participation in the interaction of EcoRI with cognate, alternate (“star”) sites and nonspecific DNA in an effort to determine the number of water molecules involved in the recognition process and the steps during which they act.
EXPERIMENTAL PROCEDURES
Proteins and DNA.
EcoRI purified essentially according to the method of Geiger et al. (39) was donated by Richard Gumport (College of Medicine, Univ. of Illinois, Urbana). EcoRI obtained from GIBCO/BRL gave equivalent results. Protein concentrations were determined by using an extinction coefficient of 8.3 at 278 nm according to Modrich and Zabel (5).
DNA oligonucleotides were synthesized by using an Applied Biosystems 381A DNA synthesizer with the trityl group on, purified by reverse phase HPLC, detritylated, and repurified by HPLC. End labeling of both single-stranded oligonucleotides with γ-32P-labeled ATP and T4 polynucleotide kinase (New England Biolabs) was carried out as described (40). The mixture was extracted with phenol–chloroform and passed over a Sephadex G-25 spin column to separate the labeled duplex from unincorporated nucleotides. Double-stranded cassettes were annealed by mixing equimolar amounts of the complementary oligonucleotides, heating to 90°C, and cooling slowly.
Sequences of the DNA cassettes used in binding and cleavage assays are as follows.
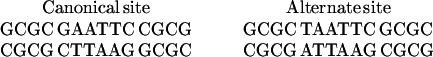 |
 |
DNA Binding Assays.
Equilibrium binding assays were performed essentially as described previously (40, 41). EcoRI was incubated with 32P-labeled DNA at 25°C for at least 2 h in a buffer containing 20 mM Tris⋅Cl (pH 7.5), 100 mM NaCl, 1 mM DTT, 0.1 mM EDTA, 100 μg/ml BSA, and 0.02% Nonidet P-40 nonionic detergent. Ethylene glycol was added to concentrations between 0 and 2 M to induce osmotic pressures between 0 and 50 atm (1 atm = 101.3 kPa) as described previously (22, 23, 29). The DNA fragments were used at a concentration of 50 fM. Protein concentrations were always at least 10-fold higher than the concentration of the DNA. A small amount of glycerol (equivalent to <5 atm osmotic pressure) was added just before loading the samples onto 8% polyacrylamide gels in 0.5× TBE buffer. Gels were dried and exposed by using Molecular Dynamics PhosphorImager screens, and band intensities quantitated with ImageQuant software by using the volume measurement utility. The fraction of DNA bound was determined as the volume of the slower migrating band (corresponding to bound DNA) divided by the sum of free and bound bands. Under the conditions used, the protein–nucleic acid complex migrated as a discrete, well resolved band. However, other buffers and conditions tested resulted in poorly resolved complexes or undetectable shifts, as described previously (23). Over the range of osmotic pressure tested, the appearance and mobility of the bound and free DNA were unchanged, and the total band intensity remained essentially constant.
The equilibrium dissociation constant (Kd) was determined essentially as described previously (42). Because EcoRI is always present in significant excess of the DNA, the concentration of free protein can be closely approximated by using the total protein concentration [Pt]. The binding isotherm simplifies to
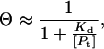 |
1 |
where Θ is the fraction of bound DNA and Kd is the equilibrium dissociation constant,
 |
Data were fit to Eq. 1 by nonlinear least-squares analysis with KaleidaGraph software running on a Macintosh computer.
DNA Cleavage Assays.
Reactions were performed at 25°C in a buffer containing 20 mM Tris⋅Cl (pH 7.5), 100 mM NaCl, 1 mM DTT, 0.1 mM EDTA, and 10 mM MgCl2 with the addition of 0–4 M ethylene glycol to induce osmotic pressures from 0 to 100 atm. At each osmotic pressure, reactions containing 1 or 10 μM 32P-labeled substrate DNA and EcoRI at concentrations between 0.1 and 1.0 μM (always at least 10 times higher than the Kd) were incubated at 25°C for 30 min to allow binding. Reactions were initiated by the addition of MgCl2 to the final concentration of 10 mM. Aliquots were removed at each time point, quenched by the addition of EDTA to 50 mM, SDS to 0.1%, and proteinase K to 0.5 mg/ml as described previously (6), and stored at −70°C until just before running on 8% acrylamide gels in 0.5× TBE buffer.
At each osmotic pressure, reaction progress curves were generated by measuring the appearance of product or disappearance of substrate at each time point with the PhosphorImager described above. Reaction velocities (vo) were determined with the initial linear part of the reaction progress curves according to vo = Fp⋅[S]o/t, where Fp is the fraction product, [S]o is the initial substrate concentration, and t is the time. Values for kcat were calculated with vo = kcat [ES], where [ES] is the concentration of the enzyme–substrate complex.
Volume and Solvation Changes.
Osmotic pressures were determined as described previously (36). Briefly, values for osmotic pressure (π) were calculated both from measured osmolalities (Os) by using a UIC 070 vapor phase osmometer and from tabulated data (43), according to the equation π = RT⋅Os (44).
Equilibrium volume changes (ΔV) and activation volumes (ΔV‡) were obtained from the slopes of plots of ln(Kd[π]/Kd[0]) vs. osmotic pressure and ln(kcat[π]/kcat[0]) vs. osmotic pressure, respectively, by applying the relationship
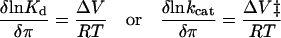 |
2 |
(where π is the osmotic pressure, R is the gas constant, and T is the temperature) according to Weber and Drickamer (45). ΔNw is defined as the number of water molecules expected to give rise to the volumes changes indicated, assuming a constant density of 18 ml/mol for all waters (30).
RESULTS
DNA Binding.
Equilibrium gel mobility shift analysis was performed to measure the affinity of EcoRI for canonical, alternate, and nonspecific DNA binding sites as a function of osmotic pressure. Binding isotherms were constructed, and the data were fit to Eq. 1 to obtain the equilibrium dissociation constant, Kd (Fig. 1). Values of Kd at ambient osmotic pressure were close to those obtained previously by other investigators (5, 46). As expected for a macromolecular process that involves a dehydration of a large surface area, binding was enhanced as osmotic pressure was increased. Under all conditions tested binding was always tightest to the cognate site, GAATTC. However, Kd for the “star” site TAATTC is more sensitive to osmotic pressure than is GAATTC. This can be seen in Fig. 1; the binding isotherm for TAATTC is moved more by 20 atm osmotic pressure than is the isotherm for GAATTC.
Figure 1.

Representative binding isotherms for EcoRI binding to DNA substrates containing the canonical recognition sequence GAATTC (circles) or the alternate sequence TAATTC (squares), at ambient osmotic pressure (filled symbols) or 20 atm osmotic pressure (open symbols). The symbols indicate the experimental data points. The lines represent the best theoretical fits to Eq. 1 as follows: GAATTC, 0 atm, Kd = 450 (±20) pM; TAATTC, 0 atm, Kd = 820 (±40) nM; GAATTC, 20 atm, Kd = 50 (±10) pM; TAATTC, 20 atm, Kd = 40 (±15) nM. In all experiments the DNA concentration was 50 fM. Binding reactions were performed at 25°C in a buffer containing 20 mM Tris⋅Cl (pH 7.5), 100 mM NaCl, 1 mM DTT, 0.1 mM EDTA, 100 μg/ml BSA, and 0.02% Nonidet P-40 nonionic detergent. Ethylene glycol was added to induce the osmotic pressure indicated.
As osmotic pressure is increased from 0 to 50 atm, the Kd for the canonical GAATTC site drops by about 200-fold (from 400 to 2 pM) (Table 1). Over the same pressure range, the Kd for the alternate TAATTC site falls by about 2000-fold (from 800 to 0.4 nM), whereas the Kd for the nonspecific site decreases by only 20-fold (Table 1). For each DNA binding site, the natural logarithm of the ratios of Kd at ambient and elevated osmotic pressures was plotted as a function of osmotic pressure, and the data were fit by linear regression (Fig. 2). From the slope of these lines, the volume changes (ΔV) for the dissociation of the EcoRI–DNA complexes were determined by using Eq. 2 (Table 2). Note that these calculations are performed with the dissociation constant, so that a ΔV less than zero indicates an increase in volume for the association reaction. Because the increase in osmotic pressure acts to promote release of water, these negative ΔV values correspond to the volume change because of solvent release. Assuming a constant volume for water molecules of 18 ml/mol (30), the number of water molecules was calculated based on these ΔV values (Table 2). ΔV for binding to the cognate GAATTC is about twice as large as for nonspecific binding, whereas ΔV for binding to TAATTC is about three times larger than nonspecific, i.e., 50% larger than for binding to the canonical site. About 70 waters are released when EcoRI binds nonspecifically, compared with nearly 150 for binding to GAATTC and over 200 for binding to TAATTC.
Table 1.
Equilibrium dissociation constants (Kd) for EcoRI–DNA complexes
| Osmotic pressure, atm | Canonical site GAATTC, pM | Alternate site TAATTC, nM | Nonspecific site TAGACG, μM |
|---|---|---|---|
| 0 | 400 (±100) | 840 (±430) | 8 (±4) |
| 10 | 100 (±30) | 300 (±150) | 5 (±3) |
| 20 | 60 (±40) | 40 (±20) | 3 (±2) |
| 30 | 20 (±10) | 9 (±6) | 1.1 (±0.8) |
| 40 | 4 (±3) | 1.0 (±0.4) | 0.9 (±0.5) |
| 50 | 2 (±1) | 0.4 (±0.2) | 0.4 (±0.2) |
Kd values are the average of at least five independent determinations from experiments like those shown in Fig. 1. Errors indicate 1 SD.
Figure 2.

Dependence of dissociation constant (Kd) on osmotic pressure for EcoRI binding to canonical GAATTC (circles), alternate TAATTC (squares), and nonspecific (triangles) DNA substrates. Symbols indicate the natural log of the ratios of Kd values at 0 and the indicated osmotic pressure (π); the lines represent the best fits by linear regression. Slopes are: GAATTC, −0.108 (±0.003) atm−1; TAATTC, −0.159 (±0.005) atm−1; nonspecific, −0.056 (±0.003) atm−1.
Table 2.
Volume and solvation changes
| Substrate | Dissociation, Kd
|
Turnover, kcat
|
||
|---|---|---|---|---|
| ΔV, ml/mol | ΔNw | ΔV‡, ml/mol | ΔNw | |
| Canonical site, GAATTC | −2640 ± 70 | −146 ± 4 | −490 ± 20 | −27 ± 1 |
| Alternate site, TAATTC | −3880 ± 120 | −216 ± 7 | 760 ± 20 | 42 ± 1 |
| Nonspecific site, TAGACG | −1370 ± 70 | −76 ± 4 | ||
Values for ΔV are the volume changes for dissociation and turnover calculated from Eq. 2 based upon the data in Fig. 2 and 4. Values for ΔNw are the calculated change in number of waters bound based upon the volume changes and a volume of 18 ml/mol per water molecule. Since Kd is the dissociation equilibrium constant, negative values indicate a positive volume change (water release) accompanying complex formation. Negative values for the kcat data indicate a negative volume change (water binding) during catalysis.
DNA Cleavage.
When the substrate contained the cognate sequence GAATTC, the reaction rate fell sharply as osmotic pressure was increased, whereas for the TAATTC substrate the reaction rate increased dramatically with increasing osmotic pressure (Fig. 3). Reaction progress curves were generated at osmotic pressures between 0 and 100 atm. The reaction velocities (vo) were determined from the slope of the initial linear portion, and the turnover number (kcat) was calculated (Table 3).
Figure 3.

Representative initial kinetic profiles for EcoRI cleavage of DNA substrates containing the canonical recognition sequence GAATTC (circles) or the alternate sequence TAATTC (squares), at ambient osmotic pressure (filled symbols) or 100 atm osmotic pressure (open symbols). The symbols indicate the experimental data points. The lines indicate the best fits by linear regression with slopes as follows: GAATTC, 0 atm, 0.49 (±0.02) μM/min; TAATTC, 0 atm, 0.11 (±0.01) μM/min; GAATTC, 100 atm, 0.073 (±0.002) μM/min; TAATTC, 100 atm, 0.192 (±0.003) μM/min. Reactions were performed at 25°C in a buffer containing 20 mM Tris⋅Cl (pH 7.5), 100 mM NaCl, 1 mM DTT, 0.1 mM EDTA, and 10 mM MgCl2. The enzyme and DNA concentrations were 0.1 and 1 μM, respectively, except for TAATTC at 0 atm, where the concentrations were 1 and 10 μM, respectively.
Table 3.
Turnover numbers (kcat) for EcoRI cleavage of canonical and alternate site DNA
| Osmotic pressure, atm | Canonical site GAATTC, min−1 | Alternate site TAATTC, min−1 |
|---|---|---|
| 0 | 5 (±2) | 0.10 (±0.05) |
| 20 | 3.3 (±1.1) | 0.21 (±0.08) |
| 40 | 2.2 (±1.0) | 0.3 (±0.1) |
| 60 | 1.2 (±0.5) | 0.6 (±0.1) |
| 80 | 0.9 (±0.3) | 1.5 (±0.3) |
| 100 | 0.89 (±0.2) | 2.0 (±0.3) |
Turnover numbers are the average of at least three independent determinations from experiments like those shown in Fig. 3. Errors indicate 1 SD.
Values of kcat at 0 atm osmotic pressure were close to those obtained previously by other investigators under similar conditions (5, 46). No discontinuities were observed in the reaction rate profiles, suggesting that for a given substrate, the rate-limiting step does not change over this range of osmotic pressures. Throughout the range of the modest osmotic pressures used in this study (0–100 atm), complete cleavage was obtained for both canonical and star sites. No evidence for enzyme degradation was found. Doubling the substrate concentration or increasing the preincubation time did not produce any change in the velocity.
From 0 to 100 atm, kcat for the GAATTC site decreases by more than 5-fold (from 5 to 0.8 min−1), although it rises 20-fold for the TAATTC site (from 0.1 to 2 min−1) (Table 3). For each substrate, the natural logarithm of the ratios of kcat at ambient and elevated osmotic pressures was plotted as a function of osmotic pressure, and the data were fit by linear regression (Fig. 4). From the slope of these lines, the activation volume (ΔV‡) for the EcoRI cleavage reaction was determined with Eq. 2 (Table 2). Cleavage of the cognate GAATTC site has a negative ΔV‡ value, corresponding to the binding of about 30 waters. Conversely, cleavage at TAATTC sites is accompanied by a large positive activation volume, corresponding to over 40 additional waters being released.
Figure 4.

Dependence of turnover number (kcat) on osmotic pressure for EcoRI cleavage of canonical GAATTC (circles) and alternate TAATTC (squares) substrates. Symbols indicate the natural log of the ratios of kcat values at 0 and the indicated osmotic pressure (π); the lines represent the best fits by linear regression. Slopes are: GAATTC, −0.020 (±0.001) atm−1; TAATTC, 0.031 (±0.001) atm−1.
DISCUSSION
Solvation Changes During Binding.
Sizable volume changes are observed when EcoRI binds to nonspecific, canonical, and alternate sequences. Accordingly, binding to all three DNA molecules involves the release of large amounts of water, and increases in osmotic pressure cause an increase in overall affinity. The crystal structure of EcoRI bound to cognate DNA indicates that over 1000 Å2 of surface area must be buried on complex formation (3), consistent with our calculation of a large negative ΔV and the release of roughly 150 waters. These values are similar to ΔVs measured by hydrostatic and osmotic pressure methods for other protein–DNA binding events and formation of other macromolecular assemblies (22, 23, 29, 45, 47, 48). It has been generally believed that for formation of macromolecular complexes, tighter binding is usually accompanied by a greater extent of dehydration (45, 49). We find that high affinity binding of EcoRI to its cognate GAATTC site is accompanied by a larger volume change and more water release than the low affinity, nonspecific binding to the TAGACG sequence. Similarly, Sidorova and Rau (23) have recently demonstrated that EcoRI binding to DNA fragments containing a GAATTC sequence releases 110 more waters than binding to poly[dI⋅dC] DNA, suggesting that a full layer of hydration is retained.
However, the surprising result of this investigation is that binding of the noncognate TAATTC site releases 70 more waters than binding to the cognate GAATTC site. Although the GAATTC site is bound most tightly under all conditions, binding to the TAATTC site releases more water. Importantly, we find that the extent of dehydration does not always correlate with the overall affinity.
Although it is possible that the change of 1 base pair results in changes in the extent to which the free DNA molecules are hydrated, we feel that the most plausible explanation for this difference is that there are approximately 70 waters in the EcoRI–GAATTC cognate complex that are not present in the complex with TAATTC (Fig. 5). This feature of the EcoRI–DNA recognition suggests a role for a population of bound waters in assisting discrimination of cognate from noncognate DNA sequences, even those differing by a single base pair.
Figure 5.

Schematic diagram of EcoRI–DNA complexes. EcoRI is depicted as black ellipses, DNA is depicted as a white box, and water molecules are depicted as Xs. Vertical displacement corresponds to the number of waters bound. Free protein and DNA are fully hydrated (Left), formation of EcoRI–DNA complexes (governed by Kd) releases small, modest, or large amounts of water depending on the DNA sequence (Center), and formation of the transition state (governed by kcat) is accompanied by binding of water for the cognate site or release of water for “star” sites (Right).
Whereas the major component of binding free energy for both sites is likely to be entropic (50, 51), in the absence of a high-resolution structural model for the EcoRI–TAATTC complex, we can only say that the relatively small differences in energetics of binding to GAATTC and TAATTC must have many (offsetting) components (1). For example, the EcoRI–TAATTC complex has nonoptimal base contacts but appears to release more solvent. As osmotic pressure increases, the energetic consequences of this feature become more pronounced so that the affinity of the TAATTC site begins to approach that of the preferred GAATTC sequence. Extrapolation of the data in Table 1 suggests that at osmotic pressures greater than 150 atm, the Kd for the TAATTC binding site would be lower than that for the cognate GAATTC site, although for practical reasons this is difficult to test.
We cannot currently distinguish between several possible structural models for origins of the water participation in binding, and the large number of waters suggests that more than one effect may be responsible. The schematic diagram in Fig. 5 is meant to reflect each of the following possible models. (i) Effects on DNA conformation and flexibility may arise from water release, including changes in hydration of the minor groove (52). These effects could be crucial to the facilitated distortion of DNA that is believed to play a major role in EcoRI–DNA recognition (53). (ii) Bound waters may mediate protein–protein contacts and dimer formation, as suggested by the recently revised model of the EcoRI–DNA complex (J. M. Rosenberg, personal communication). (iii) Water located in the protein–DNA interface may participate in an “indirect readout” of the DNA base sequence (13). The crystal structure of the EcoRI–DNA complex does reveal two bound waters mediating hydrogen-bonding interactions between EcoRI and the guanine base in each half-site (3). However, such waters are likely to be among the most tightly bound and thus the most difficult to release by osmotic pressure. (iv) The complex of EcoRI with TAATTC may have less solvent-accessible surface area relative to GAATTC. This could result from significant changes in the protein and/or DNA conformation or from an increase in the extent of local folding, which occurs for binding at the TAATTC site relative to binding at GAATTC. Changes in the heat capacity associated with EcoRI binding to its canonical recognition sequence (54) indicate that a coupling exists between local folding of EcoRI, water release, and DNA binding (55). The results presented here are consistent with a model in which either binding or local folding, or both, removes hydrophobic surfaces from water and releases solvent molecules.
Intriguingly, preliminary reports of the structure of EcoRI bound to DNA containing a noncanonical site describe important conformational differences in the structure of the EcoRI dimer and its orientation on DNA, which could be accommodated by many single base substitutions (J. M. Rosenberg, personal communication).
How do these features of the EcoRI system affect its function? EcoRI is believed to search for its cognate sequence by a processive one-dimensional translation along DNA, termed “linear diffusion.” Analysis of EcoRI during linear diffusion on the DNA helix suggests that the diffusing species corresponds to the nonspecifically bound complex (56). Taken together, these studies suggest that EcoRI can bind to DNA in three distinct modes: a loose complex competent for diffusion that releases less than 100 waters, a tight complex at GAATTC sites that releases about 150 waters, and an alternate complex formed at TAATTC and other possibly other “star” sites that releases over 200 waters and is competent for catalysis of the endonuclease reaction.
Solvation Changes During Catalysis.
The question of whether protein conformational changes occur during EcoRI catalysis has been unresolved for several years (10, 57, 58). The magnitude of the activation volumes for catalysis at both GAATTC and TAATTC sites clearly indicates that significant conformational changes do occur during the catalytic steps of the reaction (48, 59). These conformational changes are accompanied by large changes in solvation and are almost certainly in the catalytic steps because the reactions are initiated with enzyme–substrate complex already formed. Conformational changes associated with the specific process of magnesium ion binding are believed to be small, because Mg2+ can be diffused into crystals of the enzyme–substrate complex without cracking the crystal (3).
Increased osmotic pressure slows the reaction at the cognate site but accelerates the rate at the alternate site. This indicates that formation of the transition state is accompanied by changes in solvation, which are of opposite directions for the two sequences. Water binding accompanies formation of the transition state for GAATTC, whereas water release is associated with formation of the transition state for TAATTC cleavage (Fig. 5). Thus it appears that additional waters participate in one or more of the catalytically important species for the GAATTC site, which are excluded from the catalytic complex at TAATTC (Fig. 5).
Previously it has been proposed that EcoRI∗ activity (i.e., cleavage by the enzyme at noncanonical sites) arose from a global increase in affinity of the enzyme for all DNA or a hyperactivation of the enzyme at all sites (60). However, the decrease in kcat at the canonical site indicates that a fundamental change in the selectivity of the enzyme is induced by osmotic pressure. The results described here indicate that water release makes EcoRI less efficient at cutting its cognate DNA sequence, although it enhances its preference for cutting alternate sequences.
Analysis of these effects is somewhat hampered by the limited understanding of the EcoRI mechanism. It is not known for certain which step in the EcoRI DNA cleavage reaction is rate-limiting: release of doubly cleaved product or release of nicked product followed by rebinding and second strand cleavage (57). The marked difference in direction of the volume changes for kcat at canonical and alternate sites might indicate a change in the rate-limiting step. For cleavage at the canonical site, the negative volume change accompanied by water binding is consistent with a rate-limiting step being product release or transition from the tightly bound to loosely bound mode. For cleavage at the TAATTC “star” site, the positive volume change accompanied by water release is consistent with the rate-limiting step being rebinding of the nicked substrate after first strand cleavage, or transition from loosely bound to tightly bound mode, or from the tight complex to the alternate complex as described earlier.
Alternatively, it may be that the nature of the rate-limiting step and the position of the transition state on the reaction coordinate are unchanged, but the structure of the macromolecular complex is different. However, the magnitudes of the ΔV‡s suggest that the conformational changes that occur are between relatively similar states, not complete binding or release of the substrate or products.
Summary.
We have shown that changes in the amount of bound water accompany DNA binding and catalysis by the EcoRI endonuclease. As depicted in Fig. 5, EcoRI binding nonspecifically to DNA releases much less water than binding to the cognate GAATTC site, whereas binding to the alternate TAATTC site releases substantially more. Even more dramatically, cleavage at the GAATTC site is inhibited by water release, whereas cleavage at TAATTC is promoted (Fig. 5). These findings suggest that in the EcoRI system the extent of bound water is critical to binding affinity, sequence-specific recognition, and site discrimination during cleavage.
For enzyme–substrate complexes, one measure of accuracy of recognition is the specificity constant kcat/Kd. The ratio of specificity constants of canonical vs. alternate site decreases by 60-fold, from 105 to 1800 over the range of 0–40 atm osmotic pressure. Although EcoRI still prefers the canonical site at 40 atm, the release of solvent does cause a dramatic change in specificity, increasing cleavage efficiency at TAATTC while decreasing the efficiency at GAATTC. Extrapolating this trend, at about 100 atm EcoRI should cut both sequences with equal efficiency; above this osmotic pressure it should actually prefer the alternate TAATTC site over its canonical site.
In addition to increased osmotic pressure, many other conditions can cause EcoRI∗ activity, such as the presence of other divalent cations or changes in ionic strength or pH. It is not known whether the molecular events leading to altered specificity under these conditions are the same as those arising from release of bound waters (61). Certainly in complex macromolecular systems, many different effectors with different atomic and molecular origins can produce similar observable effects, as is the case with oxygen binding by hemoglobin.
Whereas release of water has been shown to accompany molecular complexation in many systems, including protein–DNA complexes, the ability of bound water to switch specificity in an enzyme–substrate or protein–DNA complex is remarkable. Moreover, it appears to be a common motif within this structural family as BamHI endonuclease displays similar behavior as EcoRI. It is hoped that further biochemical and biophysical characterizations and more detailed structural analysis will reveal the exact location and structural and energetic role of these waters. In turn, such studies should elucidate the mechanism by which water binding and release exert control over critical aspects of the molecular recognition process.
Acknowledgments
We thank Profs. Richard Gumport, Gregorio Weber, and Pierre Douzou, Drs. Anne Skaja Robinson and Eric Fisher, and Mr. Jason Rockhill for helpful discussions and advice. We also thank the laboratory of Prof. Richard Gumport for the generous gift of purified EcoRI endonuclease, Profs. Robert Sauer and Steven Zimmerman for the use of equipment, and Ms. Aretta Weber for assistance in preparation of this manuscript. This work was supported by National Institutes of Health Grants GM31756 and GM33775 (to S.G.S.) and National Institutes of Health National Research Service Award in Molecular Biophysics Grant GM08276 (to C.R.R.).
References
- 1.Lesser D R, Kurpiewski M R, Jen-Jacobson L. Science. 1990;250:776–786. doi: 10.1126/science.2237428. [DOI] [PubMed] [Google Scholar]
- 2.Kim Y, Grable J C, Love R, Greene P J, Rosenberg J M. Science. 1990;249:1307–1309. doi: 10.1126/science.2399465. [DOI] [PubMed] [Google Scholar]
- 3.Rosenberg J. Curr Opin Struct Biol. 1991;1:104–113. [Google Scholar]
- 4.Hedgpeth J, Goodman H M, Boyer H W. Proc Natl Acad Sci USA. 1972;69:3448–3452. doi: 10.1073/pnas.69.11.3448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Modrich P, Zabel D. J Biol Chem. 1976;251:5866–5874. [PubMed] [Google Scholar]
- 6.Thielking V, Alves J, Fleiss A, Maass G, Pingoud A. Biochemistry. 1990;29:4682–4691. doi: 10.1021/bi00471a024. [DOI] [PubMed] [Google Scholar]
- 7.Hsu M-T, Berg P. Biochemistry. 1978;17:131–138. doi: 10.1021/bi00594a019. [DOI] [PubMed] [Google Scholar]
- 8.Gardner R C, Howarth A J, Messing J, Shepherd R J. DNA (N Y) 1982;1:109–115. doi: 10.1089/dna.1.1982.1.109. [DOI] [PubMed] [Google Scholar]
- 9.Rosenberg J M, Greene P. DNA (N Y) 1982;1:117–124. doi: 10.1089/dna.1.1982.1.117. [DOI] [PubMed] [Google Scholar]
- 10.Bennet S P, Halford S E. Curr Top Cell Regul. 1989;30:57–104. doi: 10.1016/b978-0-12-152830-0.50005-0. [DOI] [PubMed] [Google Scholar]
- 11.Carey J, Lewis D E A, Lavoie T A, Yang J. J Biol Chem. 1991;266:24509–24513. [PubMed] [Google Scholar]
- 12.Lawson C L, Carey J. Nature (London) 1993;366:178–182. doi: 10.1038/366178a0. [DOI] [PubMed] [Google Scholar]
- 13.Otwinowski Z, Schevitz R W, Zhang R-G, Lawson C L, Joachimiak A, Marmorstein R Q, Luisi B F, Sigler P B. Nature (London) 1988;335:321–329. doi: 10.1038/335321a0. [DOI] [PubMed] [Google Scholar]
- 14.Chuprina V P, Rullmann J A, Lamerichs R M, van Boom J H, Boelens R, Kaptein R. J Mol Biol. 1993;234:446–462. doi: 10.1006/jmbi.1993.1598. [DOI] [PubMed] [Google Scholar]
- 15.Raumann B E, Rould M A, Pabo C O, Sauer R T. Nature (London) 1994;367:754–757. doi: 10.1038/367754a0. [DOI] [PubMed] [Google Scholar]
- 16.Luisi B F, Xu W X, Otwinowski Z, Freedman L P, Yamamoto K R, Sigler P B. Nature (London) 1991;352:497–505. doi: 10.1038/352497a0. [DOI] [PubMed] [Google Scholar]
- 17.Gewirth D T, Sigler P B. Nat Struct Biol. 1995;2:386–394. doi: 10.1038/nsb0595-386. [DOI] [PubMed] [Google Scholar]
- 18.Feng J A, Johnson R C, Dickerson R E. Science. 1994;263:348–355. doi: 10.1126/science.8278807. [DOI] [PubMed] [Google Scholar]
- 19.Colombo M F, Rau D C, Parsegian V A. Science. 1992;256:655–659. doi: 10.1126/science.1585178. [DOI] [PubMed] [Google Scholar]
- 20.Kornblatt J A, Hui Bon Hoa G. Biochemistry. 1990;29:9370–9376. doi: 10.1021/bi00492a010. [DOI] [PubMed] [Google Scholar]
- 21.Rand R P, Fuller N L, Butko P, Francis G, Nicholls P. Biochemistry. 1993;32:5925–5929. doi: 10.1021/bi00074a001. [DOI] [PubMed] [Google Scholar]
- 22.Garner M M, Rau D C. EMBO J. 1995;14:1257–1263. doi: 10.1002/j.1460-2075.1995.tb07109.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sidorova N Y, Rau D C. Proc Natl Acad Sci USA. 1996;93:12272–12277. doi: 10.1073/pnas.93.22.12272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Santoro M M, Liu Y, Khan S M A, Hou L-X, Bolen D W. Biochemistry. 1992;31:5278–5283. doi: 10.1021/bi00138a006. [DOI] [PubMed] [Google Scholar]
- 25.Arakawa T, Timasheff S A. Biochemistry. 1982;21:6536–6544. doi: 10.1021/bi00268a033. [DOI] [PubMed] [Google Scholar]
- 26.Kornblatt J A, Kornblatt M J, Hui Bon Hoa G, Mauk A G. Biophys J. 1993;65:1059–1065. doi: 10.1016/S0006-3495(93)81168-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DiPrimo C, Sligar S G, Hui Bon Hoa G, Douzou P. FEBS Lett. 1992;312:252–254. doi: 10.1016/0014-5793(92)80946-e. [DOI] [PubMed] [Google Scholar]
- 28.Dzingeleski G D, Wolfenden R. Biochemistry. 1993;32:9143–9147. doi: 10.1021/bi00086a020. [DOI] [PubMed] [Google Scholar]
- 29.Robinson C R, Sligar S G. Protein Sci. 1996;5:2119–2124. doi: 10.1002/pro.5560051019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rand R P. Science. 1992;256:618. doi: 10.1126/science.256.5057.618. [DOI] [PubMed] [Google Scholar]
- 31.Timasheff S N. Biochemistry. 1992;31:9857–9864. doi: 10.1021/bi00156a001. [DOI] [PubMed] [Google Scholar]
- 32.Kuhn L A, Siani M A, Pique M E, Fisher C L, Getzoff E D, Tainer J A. J Mol Biol. 1992;228:13–22. doi: 10.1016/0022-2836(92)90487-5. [DOI] [PubMed] [Google Scholar]
- 33.Sidorova N Y, Rau D. Biopolymers. 1995;35:377–384. doi: 10.1002/bip.360350405. [DOI] [PubMed] [Google Scholar]
- 34.Zhang X-J, Matthews B W. Protein Sci. 1994;3:1031–1039. doi: 10.1002/pro.5560030705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parsegian V A, Rand R P, Fuller N L, Rau D C. Methods Enzymol. 1986;127:400–437. doi: 10.1016/0076-6879(86)27032-9. [DOI] [PubMed] [Google Scholar]
- 36.Robinson C R, Sligar S G. J Mol Biol. 1993;234:302–306. doi: 10.1006/jmbi.1993.1586. [DOI] [PubMed] [Google Scholar]
- 37.Robinson C R, Sligar S G. Proc Natl Acad Sci USA. 1995;92:3444–3448. doi: 10.1073/pnas.92.8.3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robinson C R, Sligar S G. Biochemistry. 1994;33:3787–3793. doi: 10.1021/bi00179a001. [DOI] [PubMed] [Google Scholar]
- 39.Geiger R, Rüter T, Alves J, Fleiss A, Wolfes H, Pingoud V, Urbanke C, Maass G, Pingoud A, Düsterhöft A, Kröger M. Biochemistry. 1989;28:2667–2677. doi: 10.1021/bi00432a046. [DOI] [PubMed] [Google Scholar]
- 40.Brown B M, Sauer R T. Biochemistry. 1993;32:1354–1363. doi: 10.1021/bi00056a022. [DOI] [PubMed] [Google Scholar]
- 41.Fried M. Electrophoresis. 1989;10:366–376. doi: 10.1002/elps.1150100515. [DOI] [PubMed] [Google Scholar]
- 42.Jen-Jacobsen L, Kurpiewski M, Lesser D, Grable J, Boyer H W, Rosenberg J M, Greene P J. J Biol Chem. 1983;258:14638–14646. [PubMed] [Google Scholar]
- 43.Weast R C, editor. CRC Handbook of Chemistry and Physics. Boca Raton, FL: CRC; 1985. pp. E368–E369. [Google Scholar]
- 44.Tombs M P, Peacocke A R. The Osmotic Pressure of Biological Macromolecules. New York: Oxford Univ. Press; 1974. [Google Scholar]
- 45.Weber G, Drickamer H G. Q Rev Biophys. 1983;16:89–112. doi: 10.1017/s0033583500004935. [DOI] [PubMed] [Google Scholar]
- 46.Halford S E, Johnson N P. Biochem J. 1981;199:767–777. doi: 10.1042/bj1990767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Silva J L, Weber G. Annu Rev Physical Chemistry. 1993;44:89–113. doi: 10.1146/annurev.pc.44.100193.000513. [DOI] [PubMed] [Google Scholar]
- 48.Robinson C R, Sligar S G. Methods Enzymol. 1995;259:395–427. doi: 10.1016/0076-6879(95)59054-4. [DOI] [PubMed] [Google Scholar]
- 49.Parsegian V A, Rand R P, Rau D C. Methods Enzymol. 1995;259:43–94. doi: 10.1016/0076-6879(95)59039-0. [DOI] [PubMed] [Google Scholar]
- 50.Foguel D, Silva J L. Proc Natl Acad Sci USA. 1994;91:8244–8247. doi: 10.1073/pnas.91.17.8244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Terry B J, Jack W E, Rubin R A, Modrich P. J Biol Chem. 1983;258:9820–9825. [PubMed] [Google Scholar]
- 52.Liepinsh E, Otting G, Wuthrich K. Nucleic Acids Res. 1992;20:6549–6557. doi: 10.1093/nar/20.24.6549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lesser D R, Kurpiewski M R, Waters T, Connolly B A, Jen-Jacobsen L. Proc Natl Acad Sci USA. 1993;90:7548–7552. doi: 10.1073/pnas.90.16.7548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ha J H, Spolar R S, Record M T., Jr J Mol Biol. 1989;209:801–816. doi: 10.1016/0022-2836(89)90608-6. [DOI] [PubMed] [Google Scholar]
- 55.Spolar R S, Record M T J. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 56.Jeltsch A, Alves J, Wolfes H, Maass G, Pingoud A. Biochemistry. 1994;33:10215–10219. doi: 10.1021/bi00200a001. [DOI] [PubMed] [Google Scholar]
- 57.Heitman J. BioEssays. 1992;14:445–454. doi: 10.1002/bies.950140704. [DOI] [PubMed] [Google Scholar]
- 58.Halford S E. Trends Biochem Sci. 1983;8:455–460. [Google Scholar]
- 59.Royer C A. Methods Enzymol. 1995;259:357–377. doi: 10.1016/0076-6879(95)59052-8. [DOI] [PubMed] [Google Scholar]
- 60.Nasri M, Thomas D. Biomed Biochim Acta. 1986;45:997–1005. [PubMed] [Google Scholar]
- 61.Pingoud A, Jeltsch A. Eur J Biochem. 1997;246:1–22. doi: 10.1111/j.1432-1033.1997.t01-6-00001.x. [DOI] [PubMed] [Google Scholar]