

Abstract
Mice deficient in c‐jun‐NH2‐terminal kinase 1 (JNK1) exhibit decreased fasting blood glucose and insulin levels, and protection against obesity‐induced insulin resistance, suggesting increased glucose disposal into skeletal muscle. Thus, we assessed whether JNK1 deficiency enhances muscle glucose metabolism. Ex vivo insulin or contraction‐induced muscle [³H]‐2‐deoxyglucose uptake was not altered in JNK1 knockout mice, demonstrating that JNK1 does not regulate blood glucose levels via direct alterations in muscle. In vivo muscle [³H]‐2‐deoxyglucose uptake in response to a glucose injection was also not enhanced by JNK1 deficiency, demonstrating that a circulating factor was not required to observe altered muscle glucose uptake in the knockout mice. JNK1 deficiency did not affect muscle glycogen levels or the protein expression of key molecules involved in glucose metabolism. This study is the first to directly demonstrate that enhanced skeletal muscle glucose metabolism does not underlie the beneficial effects of JNK1 deficiency in lean mice.
Keywords: knockout, mouse, glucose uptake, c‐jun‐NH2‐terminal kinase, glycogen
INTRODUCTION
Type 2 diabetes is associated with a chronic inflammatory response characterized by the increased production of stress‐inducing molecules, such as tumor necrosis factor alpha (TNF‐α) and free fatty acids [1–3]. Studies have suggested that this increase in inflammatory mediators may underlie the development of insulin resistance in type 2 diabetes [4;5] as the activation of stress responsive kinases has been shown to negatively impact the insulin‐induced activation of signaling proteins such as insulin receptor substrate‐1 (IRS‐1) [3;6] and Akt [3], and impair glucose uptake [3;7]. The mechanisms and/or signaling proteins involved in the impairment of insulin signaling by inflammatory mediators are still unclear.
The c‐jun‐NH2‐terminal kinase (JNK) is a member of the stress‐activated protein kinase family that is stimulated by a wide variety of cellular stresses including stretch [8;9], cytokines [10–16] and free fatty acids [3]. Studies from JNK1 knockout (JNK1 KO) mice [6], and mice overexpressing the endogenous JNK‐interacting protein‐1 (JIP‐1) [17], have suggested that JNK1 may play an important role in the development of obesity‐induced insulin resistance in inflammatory states. In lean mice, JNK1 deficiency and/or inhibition of JNK activity results in a significant decrease in fasted blood glucose [6], non‐fasted blood glucose [17], and fasted blood insulin levels [6;17]. In addition, both JNK1 KO and JIP‐1 overexpressing mice fed a high fat diet were protected against high fat diet‐induced insulin resistance [6;17]. Collectively, these results suggest that inhibition of JNK1 signaling prevents the dysregulation of blood glucose levels by increasing blood glucose disposal. Direct assessment of glucose disposal in JIP‐1 overexpressing mice demonstrates that inhibition of JNK activity increases the rate of glucose disappearance from the blood [17], demonstrating enhanced glucose disposal into peripheral tissues with JNK inhibition.
Skeletal muscle is the primary tissue responsible for blood glucose disposal, and JNK1 is expressed in skeletal muscle. Thus, it seems likely that the primary mechanism underlying the beneficial effects of JNK1 deficiency and/or JIP‐1 overexpression on the regulation of blood glucose levels is the enhancement of skeletal muscle glucose metabolism. However, to date, no studies have directly examined the effect of JNK1 ablation or inhibition of JNK activity on skeletal muscle glucose metabolism. Thus, the goal of this study was to determine whether glucose metabolism was enhanced in skeletal muscle from lean JNK1 KO mice.
MATERIALS AND METHODS
JNK1 knockout mice
All experiments were performed in accordance with the Institutional Animal Care and Use Committee of the Joslin Diabetes Center and the National Institutes of Health guidelines for the care and use of laboratory animals. The generation of jnk1−/− mice was previously described [18]. Heterozygous JNK1 mice were intercrossed to produce jnk1+/+ and jnk1−/− mice. To produce sufficient numbers of knockout mice, jnk1−/− × jnk1−/− intercrosses were also performed. In these cases, age‐ and sex‐matched wild‐type mice were used as controls. All experimental animals were backcrossed six generations to C57BI/6J mice. Mice were housed at a constant temperature (20–22°C) with a 12 hr light/dark cycle. LabDiet® rodent chow (Purina Mills Inc, St. Louis, MO) and water were available ad libitum.
Body weight and fasting blood glucose measurements were obtained in the morning following a 12–14 hr fast. Body weight values were obtained using a digital scale (Model CS200, Ohaus Corporation USA). Blood samples were obtained from the tail of fully conscious mice, and blood glucose levels were determined using a One Touch Ultra portable glucometer (Lifescan Inc, Mipitas, CA ).
Measurement of Skeletal Muscle [³H]‐2‐Deoxyglucose Uptake Ex Vivo
Ex vivo skeletal muscle incubation experiments were performed on male mice, aged 22–24 weeks, as previously described [19;20]. Briefly, mice were fasted overnight (12–14 hrs), and sacrificed by cervical dislocation. The extensor digitorum longus (EDL) and soleus muscles were rapidly removed and placed in 6 ml of oxygenated Krebs‐Ringer‐Bicarbonate (KRB) solution containing (in mM): 117 NaCl, 4.7 KCl, 2.5 CaCl2•2H2O, 1.2 KH2PO4, 1.2 MgSO4•7H2O, 24.6 NaHCO3, pH 7.5 supplemented with 2 mM pyruvic acid.
For insulin experiments, muscles were incubated for 40 min in KRB + pyruvic acid prior to a 20 min incubation with insulin (300, 600, or 50,000 µU/ml). For contraction experiments, muscles were incubated for 30 min in KRB + pyruvic acid and then electrically stimulated for 10 min. For contraction, muscles were transferred to a tissue support with stimulating electrodes (Harvard Apparatus, Holliston, MA), and resting tension was set to 0.5 g. Muscles were electrically stimulated with a Grass stimulator (Model S88, Grass Instruments, Quincy, MA) set to the following parameters: train rate = 2/min; train duration = 10 s; pulse rate = 100 pulses/s; duration = 0.1 ms; volts = 100 V. Force production during the contraction was monitored using an isometric force transducer (Kent Scientific, Litchfield, CT) and the converted digital signal captured by a data acquisition system (iWorx114, CB Sciences, Dover, NH). Force production was assessed with data analysis software (Labscribe, CB Sciences, Dover, NH).
For glucose uptake measurements, muscles were transferred to vials containing 2 ml KRB solution supplemented with 1.5 µCi/ml [³H]‐2‐deoxyglucose, 1 mM glucose, 0.45 µCi/ml [14C]‐mannitol, and 7 mM cold‐mannitol, and the appropriate amount of insulin. Muscles were dipped in ice‐cold KRB solution to terminate the glucose uptake, and frozen in liquid nitrogen. Frozen muscles were weighed and solubilized in 1M NaOH at 80°C. Solubilized muscles were neutralized with 1M HCl. Non‐soluble particulates precipitated by centrifugation at 13,000 × g for 1 min. Radioactivity in the samples was assessed by liquid scintillation counting for the dual labels, and the extracellular and intracellular spaces calculated to determine glucose uptake.
Glucose uptake in vivo
Skeletal muscle glucose uptake in vivo was measured as previously described [21]. Briefly, mice were fasted overnight and then anesthetized with nembutal sodium (100 mg/kg mouse body weight, intraperitoneal injection). After 30 min blood was taken from the tail to assess basal glucose and background radioactivity levels. A bolus of 1 mg glucose (0.33 µCi [³H]‐2‐dexoyglucose) / g mouse body weight, was administered via a retro‐orbital injection and blood samples taken 5, 10, 15, 25, 35 and 45 min later for the determination of glucose and [³H] levels. After the last blood draw, animals were sacrificed by cervical dislocation and the tibialis anterior muscles harvested. Muscles were immediately frozen in liquid nitrogen.
Accumulation of [³H]‐2‐dexoyglucose was assessed in muscles using a perchloric acid precipitation procedure modified from Ferre et al. [22]. In brief, frozen muscles were pulverized and then homogenized in ice‐cold buffer containing (in mM): 20 Tris‐HCl, pH 7.5, 5 EDTA, 10 Na4P2O7, 100 NaF, 2 NaVO4, 0.01 leupeptin, 3 benzamidine, 1 phenylmethylsulfonyl fluoride, and 10 µg/ml aprotinin. Aliquots (150 µl) were added to either 6% perchloric acid (600 µl) or Ba(OH2)/ZnSO4 (600 µl) and centrifuged at 13,000 × g for 4 min. Radioactivity in supernatants (500 µl) was assessed by liquid scintillation counting for the [³H] label. Phosphorylated 2‐deoxyglucose was calculated as the difference between the radioactivity in the perchloric acid and the Ba(OH2)/ZnSO4 supernatants and used to calculate the rates of glucose uptake.
Skeletal muscle glycogen analysis
Skeletal muscle glycogen levels were assessed by a hexokinase enzymatic reaction as previously described [23;24]. For glycogen measurements from EDL and soleus muscles, the muscles were weighed and then solubilized in 250 µl of 1.0 N NaOH at 80°C. Samples were neutralized with 250 µl of 1.0 N HCl, and centrifuged at 10,000 × g for 1 min. A 150 µl aliquot of 3.3 N HCl was added to the sample, and then boiled for 2 hrs at 95°C. Samples were neutralized by the addition of 250 µl of 2.0 N NaOH, vortexed, and then centrifuged at 13,000 × g for 30 sec. For glycogen measurements from tibialis anterior muscles, muscles were pulverized, weighed, and then hydrolyzed in 2.0 N HCl at 95°C for 2 hrs. Samples were neutralized with 2.0 N NaOH, vortexed, and then centrifuged at 13,000 × g. For all muscles, muscle glucose levels were assessed using hexokinase reagent (CIMA Scientific, De Soto, TX).
Immunoblot analysis
Immunoblot analyses were performed using standard procedures. Briefly, frozen muscles were homogenized on ice in a lysis buffer containing (in mM): 20 Tris‐HCl (pH 7.4), 5 EDTA, 10 sodium pyrophosphate, 100 NaF, 2 Na3VO4, 3 benzamidine, 1 phenylmethylsulfonyl fluoride, 1% NP‐40, 10 µg/ml aprotinin and 10 µg/ml leupeptin, and then centrifuged at 13,000 × g for 30 min. Total protein content was assessed via the Bradford assay. Skeletal muscle lysates (20–40 µg) were resolved by SDS‐PAGE on 10% acrylamide gels, and proteins transferred onto a nitrocellulose membranes. Membranes were blocked with 5% bovine serum albumin or 5% non‐fat dry milk. Primary antibodies were incubated with membranes overnight at 4°C. Horseradish peroxide‐conjugated secondary antibodies were incubated with the membrane at room temperature, and detected using chemiluminescence detection reagents (PerkinElmer Life Sciences, Inc, Boston, MA). Densitometric analysis of immunoblots was performed using FluorChem software (Alpha Innotech Corporation, San Leandro, CA).
Primary antibodies were obtained from commercial sources as follows: glycogen synthase (GS), glucose transporter 1 (GLUT1) and glucose transporter 4 (GLUT4) from Chemicon International, Inc., Temecula, CA; acetyl‐CoA‐carboxylase (ACC; streptavidin‐HRP antibody) from Pierce Biotechnology, Inc., Rockford, IL; JNK1/2 Santa Cruz Biotechnology, Santa Cruz, CA; Akt, AS160, glycogen synthase kinase‐3α/β (GSK‐3α/β) and insulin receptor substrate‐1 (IRS‐1) from Upstate Biotechnology, Inc., Charlottesville, VA; AMPK‐activated protein kinase α1/2 (AMPKα1/2) antibody was generated from amino acids 2–16 (CAEKQKKHDGRVKIGHY) of rat AMPK (Covance Inc., Princeton, NJ).
Statistical analysis
The data are presented as the mean ± standard error of the mean. Statistical significance was defined as P<0.05 and determined by t‐tests, or two‐way analysis of variance and Student‐Newman‐Keuls post hoc analysis. The number of animals, or muscles, utilized to determine statistical significance is indicated in the figure captions.
RESULTS AND DISCUSSION
Phenotypic characteristics of JNK1 knockout mice
A previous study using JNK1 KO mice demonstrated that lean, male JNK1 KO mice have decreased body weights, fasting blood glucose levels, and fasting blood insulin levels compared to their wild‐type controls [6]. To confirm the presence of this phenotype in our experimental animals, body weight and fasting blood glucose measurements were taken every two weeks. JNK1 KO mice had significantly lower body weights at all ages (Fig 1A), and fasting blood glucose levels were significantly decreased in JNK1 KO mice at ages ≥16 weeks compared to age‐matched, wild‐type control mice (Fig 1B). At 24 weeks of age, mice were given an intravenous injection of a glucose solution (1 mg glucose/g mouse body weight), and blood glucose levels assessed at 5, 10, 15, 25, 35 and 45 min. Glucose tolerance, as assessed by the rate of blood glucose decline following the glucose bolus, was significantly increased in JNK1 KO mice (Fig 1C). This finding was consistent with the results of Hirosumi et al. [6], and suggests that JNK1 KO mice fed a standard chow diet have enhanced glucose disposal into peripheral tissues. Since skeletal muscle is the main target for blood glucose disposal, we focused our studies on the role of JNK1 in the regulation of skeletal muscle glucose uptake and glycogen metabolism.
Figure 1. Phenotypic characteristics of JNK1 knockout mice.
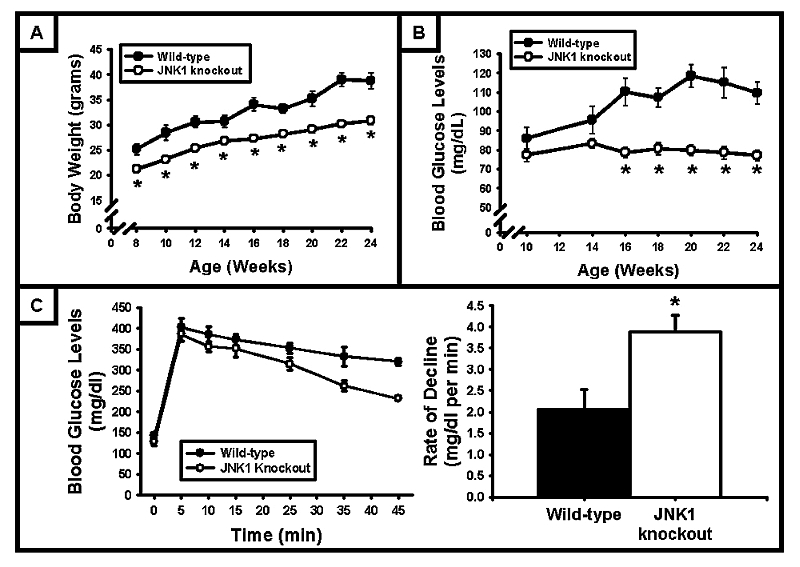
(A) At all ages studied, JNK1 knockout mice had significantly lower body weights compared to wild‐type controls (N = 15–26 mice). (B) Following a 12 hr fast, blood glucose levels were significantly lower in JNK1 knockout mice at age ≥16 weeks compared to wild‐type control mice (N = 18–26 mice). (C) Following an intravenous injection of glucose (1 mg glucose / g mouse body weight), blood glucose levels decreased faster in JNK1 knockout mice. Glucose tolerance, assessed as the rate of decline following the glucose bolus, was enhanced by JNK1 deficiency (N = 3–5 mice/group). (* = P<0.05 vs wild‐type)
Ex vivo skeletal muscle glucose uptake and muscle glycogen levels
The effect of JNK1 deficiency on skeletal muscle glucose uptake was assessed by isolated muscle incubation experiments performed on two different hindlimb muscles, the soleus and EDL, to examine the possibility of fiber‐type specific differences in JNK1 deficiency. To determine whether JNK1 KO mice have enhanced insulin‐stimulated skeletal muscle glucose uptake, muscles were incubated in KRB buffer containing 300, 600 µU/ml insulin (submaximal concentrations) or 50,000 µU/ml insulin (maximal concentration), and [³H]‐2‐deoxyglucose uptake assessed. As shown in Fig 2A and Fig 2B, insulin‐stimulated glucose uptake into the soleus and EDL muscles was not different between the wild‐type and JNK1 KO mice at any of the insulin concentrations. These data demonstrate that neither insulin‐stimulated glucose uptake, nor insulin sensitivity, was altered in the skeletal muscle from the JNK1 KO mice.
Figure 2. Ex vivo skeletal muscle glucose uptake was not altered by JNK1 deficiency.
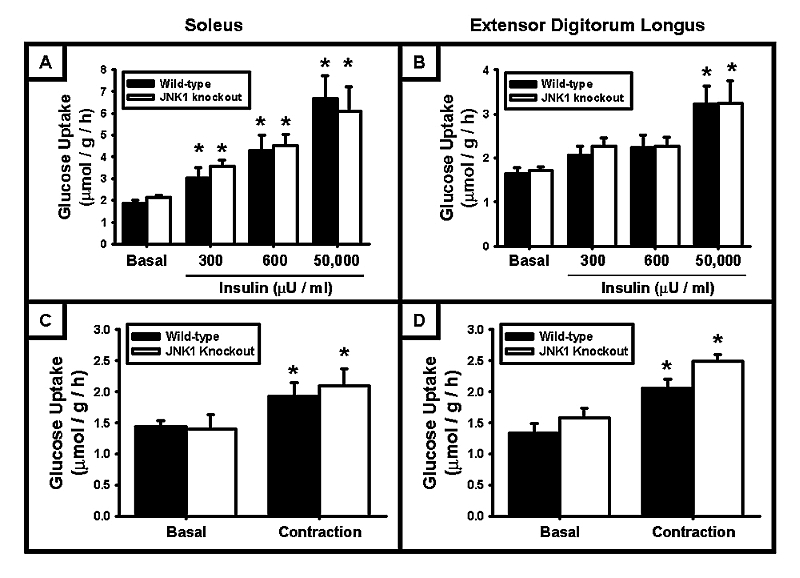
Ex vivo muscle incubation experiments were performed on soleus and extensor digitorum longus muscles excised from 20–24 week old male mice. (A + B) JNK1 deficiency had no significant effect on the insulin‐stimulated accumulation of glucose in the soleus or the extensor digitorum longus muscle. (C + D) JNK1 deficiency had no significant effect on contraction‐stimulated glucose uptake in the soleus or the extensor digitorum longus muscle. (N = 3–20 muscles/group) (* = P<0.05 vs basal)
Physical exercise and muscle contraction increase glucose uptake via non‐insulin‐dependent signaling pathways, and exercise/muscle contraction in rodents has been shown to stimulate JNK1/2 phosphorylation and activity [25]. Thus, JNK1 may play a role in the regulation of contraction‐stimulated muscle glucose uptake. To determine whether JNK1 KO mice have enhanced skeletal muscle glucose uptake in response to muscle contraction, isolated muscle incubation experiments were performed in conjunction with electrical stimulation. Soleus and EDL muscles were maximally contracted for 10 min and force production monitored throughout the contraction. As shown in Fig 2C and Fig 2D, contraction‐stimulated skeletal muscle glucose uptake was not different between the wild‐type and JNK1 KO mice, in both the soleus and the EDL. In addition, there was no significant difference in force production measured during the contraction between the wild‐type and JNK1 KO mice (data not shown).
Muscle glycogen levels can significantly impact skeletal muscle glucose uptake. Thus, to determine whether alterations in muscle glycogen levels affected our ability to detect differences in glucose uptake between the wild‐type and JNK1 KO mice, muscle glycogen levels were assessed in the soleus and EDL following a 12 hr fast. JNK1 deficiency did not alter muscle glycogen levels in the soleus (WT: 5.96±0.45 nmol/mg, n=13; JNK1 KO: 5.80±0.41 nmol/mg, n=20) or EDL (WT: 6.97±0.52 nmol/mg, n=13; JNK1 KO: 6.80±0.51 nmol/mg, n=20).
In vivo skeletal muscle glucose uptake
Circulating adipokines, such as adiponectin, have been shown to positively regulate skeletal muscle glucose uptake [26;27]. In JNK1 KO mice, serum adiponectin levels are increased ∼2‐fold [6]. Thus, circulating adipokines such as adiponectin could be required in order to observe alterations in skeletal muscle glucose uptake in the JNK1 KO mice. To determine whether circulating factors are required to observe increased insulin‐stimulated glucose uptake in the muscles from the JNK1 KO mice, [³H]‐2‐deoxyglucose was assessed in vivo following a retro‐orbital injection of a glucose bolus [1 mg glucose (0.33 µCi [³H]‐2‐deoxyglucose) / g mouse body weight]. Consistent with the ex vivo skeletal muscle glucose uptake data, in vivo glucose uptake was not significantly increased in the tibialis anterior muscle of JNK1 KO mice (Fig 3A). Glycogen levels were also not significantly different between the wild‐type and JNK1 KO mice in the same muscles (Fig 3B).
Figure 3. In vivo skeletal muscle glucose uptake, and muscle glycogen levels, were not affected by JNK1 deficiency.
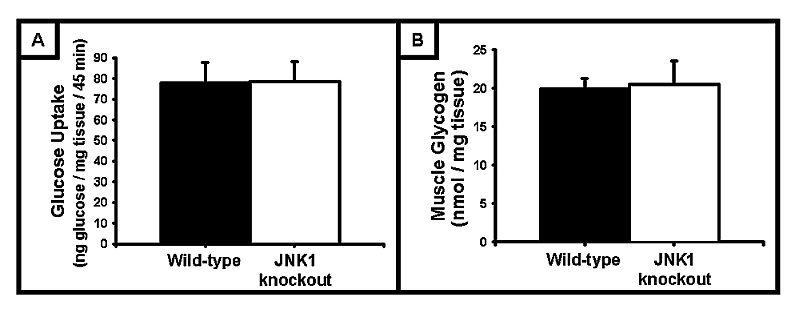
(A) In vivo [3H]‐2‐deoxyglucose uptake into the tibialis anterior muscle was not affected by JNK1 deficiency. (B) Glycogen levels in the tibialis anterior muscle were not different between wild‐type and JNK1 knockout mice. (N = 3–5 mice/group)
Intracellular signaling proteins involved in skeletal muscle glucose metabolism
To determine whether JNK1 deficiency altered the protein expression of key signaling molecules known to be involved in regulating skeletal muscle glucose metabolism, immunoblot analyses were performed in gastrocnemius muscles for JNK1/2, insulin receptor substrate‐1 (IRS‐1), Akt, AS160, glycogen synthase kinase‐3 (GSK‐3), glycogen synthase (GS), AMP‐activated protein kinase (AMPK), acetyl‐CoA carboxylase (ACC), glucose transporter 1 (GLUT1) and glucose transporter 4 (GLUT4). There was no significant change in the expression of any of these proteins (Fig 4). Importantly, since we did not detect an increase in JNK2 protein expression, the lack of alterations in skeletal muscle glucose metabolism cannot be attributed to a compensatory increase in JNK2 protein under these experimental conditions. However, recent work has shown that total JNK activity is not altered in muscle from JNK1 KO mice [28], suggesting an upregulation of JNK2 activity in the absence of changes in protein expression. Although surprising, this data cannot explain the lack of changes in skeletal muscle glucose metabolism between the wild‐type and JNK1 KO mice, since mice lacking JNK1 protein still exhibit a phenotype consistent with enhanced glucose disposal into peripheral tissues.
Figure 4. JNK1 deficiency did not alter the protein expression of intracellular signaling proteins involved in skeletal muscle glucose metabolism.
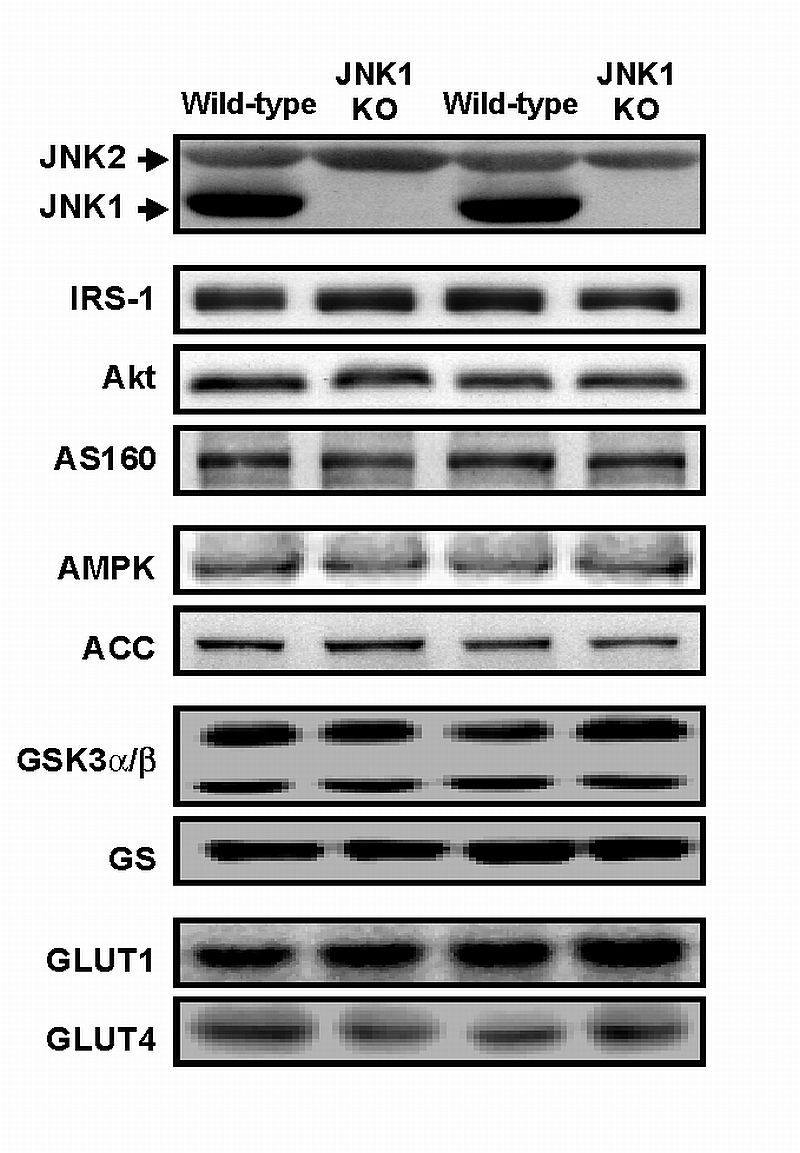
In mouse gastrocnemius muscle, there was no significant change in the protein expression of c‐jun‐N‐terminal kinase 2 (JNK2), insulin receptor substrate‐1 (IRS‐1), Akt, AS160, AMP‐activated protein kinase (AMPK), acetyl‐CoA‐carboxylase (ACC), glycogen synthase kinase‐3α/β (GSK3α/β), glycogen synthase (GS), glucose transporter 1 (GLUT1) and glucose transporter 4 (GLUT4) between wild‐type and JNK1 knockout (JNK1 KO) mice.
The main findings from this study are an important contribution towards understanding the role that JNK1 plays in preventing both high fat diet‐ and genetically‐induced insulin resistance, since studies using both JNK1 KO mice and JIP‐1 overexpressing mice have suggested that loss of JNK1 protein and/or activity significantly enhances blood glucose disposal. Since skeletal muscle is the primary site for blood glucose disposal, and also a primary target tissue for type 2 diabetes treatments, it was critical to determine the effects of JNK1 inhibition on muscle glucose metabolism. Contrary to our hypothesis that enhanced skeletal muscle glucose metabolism was the primary mechanism by which JNK1 ablation prevented blood glucose dysregulation, our study is the first to directly demonstrate that the beneficial effects of JNK1 deficiency are not mediated via alterations in muscle glucose uptake and/or glycogen metabolism. These findings suggest that pharmacological interventions designed to inhibit JNK1 can be utilized in combination with other drugs targeting skeletal muscle glucose metabolism to enhance blood glucose disposal in the growing number individuals afflicted with insulin resistance and type 2 diabetes.
ACKNOWLEDGMENTS
This work was supported by grants from the National Institutes of Health to LJ Goodyear (R01AR42238 and R01AR45670), to CA Witczak (F32AR051663), to the Joslin Diabetes Center (T32DK07260), and Diabetes Endocrinology Research Grant (DK36836). Additional funds to support N. Jessen came from the University of Aarhus, Denmark. We would like to thank David E. Arnolds, Laura E. Holton, Yangfeng Li, and Lauren E. Peter for technical assistance.
Footnotes
JNK1= c‐jun‐NH2‐terminal kinase 1
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sethi JK, Hotamisligil GS. The role of TNF alpha in adipocyte metabolism. Semin. Cell Dev. Biol. 1999;10:19–29. doi: 10.1006/scdb.1998.0273. [DOI] [PubMed] [Google Scholar]
- 2.Hotamisligil GS, Spiegelman BM. In: Diabetes Mellitus. LeRoith D, Taylor SI, Olefsky JM, editors. Philadelphia: Lippincott Williams & Wilkins; 2000. [Google Scholar]
- 3.Nguyen MT, Satoh H, Favelyukis S, Babendure JL, Imamura T, Sbodio JI, Zalevsky J, Dahiyat BI, Chi NW, Olefsky JM. JNK and tumor necrosis factor‐alpha mediate free fatty acid‐induced insulin resistance in 3T3‐L1 adipocytes. J Biol. Chem. 2005;280:35361–35371. doi: 10.1074/jbc.M504611200. [DOI] [PubMed] [Google Scholar]
- 4.Wellen KE, Hotamisligil GS. Inflammation, stress, and diabetes. J Clin. Invest. 2005;115:1111–1119. doi: 10.1172/JCI25102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hotamisligil GS. Role of endoplasmic reticulum stress and c‐Jun NH2‐terminal kinase pathways in inflammation and origin of obesity and diabetes. Diabetes. 2005;54 Suppl 2:S73–S78. doi: 10.2337/diabetes.54.suppl_2.s73. [DOI] [PubMed] [Google Scholar]
- 6.Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, Hotamisligil GS. A central role for JNK in obesity and insulin resistance. Nature. 2002;420:333–336. doi: 10.1038/nature01137. [DOI] [PubMed] [Google Scholar]
- 7.Fujishiro M, Gotoh Y, Katagiri H, Sakoda H, Ogihara T, Anai M, Onish Y, Ono H, Abe M, Shojima N, Fukushima Y, Kikuchi M, Oka Y, Asano T. Three mitogen‐activated protein kinases inhibit insulin signaling by different mechanisms in 3T3‐L1 adipocytes. Mol. Endocrinol. 2003;17:487–497. doi: 10.1210/me.2002-0131. [DOI] [PubMed] [Google Scholar]
- 8.Hamada K, Takuwa N, Yokoyama K, Takuwa Y. Stretch activates Jun N‐terminal kinase/stress‐activated protein kinase in vascular smooth muscle cells through mechanisms involving autocrine ATP stimulation of purinoceptors. J Biol. Chem. 1998;273:6334–6340. doi: 10.1074/jbc.273.11.6334. [DOI] [PubMed] [Google Scholar]
- 9.MacKenna DA, Dolfi F, Vuori K, Ruoslahti E. Extracellular signal‐regulated kinase and c‐ Jun NH2‐terminal kinase activation by mechanical stretch is integrin‐dependent and matrix‐specific in rat cardiac fibroblasts. J Clin. Invest. 1998;101:301–310. doi: 10.1172/JCI1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sluss HK, Barrett T, Derijard B, Davis RJ. Signal transduction by tumor necrosis factor mediated by JNK protein kinases. Mol. Cell Biol. 1994;14:8376–8384. doi: 10.1128/mcb.14.12.8376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Raingeaud J, Gupta S, Rogers JS, Dickens M, Han J, Ulevitch RJ, Davis RJ. Pro‐inflammatory cytokines and environmental stress cause p38 mitogen‐activated protein kinase activation by dual phosphorylation on tyrosine and threonine. J. Biol. Chem. 1995;270:7420–7426. doi: 10.1074/jbc.270.13.7420. [DOI] [PubMed] [Google Scholar]
- 12.Westwick JK, Weitzel C, Minden A, Karin M, Brenner DA. Tumor necrosis factor alpha stimulates AP‐1 activity through prolonged activation of the c‐Jun kinase. J Biol. Chem. 1994;269:26396–26401. [PubMed] [Google Scholar]
- 13.Chang L, Karin M. Mammalian MAP kinase signalling cascades. Nature. 2001;410:37–40. doi: 10.1038/35065000. [DOI] [PubMed] [Google Scholar]
- 14.Shin EA, Kim KH, Han SI, Ha KS, Kim JH, Kang KI, Kim HD, Kang HS. Arachidonic acid induces the activation of the stress‐activated protein kinase, membrane ruffling and H2O2 production via a small GTPase Rac1. FEBS Lett. 1999;452:355–359. doi: 10.1016/s0014-5793(99)00657-2. [DOI] [PubMed] [Google Scholar]
- 15.Rizzo MT, Leaver AH, Yu WM, Kovacs RJ. Arachidonic acid induces mobilization of calcium stores and c‐jun gene expression: evidence that intracellular calcium release is associated with c‐jun activation, Prostaglandins Leukot. Essent. Fatty Acids. 1999;60:187–198. doi: 10.1054/plef.1999.0024. [DOI] [PubMed] [Google Scholar]
- 16.Kyriakis JM, Avruch J. Sounding the alarm: protein kinase cascades activated by stress and inflammation. J Biol. Chem. 1996;271:24313–24316. doi: 10.1074/jbc.271.40.24313. [DOI] [PubMed] [Google Scholar]
- 17.Kaneto H, Nakatani Y, Miyatsuka T, Kawamori D, Matsuoka TA, Matsuhisa M, Kajimoto Y, Ichijo H, Yamasaki Y, Hori M. Possible novel therapy for diabetes with cell‐permeable JNK‐inhibitory peptide. Nat. Med. 2004;10:1128–1132. doi: 10.1038/nm1111. [DOI] [PubMed] [Google Scholar]
- 18.Sabapathy K, Kallunki T, David JP, Graef I, Karin M, Wagner EF. c‐Jun NH2‐terminal kinase (JNK)1 and JNK2 have similar and stage‐dependent roles in regulating T cell apoptosis and proliferation. J Exp. Med. 2001;193:317–328. doi: 10.1084/jem.193.3.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fujii N, Hirshman MF, Kane EM, Ho RC, Peter LE, Seifert MM, Goodyear LJ. AMP‐activated Protein Kinase {alpha}2 Activity Is Not Essential for Contraction‐ and Hyperosmolarity‐induced Glucose Transport in Skeletal Muscle. J Biol. Chem. 2005;280:39033–39041. doi: 10.1074/jbc.M504208200. [DOI] [PubMed] [Google Scholar]
- 20.Kramer HF, Witczak CA, Fujii N, Jessen N, Taylor EB, Arnolds DE, Sakamoto K, Hirshman MF, Goodyear LJ. Distinct signals regulate AS160 phosphorylation in response to insulin, AICAR, and contraction in mouse skeletal muscle. Diabetes. 2006;55:2067–2076. doi: 10.2337/db06-0150. [DOI] [PubMed] [Google Scholar]
- 21.Ho RC, Alcazar O, Fujii N, Hirshman MF, Goodyear LJ. p38{gamma} MAPK regulation of glucose transporter expression and glucose uptake in L6 myotubes and mouse skeletal muscle. Am. J Physiol Regul. Integr. Comp Physiol. 2004;286:R342–R349. doi: 10.1152/ajpregu.00563.2003. [DOI] [PubMed] [Google Scholar]
- 22.Ferre P, Leturque A, Burnol A‐F, Peincaud L, Girard J. A method to quantify glucose utilization in vivo in skeletal muscle and white adipose tissue of the anaethetized rat. Biochem. J. 1985;228:103–110. doi: 10.1042/bj2280103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stein MW. Determination with Hexokinase and Glucose‐6‐Phosphate Dehydrogenase. In: Bergmeyer H, editor. Methods of Enzymatic Analysis. New York: Academic Press; 1963. p. 117. [Google Scholar]
- 24.Bondar RJ, Mead DC. Evaluation of glucose‐6‐phosphate dehydrogenase from Leuconostoc mesenteroides in the hexokinase method for determining glucose in serum. Clin. Chem. 1974;20:586–590. [PubMed] [Google Scholar]
- 25.Goodyear LJ, Chung P‐Y, Sherwood D, Dufresne SD, Moller DE. Effects of exercise and insulin on mitogen‐activated protein kinase signaling pathways in rat skeletal muscle. Am. J. Physiol. 1996;271:E403–E408. doi: 10.1152/ajpendo.1996.271.2.E403. [DOI] [PubMed] [Google Scholar]
- 26.Bruce CR, Mertz VA, Heigenhauser GJ, Dyck DJ. The stimulatory effect of globular adiponectin on insulin‐stimulated glucose uptake and fatty acid oxidation is impaired in skeletal muscle from obese subjects. Diabetes. 2005;54:3154–3160. doi: 10.2337/diabetes.54.11.3154. [DOI] [PubMed] [Google Scholar]
- 27.Ceddia RB, Somwar R, Maida A, Fang X, Bikopoulos G, Sweeney G. Globular adiponectin increases GLUT4 translocation and glucose uptake but reduces glycogen synthesis in rat skeletal muscle cells. Diabetologia. 2005;48:132–139. doi: 10.1007/s00125-004-1609-y. [DOI] [PubMed] [Google Scholar]
- 28.Tuncman G, Hirosumi J, Solinas G, Chang L, Karin M, Hotamisligil GS. Functional in vivo interactions between JNK1 and JNK2 isoforms in obesity and insulin resistance. Proc. Natl. Acad. Sci. U.S.A. 2006;103:10741–10746. doi: 10.1073/pnas.0603509103. [DOI] [PMC free article] [PubMed] [Google Scholar]