

Abstract
We present a single virion method to determine absolute distributions of copy number in the protein composition of viruses and apply it to herpes simplex virus type 1. Using two-color coincidence fluorescence spectroscopy, we determine the virion-to-virion variability in copy numbers of fluorescently labeled tegument and envelope proteins relative to a capsid protein by analyzing fluorescence intensity ratios for ensembles of individual dual-labeled virions and fitting the resulting histogram of ratios. Using EYFP-tagged capsid protein VP26 as a reference for fluorescence intensity, we are able to calculate the mean and also, for the first time to our knowledge, the variation in numbers of gD, VP16, and VP22 tegument. The measurement of the number of glycoprotein D molecules was in good agreement with independent measurements of average numbers of these glycoproteins in bulk virus preparations, validating the method. The accuracy, straightforward data processing, and high throughput of this technique make it widely applicable to the analysis of the molecular composition of large complexes in general, and it is particularly suited to providing insights into virus structure, assembly, and infectivity.
INTRODUCTION
Viruses are large and sophisticated macromolecular complexes, whose assembly is still poorly understood and whose composition may vary from one particle to another. The variation in the protein composition of virions may affect the biological characteristics of different populations and may determine important properties such as infectivity and the ability to neutralize the virus with antibodies. Furthermore, characterizing variability in the protein composition of populations of virions produced during virus replication is fundamental to understanding mechanisms of virus assembly and packaging. To date, it has not been possible to explore these relationships due to the lack of suitable methods for assessing compositional heterogeneity at the single-particle level. However, recently developed single-molecule fluorescence methods (1) have offered the opportunity to study such variation for large numbers of individual molecules or assemblies of molecules. Applying these techniques to virions enables us to measure the extent to which individual virus particles vary in terms of their compositional makeup. Such information is not available from ensemble methods and extends the application of single-molecule methods to the virion-by-virion measurement of the variation in protein distribution. We demonstrate here a single-virion fluorescence spectroscopy method that facilitates the analysis of tens of thousands of individual virus particles to accurately examine both the mean number of proteins present per virion and the variation of this protein composition in the population. This method is applied to herpes simplex virus type 1 (HSV1), one of the largest and most complex virus structures known.
Herpesviruses are enveloped viruses containing asymmetric structural features including an outer membrane and an amorphous protein layer (tegument) inside the envelope that makes analysis by x-ray crystallography or cryoelectron microscopy difficult. The human herpesviruses are associated with a wide range of diseases, including cold sores, genital lesions, chicken pox, and glandular fever. All herpesviruses share a common virion morphology: a double-stranded DNA genome is contained within an icosahedral protein shell (capsid), and this is surrounded by a layer of ∼20 types of tegument proteins and a lipid bilayer envelope containing 11 types of embedded viral glycoproteins (2,3). The 125-nm-diameter capsid of HSV1 has been studied in detail by cryoelectron microscopy, and its structure is known to 8.5-Å resolution (4). The tegument and envelope layers of HSV1 virions, 225 nm in diameter, have been observed to ∼70-Å resolution by cryoelectron tomography (2). At this resolution, 600–750 spikes could be observed on the virion surface but could not be identified as specific glycoproteins. Proteins within the irregular tegument layer could not be resolved (2). At present it is unclear how the assembly process of HSV is regulated and how the virus ensures that all the virion components are incorporated at the correct stoichiometries into the mature virus particle. Due to the lack of detailed structural information available for the irregular tegument and envelope layers of the virus, there are currently no data available on whether the protein composition of these layers can vary considerably between virus particles or whether tight regulation of copy number is important for virus infectivity.
The fluorescently tagged capsid protein VP26 has previously been shown by confocal microscopy to be present at homogenous levels within herpes virions (5,6). Homogeneous expression of capsid proteins is expected because of the regular geometry of the capsid, which can incorporate only a fixed number of its component proteins. Only one previous study, however, has directly examined virion-to-virion variability in levels of a noncapsid protein. del Rio et al. (5) measured the total fluorescence from 200 individual purified virions expressing green fluorescent protein (GFP)-VP22 (a tegument protein of the α-herpesvirus, pseudorabies virus) by confocal microscopy. The authors make some incisive conclusions about the distributions of tegument proteins, which are supported by our measurements and which we discuss later in more detail. However, they do not attempt to define these distributions in terms of absolute copy numbers, and their sample sizes are quite limited by the labor-intensive method used to collect data (in contrast to our method, which can easily sample 10,000 virions, as detailed below).
The single-virion technique that we have developed allows analysis of heterogeneity in the protein composition of HSV1 virions by quantitative measurement of relative numbers of fluorescently labeled proteins present in individual particles and can easily generate sample sizes of the order of 10,000 virions; at a rate of 5–10 virions sampled per second, 10,000 virions are measured in ∼22 min. The schematic of the apparatus is shown in Fig. 1. This method involves detection of particles labeled with two different fluorophores that are excited independently in the focus of two overlapped laser beams and the fluorescence detected simultaneously, as we recently demonstrated for single molecules (7,8). Because the laser beam has a Gaussian intensity profile, there is significant variation in fluorescence measured from fluorophores passing through the beam at different places. Passage of fluorophores through the beam center results in the brightest intensity measurements, but it occurs less frequently than trajectories that pass through the edges of the beam. Analyzing the ratio of fluorescence signals from two overlapping beams with similar intensity profiles reduces the contribution of this spatial variation in laser intensity to the measurements, allowing the relative intensities of fluorescence recorded from two different color fluorophores present in the same molecule, or complex particle, to be compared (9,10). This is done by identifying coincident fluorescence bursts and then forming a histogram of the ratio of fluorescence for each burst. At the single-molecule level, dimers and trimers can be distinguished by the position of the peak in the ratio histogram (9,10). This ratiometric method is similar to fluorescence cross correlation spectroscopy that also uses dual laser excitation (11,12) but is performed on individual molecules and is based on direct counting; the data are thresholded and the method allows the stoichiometry and its variation to be determined by fitting of the ratio histograms.
FIGURE 1.

Schematic of experimental apparatus.
By examining whole virions, we now extend this ratiometric approach to analysis of much larger and more complex associations of labeled molecules. To analyze relative protein levels, we made use of dual-labeled HSV1 particles, previously constructed for studies on HSV1 morphogenesis (13). For the first color channel we used virions expressing either enhanced yellow fluorescent protein (EYFP)-tagged capsid protein VP26 or enhanced green fluorescent protein (EGFP)-tagged tegument proteins VP16 or VP22. For the second color channel, virions were bound by Alexa-647-labeled antibodies (LP2) directed against envelope glycoprotein D (gD). By detecting only coincident events, free antibodies are automatically distinguished from those bound to virions. Representative data for the signals observed in the two channels are shown in Fig. 2. We used the peak positions in the ratio histograms derived from such data to determine the number of proteins per virion and the widths of the histograms to determine the variation in the proteins per virion. This method is general and offers the potential for studying variation in the protein composition of other viruses and other complex molecular assemblies.
FIGURE 2.

Representative fluorescence bursts due to single virions in the two color channels: Coincident events are marked by horizontal bars. The thresholds used to analyze the data are also indicated (green and black lines). Note that due to incomplete overlap of the confocal volumes excited by the red and blue lasers the coincidence detection efficiency is ∼20%, so that not all bursts are coincident (7). In addition, both unenveloped capsids and light particles, which do not have a capsid but have a tegument and envelope, will be present and give rise to fluorescent bursts on only one channel.
MATERIALS AND METHODS
Purification of HSV1 virions
Recombinant HSV1 viruses that had been engineered to express fluorescently tagged capsid (VP26-YFP) (14) and tegument (VP16-GFP, VP22-GFP) proteins (15,16) were used in all dual-labeling experiments. These recombinant viruses all replicate as efficiently as wild-type virus and incorporate the GFP-tagged proteins into virions normally (15–17). Virions were purified according to the following procedure. Medium from infected rodent fibroblast (baby hamster kidney cells) was collected 2–3 days postinfection and clarified by centrifugation at 2500 rpm in a Mistral 6000 rotor (MSE(UK) Limited, London, United Kingdom). Virus particles were pelleted from the supernatant by centrifugation for 2 h at 16,000 rpm in a Beckman 19R rotor (Beckman, Fullerton, CA). Pellets were resuspended in 2 mL phosphate buffer saline (PBS) (NaCl, 150 mM; Na2HPO4, 10 mM; NaN3, 2 mM; adjusted to pH 7.2; filtered by a 0.02-μm mesh Anotop 25 inorganic membrane filter from Whatman International, Maidstone, England), sonicated, and layered onto 30-mL Ficoll gradients (5%–15%) in PBS by centrifugation for 1.5 h at 12,000 rpm in a Beckman SW28 rotor. The banded virus was harvested from the gradient, diluted to 30 mL in PBS, and repelleted by centrifugation for 2 h at 20,000 rpm in a Beckman SW28 rotor. Pellets were resuspended in 500 μL of PBS and frozen at −70°C in 50-μL aliquots. After purification, virus concentrations were estimated by counting negatively stained particles in an electron microscope as previously described (18).
Preparation of fluorescently labeled antibodies
Monoclonal antibody LP2 (which recognizes HSV glycoprotein D) was purified from the tissue culture supernatant of hybridoma cells by immunoaffinity chromatography on a protein A sepharose column as previously described (19). LP2 effectively neutralizes HSV1 (19). Purified antibody concentrations were determined by measuring the optical density at 280 nm. LP2 was labeled with the red fluorophore Alexa-647 (Molecular Probes, Eugene, OR) according to the protocol provided by the manufacturer (8). The number of dye molecules per antibody was calculated from the optical density at 650 nm. Average labeling levels were 8.9 dye molecules per antibody for LP2-Alexa-647 so that with multiple fluorophores per antibody any possible environmental changes on binding to the virus are minimized.
Virus-antibody binding reactions
Purified virus particles were diluted with PBS to concentrations between 2 and 20 pM (∼109–1010 particles/mL) and total reaction volumes of 500 μL. Virus samples were then sonicated for 20 s to prevent aggregation, and debris was removed by pulse spinning in a microcentrifuge. Alexa-647-labeled LP2 was added to virus particles at antibody/virion molar ratios of 1000:1. Reactions were incubated for 1 h at room temperature and then fixed by the addition of 37% formaldehyde (1% final concentration) for 10 min at room temperature. These conditions were confirmed by plaque assay to inactivate the virus, and the subsequent fluorescence measurements were made under containment level 1 conditions. Samples were transported between laboratories on ice and either used immediately (see below) or stored frozen at −80°C overnight.
Anti-gD binding curve
Virions were diluted to 10 pM in PBS, and different amounts of Alexa-647-labeled LP2- immunoglobulin G (IgG) were added, ranging from 50 pM to 6 nM (5–600 times virion concentration). The fluorescence background at each antibody concentration was determined independently in a separate experiment where no virus was added. All measurements were carried out at room temperature. The mean diffusion time of a single virion was measured to be 3 ms and the mean diffusion time for an antibody was 500 μs. We collected data for 180 min with a 10-ms integration time longer than the diffusion time. A threshold of 10 times higher than average background was used at all antibody concentrations to identify single virions. For each concentration of antibody, the brightness of labeled virions was estimated by averaging the 1000 fluorescence bursts of highest intensity. In this experiment, a replication-defective gH-negative mutant of HSV1, named HFEMdelUL22Z (20), was used. This virus was propagated in a helper cell line, CR1, which supplies gH in trans (21). Previous studies have shown that the absence of gH from HSV1 virions does not influence the amounts of other glycoproteins, including gD, that are incorporated into virus particles (20). Therefore, a gH-null virus was considered to be an appropriate mutant to use for this analysis, which allowed these experiments to be carried out under category 1 containment conditions.
Western blot analysis of the average number of gD molecules per virion
A fusion protein (gDFc) consisting of the ectodomain of HSV1 gD (amino acids 1–318) fused to the Fc region of the human IgG was generated as described in Stura et al. (22). The protein was expressed in Chinese hamster ovary cells and purified from tissue culture supernatant by affinity chromatography on protein A sepharose. A Western blot analysis of known numbers of virus particles was carried out in comparison with known amounts of purified HSV1 gDFc fusion protein. This analysis was performed on two independently prepared and particle-counted virus stocks. The Western blots were semiquantified using a Flowgen ChemiImager 4000 phosphoimager (Flowgen Bioscience, Wilford, UK), and from the data obtained in the linear region of these histograms it was possible to calculate a mean value for the number of gD molecules per virion.
Laser setup
Samples were excited using two laser beams with overlapping focal volumes of 30%, as described previously (7). A red laser beam (633 nm, He-Ne, model 25LHP151, Melles Griot, Carlsbad, CA) and blue laser beam (488 nm, argon ion, model 35LAP321-230, Melles Griot) were directed through a dichroic mirror (FITC/CY5, AHF Analysentechnik, Tübingen, Germany) and the oil immersion objective (Plan Apo TIRF 60×, numerical aperture 1.45, Nikon, Tokyo, Japan) of a TE2000U optical microscope to be focused into the sample solutions. To achieve maximum overlap of the beams, the red beam was adjusted to be parallel and the blue beam was tuned to be slightly convergent (7). Fluorescence was collected by the same objective and imaged onto a 50-μm pinhole (Newport, Irvine, CA) to reject out of focus fluorescence and other background. Red and blue fluorescence were then separated using a second dichroic mirror (585DRLP, Omega Optical Filters, Brattleboro, VT). Red fluorescence was filtered by a long-pass filter with cutoff 565 nm and a band-pass filter centered at 695 nm (565ALP and 695AF55, Omega Optical Filters), and blue fluorescence was filtered by a long-pass filter with cutoff 510 nm and a band-pass filter centered at 535 nm (510ALP and 535AF45, Omega Optical Filters). Each color fluorescence was then focused onto a separate avalanche photodiode (APD) (SPCM AQR-141, EG&G, Quebec, Canada), and output from the APDs was coupled to two PC-implemented multi-channel scalar cards (MCS-Plus, Ortec, Oak Ridge, TN). The synchronous start output of one MCS card was used to trigger the second, with negligible time delay between the two cards (7).
Virus measurements
Each virus-antibody preparation was tested separately by placing a 20-μl drop of the solution on a cover glass above the oil-immersion objective of the inverted microscope. The droplet was then covered with a 4-mm-high plastic lid to reduce evaporation and any possible contamination. The microscope was focused 5 μm into the solution above the glass. The laser powers at the microscope were 0.391 μW at 633 nm (red) and 1.355 μW at 488 nm (blue). Extremely low laser powers had to be used to keep the count rates within the working range of the APD apparatus (up to MHz). The laser power is <1 kWcm−2 so that saturation effects are negligible. Upon illumination of the sample, the droplet was scanned at a rate of ∼1 mm s−1 over the objective by slowly adjusting the position of the stage, resulting in virus particles in the solution crossing the focal volume. At these low concentrations we found no evidence of two or more virions in the probe volume at one time. The number of photons incident in intervals of 1 ms upon each APD were counted by multi-channel scalar cards. This information was stored in files 10,000 intervals long. For each virus sample ∼100–200 files of data were obtained, representing 25 min of data acquisition time on average. The thresholds used to analyze the data were the averages for the two channels of those calculated for each set of data by optimizing the association quotient (the ratio of the significant rate of coincident events to the total rate of events) as a function of the thresholds (23). These were 25 per interval in the red channel and 17 per interval in the blue channel.
Data analysis
For each coincident event the ratio of the red count per interval to the blue count per interval was calculated, and normalized histograms were formed of the logarithms (to base e) of these numbers. There were typically 10,000 coincident events per sample analyzed. Taking logarithms of the ratios before forming the histograms ensures that homogenous subpopulations appear as Gaussian distributions in the probability density function (10). The three histograms for each type of sample were then averaged together and the resultant histograms fitted by Gaussian peaks. The central positions and widths of these peaks were then used to calculate the relative size and variation of the populations of tegument and envelope proteins as follows:
The average brightnesses of coincident events (100 photons per interval in the red channel; 124 photons per interval in the blue channel) were used to estimate the proportion of the width of the peaks due to photon shot noise according to the formula
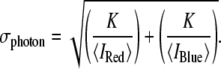 |
In this formula the parameter K is the square of the factor by which this contribution to the peak width exceeds that due to shot noise alone (10). For this work this parameter is set equal to unity because the extremely low laser powers used in these experiments ensure that all photophysical paths apart from fluorescence emission from the excited state are negligible. This means that the distributions of the dyes' fluorescence emissions from each point in the confocal probe volume are expected to be perfectly Poissonian. At the brightnesses specified above, the shot noise width is estimated to be 0.1344. The actual photon shot noise will be slightly higher than this estimate because of imperfect beam overlap. This may be accurately characterized in the future by using a virion strain with dual-labeled capsids.
The width of the VP26-gD peak after subtraction of the width estimated to be due to shot noise was used to estimate the width of the other peaks due to variation in the amount of gD envelope protein in each virion. The widths of the VP22-gD and VP16-gD peaks after subtraction of both the shot noise width and the gD width were then attributed to variation in the levels of VP22 and VP16 expressed in these strains of the virus. Thus, the slight underestimation of photon shot noise mentioned above does not affect the variation derived for VP22 and VP16, and this means that the calculated variation in gD number is marginally overestimated.
All of the virus data were taken at the same laser powers except, to explore the variation of the data with these parameters, two of the VP26-gD measurements were performed at higher combinations of laser powers (0.40 μW at 633 nm (red) and 1.50 μW at 488 nm (blue), and 0.50 μW at 633 nm (red), and 1.51 μW at 488 nm (blue)) and the photon counts of coincident events for these sets of data were rescaled: For each coincident event, the individual photon count in each channel was multiplied by the ratio of the average brightness of coincident events for that sample to the average brightness of coincident events for the VP26 sample measured with the lower laser powers. This simple rescaling procedure immediately resulted in good overlap of the histograms for the VP26 data (Fig. 3), indicating that the technique is resilient to variations in laser power.
FIGURE 3.

Logarithmic ratio histograms of individual virions for labeled VP26, VP16, and VP22. The lower right panel shows an overlay of the fitted peaks, shown as diamond symbols in the other three panels.
Estimation of individual brightnesses
To estimate the brightnesses of the individual fluorophores, data were collected from 10- and 100-pM solutions of GFP, YFP, and LP2-Alexa-647 in the same buffer solution as the main experiments. The presence of formaldehyde was found to have a negligible effect on the brightnesses measured. The laser powers at the microscope were 47.3 μW at 633 nm (red) and 209 μW at 488 nm (blue). The brightnesses were assumed to scale linearly with laser power. This is a reasonable assumption because the laser powers were low enough not to saturate the fluorophores for the brightness calibration measurements and very significantly lower than would have been needed to do this in the virus experiments. We also assumed that the variation in diffusion time between the different species (all approximately the same size) would be small and therefore would not affect the brightnesses. The thresholds used to analyze these data were those calculated for the virus data, corrected by a constant arbitrary scaling factor (0.005) and multiplied by the ratio of laser powers used in the brightness calibration to the virus measurement (yielding 15 per interval in the red channel and 13 per interval in the blue channel). The brightnesses measured in this way were 24.0 per interval for YFP, 23.7 per interval for GFP (on average 23.9 per interval for YFP and GFP), and 50.2 per interval for the Alexa-647-labeled LP2 antibody. Therefore the expected brightnesses at the laser powers used in the virus experiments were, for a single fluorescent protein, 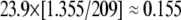 and for a single antibody,
and for a single antibody,  The ratio red/blue of these brightnesses was therefore found to be 2.68, allowing the position of the peaks in the ratio histograms to be quantitatively related to the numbers of proteins.
The ratio red/blue of these brightnesses was therefore found to be 2.68, allowing the position of the peaks in the ratio histograms to be quantitatively related to the numbers of proteins.
RESULTS
Ratio histograms for VP26, VP22, and VP16
In these experiments multiple copies of the individual proteins that make up a virion are produced in the host cell and then assembled together to produce complete virions that are then analyzed. Plaque forming unit (pfu) ratios were determined by comparing the concentration of virus particles in each virus stock with the concentration of infectious virions as measured by plaque assays giving a particle/pfu ratio of ∼20 (24). There are ∼100 pfu/cell released (data not shown), which equates to ∼2000 virions per cell. This means that any measured variation is not due to differences in expression levels but differences in assembly. The ratio histograms and average fits for the repeats of experiments performed on viruses expressing VP26yfp, VP22gfp, and VP16gfp are shown in Fig. 3. In all cases the virions were labeled with antibody to gD (LP2) that was tagged with a red fluorophore. Each sample was fixed in 1% formaldehyde. Having added a droplet of the solution to a microscope slide, the sample was scanned through the two overlapped laser beams for analysis. This raised the encounter rate of the virions with the probe volume by more than two orders of magnitude compared with the encounter rate achievable by their free diffusion without scanning. This method showed discrete bursts of fluorescence on both channels above a low background, as shown in Fig. 2. Any aggregation of the virus sample was readily detectable since this resulted in an absence of bursts and then one much longer intense burst. Data were only taken when discrete bursts were detected. The results for different preparations are highly reproducible, as shown by the close agreement of histograms corresponding to different preparations of the same types of sample in Fig. 3. The fits to all the repeats for these experiments are overlaid and shown in the lower right-hand panel of this figure. This shows small differences in peak position but marked differences between the widths of the histograms.
To convert these histograms into the mean number of proteins and their variation, we assumed that the number of capsid proteins was the same for all virions, with 900 copies of VP26 per virion (25–27). The widths of the histograms are then due to photon shot noise and the variation in the numbers of the tegument proteins and envelope gD antigen. In principle the variation in the number of fluorophores on each antibody could affect our results. However since 330 antibodies bind on average to a virion, as shown below, this variation is reduced by a factor of (330)0.5, i.e., a factor of 18, and hence makes a negligible contribution to the measured widths. We also needed to measure the relative brightness of EGFP, EYFP, and the anti-gD antibodies in a separate control experiment using individual proteins (see Materials and Methods for details) and assume that the relative brightness did not change when these components are in the virus and scaled linearly with laser power. This allows us to convert the peak position in the histogram into relative number of proteins. As shown below, these assumptions give the same mean number of proteins per virion as those obtained by other methods. We can then calculate the variation in protein copy number. The results of this analysis are shown in Table 1.
TABLE 1.
Variation in protein copy number
 |
Estimation of the number of molecules present in HSV1 virions
We used two different methods to independently measure the number of antibody-accessible gDs per virion for comparison with the results obtained above and to validate the assumptions made. We first used single-color fluorescence to study the binding of an Alexa-647-labeled antibody (LP2) bound to HSV virions freely diffusing in solution. Increasing concentrations of antibody were added to a solution of virions, and the average intensity of the top 1000 fluorescence bursts was calculated for each concentration. This intensity reflects the average number of labeled LP2 molecules bound to individual virions. The average intensity increased with the antibody/virus molar ratio until it reached a plateau above a ratio of 300:1 (Fig. 4). Note that the photon counting is still in a linear regime for the brightest virions detected. This result indicates that the maximum number of sites available for LP2 on the HSV1 envelope is ∼300. Two control experiments confirmed that the observed LP2 binding to gD is specific. First, HSV1 virions were incubated with pooled Alexa-647-labeled rabbit IgG. Second, a gD-negative mutant of HSV1 (28) was incubated with the LP2 antibody. For both control experiments, 10 pM virus and 1 nM antibody were tested under identical experimental conditions to those used for analyzing LP2 binding to gD-positive virions, and in both cases we detected no brighter fluorescence bursts than those for the antibody-alone background (data not shown).
FIGURE 4.

Average fluorescence photon counts per 10 ms for individual HSV1 virions as a function of the molar ratio of fluorophore-labeled LP2-IgG antibody to HSV1. All measurements were performed at a concentration of 10 pM HSV1. The number of counts increases linearly and then plateaus at a molar ratio of ∼300. A threshold 10 times higher than background was used for all points. The error bars indicate one standard deviation.
Second an independent measurement of the number of gD molecules present in HSV virions was made by comparing the intensity of a known quantity of purified gD to the intensity of the gD band from a preparation of virions in a Western blot analysis (Fig. 5). The signal obtained from 15.6 ng of gDFc was equivalent to the signal from ∼3.3 × 108 virus particles in experiment A and ∼5 × 108 virions in experiment B. Using the molecular weight of the gDFc fusion protein whose amino acid sequence is known (1.5 × 105 amu), we estimate that HSV virions contain 378 (experiment A) or 250 (experiment B) molecules of gD per particle, giving a mean value of 310. The agreement between these values for the Western blot and fluorescence experiments implies that antibody binding to gD is monovalent and that the relative fluorescence intensity measured from single virions is an accurate indicator of the relative number of gD molecules per virion. Both these results are in agreement with the value obtained from the ratio histograms.
FIGURE 5.

Quantification of the amount of gD per HSV virion. Two independently prepared stocks (A and B) of HSV1 virions were analyzed by Western blot using an antibody to gD. The top panel in each case shows the Western blot, and the histograms show the densitometry readings for the range of concentrations of gDFc protein compared to the range of dilutions of virions.
DISCUSSION
Our studies have focused largely on using viruses expressing fluorophore-tagged virion components. Despite the addition of GFP or YFP tags to the VP26 capsid protein or to VP22 or VP16 tegument proteins, these recombinant viruses replicate just as efficiently as wild-type nontagged HSV1 (14–16). This strongly supports the view that they incorporate these tagged proteins into the virus particle as efficiently as nontagged versions and are therefore valid reagents to use as a model system for studying populations of wild-type HSV. Our technique provides quantitative information about viral protein copy number in these viruses that is consistent with measurements that we have made using the more traditional Western blot method that does not use GFP tags. A previous report claimed that there are 3.4 × 104 gD molecules per HSV virion (29). However, this seems an unlikely value since this number of gD molecules would weigh more than the mass of an entire HSV virion. The assumption that the fluorescence intensity of the GFP is not altered in the virus is justified by the agreement with the Western blot analysis and by the protective barrel structure of the fluorophore, which makes it environmentally insensitive. Our two-color coincident detection method also allows us to distinguish between antibody bound to virus and free antibody so that the method does not require dual expression of GFP tagged proteins and provides information about the number of antibody-accessible binding sites, an important parameter for interpretation of antibody-mediated neutralization. All the gD sites appear accessible to the LP2 antibodies.
Important to inferences about the assembly of the virus by performing virion-by-virion analysis, this technique allows the calculation of variations in copy numbers of the envelope glycoproteins (and the tegument proteins), neither of which has been measured to date. The variation in gD number measured here was 235–480. This is in reasonable agreement with a measurement of the variation in the number of glycoprotein spikes (2), 600–750, which was performed on a much smaller number of virus particles and could not identify the glycoproteins present. It also suggests that not all the glycoprotein spikes contain gD. The data we present also indicate that there is large variation in the copy numbers of VP16 and VP22 tegument proteins between individual HSV virions, in agreement with del Rio et al. (5). Furthermore, our estimates for the absolute copy numbers are comparable to previous estimates of the average numbers of VP16 and VP22 proteins of between 1000–1500 per virion, although the methods used (i.e., radiochemical labeling and Coomassie staining) to determine these earlier values reportedly suffered from some inherent sources of error (30).
Our data indicate that the distributions of tegument proteins are highly skewed and approximately lognormal, with significant standard deviations in the statistic log(N). Such distributions are common in biology because they result from multiplicative processes, the effective multiplication and division of a range of independently distributed factors (31). This is a purely statistical effect that is analogous to the case proved by the central limit theorem, in which the addition and subtraction of independent variables results in a Gaussian distribution. For the case of tegument proteins, there are many possibilities for the multiplicative factors that include, for example, the rates of transcription and translation and the rates of diffusion or transport through the cytoplasm and incorporation into vesicles, etc. The distribution of VP26 capsid protein, on the other hand, is highly homogenous and even the variation of the gD copy number only results in relatively small widths of the histograms in Fig. 3 (top left panel). This is, of course, expected and is because the process of virion formation is evidently nucleated by the formation of complete capsids. This interpretation of our results broadly agrees with del Rio et al. (5), who fit a histogram of relative fluorescence emission from individual purified virions expressing GFP-VP22 with a Poisson distribution, rather than the Gaussian distribution used for control virions expressing fluorescently tagged VP26. They conclude that incorporation of VP22 into virions is much more heterogeneous than incorporation of the capsid protein VP26.
This technique offers the potential for addressing a number of fundamental questions relating to virus infectivity, antibody neutralization mechanisms, and virion assembly. We have demonstrated the feasibility of the method by measuring the number of an envelope glycoprotein (gD) and two tegument proteins (VP16 and VP22) in individual HSV1 virus particles and the variation in the copy number of these proteins for the first time, to our knowledge. This may now be extended to determine the relative stoichiometries and variation in copy numbers of many of the other herpesvirus virion components in different populations of viruses. Assessing the extent to which such compositional heterogeneity exists in preparations of different HSV strains in populations of virions produced by different cell types (e.g., epithelial cells and neurons) and in clinical isolates of HSV will provide insight into the biological functions of virion components in HSV pathogenesis. Furthermore, the morphogenesis of herpesviruses involves multiple, yet still poorly understood, interactions between tegument proteins and glycoproteins (32). The application of two-color fluorescence analysis to examine compositional variation in viruses that lack specific virion components that are thought to play a role in virus egress is likely to shed further light on these complex assembly routes. Finally, the ability to detect and measure both envelope proteins and structural proteins of individual virus particles can now also be applied to the study of populations of other clinically important virus pathogens, such as influenza virus and human immunodeficiency virus.
Acknowledgments
We are grateful to Susanne Bell and Birgitte Bruun for excellent technical assistance and to Tim Craggs for solutions of GFP and YFP.
This work was funded by the Biotechnology and Biological Sciences Research Council (BBSRC), UK, and the Wellcome Trust.
Editor: Thomas Schmidt.
References
- 1.Weiss, S. 1999. Fluorescence spectroscopy of single biomolecules. Science. 283:1676–1683. [DOI] [PubMed] [Google Scholar]
- 2.Grunewald, K., P. Desai, D. C. Winkler, J. B. Heymann, D. M. Belnap, W. Baumeister, and A. C. Steven. 2003. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science. 302:1396–1398. [DOI] [PubMed] [Google Scholar]
- 3.Subak-Sharpe, J. H., and D. J. Dargan. 1998. HSV molecular biology: general aspects of herpes simplex virus molecular biology. Virus Genes. 16:239–251. [DOI] [PubMed] [Google Scholar]
- 4.Zhou, Z. H., M. Dougherty, J. Jakana, J. He, F. J. Rixon, and W. Chiu. 2000. Seeing the herpesvirus capsid at 8.5 angstrom. Science. 288:877–880. [DOI] [PubMed] [Google Scholar]
- 5.del Rio, T., T. H. Ch'ng, E. A. Flood, S. P. Gross, and L. W. Enquist. 2005. Heterogeneity of a fluorescent tegument component in single pseudorabies virus virions and enveloped axonal assemblies. J. Virol. 79:3903–3919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith, G. A., S. P. Gross, and L. W. Enquist. 2001. Herpesviruses use bidirectional fast-axonal transport to spread in sensory neurons. Proc. Natl. Acad. Sci. USA. 98:3466–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li, H. T., L. M. Ying, J. J. Green, S. Balasubramanian, and D. Klenerman. 2003. Ultrasensitive coincidence fluorescence detection of single DNA molecules. Anal. Chem. 75:1664–1670. [DOI] [PubMed] [Google Scholar]
- 8.Li, H. T., D. J. Zhou, H. Browne, S. Balasubramanian, and D. Klenerman. 2004. Molecule by molecule direct and quantitative counting of antibody-protein complexes in solution. Anal. Chem. 76:4446–4451. [DOI] [PubMed] [Google Scholar]
- 9.Ren, X. J., G. Gavory, H. T. Li, L. M. Ying, D. Klenerman, and S. Balasubramanian. 2003. Identification of a new RNA center dot RNA interaction site for human telomerase RNA (hTR): structural implications for hTR accumulation and a dyskeratosis congenita point mutation. Nucleic Acids Res. 31:6509–6515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ren, X. J., H. T. Li, R. W. Clarke, D. A. Alves, L. M. Ying, D. Klenerman, and S. Balasubramanian. 2006. Analysis of human telomerase activity and function by two color single molecule coincidence fluorescence spectroscopy. J. Am. Chem. Soc. 128:4992–5000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bacia, K., S. A. Kim, and P. Schwille. 2006. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods. 3:83–89. [DOI] [PubMed] [Google Scholar]
- 12.Schwille, P., F. J. Meyer-Almes, and R. Rigler. 1997. Dual-color fluorescence cross-correlation spectroscopy for multicomponent diffusional analysis in solution. Biophys. J. 72:1878–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Naldinho-Souto, R., H. Browne, and T. Minson. 2006. Herpes simplex virus tegument protein VP16 is a component of primary enveloped virions. J. Virol. 80:2582–2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hutchinson, I., A. Whiteley, H. Browne, and G. Elliott. 2002. Sequential localization of two herpes simplex virus tegument proteins to punctate nuclear dots adjacent to ICP0 domains. J. Virol. 76:10365–10373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Elliott, G., and P. O'Hare. 1999. Live-cell analysis of a green fluorescent protein-tagged herpes simplex virus infection. J. Virol. 73:4110–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.La Boissiere, S., A. Izeta, S. Malcomber, and P. O'Hare. 2004. Compartmentalization of VP16 in cells infected with recombinant herpes simplex virus expressing VP16-green fluorescent protein fusion proteins. J. Virol. 78:8002–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desai, P., and S. Person. 1998. Incorporation of the green fluorescent protein into the herpes simplex virus type 1 capsid. J. Virol. 72:7563–7568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Watson, D. H., W. C. Russel, and P. Wildy. 1963. Electron microscopic particle counts on herpes virus using the phosphotungstate negative staining technique. Virology. 19:250–260. [DOI] [PubMed] [Google Scholar]
- 19.Minson, A. C., T. C. Hodgman, P. Digard, D. C. Hancock, S. E. Bell, and E. A. Buckmaster. 1986. An analysis of the biological properties of monoclonal-antibodies against glycoprotein-d of herpes-simplex virus and identification of amino-acid substitutions that confer resistance to neutralization. J. Gen. Virol. 67:1001–1013. [DOI] [PubMed] [Google Scholar]
- 20.Rodger, G., J. Boname, S. Bell, and T. Minson. 2001. Assembly and organization of glycoproteins B, C, D, and H in herpes simplex virus type 1 particles lacking individual glycoproteins: no evidence for the formation of a complex of these molecules. J. Virol. 75:710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boursnell, M. E. G., C. Entwisle, D. Blakeley, C. Roberts, I. A. Duncan, S. E. Chisholm, G. M. Martin, R. Jennings, D. N. Challanain, I. Sobek, S. C. Inglis, and C. S. McLean. 1997. A genetically inactivated herpes simplex virus type 2 (HSV-2) vaccine provides effective protection against primary and recurrent HSV-2 disease. J. Infect. Dis. 175:16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stura, E. A., M. J. Taussig, B. J. Sutton, S. Duquerroy, S. Bressanelli, A. C. Minson, and F. A. Rey. 2002. Scaffolds for protein crystallisation. Acta Crystallogr. D. 58:1715–1721. [DOI] [PubMed] [Google Scholar]
- 23.Clarke, R. W., A. Orte, and D. Klenerman. 2007. Optimized threshold selection for single-molecule two-color fluorescence coincidence spectroscopy. Anal. Chem. 79:2771–2777. [DOI] [PubMed] [Google Scholar]
- 24.Griffiths, A., S. Renfrey, and T. Minson. 1998. Glycoprotein C-deficient mutants of two strains of herpes simplex virus type 1 exhibit unaltered adsorption characteristics on polarized or non-polarized cells. J. Gen. Virol. 79:807–812. [DOI] [PubMed] [Google Scholar]
- 25.Booy, F. P., B. L. Trus, W. W. Newcomb, J. C. Brown, J. F. Conway, and A. C. Steven. 1994. Finding a needle in a haystack: detection of a small protein (the 12-kDa VP26) in a large complex (the 200-MDa capsid of herpes simplex virus). Proc. Natl. Acad. Sci. USA. 91:5652–5656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trus, B. L., F. L. Homa, F. P. Booy, W. W. Newcomb, D. R. Thomsen, N. Cheng, J. C. Brown, and A. C. Steven. 1995. Herpes simplex virus capsids assembled in insect cells infected with recombinant baculoviruses: structural authenticity and localization of VP26. J. Virol. 69:7362–7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhou, Z. H., J. He, J. Jakana, J. D. Tatman, and F. Rixon. 1995. Assembly of VP26 in herpes simplex virus-1 inferred from structures of wild-type and recombinant capsids. Nat. Struct. Biol. 2:1026–1030. [DOI] [PubMed] [Google Scholar]
- 28.Johnson, D. C., and M. W. Ligas. 1988. Herpes simplex viruses lacking glycoprotein D are unable to inhibit virus penetration: quantitative evidence for virus-specific cell surface receptors. J. Virol. 62:4605–4612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Handler, C. G., R. J. Eisenberg, and G. H. Cohen. 1996. Oligomeric structure of glycoproteins in herpes simplex virus type 1. J. Virol. 70:6067–6075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Heine, J. W., R. W. Honess, E. Cassai, and B. Roizman. 1974. Proteins specified by herpes-simplex virus. 2. Virion polypeptides of type 1 strains. J. Virol. 14:640–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Limpert, E., W. A. Stahel, and M. Abbt. 2001. Log-normal distributions across the sciences: keys and clues. Bioscience. 51:341–352. [Google Scholar]
- 32.Mettenleiter, T. C., B. G. Klupp, and H. Granzow. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9:423–429. [DOI] [PubMed] [Google Scholar]