

Abstract
Misfolding and aggregation of the prion protein (PrP) is responsible for the development of transmissible spongiform encephalopathies (TSE). To gain insights into possible aggregation-prone intermediate states, we construct the free energy surface of the C-terminal globular domain of the PrP from enhanced sampling of replica exchange molecular dynamics. This cellular domain is characterized by three helices H1–H3 and a small β-sheet. In agreement with experimental studies, the partially unfolded states display a stable core built from the central portions of helices H2 and H3 and a high mobility of helix H1 from the core. Among all identified conformational basins, a marginally populated state appears to be a very good candidate for aggregation. This intermediate is stabilized by four TSE-sensitive key interactions, displays a longer helix H1 with both a dry and solvated surface, and is featured by a significant detachment of helix H1 from the PrP-core.
INTRODUCTION
Prion protein (PrP) is a glycoprotein present in a wide range of organisms. The mature cellular prion (PrPC, ∼210 amino acids in mammalians) is characterized by a flexible, unstructured N-terminal tail and a globular C-terminal domain (1) of three α-helices, H1 (144–153), H2 (172–192), and H3 (200–225), and a double-stranded antiparallel β-sheet, S1 (129–130) and S2 (162–163).
In contrast to many proteins, PrPC misfolds into a PrP scrapie form (PrPSc) that readily aggregates into insoluble fibrils of unknown structure (2). This transition is a key event in the development of fatal neurodegenerative disorders better known as transmissible spongiform encephalopathies (TSE) or prion diseases (3). Despite extensive experimental and theoretical studies, we still do not know whether PrPC folding/unfolding follows a two-state (4) or three-state (5) model under physiological conditions and what leads prion protein to become misfolded. It is, yet, firmly established that folding to the PrPC isoform is under kinetic control, (6) and, within the nucleation polymerization model (7), amyloid fibril formation involves transient intermediates of higher quaternary structure complexity. There is also strong evidence that early formed oligomers are themselves cytotoxic (8). In the context of prion propagation, the β-nucleation model suggests that a highly specific change in PrPC structure and notably the unraveling of helix H1, induced by PrPSc, comprise the key event in PrPSc propagation (9). The β-nucleation model was shown to be consistent with infectivity data (10,11), prion-species barrier (12), and is also supported by recent experimental results (13,14) and hypotheses (15).
In this article, we describe the conformational free energy surface of the C-terminal domain of PrPC (residues 125–230) with full atomic details of the protein and the solvent using replica exchange molecular dynamics (REMD) simulations, as done recently for the 46-residue protein A (16). The improved sampling provided by REMD with respect to a single-temperature MD simulation allows us to characterize the partially unfolded states of the PrP. Identification of monomeric unfolded states is critical because they may represent transient aggregation-prone species for prion propagation. In addition to the conformational aspect, our study focuses on the hydration properties of PrP unfolded states. There is increasing evidence that both bulk solvent and protein hydration contribute to PrP misfolding; i.e., high-pressure calorimetry studies revealed variations in hydration properties between PrPC and PrPSc (17), and MD simulations pointed out a dual behavior of water at the PrPC surface (18). Nevertheless, the hydration properties of the PrP unfolded states and the resulting impact on the aggregation propensity still remain to be elucidated.
Here, we demonstrate that a marginally populated intermediate state, characterized by a full detachment of helix H1 from the region H2–H3, is a very good candidate for PrP aggregation. The structural determinants on the observed pathway provide new insights into the early misfolding events of PrPC.
MATERIAL AND METHODS
Simulations
REMD simulates several copies (replicas) of the same system evolving independently at different temperatures. Exchanges between neighboring replicas are attempted every Tswap on the basis of the Metropolis criterion (Eq. 1):
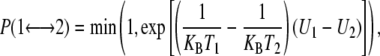 |
(1) |
where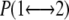 is the exchange probability, KB is Boltzmann's constant, U1 and U2 are the instantaneous potential energies, and T1 and T2 are the reference temperatures. Because Eq. 1 is suited in the NVT ensemble, an additional term for the isobaric-isothermal (NPT) ensemble, here adopted, should be introduced (19). This term is, however, negligible when the volumetric fluctuations are small (20), as in our case. Herein, a Tswap time of 2 ps was chosen to ensure potential energy relaxation before the following exchange is attempted. Two REMD runs of the PrPC domain (residues 125–230) have been performed in explicit solvent and under periodic condition. Run 1 is composed of 24 replicas at 320.0, 322.0, 324.0, 326.0, 328.1, 330.2, 332.3, 334.4, 336.5, 338.6, 340.8, 343.0, 345.2, 347.4, 349.6, 351.8, 354, 356.2, 358.5, 360.8, 363.1, 365.4, 367.7, and 370.0 K. Run 2 includes 16 replicas at 370.0, 372.4, 374.8, 377.2, 379.7, 382.2, 384.7, 387.2, 389.7, 392.3, 394.9, 397.5, 400.1, 402.7, 405.4, and 408.0 K. All replicas were equilibrated for 2 ns without exchanging temperatures and extended for 30 ns (1.2 μs of total simulation time). The last 20 ns (40,000 configurations) of the 40 trajectories were used to calculate all the averages reported here.
is the exchange probability, KB is Boltzmann's constant, U1 and U2 are the instantaneous potential energies, and T1 and T2 are the reference temperatures. Because Eq. 1 is suited in the NVT ensemble, an additional term for the isobaric-isothermal (NPT) ensemble, here adopted, should be introduced (19). This term is, however, negligible when the volumetric fluctuations are small (20), as in our case. Herein, a Tswap time of 2 ps was chosen to ensure potential energy relaxation before the following exchange is attempted. Two REMD runs of the PrPC domain (residues 125–230) have been performed in explicit solvent and under periodic condition. Run 1 is composed of 24 replicas at 320.0, 322.0, 324.0, 326.0, 328.1, 330.2, 332.3, 334.4, 336.5, 338.6, 340.8, 343.0, 345.2, 347.4, 349.6, 351.8, 354, 356.2, 358.5, 360.8, 363.1, 365.4, 367.7, and 370.0 K. Run 2 includes 16 replicas at 370.0, 372.4, 374.8, 377.2, 379.7, 382.2, 384.7, 387.2, 389.7, 392.3, 394.9, 397.5, 400.1, 402.7, 405.4, and 408.0 K. All replicas were equilibrated for 2 ns without exchanging temperatures and extended for 30 ns (1.2 μs of total simulation time). The last 20 ns (40,000 configurations) of the 40 trajectories were used to calculate all the averages reported here.
The simulated system consists of the C-terminal domain of the sheep prion protein (x-ray structure, Protein Data Bank (PDB) code 1UW3 (21)) immersed in a box filled with ∼10,000 extended single-point-charge (SPCE) waters. The simulations have been performed with the GROMACS (22) package by using the GROMOS96 force field with an integration time step of 2 fs. Nonbonded interactions were accounted for by using the particle mesh Ewald method (grid spacing 0.12 nm) (23) for the electrostatic contribution and cutoff distances of 1.4 nm for Van der Waals contribution. A neutral pH was ensured by selecting the appropriate protonation states of the pH-sensitive side chains. It is worth noting that the starting structure (PDB code 1UW3 (21)) features the R148C mutation, which has been introduced to covalently link the protein to a glutathione molecule (21). To correctly account for PrPC conformational properties, we restored the original sequence (R148). Throughout this article the huPrP residue numbering is adopted consistently with the crystal structure (PDB code 1UW3 (21)).
Hydration analyses
Hydration maps are produced for representative basins of the REMD samplings by two 50-ns NPT MD simulations at 300 K and 1 atm using either GROMOS96 force field with SPCE waters or OPLS force field with TIP5P waters. Both runs showed similar water distributions around the protein; therefore, we present the results using the OPLS-TIP5P force field.
Water density function
Our hydration analysis is largely based on the solvent density map (18,24–26), whose maxima are assumed to be the molecular dynamics hydration sites (MDHS). The space surrounding the protein is divided in two shells: the first shell describes the water within a distance of 0.6 nm from the protein surface. The second shell extends from 0.6 nm to 0.8 nm from the protein surface and represents the bulk solvent shell. The solvent density calculation is grid based (step size 0.05 nm). To eliminate protein translation and rotation, the coordinates of each frame are transformed by superimposing the current model onto a reference one. To prevent sweeping effects from backbone flexibility, a selection of frames based on Cα-RMSD is adopted; therefore, only structures with a Cα-RMSD, from the reference set, lower than the cutoff of 1.2 Å are considered.
Solvent entropy map
The solvent entropy is evaluated from the spatial distribution of the water oxygen in the protein's surrounding space (18). The map is a grid-based calculation (mesh spacing of 0.1 nm). Each grid node is connected to a narrower grid of 0.02 nm mesh size, which takes into account the water distribution around the node. The solvent entropy for a generic node of the map is calculated according to the equation
 |
(2) |
where Pl,m,n is the probability of finding a water in the subvolume l,m,n of the space surrounding the node, and R is the gas constant.
RESULTS
Replica exchange molecular dynamics
REMD includes different copies (replicas) of the system evolving independently at different temperatures (T) and exchanging periodically. High Ts enable the system to cross the energy barriers, whereas low Ts allow for an efficient exploration of local minima. Run 1 includes 24 replicas with T ranging from 320 to 370 K, i.e., across the experimental (340 K (27)) and the calculated (342 K) melting T. In contrast, run 2 (16 replicas) explores T ranging from 370 to 408 K and does not greatly exceed 400 K for avoiding biases in the thermodynamics and kinetics of the system (28). A total sampling of 1.2 μs has been carried out (30 ns per replica). Potential energy distributions and exchange frequencies between neighboring replicas are reported in Fig. S1 in the Supplementary Material (panels A and B). Both runs are featured by significant random walks (Fig. S1 D) in temperature space consistent with high exchange frequencies (between 15% and 20%) (Fig. S1 C). As a result, each replica explores the whole temperature space by passing repeatedly throughout all the thermal baths.
We find that REMD explores short- and large-scale motions. Small fluctuations are detected within the loops and specific regions of secondary structure, whereas large-scale motions involve the displacement of helix H1 and its flanking residues with respect to the structural core of the protein. PrP-core is formed by the central portions of helices H2 and H3 and is stabilized by a disulfide bridge (RMSDs in Table S1 in the Supplementary Material). The secondary structure is well conserved in the unfolded states. The fragment covering the β-sheet and helix H1 fully maintains its secondary structure (Table S2 in the Supplementary Material) despite large H1 displacements. In contrast, the H2 and H3 are extremely stable around the disulfide bridge but display large fluctuations at their C- and N-extremities, respectively. All these findings are consistent with NMR relaxation dynamics (29). In what follows, we adopt a subdivision of the PrPC domain (Fig. S2 A): the S1H1S2 subdomain (residues 125–171) and the H2H3 subdomain (residues 172–230). The S1H1S2 subdomain includes strands S1 and S2, helix H1, and their connecting loops as well as the loop S2-H2, whereas the H2H3 subdomain includes helices H2 and H3 and the loop H2-H3.
Because the coexistence of both large and small fluctuations increases the complexity of the free energy surface, we use two distinct sets of reaction coordinates to project the free energy: the first set (large free energy, LFE) accounts for large fluctuations and individuates basins of different “packing states”, whereas the second set (fine free energy, FFE) focuses on subtle conformational variations within the same packing state. Both LFE and FFE free energy surfaces are histogram-based calculations. In what follows, basin i in LFE is denoted by Bi, and basin i in FFE is denoted by bi.
LFE: large free energy surface
Building LFE from warm baths is optimal for exploring large motions and identifying partially unfolded states. To distinguish the conformations, we use the principal components of three independent sets of parameters (Fig. S2 in the Supplementary Material). The first set is sensitive to variations in the relative orientations of the subdomains; the second set uses native and nonnative contacts between the subdomains; and the third set accounts for shape-related parameters of the protein.
LFE calculated at 408.0 K (16th replica of run 2) shows nine basins connected to different packing states (Fig. 1). Four basins (B1, B2, B4, and B5) display compact packing states with helix H1 anchored to helix H3; these minima are close in the LFE space. B1 has native packing features, whereas B2, B4, and B5 show distinct, moderate displacements of helix H1 with respect to the PrP core (Fig. S3 A in the Supplementary Material). In contrast, B3 and B6 are characterized by noncompact states with a detachment of H1 from the native contact region (Fig. 1 C). B3 features an elongated shape with a shift of helix H1 along the H3 N-terminal direction. This state does not display any significant variation in the exposure of the native buried residues except within the amyloidogenic fragment 138–141 of sequence LIHF (30) (280.3 Å2 and 381.8 Å2 of solvent-accessible surface area in the native state and B3, respectively). As seen in the LFE surface and the disconnectivity tree, B3 is connected to the compact packing basins through the less populated basin B5, which shows similar, but minor, H1 displacement from the native packing area.
FIGURE 1.

LFE projection at 408 K. (A) Three-dimensional free energy surface and local minima. The free energy is calculated in kJ/mol. The X dimension is the principal component of the H1/PrP-core orientation parameters (see Fig. S2 of the Supplementary Material). The Y dimension is the principal component of the native and nonnative contacts. The Z dimension is the principal component of the set of the radius of gyration and the maximum value of the pair distances between any atoms of helix H1 and the disulfide bridge. The location of the nine minima is marked with black spheres. Two level curves enclose regions below 16 kJ/mol (gray lines) and 18 kJ/mol (orange lines), respectively. (B) Disconnectivity tree calculated from the energy values of minima and the connecting saddle points (s1–s6) of LFE projection. (C) Representative structure of B1, B2, B3, and B6.
Basin B6 goes one step beyond basin B3 by revealing a complete detachment of helix H1, which is now totally protruded into the bulk solvent and rotated with respect to the H2H3 subdomain (Fig. S3 B in the Supplementary Material). B6 is explored in two independent replicas of run 2, one of them experiencing an unpacking-repacking event within 30 ns (Fig. 2). Although the folding/unfolding paths cannot be extracted from REMD, this replica, along with the disconnectivity tree (Fig. 1 B), provides strong evidence that B6 is an intermediate between two native-like packing states. The temporal evolution of such a transition can be followed from the maximum distance between helix H1 and the disulfide bond (Fig. 2 D). Remarkably, despite a significant loss of tertiary interactions (16→3 contacts between helix H1 and the H2-H3 subdomain in going from the native state to B6), H1 remains elongated and very stable (see below). In addition, only four anchoring interactions are found at the interface between the S1H1S2 and H2H3 subdomains. These include D178–R164 (and partially D178–Y128) (Fig. 2B-1), T183–Y162 amide (Fig. 2B-2), H187–R156 carbonyl (Fig. 2B-3), and R156–D202 (Fig. 2 B-4). Strikingly, all of these interactions involve TSE pathological mutations: D178N (Fatal Familial Insomnia (FFI), (Creutzfeldt-Jakob Disease (CJD)), T183A (CJD), H187R (Gerstmann-Sträussler-Scheinker syndrome (GSS)), and D202N (GSS).
FIGURE 2.

H1 unpacking/repacking event. The sixth replica of run 2 explores three states. (A) Packed native-like state. (B) Unpacked intermediate state, B6. The marked interactions are: D178–R164 (1), T183–Y162 amide (2), H187–R156 carbonyl (3), and R156–D202 (4). (C) Native-like packed state. (D) Reaction coordinate followed by the maximum distance between H1 and the disulfide bridge as a function of time.
Finally, LFE indicates the existence of two marginally populated basins, B7 and B8, associated with B6 by the disconnectivity graph (Fig. 1 B). Similarly to B6, these basins display the T-shape conformation resulting from an analogous large-scale motion, but they are located in different regions of the LFE surface because of differences in H1 position and orientation. The least-populated basin, B9, is again a compact state and presents a unique shift of H1 along the H3 C-terminal direction.
It is difficult to extract energy barriers from 3D projections, and the choice of the reaction coordinates, optimized to separate conformational clusters, is likely to bias the energies. In addition, because high T sampling might also flatten the energetic profiles, the next energies are only qualitative and indicative. Here, LFE projection reveals energy barriers of 5–6 kJ/mol between the compact states and 10–11 kJ/mol between the compact and noncompact states (Fig. 1 B).
FFE surface
As for LFE, a 3D projection has been set-up for FFE. The first coordinate is associated with the Cα-RMSD from the starting crystal structure (PDB code 1UW3). The second is the principal component of the set of native and nonnative contacts. The third is the principal component of a set of parameters describing the radius of gyration, total content of secondary structure, and contents of α-helix, turns, and coils. FFE is here applied with the aim of determining local fluctuations within the native state; therefore, it has been computed on the conformations sampled by run 1. The free energy surface projected on the conformations of the 11th bath of run 1 at 340.8 K (nearest thermal bath to the melting temperature) reveals five basins (Fig. 3 A). The lowest free energy basin (b1, Fig. 3 C) essentially represents the native ensemble, preserving native packing and secondary structure elements, whereas b2 and b3 (Fig. 3 C) are featured by a progressive unscrewing of the C-terminal turns of helix H2. These two basins are close in the FFE surface and are similarly populated. A small energy barrier of 2.5 kJ/mol separates b2 and b3, whereas an energy barrier of 9 kJ/mol separates b1 and b2 (Fig. 3 B). Accordingly, unfolding the first helical turn (going from C- to N- term) of H2 (b1→b2) is therefore more costly than unfolding the second helical turn of H2 (b2→b3), indicative of a cooperative effect in unfolding the H2 C-terminal turns.
FIGURE 3.

FFE projection at 340.8 K. (A) Three-dimensional free energy surface and local minima. The free energy is calculated in kJ/mol. The location of the five minima is marked with black spheres. Two level curves enclose regions below 13 kJ/mol (orange curves) and 9 kJ/mol (gray curves). (B) Disconnectivity tree calculated from the energy of minima and connecting saddle points. (C) Representative structure of the basins b1–b5.
The less populated basin b5 (Fig. 3 C) shows a comparable loss of secondary structure as depicted by b3. Basin b5 is located in a different region of the FFE surface because it displays an additional displacement of helix H1, which is shifted along helix H3. Finally, basin b4 reveals the beginning of H1-detachment, recalling the structural characteristics of basin B6 (Fig. 3 C). This indicates a high similarity between our two independent REMD runs. Of course, basin b4, explored at low T, shows less H1 detachment than basin B6, sampled at high T, but b4 and B6 are certainly on the same unpacking path (Fig. S3 B in the Supplementary Material).
Stability of the β-sheet
Secondary structure composition in selected baths is reported in Table S2 in the Supplementary Material. It is a matter of debate whether the increase in β-structure from PrPC to PrPSc is (or is not) an intrinsic characteristic of the cellular PrP fold. Previous MD simulations have reported conflicting conclusions on the propensity of the native β-sheet to elongate (31–35). Parrinello and co-workers have shown, by means of metadynamics free energy reconstruction (36), that the antiparallel β-sheet is likely to undergo disruption rather than growth under pathogenic conditions (37).
Here, we do not find any significant increase in β-sheet content except for two additional transient H bonds: one between the G131 carbonyl and V161 amide, the other between the H159 carbonyl and M134 amide. Interestingly, this slight elongation of the β-sheet results from the thermal disruption of the β-bulge after strand S1; such a bulge, conserved in all PrP structures known, was previously suggested (24) as a “negatively designed” element (38) to avoid an edge-to-edge intermolecular β-sheet. The disruption of the β-bulge increases the number of unsaturated backbone amides and carbonyls on the edge strand S1; such a conformational change enhances the probability of a dimerization event, a scenario that is also strongly supported by the crystal structure of sheep PrP, where an intermolecular β-sheet is formed because of the crystal packing (21). Consistent with such an intermolecular β-sheet aggregation pathway (39), the point mutations G131V and M129V increase the β-sheet propensity of the edge strand S1 (35).
Stability of helices H2 and H3
A central topic is the stability of the PrPC helices because a marked decrease in α-helical content is measured on prion amyloid formation (2,40). In our simulations, major loss of helix content is found at the N- and C-extremities of helix H3 and at the C-terminal extremity of H2. The C-terminal residues in H3 are affected because they are free to move, whereas the C-extremity of H2 and the N-extremity of H3 are perturbed by their highly flexible connecting loop.
Among these regions, the C-terminal end of helix H2 has aroused much attention. Its local instability observed in the FFE and LFE projections is in line with NMR spectroscopy (41,42), MD simulations (31,35,43), and the suggestion that the fragment of sequence THTTTT at positions 188–193 is frustrated in a helical state (43).
Stability of helix H1
It is well established that H1 presents a unique pattern of ion pairs (salt-bridges) resulting in an extended network covering almost the entire helix length (9). This network confers an intrinsic high stability to helix H1 as shown by both theoretical and experimental studies (9,12,43–45). In the native state, the network extends from D144 to E152 (Fig. S4 A in the Supplementary Material) including three salt-bridges: D144-R148, R148-E152, and D147-R151. In addition, the ordering of negatively (D144-D147) and positively (R148-R152) charged residues produces a favorable electrostatic interaction with the intrinsic helix dipole (9).
In B6 (and b4), R156 is recruited by the salt-bridge pattern, which in turn is rearranged into four stable salt bridges: D144-R148, D147-R151, E152-R156, and R156-D202 (Fig. S4 B in the Supplementary Material). It is noteworthy that all the ion pairs on H1 are now antiparallel to the helix dipole (energetically favorable orientation), whereas in the native state, the pair R148-E152 is parallel to the helix dipole and antiparallel to both D144-R148 and D147-R151 pairs. As a result of the more extended salt-bridge pattern, helix H1 is elongated by an additional helical turn. This finding is consistent with NMR measurements on the H1 peptide in solution where, because of the lack of PrPC tertiary structure constraints, the same elongation and improved salt-bridge pattern was observed (12).
Hydration of partially unfolded states
As a first step, we focused on the hydration properties of PrPC partially unfolded states generated from large-amplitude motions and presenting significant conformational rearrangements from the native state (B3 and B6). Water density function of B6 generated by a classical 50-ns MD simulation at 300 K (see Methods) is reported in Fig. 4 A. We find a significant redistribution of the hydration sites (peaks in the density function) compared to the native state. The major perturbations occur in the regions subjected to large conformational changes. In particular, helix H1 and the loop β1-H1, now protruded in the bulk phase, are asymmetrically solvated with both a dry and fully hydrated surface (Fig. 4 A). The dry surface is rather buried in the native state and encompasses four aromatic side chains (F141, Y145, Y149, and Y150) and three aliphatic residues (P137, I139, and M154). The solvent-accessible areas of this surface are 383.6 Å2 and 477.2 Å2 in the native and B6 states, respectively. Conversely, the hydrated surface covers the charged and hydrophilic residues D144, D147, R148, R151, E152, and R156. It is worth noting that a local minor dewetting was reported by 300 K MD simulations of the native state (18,24). However, the large motion of helix H1 leading to B6 perturbed the hydration properties of the system and, more importantly, changed the aggregation propensity by producing a massive dry surface (see Discussion).
FIGURE 4.

Solvent density maps of the B6 and B3. The maps have been calculated from 50-ns MD trajectories at 300 K. (A) B6 MD hydration sites. The cyan level curves encompass region at high water density and are assumed to be the molecular dynamics hydration sites. Yellow ribbons locate the position of the dry surface (top of the fragment). (B) B3 hydration sites. The protein is rotated by 180° around a vertical axis in the two panels.
A different scenario emerges from the solvent density map of B3. Indeed, this state is associated with a homogeneous distribution of hydration sites covering the entire protein surface consistent with the lack of significant hydrophobic surfaces exposed to the solvent. Thus, in contrast to B6, B3 hydration does not suggest any particular propensity for aggregation.
We also focused on the local hydration of the C-terminal region of helix H2, which displays high mobility. This region was found to have a large number of defectively wrapped H bonds (46) and a high content of water distributional entropy (18). Maps of solvent entropy have been calculated on the structures of the three basins showing progressive unfolding of the C-terminal helical turns of H2. Interestingly, unfolding the helix H2 C-terminus (Fig. 5, A→B→C) leads to a decrease in water entropy content, probably via an increase in the solvent exposure of the main-chain amides and carbonyls previously engaged in the helix H bonds. Thus, increase in the backbone entropy is compensated by a decrease in solvent entropy; similarly, the loss of protein-protein interactions is restored by protein-solvent interactions. Such opposite effects reduce the free energy to unfold the extremity of H2, thereby accounting for its local structural weakness.
FIGURE 5.

Water entropy maps. High-entropy contours are drawn in blue. The maps have been calculated on the three basins displaying progressive unfolding of the C-terminal extremity of helix H2. (A) FFE-b1. (B) FFE-b2. (C) FFE-b3.
DISCUSSION AND CONCLUSIONS
The identification of the regions promoting and/or propagating PrP aggregation is of high importance. PrP(106–147) (47) and PrP(106–126) (48) show high fibrillogenic properties in vitro. Murine PrP(121–231) is also sufficient to form amyloid fibrils in vitro (49). There is a large body of experimental data suggesting the occurrence of two PrPC subdomains (here denoted as S1H1S2 and H2H3). NMR relaxation dynamics showed that the β-sheet displays slow conformational fluctuations on a millisecond to microsecond time scale, whereas the central portions of helices H2 and H3 form a very stable core (29). The stability of this core is also supported by hydrogen/deuterium exchange measurements under unfolding conditions (41). Subdomains also show a different response to pH variations (50) and pressure (51,52), and their mutual intermotion has been hypothesized to seed prion misfolding (15). In terms of prion misfolding, kinetic and structural analyses on sheep PrP revealed that, before oligomerization, separation of contacts occurs between S1H1S2 and H2H3, implicating a conformational change of the relative connection loop (53). In the context of PrPSc fibrils, Govaerts et al. accommodated the fragment 89–175 in a β-helix (constituting the repetitive motif of the fibril) with the H2-H3 subdomain retaining its native conformation (54). This model has been recently questioned by hydrogen/deuterium exchange experiments, which rather suggested a major rearrangement of the H2-H3 region (55). Interestingly, both models require an early misfolding event with detachment of the two subdomains.
Our REMD simulations clearly show that PrP unfolded states are characterized by a stable H2H3 core around the disulfide bridge (Table S1 in the Supplementary Material) and a very stable helix H1 moving around the H2H3 core. The free energy surfaces point to a limited number of basins ranging from compact to noncompact packing states. The group (B1, B2, B4, and B5) of compact states is fully consistent with the variable orientations of helix H1 observed in the structures of PrP with low sequence identity (i.e., from frog, chicken, and turtle (56); Fig. S3 A in the Supplementary Material). This strongly indicates that H1 packing variability is an intrinsic dynamics feature of the PrPC-fold.
In the group of noncompact states, B6 and b4 are on a unique path describing the onset of the separation between the subdomains. Our free energy sampling shows that this motion, seeded by S1-H1 loop fluctuations, involves H1 detachment and rotation from the native packing area (Fig. S3 B). Structural and dynamics features along this path are in agreement with experimental data. The enhanced solvent exposure of the S1-H1, H1-S2, and H2-H3 loops we observe correlates well with the NMR-derived protection factors (41). NMR studies showed that the S1-H1 loop is more mobile than the H1-S2 loop (29), and when the chemical denaturant guanidine hydrochloride is present, the fragment 125–146 spends a proportion of time detached from the remainder of the protein (57). Moreover, pressure and heat unfolding experiments monitored by tryptophan fluorescence of two PrP mutants, F141W and Y150W, have revealed a substantial rearrangement within the region 132–160. Consistent with the B6 basin, they observed a change in the exposure of the engineered tryptophans (52). Similarly, high-pressure NMR studies have revealed that the most pressure-sensitive region is within the loop S1-H1 (residues 139–141), suggesting that this region might be the first entry point for the infectious conformer to convert the cellular protein (58). The B6 pathway is also consistent with recent studies showing that oligomerization of the human prion protein proceeds via a molten globule intermediate with almost intact α-helical organization and loss of tertiary contacts compatible with the subdomain detachment (59). Interestingly, such a state is able to bind ANS, suggesting a partial opening of the hydrophobic core. This feature is well accounted for by our hydration analysis of B6.
Overall, B6 presents four interesting features in terms of aggregation propensity. First, B6 displays a dry surface composed of four aromatic side chains (F141, Y145, Y149, and Y150) and three aliphatic residues (P137, I139, and M154) (Fig. 4 A). Such a dry surface might promote hydrophobic collapse (60), thereby representing a hot spot for aggregation. It is worth recalling that, according to the steric zipper model (61), a dry surface is a stabilizing factor of amyloid-like fibrils. Remarkably, a variant of steric zipper has been recently resolved for the fragment 170–175 of the human PrP (PDB code 2OL9 (62)). This fragment connects the two subdomains and includes both final and initial part of S1H1S2 and H2H3, respectively. It is very likely that a massive detachment of subdomains might trigger significant conformational variations of the fragment, also enhancing its accessibility to other molecules.
Second, B6 is an on-pathway, marginally populated intermediate between two native-like packing states (Fig. 2). This is shown, for the first time at atomic detail, by one of the replicas of our ensemble and our disconnectivity tree. This is an intrinsic, rare breathing motion of the PrPC domain that might be amplified under misfolding conditions or in the early steps of aggregation.
Third, the interface between the two subdomains within the B6 state is essentially stabilized by four TSE-sensitive anchoring interactions (Fig. 3 B). It is possible that the pathogenic mutations D178N (FFI, CJD), T183A (CJD), H187R (GSS), and D202N (GSS) may affect the stability of B6 and the probability of a repacking event and reduce the free energy barrier for a total detachment of the subdomains.
Finally, B6 is characterized by a longer helix H1 with a rearranged, more stable salt-bridge pattern. This finding supports a recent view that helix H1 unfolding is likely to represent an energetic barrier in PrP interconversion (43,44). This result is also significant in the context of the β-nucleation model, which proposes ionic interactions between helices of adjacent PrP molecules to have a role in the PrPSc conversion (9). In addition, our model also proposes the possibility that intermolecular H1 interactions may take place from the dry surface.
This study reports the free energy surface of the prion protein, which has been sampled for the first time by means of replica exchange molecular dynamics. We provide an atomic description of the partially unfolded states. Among all the basins identified, basin B6 displays very interesting structural and hydration features that make it an ideal precursor for aggregation. Its Boltzmann population is rather marginal in solution under physiological conditions, and it remains to be determined whether pH acidic conditions, which favor PrP misfolding, and the pathological mutations (D178N, T183A, H187R, and D202N) impact the population of this transient intermediate.
SUPPLEMENTARY MATERIAL
To view all of the supplemental files associated with this article, visit www.biophysj.org.
Supplementary Material
Acknowledgments
We thank Dr. P. Lucignano, Prof. A. Tagliacozzo, Prof. L. De Arcangelis, and Dr. A. De Candia for computational support. A.D. and A.Z. thank F. Fraternali, and A.D. thanks I. Coluzza and L. Vitagliano for stimulating discussions.
The calculations have been performed at CINECA supercomputing center (project: cne0fm2b) and CNISM (cluster address: clues.na.infn.it).
Editor: Ruth Nussinov.
References
- 1.Wuthrich, K., and R. Riek. 2001. Three-dimensional structures of prion proteins. Adv. Protein Chem. 57:55–82. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner, S. B. 1998. Prions. Proc. Natl. Acad. Sci. USA. 95:13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chiti, F., and C. M. Dobson. 2006. Protein misfolding, functional amyloid, and human disease. Annu. Rev. Biochem. 75:333–366. [DOI] [PubMed] [Google Scholar]
- 4.Wildegger, G., S. Liemann, and R. Glockshuber. 1999. Extremely rapid folding of the C-terminal domain of the prion protein without kinetic intermediates. Nat. Struct. Biol. 6:550–553. [DOI] [PubMed] [Google Scholar]
- 5.Apetri, A. C., K. Maki, H. Roder, and W. K. Surewicz. 2006. Early intermediate in human prion protein folding as evidenced by ultrarapid mixing experiments. J. Am. Chem. Soc. 128:11673–11678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Baskakov, I. V., G. Legname, S. B. Prusiner, and F. E. Cohen. 2001. Folding of prion protein to its native alpha-helical conformation is under kinetic control. J. Biol. Chem. 276:19687–19690. [DOI] [PubMed] [Google Scholar]
- 7.Kelly, J. W. 1998. The alternative conformations of amyloidogenic proteins and their multi-step assembly pathways. Curr. Opin. Struct. Biol. 8:101–106. [DOI] [PubMed] [Google Scholar]
- 8.Bucciantini, M., E. Giannoni, F. Chiti, F. Baroni, L. Formigli, J. Zurdo, N. Taddei, G. Ramponi, C. M. Dobson, and M. Stefani. 2002. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature. 416:507–511. [DOI] [PubMed] [Google Scholar]
- 9.Morrissey, M. P., and E. I. Shakhnovich. 1999. Evidence for the role of PrPC helix 1 in the hydrophilic seeding of prion aggregates. Proc. Natl. Acad. Sci. USA. 96:11293–11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Speare, J. O., T. S. Rush 3rd, M. E. Bloom, and B. Caughey. 2003. The role of helix 1 aspartates and salt bridges in the stability and conversion of prion protein. J. Biol. Chem. 278:12522–12529. [DOI] [PubMed] [Google Scholar]
- 11.Apetri, A. C., and W. K. Surewicz. 2003. Atypical effect of salts on the thermodynamic stability of human prion protein. J. Biol. Chem. 278:22187–22192. [DOI] [PubMed] [Google Scholar]
- 12.Megy, S., G. Bertho, S. A. Kozin, P. Debey, G. H. Hoa, and J. P. Girault. 2004. Possible role of region 152–156 in the structural duality of a peptide fragment from sheep prion protein. Protein Sci. 13:3151–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Norstrom, E. M., and J. A. Mastrianni. 2006. The charge structure of helix 1 in the prion protein regulates conversion to pathogenic PrPSc. J. Virol. 80:8521–8529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Watzlawik, J., L. Skora, D. Frense, C. Griesinger, M. Zweckstetter, W. J. Schulz-Schaeffer, and M. L. Kramer. 2006. Prion protein Helix1 promotes aggregation but is not converted into beta-sheet. J. Biol. Chem. 281:30242–30250. [DOI] [PubMed] [Google Scholar]
- 15.Schwarzinger, S., A. H. Horn, J. Ziegler, and H. Sticht. 2006. Rare large scale subdomain motions in prion protein can initiate aggregation. J. Biomol. Struct. Dyn. 23:581–590. [DOI] [PubMed] [Google Scholar]
- 16.Garcia, A. E., and J. N. Onuchic. 2003. Folding a protein in a computer: an atomic description of the folding/unfolding of protein A. Proc. Natl. Acad. Sci. USA. 100:13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cordeiro, Y., J. Kraineva, R. Ravindra, L. M. Lima, M. P. Gomes, D. Foguel, R. Winter, and J. L. Silva. 2004. Hydration and packing effects on prion folding and beta-sheet conversion. High pressure spectroscopy and pressure perturbation calorimetry studies. J. Biol. Chem. 279:32354–32359. [DOI] [PubMed] [Google Scholar]
- 18.De Simone, A., G. G. Dodson, C. S. Verma, A. Zagari, and F. Fraternali. 2005. Prion and water: tight and dynamical hydration sites have a key role in structural stability. Proc. Natl. Acad. Sci. USA. 102:7535–7540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okabe, T., M. Kawata, Y. Okamoto, and M. Mikami. 2001. Replica-exchange Monte Carlo method for the isobaric-isothermal ensemble. Chem. Phys. Lett. 335:435–439. [Google Scholar]
- 20.Seibert, M. M., A. Patriksson, B. Hess, and D. van der Spoel. 2005. Reproducible polypeptide folding and structure prediction using molecular dynamics simulations. J. Mol. Biol. 354:173–183. [DOI] [PubMed] [Google Scholar]
- 21.Haire, L. F., S. M. Whyte, N. Vasisht, A. C. Gill, C. Verma, E. J. Dodson, G. G. Dodson, and P. M. Bayley. 2004. The crystal structure of the globular domain of sheep prion protein. J. Mol. Biol. 336:1175–1183. [DOI] [PubMed] [Google Scholar]
- 22.Berendsen, H. J. C., D. van der Spoel, and R. van Drunen. 1995. GROMACS: “A message-passing parallel molecular dynamics implementation.” Comp. Phys. Comm. 91:43–56. [Google Scholar]
- 23.Darden, T., L. Perera, L. Li, and L. Pedersen. 1999. New tricks for modelers from the crystallography toolkit: the particle mesh Ewald algorithm and its use in nucleic acid simulations. Struct. Fold. Des. 7:R55–R60. [DOI] [PubMed] [Google Scholar]
- 24.De Simone, A., G. G. Dodson, F. Fraternali, and A. Zagari. 2006. Water molecules as structural determinants among prions of low sequence identity. FEBS Lett. 580:2488–2494. [DOI] [PubMed] [Google Scholar]
- 25.De Simone, A., R. Spadaccini, P. A. Temussi, and F. Fraternali. 2006. Toward the understanding of MNEI sweetness from hydration map surfaces. Biophys. J. 90:3052–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lounnas, V., and B. M. Pettitt. 1994. A connected-cluster of hydration around myoglobin: correlation between molecular dynamics simulations and experiment. Proteins. 18:133–147. [DOI] [PubMed] [Google Scholar]
- 27.Rezaei, H., D. Marc, Y. Choiset, M. Takahashi, G. Hui Bon Hoa, T. Haertle, J. Grosclaude, and P. Debey. 2000. High yield purification and physico-chemical properties of full-length recombinant allelic variants of sheep prion protein linked to scrapie susceptibility. Eur. J. Biochem. 267:2833–2839. [DOI] [PubMed] [Google Scholar]
- 28.Walser, R., A. E. Mark, and W. F. van Gunsteren. 2000. On the temperature and pressure dependence of a range of properties of a type of water model commonly used in high-temperature protein unfolding simulations. Biophys. J. 78:2752–2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viles, J. H., D. Donne, G. Kroon, S. B. Prusiner, F. E. Cohen, H. J. Dyson, and P. E. Wright. 2001. Local structural plasticity of the prion protein. Analysis of NMR relaxation dynamics. Biochemistry. 40:2743–2753. [DOI] [PubMed] [Google Scholar]
- 30.Ziegler, J., C. Viehrig, S. Geimer, P. Rosch, and S. Schwarzinger. 2006. Putative aggregation initiation sites in prion protein. FEBS Lett. 580:2033–2040. [DOI] [PubMed] [Google Scholar]
- 31.Langella, E., R. Improta, and V. Barone. 2004. Checking the pH-induced conformational transition of prion protein by molecular dynamics simulations: effect of protonation of histidine residues. Biophys. J. 87:3623–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Alonso, D. O., S. J. DeArmond, F. E. Cohen, and V. Daggett. 2001. Mapping the early steps in the pH-induced conformational conversion of the prion protein. Proc. Natl. Acad. Sci. USA. 98:2985–2989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sekijima, M., C. Motono, S. Yamasaki, K. Kaneko, and Y. Akiyama. 2003. Molecular dynamics simulation of dimeric and monomeric forms of human prion protein: insight into dynamics and properties. Biophys. J. 85:1176–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Colacino, S., G. Tiana, R. A. Broglia, and G. Colombo. 2006. The determinants of stability in the human prion protein: insights into folding and misfolding from the analysis of the change in the stabilization energy distribution in different conditions. Proteins. 62:698–707. [DOI] [PubMed] [Google Scholar]
- 35.Santini, S., J. B. Claude, S. Audic, and P. Derreumaux. 2003. Impact of the tail and mutations G131V and M129V on prion protein flexibility. Proteins. 51:258–265. [DOI] [PubMed] [Google Scholar]
- 36.Laio, A., and M. Parrinello. 2002. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA. 99:12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barducci, A., R. Chelli, P. Procacci, V. Schettino, F. L. Gervasio, and M. Parrinello. 2006. Metadynamics simulation of prion protein: beta-structure stability and the early stages of misfolding. J. Am. Chem. Soc. 128:2705–2710. [DOI] [PubMed] [Google Scholar]
- 38.Richardson, J. S., and D. C. Richardson. 2002. Natural beta-sheet proteins use negative design to avoid edge-to-edge aggregation. Proc. Natl. Acad. Sci. USA. 99:2754–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bemporad, F., G. Calloni, S. Campioni, G. Plakoutsi, N. Taddei, and F. Chiti. 2006. Sequence and structural determinants of amyloid fibril formation. Acc. Chem. Res. 39:620–627. [DOI] [PubMed] [Google Scholar]
- 40.Pan, K. M., M. Baldwin, J. Nguyen, M. Gasset, A. Serban, D. Groth, I. Mehlhorn, Z. Huang, R. J. Fletterick, F. E. Cohen, and othersl. 1993. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA. 90:10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hosszu, L. L., N. J. Baxter, G. S. Jackson, A. Power, A. R. Clarke, J. P. Waltho, C. J. Craven, and J. Collinge. 1999. Structural mobility of the human prion protein probed by backbone hydrogen exchange. Nat. Struct. Biol. 6:740–743. [DOI] [PubMed] [Google Scholar]
- 42.Tizzano, B., P. Palladino, A. De Capua, D. Marasco, F. Rossi, E. Benedetti, C. Pedone, R. Ragone, and M. Ruvo. 2005. The human prion protein alpha2 helix: a thermodynamic study of its conformational preferences. Proteins. 59:72–79. [DOI] [PubMed] [Google Scholar]
- 43.Dima, R. I., and D. Thirumalai. 2004. Probing the instabilities in the dynamics of helical fragments from mouse PrPC. Proc. Natl. Acad. Sci. USA. 101:15335–15340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Santini, S., and P. Derreumaux. 2004. Helix H1 of the prion protein is rather stable against environmental perturbations: molecular dynamics of mutation and deletion variants of PrP(90–231). Cell. Mol. Life Sci. 61:951–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ziegler, J., H. Sticht, U. C. Marx, W. Muller, P. Rosch, and S. Schwarzinger. 2003. CD and NMR studies of prion protein (PrP) helix 1. Novel implications for its role in the PrPC→PrPSc conversion process. J. Biol. Chem. 278:50175–50181. [DOI] [PubMed] [Google Scholar]
- 46.Fernandez, A., and H. A. Scheraga. 2003. Insufficiently dehydrated hydrogen bonds as determinants of protein interactions. Proc. Natl. Acad. Sci. USA. 100:113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tagliavini, F., F. Prelli, L. Verga, G. Giaccone, R. Sarma, P. Gorevic, B. Ghetti, F. Passerini, E. Ghibaudi, G. Forloni, and others. 1993. Synthetic peptides homologous to prion protein residues 106–147 form amyloid-like fibrils in vitro. Proc. Natl. Acad. Sci. USA. 90:9678–9682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Gioia, L., C. Selvaggini, E. Ghibaudi, L. Diomede, O. Bugiani, G. Forloni, F. Tagliavini, and M. Salmona. 1994. Conformational polymorphism of the amyloidogenic and neurotoxic peptide homologous to residues 106–126 of the prion protein. J. Biol. Chem. 269:7859–7862. [PubMed] [Google Scholar]
- 49.Martins, S. M., D. J. Frosoni, A. M. Martinez, F. G. De Felice, and S. T. Ferreira. 2006. Formation of soluble oligomers and amyloid fibrils with physical properties of the scrapie isoform of the prion protein from the C-terminal domain of recombinant murine prion protein mPrP-(121–231). J. Biol. Chem. 281:26121–26128. [DOI] [PubMed] [Google Scholar]
- 50.Calzolai, L., and R. Zahn. 2003. Influence of pH on NMR structure and stability of the human prion protein globular domain. J. Biol. Chem. 278:35592–35596. [DOI] [PubMed] [Google Scholar]
- 51.Kuwata, K., H. Li, H. Yamada, G. Legname, S. B. Prusiner, K. Akasaka, and T. L. James. 2002. Locally disordered conformer of the hamster prion protein: a crucial intermediate to PrPSc? Biochemistry. 41:12277–12283. [DOI] [PubMed] [Google Scholar]
- 52.Torrent, J., M. T. Alvarez-Martinez, J. P. Liautard, C. Balny, and R. Lange. 2005. The role of the 132–160 region in prion protein conformational transitions. Protein Sci. 14:956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eghiaian, F., T. Daubenfeld, Y. Quenet, M. van Audenhaege, A. P. Bouin, G. van der Rest, J. Grosclaude, and H. Rezaei. 2007. Diversity in prion protein oligomerization pathways results from domain expansion as revealed by hydrogen/deuterium exchange and disulfide linkage. Proc. Natl. Acad. Sci. USA. 104:7414–7419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Govaerts, C., H. Wille, S. B. Prusiner, and F. E. Cohen. 2004. Evidence for assembly of prions with left-handed beta-helices into trimers. Proc. Natl. Acad. Sci. USA. 101:8342–8347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lu, X., P. L. Wintrode, and W. K. Surewicz. 2007. Beta-sheet core of human prion protein amyloid fibrils as determined by hydrogen/deuterium exchange. Proc. Natl. Acad. Sci. USA. 104:1510–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Calzolai, L., D. A. Lysek, D. R. Perez, P. Guntert, and K. Wuthrich. 2005. Prion protein NMR structures of chickens, turtles, and frogs. Proc. Natl. Acad. Sci. USA. 102:651–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hosszu, L. L., M. A. Wells, G. S. Jackson, S. Jones, M. Batchelor, A. R. Clarke, C. J. Craven, J. P. Waltho, and J. Collinge. 2005. Definable equilibrium states in the folding of human prion protein. Biochemistry. 44:16649–16657. [DOI] [PubMed] [Google Scholar]
- 58.Kachel, N., W. Kremer, R. Zahn, and H. R. Kalbitzer. 2006. Observation of intermediate states of the human prion protein by high pressure NMR spectroscopy. BMC Struct. Biol. 6:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gerber, R., A. Tahiri-Alaoui, P. J. Hore, and W. James. 2007. Oligomerization of the human prion protein proceeds via a molten globule intermediate. J. Biol. Chem. 282:6300–6307. [DOI] [PubMed] [Google Scholar]
- 60.Ball, P. 2003. Chemical physics: How to keep dry in water. Nature. 423:25–26. [DOI] [PubMed] [Google Scholar]
- 61.Nelson, R., M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe, and D. Eisenberg. 2005. Structure of the cross-beta spine of amyloid-like fibrils. Nature. 435:773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sawaya, M. R., S. Sambashivan, R. Nelson, M. Ivanova, S. A. Seivers, M. I. Apostol, M. J. Thompson, M. Balbirnie, J. J. Wiltzius, H. McFarlane, A. O. Madsen, C. Reikel, and D. Eisenberg. 2007. Atomic structures of amyloid cross-beta spines reveal varied steric zippers. Nature. 447:453–457. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.