

Abstract
Delayed recurrences, common in breast cancer, are well explained by the concept of tumour dormancy. Numerous publications describe clinical times to disease recurrence or death, using mathematical approaches to infer mechanisms responsible for delayed recurrences. However, most of the clinical literature discussing tumour dormancy uses data from over a half century ago and much has since changed. This review explores how current breast cancer treatment could change our understanding of the biology of breast cancer tumour dormancy, and summarizes relevant experimental models to date. Current knowledge gaps are highlighted and potential areas of future research are identified.
Introduction
Breast cancer remains the leading cancer diagnosis among women in the western world. With ever-improving chemotherapeutic, radiation and hormonal treatments, clinicians have seen an improvement in overall survival, leading to increasing numbers of patients living with breast cancer [1]. Due in part to these advances, breast cancer, and in particular early breast cancer, appears to behave increasingly akin to a chronic disease [2]. Patients who have distant or regional metastasis at the time of diagnosis have poor clinical outcomes in terms of early distant disease and subsequent death from breast cancer. However, scientists and clinicians alike have become intrigued with the finding that many patients who are diagnosed at an early stage, with small tumours and no evidence of regional lymph node metastases, can have a high level of recurrence when followed for greater than 10 to 15 years, in excess of 25% to 30% [2,3]. In such patients who are estrogen receptor positive, for example, more patients recur after completing five years of hormonal therapy, such as tamoxifen, than they do in the first five years [1]. These findings suggest that perhaps our understanding of the biology of breast cancer recurrence and the implications for tumour dormancy research should be reviewed in the setting of current clinical treatments for breast cancer, and the biology of tumour response to these treatments.
Clinical dormancy in breast cancer
When using the term 'tumour dormancy' in this review, we are referring to breast cancers that become clinically evident following a prolonged disease-free interval. Although there is no strict definition to the time interval between initial treatment and disease recurrence, many publications have noted disease-free intervals in excess of five to six years. In the literature published to date in the field of tumour dormancy in breast cancer, two prominent authors have contributed significantly to the field: Demicheli [4-7] and Karrison [8,9].
Demicheli and colleagues [7] have studied the timeline of disease recurrence based on a retrospective database (initiated in the 1940s in Italy) of 1,173 patients treated by the accepted treatments of that era, radical or modified radical mastectomy (Figure 1). Their findings of disease recurrences up to 20 years after diagnosis were felt to support the concept of tumour dormancy by breast cancer cells. It is important to note that only a minority of those patients received any adjuvant chemotherapy and/or radiation, and there was no mention of adjuvant hormonal therapy, which would likely not have been considered standard of care at that time. Demicheli and colleagues [10] subsequently presented further data on these patients, comparing chemotherapy-treated patients versus none and determined that chemotherapy was beneficial in reducing early recurrences but not late recurrences. Karrison and colleagues [8] also retrospectively studied 1,547 patients diagnosed between 1945 and 1987. Demicheli and colleagues demonstrated that plotting the hazard rate (or conditional probability of outcome) from time of surgery to death revealed a peak incidence of death at 1 to 2 years and again at 5 years (60 months) [7,9-11]. These studies have led to speculation that this peak in death at five years likely represents the awakening of dormant tumour cells unaffected by adjuvant regional or systemic treatment [6-11].
Figure 1.
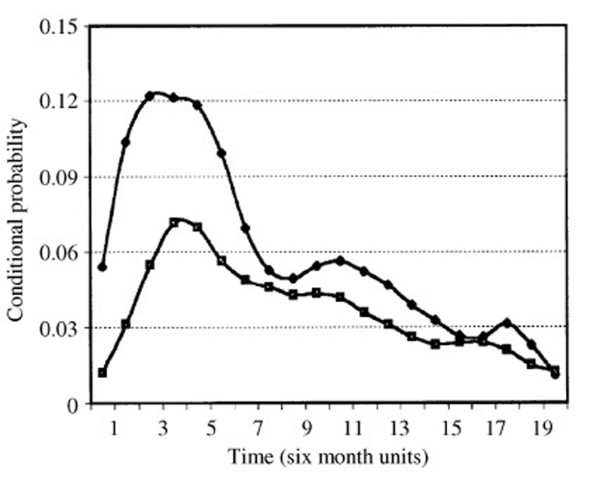
Comparison of the hazard function for development of distant metastases among axillary lymph node positive breast cancer patients treated with mastectomy without (upper curve) or with (lower curve) adjuvant chemotherapy (cyclophosphamide, methotrexate and fluorouracil (CMF)). Note that the x-axis is displayed in six month units. Reprinted from [7], with permission from Elsevier.
In Figure 1, use of cytotoxic chemotherapy (cyclophosphamide, methotrexate and fluorouracil (CMF)) post-mastectomy was shown to result in a dampening of the first, early peak of recurrences, with little effect on later recurrences, suggesting that this chemotherapy, while improving survival for the group, was having minimal effect on the development of late recurrences, seen to peak at five years post-treatment.
The theory of tumour dormancy, rather than steady re-growth of remaining cancer in these patients, is supported by growth kinetic theory utilizing mathematical models to estimate residual tumour cell doubling times [5], because recurrences were found to develop considerably later than would be expected based on growth kinetics alone. Additionally, the rate of tumour growth from clinically undetectable to clinically evident recurrence further supports this theory [4]. This theory is consistent with multiple clinical datasets, but the mechanisms responsible remain poorly elucidated.
Although delayed recurrences and graphical representation to suggest a peak in recurrence several years after initial treatments is of great clinical and scientific interest, there is no doubt that the patients included in the above studies were treated by what would be considered now to be substandard therapy. Additionally, findings obtained from early studies initiated in the 1940s are difficult to apply to current patient populations due to changes in overall life expectancies. Thus, it will be of great interest to learn if current therapies are able to reduce late-occurring recurrences, in addition to improving overall and disease-specific survival.
Evaluations to date of current randomized clinical trials data with long term follow-up have consistently demonstrated a smooth cumulative incidence curve for disease recurrence (cumulative frequency of recurrence with each subsequent time point) with a gradual decline in slope after 10 years [1,3,12-14]. This differs from the double recurrence peaks proposed by Demicheli (Figure 1), where the time to recurrence is plotted as a hazard function (incidence of recurrence for each time point). If a peak in recurrence at the five year mark had remained, as was seen by Demicheli and Karrison, one would have expected to see an increase in the slope of cumulative incidence over that time in the more recent studies described above. The majority of current publications of international trials display their results as cumulative incidence functions rather than hazard rates (otherwise termed conditional probability plots), subdivided by various treatments (chemotherapy with or without tamoxifen, mastectomy with or without radiation, aromatase inhibitor versus not, high risk phenotype versus low risk, and so on; for examples, see Figure 2 in [14], Figure 4 in [3] and Figure 8 in [1]). Thus, even when therapies are effective overall, steady increases in numbers of late recurrences are seen for both early and late stage tumours. Our intent in highlighting this difference is simply to demonstrate that the peaks in cancer recurrences demonstrated by Demicheli are not readily seen in long-term follow-up data in patients treated with current treatment regimes. As such, our understanding of tumour dormancy should be updated in light of current treatment methods (radiation, hormonal therapy and tumour targeted antibody treatments), which are likely influencing tumour dormancy in a different way now compared to patients in the era of Demicheli's database, as shown by vast differences in breast cancer mortality rates and recurrence patterns.
Figure 2.
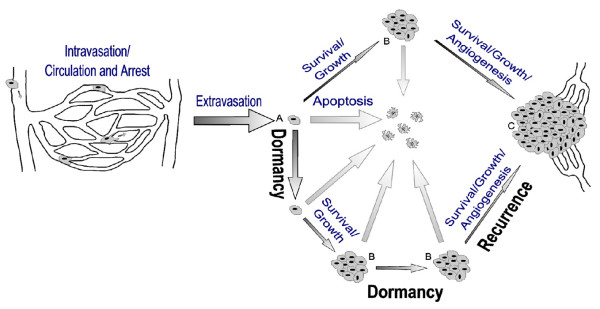
Upon arriving in a secondary site the majority of metastatic cells extravasate and proceed to one of three possible fates: they can undergo cell death (apoptosis), remain dormant or begin proliferating. In order to form a large, vascularized and clinically relevant metastatic tumour (C), a single cell must begin and continue proliferating as well as acquire a vasculature by angiogenesis. Dormant single cells (A) and micrometastases (B) are possible sources of recurrent cancer.
Currently in North America and Europe, the vast majority of breast cancers are treated by lumpectomy, also referred to as segmental mastectomy, with adjuvant radiation. For patients considered by phenotypic tumour markers to be at high risk of recurrence (node positive, lymphatic or vascular invasion present, tumour size and grade, pre-menopausal status, Her2/neu over-expression), systemic chemotherapy is offered. Standard North American and European chemotherapy regimens are anthracycline-based, often with the addition of a taxane [1]. Regimens are intravenous q3 weekly for six to eight cycles, typically lasting almost six months. Patients who are Her2/neu over-expressers are then offered intravenous trastuzumab (Herceptin®; Her2-targeted monoclonal antibody therapy) for one year. Concurrently, they are also offered 5,000 Gy of tangential radiation over 25 to 30 fractions [12]. Patients with estrogen and/or progesterone receptor-positive tumours are additionally offered five to ten years of hormonal therapy (estrogen receptor blockade or aromatase inhibition) [13].
Overall, these therapies are improving patient disease-specific and overall survival [12]. Nonetheless, late recurrences can and do occur in patients treated with various current therapies. This fact emphasizes the concept that current (as well as past) therapies may be ineffective in reaching or killing some subset of cancer cells, such as those that are dormant, which may be responsible for late recurrences. This concept is consistent with the theory that some disseminated cancer cells are resistant to various treatments, especially those therapies that target actively dividing cells [15-17]. As noted below, this idea is further supported by results from experimental models in which dormancy can exist in two states: solitary dormant cells [17-21] and active but pre-angiogenic micrometastases [22]. It has been shown that solitary, dormant cancer cells can exist in a quiescent state, and can be spared by cytotoxic chemotherapy that is effective against the same cells when they are in an actively dividing state [15]. It will thus be of great interest to follow patients being treated with current treatment protocols to assess how effective they will be at preventing late recurrences.
Insights into tumour dormancy from pre-clinical models
To better understand how changes in breast cancer treatment may affect tumour recurrence, an improved understanding of the source of recurrent cancer is required. Assuming successful removal of the primary tumour, metastatic recurrence is the result of tumours formed by malignant cells that have spread from the primary tumour to a secondary tissue. While often lethal, metastasis, the process by which cancer cells spread, is an extremely inefficient process and most cells shed from a primary tumour behave in a non-metastatic fashion [17,18,21,23-25]. For a cancer cell to successfully form a metastatic tumour, it must detach from the primary tumour, intravasate into a blood or lymphatic vessel, survive transit within the vasculature, arrest in a secondary site, extravasate into the secondary tissue and commence and continue growth in this new micro-environment [25]. As failure at any of these numerous steps in the process will result in failure of the cell to form a clinically relevant metastatic tumour, it is perhaps not surprising that metastasis is an extremely inefficient process. Indeed, in experimental models of hematogenous metastasis only a very small proportion (for example, 0.02% or less, depending on the model) of cells successfully forms large vascularized metastases [17] despite more than 80% of cells successfully extravasating from the vasculature [18,26]. Similar metastatic inefficiency and failure of most circulating tumour cells to form metastases has also been reported in human cancer [27].
Experimental metastasis models have also revealed that a variable, but often significant, proportion of cancer cells that are delivered to a secondary organ can remain as solitary dormant cells for prolonged periods [18,19,21,24]. Metastatic inefficiency then can be attributed to two main causes, cell death and dormancy (Figure 2). While cells that undergo cell death, due to apoptosis or other mechanisms, are of no further clinical relevance, cells or micrometastases that remain dormant are a possible source of later tumour formation and, therefore, recurrence. This model is entirely consistent with the clinical model of dormancy proposed by Demicheli (see Figure 4 in [7]).
Following extravasation into a secondary organ, metastatic cells can undergo one of three possible fates (Figure 2): they can undergo cell death (apoptosis), remain dormant or begin proliferating. Cells that begin proliferating, however, are not necessarily destined to become large vascularized metastases, as a large proportion of micrometastases either die off or become dormant. Although cells that commence proliferating and continue growing as metastases may be of primary concern clinically, dormant single cells and micrometastases also have clinical implications, as they can be a source of cells responsible for recurrence.
While dormancy may occur at both the single cell and micrometastasis stages of metastasis formation, these two types of dormancy differ in several significant ways, including the mechanisms controlling their dormancy, their proliferation status, and likely their response to treatment. Solitary cell dormancy is a quiescent state defined by lack of proliferation and apoptosis (currently as assessed by histology) and retention of inert fluorescent or other markers that are diluted and unobservable after a few cycles of cell division [15,19,21]. This indicates solitary cell dormancy is due to cell cycle arrest. In contrast, micrometastasis dormancy is characterized not by the absence of proliferation and apoptosis, but by the balance of the two such that the tumour does not increase in size [22]. Although these two modes of dormancy are thus distinct, one parallel between the two types of metastatic dormancy is the limited number of experimental models, and difficulty imaging those available in vivo complicates their study.
Experimental models and possible mechanisms of metastatic dormancy
Several inherent difficulties have restricted, and continue to restrict, the study of metastatic dormancy in vivo. The primary impediments are the limited number of metastasis dormancy models and the technical difficulty of imaging a single cell or small group of cells in vivo, particularly in internal organs. Solitary cell and micrometastasis models of tumour dormancy, as well as the techniques used to study them and possible molecular mechanisms that have been revealed by these experiments, have recently been reviewed [16,28,29]. A commonality between both types of metastatic dormancy is that when studying metastasis experimentally, cell lines are classified as metastatic due to their ability to proliferate shortly after arrival in a secondary tissue. This has had a significant role in limiting the number of dormancy models available, because the rapid growth of a small proportion of proliferating cells to form metastases minimizes the time in which dormant cells and micrometastases can be observed and studied. This situation has been identified in several cell lines in which solitary cells and micrometastases are observed in the same tissue in which metastases are actively growing [18-21,24]. It is also likely that by selecting for cell lines that grow soon after injection, cell lines that may be ideal for studying dormancy have been misclassified as non-metastatic. A similar situation was clearly demonstrated by Naumov and colleagues [30] in experiments in which many "non-tumourigenic" cell lines were actually able to form tumours, but only after significant periods of time outside the range of observation for most in vivo experiments.
Ideally, models of metastatic dormancy would consist of a population of cells that would become dormant, either as single cells or micrometastases, upon arrival in a secondary tissue, and emerge from dormancy after a predictable period of time and/or in response to experimental stimuli. As breast cancer commonly metastasizes to internal organs such as liver, lung, bone and brain, the ability to observe dormant cells experimentally in a realistic microenvironment is complicated by difficulty in longitudinally imaging cells in these internal tissues. Even in a model in which dormant cells or micro-metastases are known to exist, locating and observing small numbers of cells in vivo is technically challenging [31]. Advances in cellular imaging techniques, such as brighter and dual-fluorescence cell markers, inorganic labels that do not photo-bleach and longitudinal single cell imaging by MRI, are slowly facilitating this process [21,32-35]. In consequence, the great majority of what is known about metastatic dormancy in vivo has been determined by histology, or by using chick chorioallantoic membrane models [36-39] or models that use superficial anatomic sites (for example, vitreous fluid/cornea, subcutaneous, and so on), which are much easier to image than internal organs.
As may be expected due to the challenges facing in vivo studies, the exact mechanisms controlling metastatic dormancy have not been fully elucidated. However, many molecules and pathways believed to be involved in solitary cell and micrometastasis dormancy have been identified. The molecules and pathways implicated in solitary cell dormancy are not, surprisingly, molecules that are capable of controlling cell cycle, either directly or through signalling pathways. These include p21, p27, Myc, uPAR, ERK and p38 [39-43]. In contrast, micrometastasis dormancy is thought to be regulated by the balance of pro-angiogenic proteins and angiogenic inhibitors produced by both tumour and stromal cells [29,30,44]. The mechanistic differences between the two types of metastatic dormancy are significant, as most treatments will likely have vastly different effects on these two distinct cell populations. This difference must, therefore, be kept in mind when considering the impact of treatment on dormancy and recurrence. Additionally, there is evidence to suggest that one mechanism of action for the class of metastasis suppressor genes is through inhibition of growth of tumour cells at metastatic sites, perhaps through induction of cellular or micrometastatic dormancy (see [31] for a review).
Clinical implications
When considering what we understand to date regarding tumour dormancy, most publications have focused on the development of delayed systemic metastases as evidence for tumour cell dormancy. However, given that the majority of patients are currently treated by breast-conserving surgery (or lumpectomy) and or sentinel lymph node sampling (rather than full axillary node dissection for lymph node negative cancers), delayed locoregional metastases should also be considered to give evidence for tumour dormancy within the breast or lymphatic parenchyma. Although most patients considered high risk for early recurrence and death (locally advanced breast cancer at diagnosis, presence of regional or distant metastases at diagnosis) die within the first two to four years (which likely represent the first peak identified by Demicheli [7]; Figure 1), patients with early breast cancer can be as much at risk of distant disease as they are of delayed locoregional disease [1].
There has been speculation that dormant tumour cells may represent cancer stem cells [17], which lack the cell receptor differentiation of breast cells, that is, they do not express estrogen or progesterone cell surface receptors and, as such, would be insensitive to hormonal therapy. They likely would also be resistant to cytotoxic chemotherapy due to their inherent drug resistance. Current findings that the majority of patients with hormonally sensitive tumours (which represents the majority of breast cancers diagnosed) recur following the completion of five years of hormonal therapy would suggest that changes in either the tumour cell or host environment do not favour recurrence until the completion of treatment. Perhaps more can be understood about this when we obtain long-term follow-up data from the National Cancer Institute of Canada Clinical Trials Group MA.17 study looking at extended adjuvant treatment for a minimum of ten years following surgery [13].
The development of metastatic disease is felt to proceed in an ordered fashion, arising from primary tumour cells having developed metastatic potential, with locoregional then distant metastases in sequence (Figure 2). This is supported clinically by the improvement of survival in patients treated with locoregional radiation [14]. With this notion, it becomes increasingly important to understand how metastatic transformation can develop with the current treatments aimed at locoregional control, particularly since most early breast cancer patients with favourable phenotypic features will undergo lumpectomy with adjuvant radiation, often without the addition of systemic therapy. Although micrometastatic cell clusters should be sensitive to systemic chemotherapy, isolated dormant tumour cells and cancer stem cells in cell cycle arrest may be resistant to such regional as well as systemic therapies, resulting in a gradual increase in delayed recurrences among these early breast cancers over time. This theory is supported by Demicheli's persistent delayed peak in recurrence despite chemotherapy (Figure 1), as well as delayed recurrences seen in the more recent literature [1,2,14]. In fact, the dampening in early recurrence demonstrated in Figure 1 due to chemotherapy may be due to the effect of systemic chemotherapy upon disseminated non-dormant or 'awakened' micrometastatic cell clusters alone [10], with no impact upon either dormant isolated tumour cells or cancer stem cells. According to the theory of disease progression, one would expect dormant cells to present later than micrometastatic cell clusters (Figure 2). Perhaps there is a promotion of dormancy initiated by local treatments and hormonal therapy (even though such tumour cells are speculated to be estrogen and progesterone insensitive), which leads to a persistent recurrence risk over the following decade or two. The influence of radiation upon the development of metastatic capability and on dormant cells is not known, and there are no good pre-clinical models examining this aspect of tumour dormancy at present.
Suggestions for future research
There has been great interest in the development of gene microarray technology as a means by which tumour behaviour can be predicted. For example, two competing products now being marketed evaluate 70 and 21 gene expression profiles in breast cancer tissue, which are said to have an ability to predict with 80% accuracy [45,46] those tumours that will behave as 'high risk' cancers, with much greater risk of metastasis development. Additionally, these gene expression profiles demonstrate a 25% to 30% discordance with current phenotypic categorization of tumour risk for metastases. A potential advantage of this diagnostic modality is that molecular phenotypic in addition to tumour phenotypic features could be included in the determination of metastatic potential and potential tumour susceptibility to systemic therapy. Critics of these gene expression profiling tools as a method for identifying cells with metastatic potential suggest that bulk tumour analysis will fail to identify the few cells within the large number of non-metastatic cells, and caution that they thus should not be used to infer molecular mechanisms. Additionally, the gene array profiles of tumour cells sampled at sites of distant recurrences have been reported to be genetically more similar to the primary tumours than they are to metastases found in other patients [47], suggesting that the similarities identified in the gene array profiles may highlight a patient's germline genetic polymorphisms rather than genetic mutations leading to the development of these metastases [48]. In other words, this is felt to mean that gene profiles predicting for and allowing metastases may indicate germline polymorphisms inherent to the host. Studies by Hunter [48] thus raise the possibility that individuals may be differentially susceptible to support or fail to support metastatic growth, depending on their germline make-up. Whether this concept can be applied to an understanding of factors that contribute to metastatic dormancy and probability of tumour cells re-awakening remains to be assessed.
Mechanisms that allow for, or lead to, tumour dormancy awakening remain very poorly understood and require further study. Questions raised by the data emerging in the setting of current clinical and biological behaviour of breast cancer include: what are the factors that can promote awakening of dormant cells – host immune factors, host polymorphisms, or other features? Is it possible to predictably influence dormancy by hormonal or other manipulations? What impact does radiation have upon dormant tumour cells in breast cancer? Will genotyping of tumours result in clinical benefit for patients and lead to further understanding of patient or tumour features that indicate high risk of harbouring dormant tumour or cancer stem cells? In order to address these sorts of questions, improved experimental models coupled with longer-term follow-up on patients treated by current therapies and the effects of these therapies on late recurrences are needed.
Conclusion
Here we have reviewed clinical concepts related to tumour dormancy in breast cancer, and have considered the potential effects of modern therapies on long-term dormancy and late recurrences. We also have discussed concepts of tumour dormancy derived from pre-clinical research models of dormancy in breast cancer. Proposed areas of future research include the need to expand our understanding of clinical tumour dormancy of breast cancer in the era of locoregional radiation, prolonged hormonal therapy and genetic profiling, as well as the need for more and better experimental models with which to study tumour dormancy, the mechanisms responsible and the factors that may be able to influence either the maintenance of dormancy on the reawakening of dormant tumour cells.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
Supported by grant #42511 from the Canadian Institutes of Health Research (to AFC). JLT is supported by a Doctoral Research Award from the Canadian Institutes of Health Research. AFC is Canada Research Chair in Oncology, and receives salary support from the Canada Research Chairs Program.
References
- Early Breast Cancer Trialists' Collaborative Group Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: an overview of the randomized trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- Fisher B, Jeong J-H, Dignam J, Anderson S, Mamounas E, Wickerham DL, Wolmark N. Findings from recent National Surgical Adjuvant Breast and Bowel Project adjuvant studies in stage 1 breast cancer. J Natl Cancer Inst Monogr. 2001;30:62–66. doi: 10.1093/oxfordjournals.jncimonographs.a003463. [DOI] [PubMed] [Google Scholar]
- Wallgren A, Bonetti M, Gelber RD, Goldhirsch A, Castiglione-Gertsch M, Holmberg SB, Lindtner J, Thurlimann B, Fey M, Werner ID, et al. Risk factors for locoregional recurrence among breast cancer patients: Results from international breast cancer study group trials I through VII. J Clin Oncol. 2003;21:1205–1213. doi: 10.1200/JCO.2003.03.130. [DOI] [PubMed] [Google Scholar]
- Demicheli R, Retsky MW, Swartzendruber DE, Bonadonna G. Proposal for a new model of breast cancer metastatic development. Ann Oncol. 1997;8:1075–1080. doi: 10.1023/A:1008263116022. [DOI] [PubMed] [Google Scholar]
- Demicheli R, Terenziani M, Bonadonna G. Estimate of tumour growth time for breast cancer local recurrences: rapid growth after wake-up? Breast Cancer Res Treat. 1998;51:133–137. doi: 10.1023/A:1005887422022. [DOI] [PubMed] [Google Scholar]
- Demicheli R, Miceli R, Valagussa P, Bonadonna G. Re: Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 2000;92:347–348. doi: 10.1093/jnci/92.4.347. [DOI] [PubMed] [Google Scholar]
- Demicheli R. Tumour dormancy: findings and hypotheses from clinical research on breast cancer. Semin Cancer Biol. 2001;11:297–305. doi: 10.1006/scbi.2001.0385. [DOI] [PubMed] [Google Scholar]
- Karrison TG, Ferguson DJ, Meier P. Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 1999;91:80–85. doi: 10.1093/jnci/91.1.80. [DOI] [PubMed] [Google Scholar]
- Karrison T, Ferguson DJ, Meier P. RESPONSE: re: dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 2000;92:348. doi: 10.1093/jnci/92.4.348. [DOI] [PubMed] [Google Scholar]
- Demicheli R, Miceli R, Moliterni A, Zambetti M, Hrushesky WJM, Retsky MW, Valagussa P, Bonadonna G. Breast cancer recurrence dynamics following adjuvant CMF is consistent with tumor dormancy and mastectomy-driven acceleration of the metastatic process. Ann Oncol. 2005;16:1449–1457. doi: 10.1093/annonc/mdi280. [DOI] [PubMed] [Google Scholar]
- Spratt JS. Re: Dormancy of mammary carcinoma after mastectomy. J Natl Cancer Inst. 2000;92:1101. doi: 10.1093/jnci/92.13.1101. [DOI] [PubMed] [Google Scholar]
- Fisher B, Jeong J-H, Dignam J, Anderson S, Mamounas E, Wickerham DL, Wolmark N. Findings from recent National Surgical Adjuvant Breast and Bowel Project adjuvant studies in stage 1 breast cancer. J Natl Cancer Inst Monogr. 2001;30:62–66. doi: 10.1093/oxfordjournals.jncimonographs.a003463. [DOI] [PubMed] [Google Scholar]
- Goss PE, Ingle JN, Martino S, Robert NJ, Muss HB, Piccart MJ, Castiglione M, Tu D, Shepherd LE, Pritchard KI, et al. Randomized trial of Letrozole following Tamoxifen as extended adjuvant therapy in receptor-positive breast cancer: Updated findings from NCIC CTG MA.17. J Natl Cancer Inst. 2005;97:1262–1271. doi: 10.1093/jnci/dji250. [DOI] [PubMed] [Google Scholar]
- Nielsen HM, Overgaard M, Grau C, Jensen AR, Overgaard J. Study of failure pattern among high-risk breast cancer patients with or without post-mastectomy radiotherapy in addition to adjuvant systemic therapy: Long-term results from the Danish Breast Cancer Cooperative Group DBCG 82 b and c randomized studies. J Clin Oncol. 2006;24:2268–2275. doi: 10.1200/JCO.2005.02.8738. [DOI] [PubMed] [Google Scholar]
- Naumov GN, Townson JL, MacDonald IC, Wilson SM, Bramwell VH, Groom AC, Chambers AF. Ineffectiveness of doxorubicin treatment on solitary dormant mammary carcinoma cells or late-developing metastases. Breast Cancer Res Treat. 2003;82:199–206. doi: 10.1023/B:BREA.0000004377.12288.3c. [DOI] [PubMed] [Google Scholar]
- Townson JL, Chambers AF. Dormancy of solitary metastatic cells. Cell Cycle. 2006;5:1744–1750. doi: 10.4161/cc.5.16.2864. [DOI] [PubMed] [Google Scholar]
- Allan AL, Vantyghem SA, Tuck AB, Chambers AF. Tumour dormancy and cancer stem cells: implications for the biology and treatment of breast cancer metastasis. Breast Disease. 2007;26:87–98. doi: 10.3233/bd-2007-26108. [DOI] [PubMed] [Google Scholar]
- Luzzi KJ, MacDonald IC, Schmidt EE, Kerkvliet N, Morris VL, Chambers AF, Groom AC. Multistep nature of metastatic inefficiency: dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am J Pathol. 1998;153:865–873. doi: 10.1016/S0002-9440(10)65628-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov GN, MacDonald IC, Weinmeister PM, Kerkvliet N, Nadkarni KV, Wilson SM, Morris VL, Groom AC, Chambers AF. Persistence of solitary mammary carcinoma cells in a secondary site: a possible contributor to dormancy. Cancer Res. 2002;62:2162–2168. [PubMed] [Google Scholar]
- Naumov GN, MacDonald IC, Chambers AF, Groom AC. Solitary cancer cells as a possible source of tumour dormancy? Semin Cancer Biol. 2001;11:271–276. doi: 10.1006/scbi.2001.0382. [DOI] [PubMed] [Google Scholar]
- Heyn C, Ronald JA, Ramadan SS, Snir JA, Barry AM, MacKenzie LT, Mikulis DJ, Palmieri D, Bronder JL, Steeg PS, et al. In vivo MRI of cancer cell fate at the single-cell level in a mouse model of breast cancer metastasis to the brain. Magn Reson Med. 2006;56:1001–1010. doi: 10.1002/mrm.21029. [DOI] [PubMed] [Google Scholar]
- Holmgren L, O'Reilly MS, Folkman J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat Med. 1995;1:149–153. doi: 10.1038/nm0295-149. [DOI] [PubMed] [Google Scholar]
- Weiss L. Metastatic inefficiency: intravascular and intraperitoneal implantation of cancer cells. Cancer Treat Res. 1996;82:1–11. doi: 10.1007/978-1-4613-1247-5_1. [DOI] [PubMed] [Google Scholar]
- Cameron MD, Schmidt EE, Kerkvliet N, Nadkarni KV, Morris VL, Groom AC, Chambers AF, MacDonald IC. Temporal progression of metastasis in lung: cell survival, dormancy, and location dependence of metastatic inefficiency. Cancer Res. 2000;60:2541–2546. [PubMed] [Google Scholar]
- Chambers AF, Groom AC, MacDonald IC. Dissemination and growth of cancer cells in metastatic sites. Nat Rev Cancer. 2002;2:563–572. doi: 10.1038/nrc865. [DOI] [PubMed] [Google Scholar]
- Koop S, MacDonald IC, Luzzi K, Schmidt EE, Morris VL, Grattan M, Khokha R, Chambers AF, Groom AC. Fate of melanoma cells entering the microcirculation: over 80% survive and extravasate. Cancer Res. 1995;55:2520–2523. [PubMed] [Google Scholar]
- Tarin D, Price JE, Kettlewell MG, Souter RG, Vass AC, Crossley B. Clinicopathological observations on metastasis in man studied in patients treated with peritoneovenous shunts. Br Med J (Clin Res Ed) 1984;288:749–751. doi: 10.1136/bmj.288.6419.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA. The problem of cancer dormancy: understanding the basic mechanisms and identifying therapeutic opportunities. Cell Cycle. 2006;5:1740–1743. doi: 10.4161/cc.5.16.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naumov GN, Akslen LA, Folkman J. Role of angiogenesis in human tumor dormancy: animal models of the angiogenic switch. Cell Cycle. 2006;5:1779–1787. doi: 10.4161/cc.5.16.3018. [DOI] [PubMed] [Google Scholar]
- Naumov GN, Bender E, Zurakowski D, Kang SY, Sampson D, Flynn E, Watnick RS, Straume O, Akslen LA, Folkman J, Almog N. Model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J Natl Cancer Inst. 2006;98:316–325. doi: 10.1093/jnci/djj068. [DOI] [PubMed] [Google Scholar]
- Hedley BD, Allan AL, Chambers AF. Tumor dormancy and the role of metastasis suppressor genes in regulating ectopic growth. Future Oncol. 2006;2:627–641. doi: 10.2217/14796694.2.5.627. [DOI] [PubMed] [Google Scholar]
- Yamamoto N, Jiang P, Yang M, Xu M, Yamauchi K, Tsuchiya H, Tomita K, Wahl GM, Moossa AR, Hoffman RM. Cellular dynamics visualized in live cells in vitro and in vivo by differential dual-color nuclear-cytoplasmic fluorescent-protein expression. Cancer Res. 2004;64:4251–4256. doi: 10.1158/0008-5472.CAN-04-0643. [DOI] [PubMed] [Google Scholar]
- Tsuji K, Yamauchi K, Yang M, Jiang P, Bouvet M, Endo H, Kanai Y, Yamashita K, Moossa AR, Hoffman RM. Dual-color imaging of nuclear-cytoplasmic dynamics, viability, and proliferation of cancer cells in the portal vein area. Cancer Res. 2006;66:303–306. doi: 10.1158/0008-5472.CAN-05-2958. [DOI] [PubMed] [Google Scholar]
- Gao X, Cui Y, Levenson RM, Chung LW, Nie S. In vivo cancer targeting and imaging with semiconductor quantum dots. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- Voura EB, Jaiswal JK, Mattoussi H, Simon SM. Tracking metastatic tumor cell extravasation with quantum dot nanocrystals and fluorescence emission-scanning microscopy. Nat Med. 2004;10:993–998. doi: 10.1038/nm1096. [DOI] [PubMed] [Google Scholar]
- Chambers AF, Shafir R, Ling V. A model system for studying metastasis using the embryonic chick. Cancer Res. 1982;42:4018–4025. [PubMed] [Google Scholar]
- Chambers AF, Schmidt EE, MacDonald IC, Morris VL, Groom AC. Early steps in hematogenous metastasis of B16F1 melanoma cells in chick embryos studied by high-resolution intravital videomicroscopy. J Natl Cancer Inst. 1992;84:797–803. doi: 10.1093/jnci/84.10.797. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Kovalski K, Ossowski L. Tumor dormancy induced by downregulation of urokinase receptor in human carcinoma involves integrin and MAPK signaling. J Cell Biol. 1999;147:89–104. doi: 10.1083/jcb.147.1.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Estrada Y, Liu D, Ossowski L. ERK(MAPK) activity as a determinant of tumor growth and dormancy; regulation by p38(SAPK) Cancer Res. 2003;63:1684–1695. [PubMed] [Google Scholar]
- Marches R, Hsueh R, Uhr JW. Cancer dormancy and cell signaling: induction of p21(waf1) initiated by membrane IgM engagement increases survival of B lymphoma cells. Proc Natl Acad Sci USA. 1999;96:8711–8715. doi: 10.1073/pnas.96.15.8711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shachaf CM, Kopelman AM, Arvanitis C, Karlsson A, Beer S, Mandl S, Bachmann MH, Borowsky AD, Ruebner B, Cardiff RD, et al. MYC inactivation uncovers pluripotent differentiation and tumour dormancy in hepatocellular cancer. Nature. 2004;431:1112–1117. doi: 10.1038/nature03043. [DOI] [PubMed] [Google Scholar]
- Aguirre-Ghiso JA, Liu D, Mignatti A, Kovalski K, Ossowski L. Urokinase receptor and fibronectin regulate the ERK(MAPK) to p38(MAPK) activity ratios that determine carcinoma cell proliferation or dormancy in vivo. Mol Biol Cell. 2001;12:863–879. doi: 10.1091/mbc.12.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ossowski L, Aguirre Ghiso J, Liu D, Yu W, Kovalski K. The role of plasminogen activator receptor in cancer invasion and dormancy. Medicina (B Aires) 1999;59:547–552. [PubMed] [Google Scholar]
- Indraccolo S, Stievano L, Minuzzo S, Tosello V, Esposito G, Piovan E, Zamarchi R, Chieco-Bianchi L, Amadori A. Interruption of tumor dormancy by a transient angiogenic burst within the tumor microenvironment. Proc Natl Acad Sci USA. 2006;103:4216–4221. doi: 10.1073/pnas.0506200103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van't Veer LJ, Dai H, van de Vijver MJ, He YK, Hart AAM, Mao M, Peterse LH, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–535. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. New Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- Weigelt B, Glas AM, Wessels LFA, Witteveen AT, Peterse JL, van't Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci USA. 2003;100:15901–15905. doi: 10.1073/pnas.2634067100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter K. Host genetics influence tumour metastasis. Nat Rev Cancer. 2006;6:141–146. doi: 10.1038/nrc1803. [DOI] [PubMed] [Google Scholar]