

Abstract
The pathophysiology of systemic inflammation and sepsis involves peripheral organs, causing gastrointestinal, renal, and cardiovascular alterations, as well as the central nervous system (CNS), affecting sleep, temperature regulation, behavior, and neuroendocrine function. The molecular basis of the CNS effects of systemic inflammation are not fully elucidated. Here we show that the CNS responds to systemic inflammation with pronounced IL-1β gene expression and limited IL-1 receptor antagonist (IL-1ra), IL-10, and IL-13 gene expression. This pattern occurs throughout the CNS, including areas such as the subfornical organ, pineal gland, neurohypophysis, and hypothalamus. In contrast, in the anterior pituitary, we found limited IL-1β gene expression but marked induction of the mRNA encoding for the secreted isoform of IL-1ra, secreted IL-1ra. We conclude that the central manifestations of peripheral inflammation are mediated by endogenous brain IL-1β synthesized during systemic inflammation in the context of limited central cytokine counter regulation of IL-1. As IL-1β is a potent stimulus for inducible nitric oxide synthase expression and activity, these findings explain our previous observation that systemic inflammation promotes inducible nitric oxide synthase gene expression in the brain and the spillover of NO metabolites into cerebrospinal fluid. The CNS transcription of the HIV-1 replication factor IL-1β in the context of limited transcription of the IL-1 replication inhibitors IL-1ra, IL-10, and IL-13 might help explain the negative impact of systemic inflammation on the clinical course of AIDS. In addition, we propose that IL-1ra may be secreted by the anterior pituitary as a systemic anti-inflammatory hormone that is released in response to IL-1β originated from multiple sources.
Keywords: lipopolysaccharide, hormone, behavior, NO, HIV-1
Inflammation originating from peripheral sites causes profound signs and symptoms mediated by the central nervous system (CNS). The central manifestations of peripheral inflammation include alterations in temperature regulation and cognition, suppression of locomotion and exploration, reductions of food gathering and sexual behavior, and increase in sleep and lethargy (1). CNS signs and symptoms of systemic illness can be reproduced by central exogenous IL-1β administration (1), and prevented if high levels of IL-1 receptor antagonist (IL-1ra) are administered in conjunction with IL-1β centrally. Moreover, IL-1β knockout mice have no fever and no alterations in ingestive behavior in response to some types of peripheral inflammation (2). Additionally, the ability of some viruses to induce fever depends on the specific bioavailability of IL-1; vaccinia viruses that express soluble IL-1 receptors do not cause fever, and vaccinia-induced fever can also be inhibited with antibodies to IL-1β (3).
The biological effects of IL-1 reflect the local ratio of IL-1β and of cytokines that inhibit IL-1 action (4); we therefore hypothesized that systemic inflammation induces IL-1β gene expression in the brain with limited effects on IL-10, IL-13, and IL-1ra, cytokines that inhibit IL-1β bioactivity. Because IL-1β is a potent stimulus for inducible nitric oxide synthase (iNOS) transcription in the brain (5), this hypothesis could help explain our recent findings that systemic inflammation causes rapid induction of iNOS gene expression in discrete areas of the rat brain, accompanied by the spillover of NO metabolites into brain parenchyma and cerebrospinal fluid (6). To test our hypothesis we studied the brains of rats during systemic inflammation caused by peripheral injection of Escherichia coli lipopolysaccharide (LPS). We assessed changes in the expression of the gene encoding for IL-1β in selected neuroanatomical structures.
Because cytokine counter regulation can be redundant, we also studied the expression of genes encoding three different cytokines that inhibit IL-1β bioactivity: IL-10, IL-13, and IL-1ra. IL-10, also known as cytokine synthesis inhibitory factor, inhibits IL-1 expression (7). IL-13 counterregulates IL-1 bioactivity by inhibiting the synthesis of IL-1 and by inducing the synthesis of IL-1ra and of the type II IL-1 receptor that is an endogenous decoy for bioactive IL-1 (8–10). IL-1ra, a neuroprotective cytokine (11–13) that we have previously localized in the brain (14), is a pure endogenous antagonist of IL-1 action (15, 16). The IL-1ra gene has two different promoters (Ps and Pic) (17, 18) that regulate the expression of secreted (sIL-1ra) (15, 16) and the intracellular (icIL-1ra) isoforms (19, 20) of IL-1ra. To determine whether the pituitary gland might secrete IL-1ra, we cloned and sequenced the IL-1ra mRNA species from the pituitary and compared it to the sequences of the secreted and the intracellular isoforms of IL-1ra mRNA.
MATERIALS AND METHODS
Animals.
Studies were carried out in accordance with animal protocols approved by the National Institutes of Health. Experiments were designed to avoid confounding variables such as infection, stress, and circadian variation in mRNA levels: we used virus- and antibody-free, male Sprague–Dawley rats (200–250 g; Harlan Breeders, Indianapolis), housed in a light- (12-h on/12-h off) and temperature-controlled environment, with food and water ad libitum. Injections were timed so that all tissue collection occurred at 10:00–11:00 h. Different groups of animals (n = 6/group) were studied 0, 2, 6, or 24 h after intraperitoneal (i.p.) injection of LPS (serotype 055:B5; Sigma), or saline (control groups), and otherwise treated under identical conditions. To prevent the confounding effects of stress on cytokine gene expression, animals were removed from their home cages by a dedicated animal handler and were decapitated within 45 sec of removal from home cages.
In Situ Hybridization Histochemistry (ISHH).
Brains were rapidly removed and stored at −70°C before processing for ISHH. Species-specific ribonucleotide probes were generated from rat IL-1β cDNA, generously provided by T. Nishida (21); rat IL-1ra cDNA, generously provided by R. Hart (22); rat IL-10 cDNA, kindly provided by R. Bell (23); and rat IL-13 cDNA, kindly provided by F. G. Lakkis (24). All probes were sequenced and characterized in our laboratory (25). Transcription of antisense and sense probes was carried out using the Riboprobe System (Promega) in the presence of [α-35S]UTP (specific activity, 1000–1500 Ci/mmol; 1 Ci = 37 GBq; New England Nuclear). IL-1β, IL-1ra, IL-10, and IL-13 mRNA levels were examined in adjacent coronal sections obtained every 1.0 mm in each animal. Each slide contained two adjacent sections. Sectioning, fixing, ISHH, autoradiography with 2 weeks of exposure, and anatomical localization of the probe were performed as described (26). To test the specificity of both antisense probes and the hybridization method, controls were generated using labeled sense probes and excess cold probe (×100). Hybridization and posthybridization treatments were concomitantly carried out on antisense and control sections.
Quantitative Densitometry.
Quantification of mRNA levels was done as described by Landau et al. (27) using a Macintosh-based image analysis program (nih image version 1.55; W. Rasband, ref. 28).
Reverse Transcriptase–PCR (RT-PCR), Cloning, and Sequencing.
Total RNA obtained from pituitaries of rats treated with LPS 6 h after i.p. injection was isolated using triZOL RNA reagent (GIBCO/BRL). Total RNA was dissolved in RNase-free water, the amount of RNA was determined by spectrophotometry, and the samples were treated with RNase-free DNase (Promega) for 30 min at 37°C. After DNase treatment, RNA was purified using RNeasy columns (Qiagen, Chatsworth, CA). Reverse transcription was done using Gene Amp RNA PCR kit (Perkin–Elmer). We did two sets of reactions, one set using random hexamer primers, and another set using oligo d(T)16 primers. RT reactions were incubated at room temperature for 10 min and then cycled once using DNA thermal Cycler 480 (Perkin–Elmer). PCRs were set up following the manufacturer’s protocol, and using three forward primers with the GenBank sequence for rat IL-1ra (22)—A (1 to 25), B (19 to 43), and C (103 to 128)—and one reverse primer (513 to 537). Primer pairs A-D and B-D were designed to amplify sIL-1ra and primer pair C-D was designed to also amplify icIL-1ra. After amplification cycles were completed (initial denaturation was carried out at 95°C for 105 sec, then 35 cycles of 95°C for 15 sec and 60°C for 30 sec, and then 72°C for 7 min), 20 μl of the product was sized on a 1% agarose gel. The gel showed a single band per reaction, and all bands were of the expected sizes. Cloning of the PCR products was done using Invitrogen TA cloning kit. The Sanger dideoxynucleotide chain termination method (29) was used for sequencing of all clones; the sequencing protocol used Sequenase 2.0 (Amersham) and [α-35S]d-ATP.
RESULTS
After i.p. administration of LPS animals showed signs of systemic inflammation: piloerection, mild shaking, and lethargy.
We found profound and widespread induction of IL-1β gene expression in the brain after LPS treatment (Figs. 1 and 2 A, D, G, J, and M). Regions of the brain lacking blood-brain barrier, such as the neurohypophysis (Figs. 1H, 2P, and 3A), the pineal body (Fig. 1I and 2D), the subfornical organ (Fig. 1 F, K, and P), and the median eminence of the hypothalamus (Fig. 4 A–C) had an early increase of IL-1β mRNA expression. Regions of blood–cerebrospinal fluid barrier (choroid plexus in the lateral, third, and fourth ventricles) (Fig. 2G) and meninges (Fig. 2M) had early and sustained induction of IL-1β gene expression. The hypothalamus, a key area that regulates temperature, food intake, and sexual behavior, had a remarkable increase of IL-1β mRNA 6 h after LPS injection (Figs. 1L, 2A, and 5). In addition, we found a widespread, diffuse, and transient increase of IL-1β mRNA throughout brain parenchyma (Fig. 1 K–O) 6 h after LPS, but 24 h after LPS administration the residual response of IL-1β gene expression is confined to the cerebral cortex (Fig. 1 P–T). We found very modest increases in IL-1ra and IL-10 gene expression in the CNS, predominantly in meninges, choroid plexus, and perivascular areas (Fig. 2 B, E, H, K, N, and Q for IL-1ra and C, F, I, L, O, and R for IL-10). Induction of IL-13 gene expression in the CNS was not detected in our experiments (data not shown).
Figure 1.

Localization of IL-1β mRNA in the rat brain by ISHH after treatment with LPS. In Figs. 1, 2, 3, 4, 5 images are representative of findings in six animals/group. A series of film autoradiographs is arranged from rostral to caudal (top to bottom), showing the regional pattern of IL-1β gene expression. Brain slices are shown in the first column (A–E) represent the hybridization of IL-1β antisense riboprobe in the brain of control rats, showing no detectable IL-1β mRNA. Two hours after a single LPS injection i.p. (5.0 mg/animal), the induction of IL-1β mRNA the brain is shown in the second column (F–J). There was induction of IL-1β mRNA in the choroid plexus (arrowhead in F) and subfornical organ (arrow in F), posterior pituitary (arrow in H), pineal (arrow in I), and meninges (arrow in J). Six hours after a single LPS injection, the induction of IL-1β throughout the brain is shown in the third column (K–O). There was a remarkable induction of IL-1β mRNA in the paraventricular nucleus of the hypothalamus (arrow in L); the induction in the choroid plexus (arrowhead in K), meninges (arrow in O), and in the subfornical organ (arrow in K) persists. Twenty-four hours after a single LPS injection the levels of IL-1β mRNA throughout the rat brain were considerably decreased (fourth column, P–T). (Bar = 1.3 cm.)
Figure 2.

Differential IL-1β, IL-1ra, and IL-10 gene expression in rat brain and pituitary following LPS treatment. A series of darkfield images show brain regions hybridized with a 35S-labeled rat antisense riboprobes for IL-1β (first column: A, D, G, J, M, and P), for IL-1ra (second column: B, E, H, K, N, and Q), and for IL-10 (third column: C, F, I, L, O, and R) after LPS treatment. Strong induction of IL-1β mRNA is evident in several brain regions, whereas the induction of IL-1ra mRNA and IL-10 mRNA is considerably less visible. Images represent the peak of mRNA induction after treatment with LPS for each of the areas shown in this figure. The following brain regions are shown: paraventricular nucleus of the hypothalamus (A–C), pineal gland (D–F), choroid plexus (G–I), vessels located in the ventral aspect of the brain (J–L), meninges (M–O), and pituitary gland (P–R). Arrow in P points to the posterior pituitary; all other arrows help discriminate cells that have increased mRNA levels of either IL-1ra or IL-10. Arrowhead in B points to vascular structure. White dots represent silver grains overlying mRNA. (Bar = 160 μm in P–R, and 80 μm in A–O.)
Figure 3.
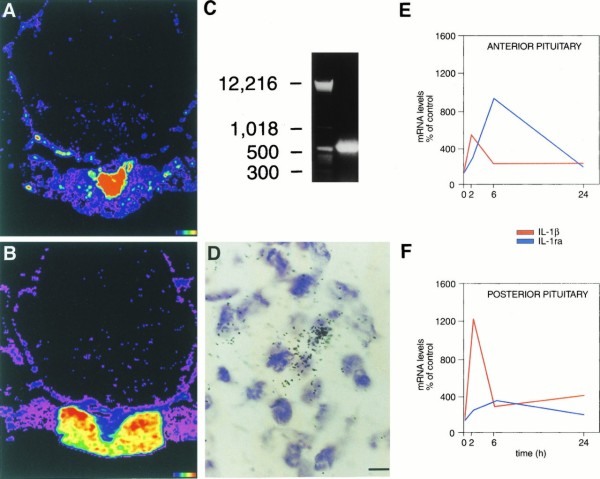
Induction of IL-1β mRNA and IL-1ra mRNA in the pituitary after LPS treatment. Computer-generated pseudocolor images are shown in A for IL-1β and in B for IL-1ra; images show peak induction of IL-1β mRNA 2 h after LPS administration (A) and of IL-1ra mRNA 6 h after LPS administration (B). Note that IL-1β mRNA induction occurs predominantly in the posterior pituitary (A) and that IL-1ra mRNA induction occurs predominantly in the anterior pituitary (B). (C) Photograph of an agarose gel containing RT-PCR product of RNA obtained from pituitaries of rats treated with LPS 6 h after i.p. injection. The gel showed a single band of the expected size (≈537 bp) for sIL-1ra; cloning and sequencing of the PCR product confirmed the specific sequence of sIL-1ra (16, 22). (D) High magnification image of IL-1ra mRNA hybridization in the anterior pituitary; black dots represent silver grains overlying IL-1ra mRNA. (E and F) Graphics show the time course for the induction of IL-1β mRNA (red line) and IL-1ra mRNA (blue line) in the anterior pituitary (E) and in the posterior pituitary (F) at 0, 2, 6, and 24 h after LPS administration, using quantitative densitometry from autoradiographic images. Using ANOVA with post hoc correction we found that both in the anterior and in the posterior pituitary increases in mRNA levels over time-matched control values were significant for IL-1β at the 0.0001 level at 2 h and at the 0.05 level at 6 and 24 h, and for IL-1ra at the 0.05 level at 2 h and at the 0.0001 level at 6 h; at 24 h IL-1ra mRNA levels were the same in LPS and saline-treated groups. (Color scale for A and B: black indicates background and red indicates areas of highest hybridization levels. Bar = 5 μm in D.)
Figure 4.
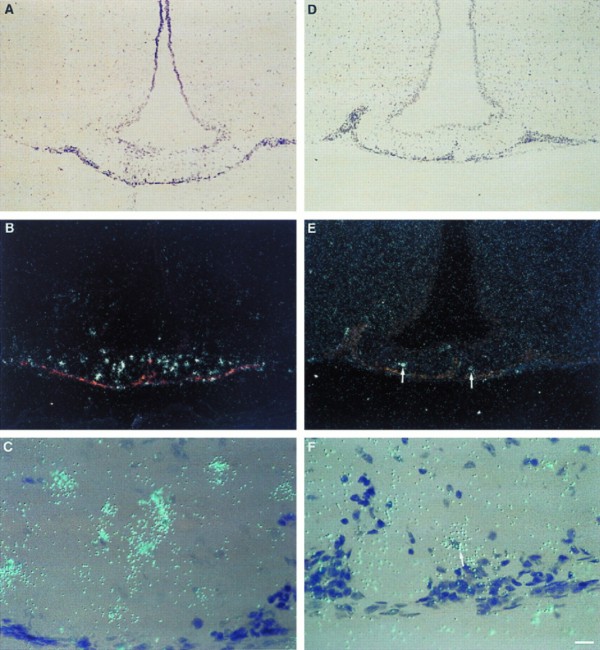
Induction of IL-1β mRNA and IL-1ra mRNA in the median eminence (ME) after treatment with i.p. LPS. IL-1β mRNA is shown in the first column (A–C), and IL-1ra mRNA is shown in the second column (D–F). There is a remarkable induction of IL-1β mRNA in the ME after LPS administration, while the induction of IL-1ra mRNA is present, but substantially less intense. The time points shown, 2 h for IL-1β (A–C) and 6 h for IL-1ra (D–F), represent the peak of mRNA induction in the ME for IL-1β and IL-1ra, respectively. Low magnification darkfield pictures of the ME are depicted in B and E, the corresponding brightfield pictures are depicted in A and D. C and F show high magnification pictures of the ME. White dots (B and E) or blue dots (C and F) represent silver grains overlying mRNA; arrow in F points to a cell overexpressing IL-1ra mRNA. (Bar = 70 μm in A, B, D, E, and 9 μm in C and F.)
Figure 5.

Induction of IL-1β mRNA in the arcuate nucleus of the hypothalamus 6 h after i.p. LPS treatment. Low magnification darkfield image (A) and high magnification image (B) are shown. White dots (A) or blue dots (B) represent silver grains overlying IL-1β mRNA. (Bar = 85 μm in A and 11 μm in B.)
In the pituitary gland we found high levels of induction of IL-1β mRNA in the posterior pituitary, with little IL-1ra, IL-10, or IL-13 gene expression. In contrast, in the anterior pituitary we found that systemic inflammation induced IL-1ra mRNA in levels that were far higher than those for IL-1β mRNA (Fig. 3 A, B, E, and F).
Hybridizations with excess cold probe (×100) and with sense riboprobes yielded a uniform, low signal, barely visible on film.
We used RT-PCR, cloning, and sequencing and verified that during peripheral inflammation the splice variant of the IL-1ra gene expressed in the pituitary and detected in our in situ hybridization experiments is the secreted isoform, sIL1-ra (Fig. 3 C and D).
DISCUSSION
We found that in the CNS, including the neurohypophysis, the gene encoding for IL-1β is expressed during systemic inflammation in higher levels than the genes encoding for the counter regulatory cytokines IL-1ra, IL-10, and IL-13. To our knowledge this is the first neuroanatomical localization of IL-10 gene expression in the brain. We conclude that the CNS manifestations of peripheral inflammation, including iNOS induction, can be mediated by IL-1β synthesized de novo within the brain.
The sequence of events and pathways leading to LPS-induced systemic inflammation, and IL-1β, IL-1ra, IL-10, and iNOS gene expression in the brain are not yet fully elucidated. The uptake of LPS probably occurs at the choroid plexus and other circumventricular organs, leading to transport to the ependyma lining of the third ventricle or tanycytes of the median eminence. Alternatively, during systemic inflammation and sepsis LPS may penetrate into the blood–brain barrier, reaching neurons. We have also recently shown that during systemic inflammation IL-1β is activated within the vascular wall in brain vasculature, acting on IL-1 type 1 receptors that are localized at the interface between vasculature and perivascular glia and causing iNOS transcription in perivascular glial cells (30). After specific glial and neuronal cells receive a signal that was initiated by LPS there is induction of IL-1β and of iNOS, an enzyme whose transcription is regulated by IL-1β. Comparing our previous findings of iNOS gene expression in the brain after LPS administration to our new findings of IL-1β gene expression, we can see that at 2 h there is induction of IL-1β mRNA and iNOS mRNA in choroid plexus, meninges, and vasculature; 6 h after LPS we observed both IL-1β and iNOS gene expression in median eminence, paraventricular nucleus, and arcuate nucleus. This pattern of IL-1β and iNOS induction first in areas without an effective blood–brain barrier and later within brain parenchyma shows a temporal course by which endotoxin-induced inflammation can affect key functions of the brain. In addition to the direct effects of IL-1β on brain functions such as temperature regulation, food intake, sleep, and neuroendocrine regulation, IL-1β can further modulate CNS functions during inflammation through IL-1β-induced iNOS, resulting in high levels of NO production that, for example, modulates release of corticotropin-releasing hormone (6, 31). We also showed IL-1ra and IL-10 gene expression in the brain during these experiments. It has been previously shown that IL-1ra can be induced by IL-1β (32); however, the stimulus for IL-10 transcription in the brain that was observed in our studies remains unknown.
Our findings of IL-1β mRNA induction in the paraventricular nucleus and arcuate nucleus, which are sites of production of the major hypothalamic and inhibiting hormones, suggest that IL-1β synthesized within the hypothalamus modulates neuroendocrine function during systemic inflammation. LPS injections in the rat have been previously shown to induce IL-1α immunoreactivity in neurons of the dorsal medial anterior hypothalamic area and preoptic region, which also contain thermosensitive cells (33). IL-1α is therefore thought to be important in the regulation neuroendocrine function during infection. Thus, during infection and inflammation, neuroendocrine function may be modulated by the induction of both IL-1α and IL-1β in the hypothalamus.
The finding of high levels of IL-1β gene expression in the CNS during systemic inflammation (Fig. 1) indicates that IL-1β might be an important neuroregulator of the central nervous system response to inflammatory stressors. We propose that the actions of brain IL-1β are so evident during systemic inflammation presumably because in the brain there is only very limited gene expression of cytokines that counter regulate IL-1 bioactivity. This is supported by the findings that exogenous IL-1β administration causes the characteristic behavioral symptoms associated with peripheral illness, and that those behavioral effects of IL-1β are inhibited by exogenous central IL-1ra administration (34–37), by a knockout of the IL-1β gene (2), or by IL-1 soluble receptors or antibodies (3). Thus, in the brain, antagonism or knock-out of IL-1β can abolish sickness-associated behavioral signs and symptoms caused by IL-1β.
In the circulation, IL-1 induction is followed by a robust response of IL-1ra, a cytokine that inhibits IL-1 function. Granowitz et al. (38) reported that the peripheral IL-1ra response to LPS is 100 fold greater than that of IL-1β. Kuhns et al. (39) have also reported that after intravenous injection of endotoxin the levels of IL-1ra in the peripheral circulation far exceed those of IL-1β. Miller et al. (4) have shown that in synovial fluid obtained from patients with Lyme arthritis the levels of IL-1ra can be 50-fold higher than those of IL-1β. The production of such high levels of endogenous IL-1ra peripherally is clinically relevant in the modulation of the inflammatory response, as patients who produce higher ratios of IL-1ra over IL-1β have rapid resolution of attacks of arthritis, whereas patients with the reverse pattern of cytokine concentrations have long intervals to recovery; interestingly, in those patients tumor necrosis factor α concentrations showed no relation to the clinical course of arthritis (4). We observed that in contrast to the bloodstream or to synovial fluid, the brain response to the endogenous IL-1β induction by LPS does not result in marked induction of IL-1ra gene expression. Because IL-1β is a stimulus for IL-1ra production in peripheral tissues (32), our data raise the possibility that the regulation of IL-1ra gene expression in the brain may be different from that described in the periphery.
In the pituitary gland we found that during systemic inflammation there is a predominant induction of IL-1β mRNA in the posterior lobe, and of IL-1ra mRNA in the anterior lobe. Embryologically and anatomically the posterior pituitary (neurohypophysis) is a downward growth or stalk from the floor of the diencephalon, and therefore a part of the CNS, while the anterior pituitary (adenohypophysis), which forms from Rathke’s pouch, an outgrowth from the roof of the embryonic mouth, is a peripheral organ (40). Thus, in the posterior pituitary we found high levels of IL-1β mRNA induction, with a limited IL-1ra mRNA response (Fig. 3 A and F), which is the same pattern we observed throughout the CNS. In contrast, in the anterior pituitary we showed a pattern of IL-1β induction that has been previously described in the bloodstream and in synovial fluid (4, 38, 39), characterized by increases in IL-1ra gene expression that far exceed the local induction of IL-1β mRNA (Fig. 3 B and E).
There are three splice variants of the IL-1ra gene, one encoding the secreted isoform of IL-1ra or sIL-1ra (15, 16) and the other two encoding nonsecreted, intracellular isoforms, or icIL-1ra (19, 20). sIL-1ra and icIL-1ra expression is regulated by two distinct promoters, Ps and Pic (17, 18). We showed that the splice variant of the IL-1ra gene expressed in the pituitary during systemic inflammation is the secreted isoform (sIL-1ra). IL-1ra is secreted in response to IL-1β (32), and in our studies we show that the anterior pituitary IL-1ra mRNA response follows that of IL-1β mRNA (Fig. 3E).
In response to systemic inflammation, the posterior lobe of the pituitary produces high levels of IL-1β mRNA, while the anterior lobe produces preferentially high levels of IL-1ra mRNA. As IL-1β mRNA appears in the anterior pituitary with a peak at 2 h, followed by a peak of IL-1ra at 6 h, it is reasonable to conclude that IL-1ra mRNA is produced as a result of the induction of IL-1β in the anterior lobe. However, as there are high levels of IL-1β induction in the posterior pituitary at 2 h with only a small increase in IL-1ra gene expression in the same area at 6 h, it is possible that the synthesis of IL-1β in the posterior pituitary is a key determinant of the induction of IL-1ra mRNA in the anterior pituitary. IL-1β originating from the posterior lobe would be transported by short portal vessels to the anterior lobe (41) and could augment the transcription of the IL-1ra gene also induced in part by the local production of IL-1β within the anterior lobe. Therefore, we propose that the induction of IL-1ra in the anterior lobe may be due to the combined action of IL-1β produced in the anterior lobe plus IL-1β synthesized in other sites, such as the posterior lobe, and transported via portal vessels.
We also show that areas of the CNS whose secretions are known to affect anterior pituitary function express the gene encoding IL-1β in high levels, with limited IL-1ra, IL-10, and IL-13 counter regulation. Those regions include the hypothalamus [paraventricular nucleus (Figs. 1L and 2A) and arcuate nucleus (Fig. 5), and the median eminence (Fig. 4 A–C)]. Moreover, during systemic inflammation, circulating IL-1β (42) is thought to modulate anterior pituitary hormone secretion (43). We hypothesize that the pituitary secretion of IL-1ra might represent a novel endogenous systemic anti-inflammatory hormonal mechanism stimulated by IL-1β reaching the anterior pituitary from multiple sources.
The different patterns of central and circulating IL-1 counter regulation may be the result of evolution. During systemic inflammation it is advantageous to counterregulate IL-1 action in peripheral body fluids, thus limiting the inflammatory response in peripheral tissues. On the other hand we suggest that it may be advantageous not to counter regulate the actions of IL-1 in the brain, as those actions cause sickness behavior, leading to sleep, decreased search for food, inhibition of reproduction, and suppression of locomotion and exploration, thereby diminishing the likelihood of confrontation with predators and facilitating recovery.
The role of endogenous factors on HIV-1 replication is a current focus of AIDS research. IL-1β is a well-known HIV-1 replication factor, and IL-1ra inhibits the inductive effects of IL-1 on HIV-1 (44). IL-10 and IL-13 are endogenous inhibitors of HIV-1 replication (45–48); IL-10 is currently being used in phase I clinical trials in AIDS. Inflammation worsens the course of HIV-1 infection (49, 50). We report here that the CNS responds to peripheral inflammation by a profound induction of the gene encoding for the HIV-1 replication factor IL-1β in the context of modest induction of IL-1ra mRNA. We also demonstrate that during peripheral inflammation there is in the CNS very limited induction of the gene encoding for the HIV-1 replication inhibitor IL-10 and no detectable induction of the gene encoding for the HIV-1 replication inhibitor IL-13. Vitkovic et al. (51) have discussed how neuroAIDS may be characterized by a vicious cycle in which HIV-infected monocytes/macrophages may induce the secretion of IL-1 by astrocytes, endothelial cells, and microglial cells: IL-1, stimulating transforming growth factor β1, and tumor necrosis factor α, may further activate HIV replication. Future studies should test the hypothesis that the CNS pattern of cytokine response to peripheral inflammation may have a detrimental effect on the clinical course of AIDS.
Acknowledgments
We are grateful to Dr. Philip W. Gold for his comments and support. This work was supported by a National Alliance for Research on Schizophrenia and Depression Young Investigator Award (M.-L.W.), and by National Institutes of Health Grant MH 51853 (S.M.M.).
Footnotes
Abbreviations: IL, interleukin; IL-1β, interleukin 1β; IL-1ra, interleukin 1 receptor antagonist; sIL-1ra, secreted IL-1ra; Ps, promoter for sIL-1ra; icIL-1ra, intracellular IL-1ra; Pic, promoter for icIL-1ra; iNOS, inducible nitric oxide synthase; CNS, central nervous system; RT-PCR, reverse transcriptase–PCR; LPS, lipopolysaccharide; ISHH, in situ hybridization histochemistry.
References
- 1.Rothwell N J, Hopkins S J. Trends Neurosci. 1995;18:130–136. doi: 10.1016/0166-2236(95)93890-a. [DOI] [PubMed] [Google Scholar]
- 2.Zheng H, Fletcher D, Kozak W, Jiang M, Hofmann K J, Conn C A, Soszynski D, Grabiec C, Trumbauer M E, Shaw A, Kostura M J, Stevens K, Rosen H, North R J, Chen H Y, Tocci M J, Klugger M J, van der Ploeg L H T. Immunity. 1995;3:9–19. doi: 10.1016/1074-7613(95)90154-x. [DOI] [PubMed] [Google Scholar]
- 3.Alcamí A, Smith G L. Proc Natl Acad Sci USA. 1996;93:11029–11034. doi: 10.1073/pnas.93.20.11029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miller L C, Lynch E A, Isa S, Logan J W, Dinarello C A, Steere A C. Lancet. 1993;341:146–148. doi: 10.1016/0140-6736(93)90006-3. [DOI] [PubMed] [Google Scholar]
- 5.Brunetti L, Volpe A R, Ragazzoni E, Preziosi P, Vacca M. Life Sci. 1996;58:PL373–PL377. doi: 10.1016/0024-3205(96)00238-x. [DOI] [PubMed] [Google Scholar]
- 6.Wong M-L, Rettori V, Al-Shekhlee A, Bongiorno P B, Canteros G, McCann S M, Gold P W, Licinio J. Nat Med. 1996;2:581–584. doi: 10.1038/nm0596-581. [DOI] [PubMed] [Google Scholar]
- 7.Vieira P, de, Waal-Malefyt R, Dang M N, Johnson K E, Kastelein R, Fiorentino D F, deVries J E, Roncarolo M G, Mosmann T R, Moore K W. Proc Natl Acad Sci USA. 1991;88:1172–1176. doi: 10.1073/pnas.88.4.1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colotta F, Re F, Muzio M, Polentarutti N, Minty A, Caput D, Ferrara P, Mantovani A. J Biol Chem. 1994;269:12403–12406. [PubMed] [Google Scholar]
- 9.Muzio M, Re F, Sironi M, Polentarutti N, Minty A, Caput D, Ferrara P, Mantovani A, Colotta F. Blood. 1994;83:1738–1743. [PubMed] [Google Scholar]
- 10.Cash E, Minty A, Ferrara P, Caput D, Fradelizi D, Rott O. J Immunol. 1994;153:4258–4267. [PubMed] [Google Scholar]
- 11.Relton J K, Rothwell N J. Brain Res Bull. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- 12.Toulmond S, Rothwell N J. Brain Res. 1995;671:261–266. doi: 10.1016/0006-8993(94)01343-g. [DOI] [PubMed] [Google Scholar]
- 13.Betz A L, Yang G Y, Davidson B L. J Cereb Blood Flow Metab. 1995;15:547–551. doi: 10.1038/jcbfm.1995.68. [DOI] [PubMed] [Google Scholar]
- 14.Licinio J, Wong M L, Gold P W. Endocrinology. 1991;129:562–564. doi: 10.1210/endo-129-1-562. [DOI] [PubMed] [Google Scholar]
- 15.Hannum C H, Wilcox C J, Arend W P, Joslin F G, Dripps D J, Heimdal P L, Armes L G, Sommer A, Eisenberg S P, Thompson R C. Nature (London) 1990;343:336–340. doi: 10.1038/343336a0. [DOI] [PubMed] [Google Scholar]
- 16.Eisenberg S P, Evans R J, Arend W P, Verderber E, Brewer M T, Hannum C H, Thompson R C. Nature (London) 1990;343:341–346. doi: 10.1038/343341a0. [DOI] [PubMed] [Google Scholar]
- 17.Butcher C, Steinkasserer A, Tejura S, Lennard A C. J Immunol. 1994;153:701–711. [PubMed] [Google Scholar]
- 18.Smith M J, Eidlen D, Brewer M T, Eisenberg S P, Arend W P, Gutierrez-Hartmann A. J Immunol. 1992;149:2000–2007. [PubMed] [Google Scholar]
- 19.Haskill S, Martin G, Van L L, Morris J, Peace A, Bigler C F, Jaffe G J, Hammerberg C, Sporn S A, Fong S, Arend W P, Ralph P. Proc Natl Acad Sci USA. 1991;88:3681–3685. doi: 10.1073/pnas.88.9.3681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Muzio M, Polentarutti N, Sironi M, Poli G, De G L, Introna M, Mantovani A, Colotta F. J Exp Med. 1995;182:623–628. doi: 10.1084/jem.182.2.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nishida T, Hirato T, Nishino N, Mizuno K, Sekiguchi Y, Takano M, Kawai K, Nakai S, Hirai Y. In: Cloning of the cDNAs for Rat Interleukin 1 Alpha and Beta. Powanda M C, Oppenheim J J, Kluger M J, Dinarello C A, editors. New York: Liss; 1988. pp. 73–78. [Google Scholar]
- 22.Eisenberg S P, Brewer M T, Verderber E, Heimdal P, Brandhuber B J, Thompson R C. Proc Natl Acad Sci USA. 1991;88:5232–5236. doi: 10.1073/pnas.88.12.5232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Goodman R E, Oblak J, Bell R G. Biochem Biophys Res Commun. 1992;189:1–7. doi: 10.1016/0006-291x(92)91516-s. [DOI] [PubMed] [Google Scholar]
- 24.Lakkis F G, Cruet E N. Biochem Biophys Res Commun. 1993;197:612–618. doi: 10.1006/bbrc.1993.2523. [DOI] [PubMed] [Google Scholar]
- 25.Wong M-L, Bongiorno P B, Gold P W, Licinio J. Neuroimmunomodulation. 1995;2:141–148. doi: 10.1159/000096884. [DOI] [PubMed] [Google Scholar]
- 26.Wong M-L, Gold P W, Licinio J. Methods Neurosci. 1993;16:81–99. [Google Scholar]
- 27.Landau D, Chin E, Bondy C, Domene H, Roberts Jr C T, Gronbaek H, Flyvbjerg A, LeRoith D. Endocrinology. 1995;136:1835–1842. doi: 10.1210/endo.136.5.7536658. [DOI] [PubMed] [Google Scholar]
- 28.Rasband W S, Bright D S. Microbeam Anal. 1995;4:137–149. [Google Scholar]
- 29.Tabor S, Richardson C C. Proc Natl Acad Sci USA. 1987;84:4767–4771. doi: 10.1073/pnas.84.14.4767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wong M-L, Bongiorno P B, Al-Shekhlee A, Esposito A, Khatri P, Licinio J. NeuroReport. 1996;7:2445–2448. doi: 10.1097/00001756-199611040-00008. [DOI] [PubMed] [Google Scholar]
- 31.Karanth S, Lyson K, McCann S M. Proc Natl Acad Sci USA. 1993;90:3383–3387. doi: 10.1073/pnas.90.8.3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lennard A C. Crit Rev Immunol. 1995;15:77–105. doi: 10.1615/critrevimmunol.v15.i1.30. [DOI] [PubMed] [Google Scholar]
- 33.Rettori V, Les Dees W, Hiney J K, Lyson K, McCann S M. Neuroimmunomodulation. 1994;1:251–258. doi: 10.1159/000097173. [DOI] [PubMed] [Google Scholar]
- 34.Kent S, Bluthé R M, Dantzer R, Hardwick A J, Kelley K W, Rothwell N J, Vannice J L. Proc Natl Acad Sci USA. 1992;89:9117–9120. doi: 10.1073/pnas.89.19.9117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plata-Salaman C. Physiol Behav. 1994;55:727–733. doi: 10.1016/0031-9384(94)90052-3. [DOI] [PubMed] [Google Scholar]
- 36.Linthorst A C, Flachskamm C, Muller P P, Holsboer F, Reul J M. J Neurosci. 1995;15:2920–2934. doi: 10.1523/JNEUROSCI.15-04-02920.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bluthé R M, Beaudu C, Kelley K W, Dantzer R. Brain Res. 1995;677:171–176. doi: 10.1016/0006-8993(95)00194-u. [DOI] [PubMed] [Google Scholar]
- 38.Granowitz E V, Santos A A, Poutsiaka D D, Cannon J G, Wilmore D W, Wolff S M, Dinarello C A. Lancet. 1991;338:1423–1424. doi: 10.1016/0140-6736(91)92725-h. [DOI] [PubMed] [Google Scholar]
- 39.Kuhns D B, Alvord W G, Gallin J I. J Infect Dis. 1995;171:145–152. doi: 10.1093/infdis/171.1.145. [DOI] [PubMed] [Google Scholar]
- 40.Matsumoto A, Ishii S. Atlas of Endocrine Organs: Vetebrates and Invertebrates. Berlin: Springer; 1992. [Google Scholar]
- 41.Asa S L, Kovacs K, Melmed S. In: The Hypothalamic-Pituitary Axis. Melmed S, editor. Cambridge, MA: Backwell Science; 1995. pp. 3–44. [Google Scholar]
- 42.Li P, Allen H, Banerjee S, Franklin S, Herzog L, Johnston C, McDowell J, Paskind M, Rodman L, Salfeld J, Towne E, Tracey D, Wardwell S, Wei F-Y, Wong W, Kamen R, Seshadri T. Cell. 1995;80:401–411. doi: 10.1016/0092-8674(95)90490-5. [DOI] [PubMed] [Google Scholar]
- 43.Bernton E W, Beach J E, Holaday J W, Smallridge R C, Fein H G. Science. 1987;238:519–521. doi: 10.1126/science.2821620. [DOI] [PubMed] [Google Scholar]
- 44.Poli G, Kinter A L, Fauci A S. Proc Natl Acad Sci USA. 1994;91:108–112. doi: 10.1073/pnas.91.1.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Montaner L J, Doyle A G, Collin M, Herbein G, Illei P, James W, Minty A, Caput D, Ferrara P, Gordon S. J Exp Med. 1993;178:743–747. doi: 10.1084/jem.178.2.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Montaner L J, Griffin P, Gordon S. J Gen Virol. 1994;75:3393–400. doi: 10.1099/0022-1317-75-12-3393. [DOI] [PubMed] [Google Scholar]
- 47.Akridge R E, Oyafuso L K, Reed S G. J Immunol. 1994;153:5782–5789. [PubMed] [Google Scholar]
- 48.Saville M W, Taga K, Foli A, Broder S, Tosato G, Yarchoan R. Blood. 1994;83:3591–3599. [PubMed] [Google Scholar]
- 49.Pantaleo G, Graziosi C, Fauci A S. N Engl J Med. 1993;328:327–335. doi: 10.1056/NEJM199302043280508. [DOI] [PubMed] [Google Scholar]
- 50.Fauci A S. Science. 1993;262:1011–1018. doi: 10.1126/science.8235617. [DOI] [PubMed] [Google Scholar]
- 51.Vitkovic L, da Cunha A, Tyor W R. Res Publ Assoc Res Nerv Ment Dis. 1994;72:203–222. [PubMed] [Google Scholar]