

Abstract
Rett syndrome (RTT) is an X-linked dominant neurodevelopmental disorder caused by mutations in MECP2, encoding methyl-CpG binding protein 2. MeCP2 is a transcriptional repressor elevated in mature neurons and is predicted to be required for neuronal maturation by regulating multiple target genes. Identifying primary gene targets in either Mecp2-deficient mice or human RTT brain has proven to be difficult, perhaps because of the transient requirement for MeCP2 during neuronal maturation. In order to experimentally control the timing of MeCP2 expression and deficiency during neuronal maturation, human SH-SY5Y cells undergoing mature neuronal differentiation were transfected with methylated MeCP2 oligonucleotide decoy to disrupt the binding of MeCP2 to endogenous targets. Genome-wide expression microarray analysis identified all four known members of the inhibitors of differentiation or inhibitors of DNA binding (ID1, ID2, ID3 and ID4) subfamily of helix-loop-helix (HLH) genes as novel neuronal targets of MeCP2. Chromatin immunoprecipitation analysis confirmed binding of MeCP2 near or within the promoters of ID1, ID2 and ID3, and quantitative RT-PCR confirmed increased expression of all four Id genes in Mecp2-deficient mouse brain. All four ID proteins were significantly increased in Mecp2-deficient mouse and human RTT brain using immunofluorescence and laser scanning cytometric analyses. Because of their involvement in cell differentiation and neural development, ID genes are ideal primary targets for MeCP2 regulation of neuronal maturation that may explain the molecular pathogenesis of RTT.
INTRODUCTION
Rett syndrome (RTT) is an X-linked dominant disorder caused by mutation in methyl CpG binding protein 2 (MECP2) located on Xq28 (1, 2). RTT primarily affects females, who are mosaic for expression of the MECP2 mutation because of random X-inactivation (3). The onset of symptoms is delayed until 6–18 months of age and includes severe mental retardation with absence of speech, stereotypic hand movements, epileptic seizures and respiratory dysfunction (4–6).
MeCP2 belongs to a family of methyl-CpG-binding proteins consisting of MBD1, MBD2, MBD3 and MBD4, all of which contain a conserved methyl-CpG-binding domain (MBD) (7, 8). Mice lacking MBD1 show deficits in adult neurogenesis and hippocampal function (9). Mice lacking MBD2 show mild maternal behavior deficits, while MBD3−/− mice die at an early embryonic stage (10). Mice deficient in MBD4, a mismatch repair enzyme, show deficits in DNA repair and increased tumor formation (11). Mecp2-deficient and mutant mouse models recapitulate the neurodevelopmental symptoms of RTT (12–14).
MeCP2, the most extensively studied member of MBD family, is a nuclear protein dynamically expressed during postnatal mammalian brain development and is a marker for neuronal maturity (15–18). Elevated MeCP2 expression is hypothesized to be required for neuronal differentiation by the regulation of multiple target genes (19). MeCP2 binds to DNA through its methyl-CpG-binding domain (MBD) and complexes with the transcriptional repressor Sin3A and histone deacetylase (HDAC) through the transcriptional repressor domain (TRD) (20–22). The involvement of MeCP2 in methylation specific transcriptional repression (20, 23) suggested that MeCP2 deficiency in RTT would result in wide spread gene dysregulation. This hypothesis was previously tested using gene expression microarray analysis with Mecp2-deficient mouse brain (24), RTT patient cell lines (25) and post-mortem RTT brain tissue (26). Subtle and non-overlapping transcriptional changes were observed in each of these studies, indicating that MeCP2 deficiency does not result in obvious high levels of genome-wide transcriptional dysregulation. The changes in gene expression observed due to MeCP2 deficiency in human and mouse brains could be representative of events downstream of direct MeCP2 target genes and therefore may be inherently noisy.
Alternative approaches have also been used to identify primary targets of MeCP2. In mammals, BDNF has been identified by a candidate gene approach (27, 28) and DLX5 was identified by cloning fragments from MeCP2 chromatin immunoprecipitation (ChIP) (29). In Xenopus laevis, the neuronal repressor xHairy2a was identified as a target of MeCP2 in differentiating neuroectoderm (30). A cDNA microarray analysis was performed on lymphoblastoid cell lines derived from RTT patients with and without MeCP2 mutations followed by ChIP to distinguish the direct targets of MeCP2 from indirect targets (31). Significantly reduced expression of UBE3A and GABRB3 genes within the human 15q11-13 region in Mecp2-deficient mice and RTT patients was shown using multiple quantitative methods (32). Finally, a recent study demonstrated increased expression of serum glucocorticoid-inducible kinase 1 (Sgk) and FK506-binding protein 5 (Fkbp5) in Mecp2-deficient mouse brain suggesting MeCP2 as a modulator of glucocorticoid-inducible gene expression (33). Each of these studies identified various genes dysregulated due to MeCP2 deficiency but none of the target genes could completely explain the primary neurodevelopmental defect observed in RTT patients.
Since the phenotypic effects of MeCP2 deficiency appears to be limited to a precise stage of postnatal neuronal maturation, we used an alternative approach to experimentally control the timing of MeCP2 expression and inhibition in a neuronal cell culture system. Microarray analysis of transcriptional changes during maturational differentiation that were specifically altered by MeCP2 inhibition identified ID1, ID2, ID3 and ID4 genes as primary targets of MeCP2. All four ID genes belong to the same class of helix loop helix transcriptional regulators, encoding known inhibitors of differentiation or inhibitors of DNA binding that block the function of tissue specific basic helix loop helix (bHLH) transcription factors involved in regulation of important neuronal differentiation genes such as NEUROD1. We report significantly increased protein expression of all four ID genes in both Mecp2-deficient mice and RTT human brain tissue compared to wild type mice or age matched controls. Further understanding the role of ID genes as neuronal targets of MeCP2 may explain the arrest in postnatal neuronal maturation seen in Rett syndrome.
RESULTS
Microarray analysis performed to identify the primary targets of MeCP2 during SH-SY5Y differentiation
In order to identify genes specifically regulated by MeCP2 at a precise stage of neuronal maturational differentiation, expression microarray experiments were conducted on human SH-SY5Y neuronal cells transfected with a methylated oligonucleotide decoy to block MeCP2 binding to endogenous target genes during phorbol 12-myristate 13-acetate (PMA) induced maturational differentiation. SH-SY5Y human neuroblastoma cells were chosen because they can be induced by PMA to produce a functional sympathetic neuronal phenotype (34), resulting in the two-fold increase in MeCP2 expression by 48 h (35). The MeCP2 decoy (MD) is a 22 mer double stranded DNA molecule containing two methylated CpG sites adjacent to AT-runs (36). A control sequence with CpG sites mutated to AT was used as a specificity and transfection control decoy (CD). The decoy approach of blocking MeCP2 binding in SH-SY5Y cells has been previously validated in our lab and the specificity to MeCP2 was demonstrated by ChIP at the SNURF/SNRPN locus (37).
Four different SH-SY5Y treatments were compared by gene expression profiling experiments: 1) Undifferentiated (UD) 2) 48 h differentiated and untransfected (D-UT) 3) 48 h differentiated and MeCP2 decoy transfected (D-MD), and 4) 48 h differentiated and control decoy transfected (D-CD). For each cell treatment, total RNA was isolated from triplicate biological experiments and labeled cRNA was hybridized to Affymetrix HG U133 plus 2.0 arrays (12 arrays in total). Data analysis was performed using dChip analysis software and significant differences between different cell treatments were identified.
As MeCP2 was hypothesized to regulate genes involved in neuronal maturation, we first chose to examine genes significantly changed following SH-SY5Y differentiation that could be potential targets of MeCP2. A Boolean logic approach was used to identify transcript levels significantly affected during differentiation by the D-MD but not the D-CD transfection. Table 1 demonstrates the pair-wise analyses that were useful in determining the genes altered specifically by the MeCP2 decoy. First, transcripts showing ≥ 2.0 or ≤ 2.0 fold significant changes (p ≤ 0.05) between UD and D-UT are selected. PMA induced differentiation of human SH-SY5Y neuronal cells resulted in up-regulation of 183 genes and down-regulation of 45 genes compared to undifferentiated cells. Of this selected list of 228 genes, 24 genes (20 increased and 4 decreased upon differentiation) were found to have significant (p ≤ 0.05) differences between undifferentiated and MeCP2 decoy but not undifferentiated and control decoy (UD vs. D-MD NOT D-CD). Interestingly, of the MeCP2 target candidate genes, expression levels of 3 out of 4 genes decreased with differentiation (ID1, ID2, and ID3) and 1 out of 12 genes increased with differentiation (ID4) were from the same family of transcriptional regulators called inhibitors of differentiation or inhibitors of DNA binding (ID) involved in cellular proliferation and differentiation (38, 39). The fold changes and P values of all four ID genes from the microarray analysis are shown in Table 2 and raw data from microarray is shown in Supplementary Figure 1. The complete lists of genes from the above analysis are shown in supplementary tables S1 to S5. The simplistic pairwise analysis of D-MD versus D-CD revealed some differentially expressed transcripts (Supplementary table, S6), but because the control decoy (D-CD) had an unexpected nonspecific effect on MBD1 and MBD2 binding (37) this comparison was less informative. The microarray data discussed in this publication have been deposited in NCBIs Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) and is accessible through GEO Series accession number GSE4600.
Table 1.
Selection for primary target genes of MeCP2 during SH-SY5Y cell differentiation
| Criteria for Selection | # of Probe sets | # of Genes |
|---|---|---|
| Significant (p ≤ 0.05) AND increased by ≥ 2.0 fold change in expression upon differentiation (UD vs. D-UT) | 261 | 183 |
| Out of 183 genes, genes significant (p ≤ 0.05) AND increased by ≥ 1.2 fold change in expression with MeCP2 decoy NOT control decoy | 14 | 12 |
| Out of 183 genes, genes significant (p ≤ 0.05) AND decreased by ≥ 1.2 fold change in expression with MeCP2 decoy NOT control decoy | 10 | 8 |
| Significant (p ≤ 0.05) AND decreased by ≤ 2.0 fold change in expression upon differentiation (UD vs. D-UT) | 60 | 45 |
| Out of 45 genes, genes significant (p ≤ 0.05) AND increased by ≥ 1.2 fold change in expression with MeCP2 decoy NOT control decoy | 5 | 4 |
| Out of 45 genes, genes significant (p ≤ 0.05) AND decreased by ≥ 1.2 fold change in expression with MeCP2 decoy NOT control decoy | 0 | 0 |
Table 2.
All members of the ID gene family are significantly increased by MeCP2 decoy
| Undifferentiated vs. Differentiated and Untransfected (UD vs. D-UT) | Differentiated and Untransfected vs. MeCP2 decoy transfected (D-UT vs. D-MD) | |||||
|---|---|---|---|---|---|---|
| Gene Description | Gene Symbol | Chromosomal Location | Fold Change | P value | Fold Change | P value |
| Inhibitor of DNA binding 1 | ID1 | 20q11 | −2.91 | 0.00054 | 1.45 | 0.00406 |
| Inhibitor of DNA binding 2 | ID2 | 2p25 | −3.22 | 0.00166 | 1.52 | 0.03498 |
| Inhibitor of DNA binding 3 | ID3 | 1p36.13-p36.12 | −2 | 0.00099 | 1.25 | 0.01702 |
| Inhibitor of DNA binding 4 | ID4 | 6p22-p21 | 2.53 | 0.00152 | 1.71 | 0.03737 |
The effect of MeCP2 deficiency and the binding sites for MeCP2 were further characterized for the ID gene family because of the common relationship between all four ID genes (40) and their known involvement in cellular differentiation (39, 41).
Validation of ID gene expression microarray results by quantitative RT-PCR in human SH-SY5Y neuronal cells
The microarray results were confirmed by performing quantitative real time reverse transcriptase polymerase chain reaction (qRT-PCR) on all four ID genes in the SH-SY5Y MeCP2 decoy experimental system. Results shown in Figure 1A are relative fold changes compared to undifferentiated SH-SY5Y cells (UD) set at 1.0, represented by the hatched bar. Consistent with the microarray results, qRT-PCR data for ID1, ID2 and ID3 genes showed a decrease in transcript level in differentiated SH-SY5Y cells (D-UT), but increased expression in differentiated cells transfected with MeCP2 decoy (D-MD) relative to D-UT. The ID4 transcript levels increased following SH-SY5Y differentiation and further increased in the MeCP2 decoy (D-MD) transfected cells also compared to D-UT.
Figure 1. Quantitative RT-PCR results for ID1-4 in SH-SY5Y, Mecp2−/y mouse brain cDNA and NeuroD1, a downstream target of ID genes.
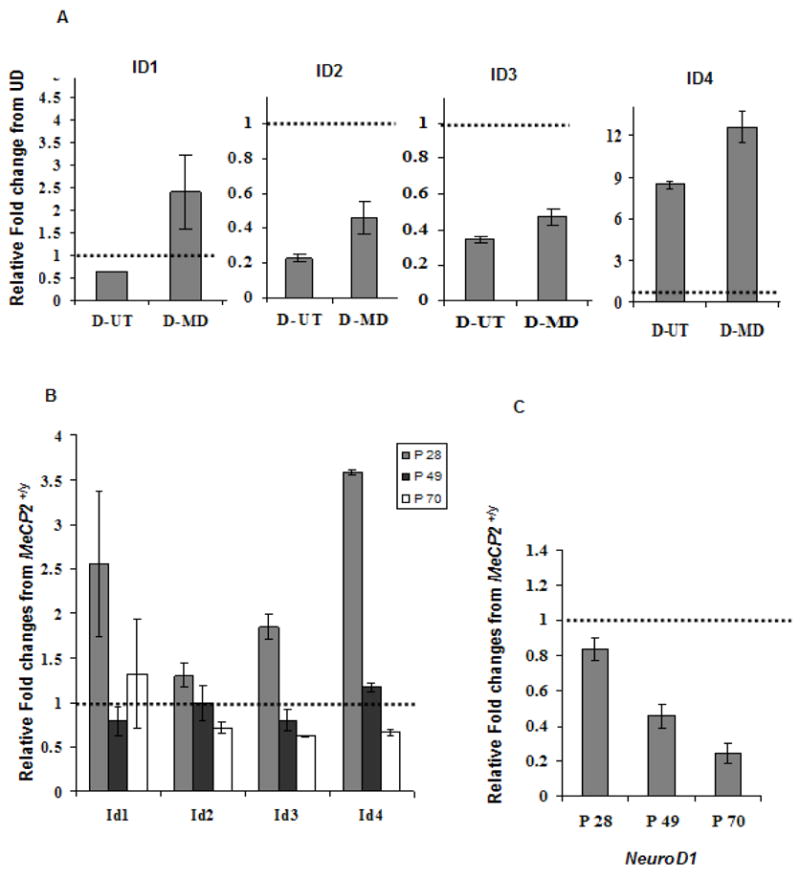
A, B and C: Graphical representation of all four ID genes qPCR data reflect fold changes relative to the control (set to 1.0, indicated by hatched bar) following normalization of the data to GAPDH house keeping control using the comparative CT method. A) The qPCR data for ID1, ID2 and ID3 genes show a decreased expression with differentiation, in untransfected SH-SY5Y cells (D-UT) and increased expression with MeCP2 decoy (D-MD). qPCR data of ID4 shows an increased expression level following SH-SY5Y differentiation. ID4 was further increased in the D-MD transfected cells compared to 48 h untransfected (D-UT). Results shown represent mean ± SEM of three replicate experiments. B) The qRT-PCR was conducted in cDNA samples from Mecp2−/yand Mecp2+/ymice brains. The relative fold change differences for all four ID genes were higher in the Mecp2−/y compared to Mecp2+/yat P28 time point, and reached significance by t-test for Id3 and Id4 (P < 0.05). No increase in cDNA was observed at later time points (P49 and P70). C) The qRT-PCR results on a downstream target of ID genes, NeuroD1, showed lower expression in all three postnatal time points (P28, P49 and P70) in Mecp2−/y brain compared to the Mecp2+/y control brain, with significantly reduced expression at P49 and P70 by t-test (P ≤ 0.05). Each time point in bar graphs B and C represents mean ± SEM of three replicate experiments from two pairs of mice per time point.
Quantitative RT-PCR of ID genes in Mecp2-deficient (Mecp2tm 1.1Bird/y) mouse brain
To test the relevance of ID genes as neuronal targets of MeCP2 in a more relevant biological system, qRT-PCR was conducted using cDNA from postnatal 28, 49 and 70 day Mecp2−/y and littermate Mecp2+/y brain. Figure 1B shows the relative fold change of Mecp2−/y compared to Mecp2+/y brain cDNA (Mecp2+/y set to 1.0, hatched bar) for all four Id genes. At the youngest time point (P28), increases in Id1 (2.5 fold), Id2 (1.3 fold), Id3 (1.9 fold) and Id4 (3.5 fold) transcripts were observed in Mecp2−/y compared to Mecp2+/y brain samples. Interestingly, in the later P70 time point increased transcript levels were observed only for Id1 in Mecp2−/y brain compared to Mecp2+/y. Decreased expression levels were observed for Id2, Id3 and Id4 by P70. These results not only show the reproducibility of the microarray results using qRT-PCR in a different experimental system for inhibiting MeCP2, but also suggest that the transcriptional changes in the ID genes caused by MeCP2 deficiency may be developmental stage specific and transient.
Quantitative RT-PCR on a downstream target of ID genes in Mecp2-deficient mouse brain
To test the hypothesis that aberrantly increased expression of ID genes has long-lived consequences on the expression of the downstream genes important for neuronal maturation, qRT-PCR was also performed on an autoregulated bHLH transcription factor, neurogenic differentiation factor 1( Neurod1) (42). Figure 1C demonstrates progressive 20 % to 75 % decreased expression of Neurod1 in Mecp2−/y brain from P28 to P70 relative to Mecp2+/y brain. It has been shown previously that ID2 in cultured HeLa and HIT cells (43) and ID2 and ID3 in Xenopus embryos (44) act as inhibitors of NEUROD1 activity. These results suggest that the transient increase in ID transcript levels associated with MeCP2 deficiency could trigger a cascade of downstream events that affect postnatal neuronal maturation.
ID protein expression changes in Mecp2-deficient mouse model using immunofluorescence and quantitative laser scanning cytometry
Interestingly, the results from qRT- PCR data for Mecp2-deficient mice showed increased transcript for all four ID genes at P28. To examine and quantitate ID protein expression, immunofluorescence followed by quantitation by laser scanning cytometry (LSC) was performed for all four ID genes on P28 day sagittal brain sections of three Mecp2tm 1.1Bird/y mice and corresponding Mecp2+/y littermate controls. The approach of immunofluorescence on tissue microarrays and brain sections followed by analysis using LSC has been previously validated in our lab in several studies (15, 32).
Briefly, immunofluorescence was performed on the 5 μm sagittal mouse tissue sections by staining the slides with each of the four ID antibodies, followed by secondary fluorescent antibody staining and detection using LSC. The fluorescently stained cells were analyzed by LSC that creates contours around the nuclei to identify individual cells. Quantitative data regarding mean fluorescence intensities (representative of total protein expression) was recorded for each cell in different fluorescent channels. Each brain region was individually gated to obtain the mean fluorescence of each ID protein. Figure 2 shows representative LSC images for all four ID protein expressions, showing differences in regional distribution in Mecp2−/y compared to Mecp2+/y. Each pixel on the LSC image represents an individual cell, colored on the basis of each of the ID protein‘max pixel’ histograms of whole brain. Max pixel is the maximum fluorescence intensity of each contoured nucleus, as described previously (15, 45). Blue colored nuclei (negative) were gated based on overlap with IgG negative control histogram. Green (low) and red (high) regions were colored based on the right half max of the major peak of the ID protein max pixel histogram. Significantly increased expression of all four ID proteins was observed in multiple brain regions (cerebrum, cerebellum, thalamus and hypothalamus, medulla oblongata and pons and hippocampus) of P28 Mecp2−/y mice brain compared to Mecp2+/y littermate control. Numerical data from separately gated brain regions for all four ID proteins at time point P28 from three pairs of mice are shown in Table 3.
Figure 2. Increased ID protein expression changes in Mecp2-deficient mice model detected by immunofluorescence and LSC.
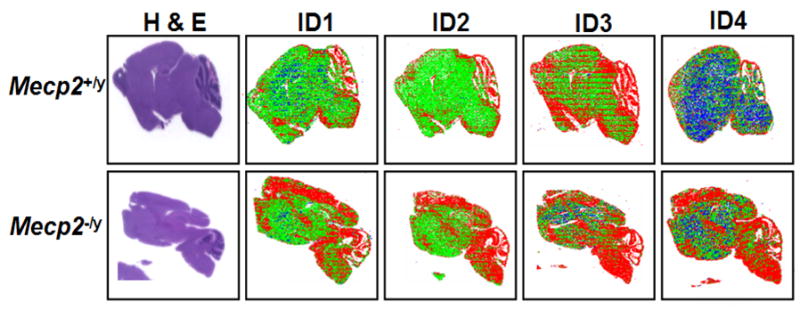
Immunofluorescence followed by analysis on LSC was performed on postnatal day 28, 5 μm sagittal brain sections of three pairs of Mecp2−/y and Mecp2+/y mice using antibodies recognizing ID1, ID2, ID3 and ID4. Representative LSC image showing protein expression differences in multiple regions of the brain of Mecp2−/y mice compared to Mecp2+/y controls for all four ID proteins. Each pixel represents individual nuclei colored blue (negative), green (low) or red (high) based on max pixel fluorescence histograms. For each section, the hemotoxylin and eosin (H&E) stained parallel slide was used to determine the tissue localization of cells and gating of individual brain regions for quantitative data shown in Table 3.
Table 3.
Significant expression differences of ID1, ID2, ID3 and ID4 proteins in different brain regions of postnatal day 28 (P28) Mecp2−/y mice compared to the Mecp2+/y mice
| Pair 1 | Pair 2 | Pair 3 | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Mecp2+/y | Mecp2−/y | Mecp2+/y | Mecp2−/y | Mecp2+/y | Mecp2−/y | |||||||
| Brain Region | Mean ± SEM | Cell # | Mean ± SEM c | Cell # | Mean ± SEM | Cell # | Mean ± SEM c | Cell # | Mean ± SEM | Cell # | Mean ± SEM c | Cell # |
| Inhibitor of differentiation 1-ID1 a | ||||||||||||
| Whole brain | 6308 ± 8 | 52657 | 7435 ± 9*** | 43790 | 5497 ± 8 | 51079 | 5852 ± 8*** | 54931 | 5057 ± 7 | 48906 | 6018 ± 7*** | 39591 |
| Cerebellum | 7466 ± 30 | 6733 | 8250 ± 29*** | 4679 | 6095 ± 24 | 4504 | 7231 ± 30*** | 5873 | 5998 ± 24 | 4705 | 6807± 32*** | 2974 |
| Cerebrum | 5946 ± 18 | 7588 | 8540 ± 26*** | 5908 | 4833 ± 13 | 10331 | 5548 ± 18*** | 8179 | 4677 ± 20 | 7547 | 6191 ± 20*** | 6504 |
| Medulla oblongata & Pons | 6716 ± 19 | 10831 | 7512 ± 17** | 9295 | 6535 ± 20 | 11919 | 6242 ± 21 | 8790 | 5802 ± 16 | 9790 | 6194 ± 19*** | 6192 |
| Thalamus & Hypothalamus | 6068 ± 21 | 5256 | 6459 ± 21*** | 5766 | 4855 ± 21 | 4407 | 5801 ± 23*** | 6926 | 4558 ± 15 | 6456 | 5776 ± 17*** | 5094 |
| Hippocampus | 5373 ± 20 | 4760 | 6660 ± 26*** | 3569 | 4572 ± 19 | 3510 | 5230 ± 28*** | 3567 | 4704 ± 24 | 3536 | 6026 ± 30*** | 2206 |
| Inhibitor of differentiation 2-ID2 b | ||||||||||||
| Whole brain | 7684 ± 9 | 47590 | 10303 ± 10*** | 44703 | 6792 ± 9 | 51113 | 8025 ± 10*** | 51882 | 8203 ± 9 | 48244 | 8720 ± 9*** | 41791 |
| Cerebellum | 9264 ± 31 | 6493 | 11948 ± 24*** | 5208 | 7451 ± 29 | 4368 | 8679 ± 32*** | 5451 | 9423 ± 27 | 4731 | 9760 ± 31*** | 3385 |
| Cerebrum | 7427 ± 22 | 5579 | 11581 ± 34*** | 4023 | 6206 ± 20 | 8494 | 8130 ± 24*** | 8093 | 7664 ± 23 | 7753 | 8172 ± 20*** | 6488 |
| Medulla oblongata & Pons | 7772 ± 22 | 9403 | 10081 ± 19*** | 7780 | 7486 ± 19 | 12563 | 7987 ± 25*** | 8805 | 9143 ± 20 | 9661 | 9211 ± 23NS | 7911 |
| Thalamus & Hypothalamus | 7480 ± 26 | 4322 | 8667 ± 24*** | 4913 | 6540 ± 27 | 5013 | 8168 ± 27*** | 6286 | 7807 ± 23 | 5587 | 8064 ± 19*** | 5813 |
| Hippocampus | 6793 ± 24 | 4210 | 9519 ± 42*** | 2119 | 5921 ± 29 | 3144 | 8110 ± 48*** | 2611 | 8052 ± 34 | 2914 | 8667 ± 33*** | 2614 |
| Inhibitor of differentiation 3 -ID3 b | ||||||||||||
| Whole brain | 5727 ± 8 | 37924 | 6625 ± 8*** | 43220 | 5265 ± 7 | 48152 | 5400 ± 8*** | 52891 | 5475 ± 7 | 48680 | 6656 ± 7*** | 37111 |
| Cerebellum | 6399 ± 22 | 4146 | 7374 ± 23*** | 4281 | 5891 ± 25 | 3907 | 6418 ± 25*** | 5869 | 6158 ± 22 | 4800 | 7600 ± 41*** | 1727 |
| Cerebrum | 7054 ± 21 | 7549 | 6648 ± 22 | 8482 | 5407 ± 18 | 8940 | 4892 ± 16 | 9054 | 5185 ± 23 | 6590 | 6816 ± 18*** | 6851 |
| Medulla oblongata & Pons | 6762 ± 15 | 8616 | 7070 ± 16*** | 8956 | 5398 ± 14 | 11080 | 5801 ± 20*** | 9235 | 6114 ± 15 | 10348 | 6812 ± 15*** | 8285 |
| Thalamus & Hypothalamus | 5612 ± 20 | 3947 | 6451 ± 25*** | 4331 | 5058 ± 20 | 4219 | 5375 ± 22*** | 6585 | 5127 ± 16 | 6560 | 6351 ± 16*** | 5117 |
| Hippocampus | 4589 ± 22 | 2725 | 5619 ± 24*** | 3595 | 4864 ± 25 | 3005 | 5019 ± 29* | 3180 | 5180 ± 29 | 3287 | 6861 ± 26*** | 2495 |
| Inhibitor of differentiation 4 -ID4 b | ||||||||||||
| Whole brain | 5247 ± 7 | 40365 | 6437 ± 8*** | 41372 | 5328 ± 8 | 47817 | 5900 ± 9*** | 50306 | 4840 ± 7 | 49215 | 6061 ± 7*** | 40687 |
| Cerebellum | 5762 ± 23 | 4189 | 6886 ± 21*** | 5167 | 5634 ± 18 | 4195 | 6738 ± 21*** | 6231 | 5572 ± 20 | 5941 | 7291 ± 34*** | 2431 |
| Cerebrum | 5476 ± 14 | 9279 | 6794 ± 21*** | 8203 | 5176 ± 19 | 8749 | 5618 ± 21*** | 8022 | 4768 ± 19 | 8157 | 6289 ± 16*** | 9627 |
| Medulla oblongata & Pons | 5290 ± 14 | 9192 | 6951 ± 15*** | 10192 | 6206 ± 23 | 10537 | 5812 ± 23 | 8560 | 5144 ± 16 | 9198 | 6063 ± 16*** | 7732 |
| Thalamus & Hypothalamus | 5016 ± 18 | 4912 | 5997 ± 21*** | 4631 | 5293 ± 25 | 5204 | 5702 ± 33*** | 6995 | 4446 ± 17 | 5432 | 5504 ± 16*** | 5860 |
| Hippocampus | 4896 ± 18 | 3618 | 5651 ± 22*** | 3726 | 5096 ± 26 | 3517 | 5790 ± 30*** | 3331 | 4706 ± 25 | 2635 | 6048 ± 27*** | 2441 |
- ID1 rabbit monoclonal antibody used is provided by Dr. Robert Benezra.
- ID2, ID3, ID4 rabbit polyclonal antibodies obtained from commercially available source (Santa Cruz Biotechnologies, Inc).
- Significance determined by chi-square and P values denoted by asterix.
P ≤ 0.05,
P ≤ 0.005,
P ≤ 0.0005.
To determine if ID protein expression changes observed in Mecp2−/y were transient or long-lived, LSC analysis was performed on a tissue microarray consisting of cerebral cortex samples from Mecp2−/y mice and corresponding Mecp2+/y littermate controls at various developmental time points ranging from embryonic day 15 (E 15) to postnatal day 70 (P70). For each time point and ID protein, fluorescence max pixel values of cells from Mecp2−/y mouse cerebral cortex tissue cores were compared to those from Mecp2+/y cores (Table 4). The protein expression of ID1 in Mecp2−/y mouse brain showed significant increases compared to controls in E15 and in four postnatal time points (P7, P28, P49 and P70), while ID2 showed significant increases at E15 and five postnatal time points (P7, P28, P49, P56 and P70 ). The protein expression of ID3 increased significantly in P7, P56 and P70, while ID4 protein expression increased significantly in four postnatal time points (P28, P49, P56 and P70) of Mecp2−/y mice compared to Mecp2+/y littermate controls.
Table 4.
Significant expression differences of ID1, ID2, ID3 and ID4 proteins in the frontal cortex of Mecp2−/y mice compared to the Mecp2+/y mice at various developmental time points
| Mecp2+/y | Mecp2−/y | ||||
|---|---|---|---|---|---|
| Brain Region | Age (days) | Mean ± SEM | Cell # | Mean ± SEM b | Cell # |
| Inhibitor of differentiation 1 - ID1 a | |||||
| Frontal Cortex | E 15 | 4381 ± 59 | 848 | 5026 ± 96** | 488 |
| P 7 | 4964 ± 19 | 2951 | 5639 ± 23*** | 2866 | |
| P 28 | 4479 ± 28 | 1943 | 5015 ± 39** | 1016 | |
| P 49 | 4441 ± 25 | 2581 | 5112 ± 43*** | 1070 | |
| P 56 | 3750 ± 20 | 2054 | 3876 ± 21 | 1918 | |
| P 70 | 4643 ± 43 | 1454 | 5097 ± 37** | 2046 | |
| Inhibitor of differentiation 2 - ID2 a | |||||
| Frontal Cortex | E 15 | 4432 ± 51 | 859 | 5078 ± 65** | 519 |
| P 7 | 5100 ± 32 | 1090 | 6152 ± 44*** | 1076 | |
| P 28 | 4387 ± 44 | 2122 | 4622 ± 48** | 947 | |
| P 49 | 3457 ± 25 | 1189 | 4228± 58*** | 523 | |
| P 56 | 3282 ± 18 | 1064 | 3552 ± 26** | 990 | |
| P 70 | 4397 ± 52 | 699 | 5665 ± 63*** | 1002 | |
| Inhibitor of differentiation 3 - ID3 a | |||||
| Frontal Cortex | E 15 | 4052 ± 48 | 1130 | 3917 ± 62 | 481 |
| P 7 | 4577 ± 38 | 2464 | 5211 ± 38*** | 2032 | |
| P 28 | 6437 ± 74 | 627 | 6494 ± 60 | 532 | |
| P 49 | 6386 ± 82 | 561 | 6687 ± 86 | 376 | |
| P 56 | 5034 ± 28 | 1122 | 5390 ± 40* | 699 | |
| P 70 | 6295 ± 70 | 581 | 6773 ± 64* | 737 | |
| Inhibitor of differentiation 4 - ID4 a | |||||
| Frontal Cortex | E 15 | 6568 ± 84 | 817 | 6295 ± 123 | 450 |
| P 7 | 5151± 45 | 566 | 5347 ± 43 | 650 | |
| P 28 | 4095 ± 38 | 1062 | 4784 ± 44* | 516 | |
| P 49 | 3653 ± 34 | 605 | 4806 ± 41** | 602 | |
| P 56 | 3645 ± 36 | 436 | 4148 ± 35** | 579 | |
| P 70 | 4607 ± 60 | 438 | 5722 ± 55*** | 557 | |
- ID1, ID2, ID3, ID4 rabbit polyclonal antibodies obtained from commercially available source (Santa Cruz Biotechnologies, Inc).
- Significance determined by chi-square and P values denoted by asterix.
P ≤ 0.05,
P ≤ 0.005,
P ≤ 0.0005.
ID protein expression changes on a tissue microarray containing human RTT and control cerebral samples using immunofluorescence and quantitative laser scanning cytometry
In order to determine whether ID protein expression is also significantly higher in MeCP2 deficient RTT human brain samples, a similar approach of immunofluorescence followed by quantiation on LSC was performed. A tissue microarray containing human post-mortem cerebral cortex samples in triplicates from different RTT individuals all with known MECP2 deficiencies (46) of different ages and corresponding age-matched control individuals was used. Figure 3 demonstrates that one male and two females with known MECP2 deficiencies (grey, filled histograms) show significantly higher ID protein expression compared to age-matched controls (black, open histogram). Increased ID protein expression was observed in all the RTT individuals compared to controls except for ID1 in RTT 4312 brain tissue. The observed dysregulation of ID protein expression in RTT brains further supports the ID gene family as relevant neuronal targets of MeCP2 and suggests a role in the pathogenesis of RTT.
Figure 3. Increased ID protein expression changes in RTT cerebrum detected by immunofluorescence and LSC.
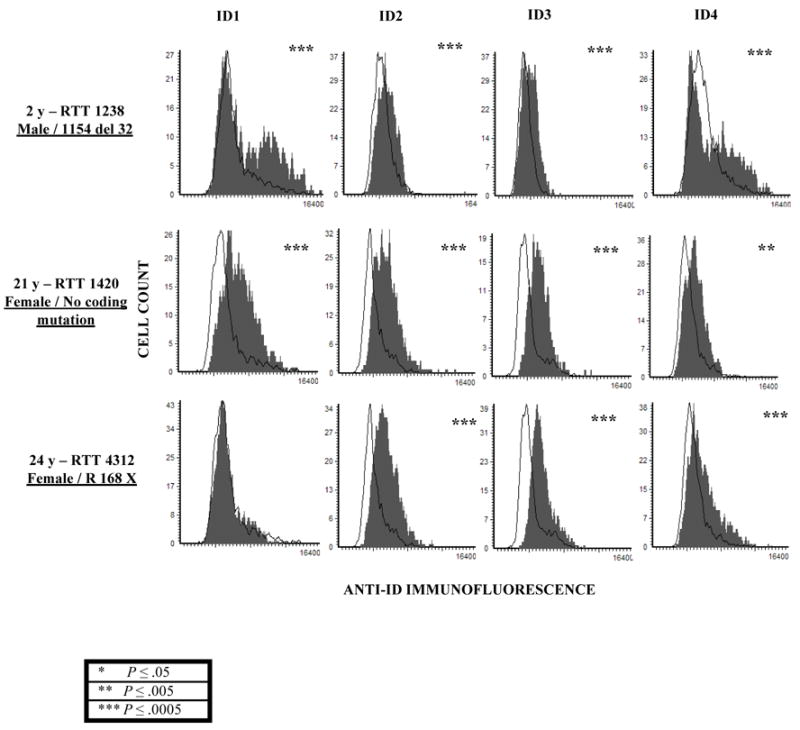
Immunofluorescence using antibodies recognizing ID1-4 was performed on postmortem cerebral cortex samples from one male and two female RTT samples with and without MECP2 coding mutations (filled, grey histograms) and age-matched controls (open, black histogram) arranged on a tissue microarray. Expression of all four ID proteins was significantly increased in RTT brain samples compared to the controls. In contrast, histone H1 expression was not significantly changed (data not shown). Significance was determined using chi-square from two replicate experiments and the P values are denoted by asterix on the top right corner of each histogram. (* P ≤ 0.05, ** P ≤ 0.005, *** P ≤ 0.0005).
These combined results demonstrate that increased expression of the ID family of transcriptional regulators is observed in Mecp2-deficient mouse brain and human RTT cortical tissue. These results validate the microarray results and suggest that the novel finding of ID genes as targets of MeCP2 is not simply an artifact of the SH-SY5Y cell culture system.
Chromatin immunoprecipitation analysis of MeCP2 binding to the ID gene promoters
In order to determine whether MeCP2 binds to the CpG islands overlapping the promoter of each ID gene, ChIP was performed on chromatin isolated from SH-SY5Y cells from all four experimental conditions used for the microarray experiment ( UD, D-UT, D-MD, D-CD). The specificity of the MD decoy in blocking the binding of MeCP2 to an endogenous target (SNRPN) has been previously validated (37), and was therefore used as a control. In Figure 4, the ChIP results for ID1, ID2 and ID3 show enriched binding for MeCP2 in differentiated SH-SY5Y cells (D-UT) and in the cells transfected with control decoy (D-CD). In contrast, the chromatin isolated from cells transfected with MeCP2 decoy (D-MD) or in undifferentiated cells (UD) did not show enriched binding of MeCP2. These results suggests that the down-regulation of ID1, ID2 and ID3 gene expression with differentiation in SH-SY5Y cells is regulated by direct binding of MeCP2 at CpG islands overlapping the promoter region of ID1, ID2 and ID3 and that MeCP2 binding is specifically reduced by MD decoy. In contrast, MeCP2 was not found to be associated with the promoter of ID4, suggesting that the MeCP2 may bind outside of the CpG island assayed. Alternatively, the effect of MeCP2 binding to the CpG island at the promoter may be more complex than simply repressing ID gene expression in cis and may instead involve long-range chromatin interactions mediated by MeCP2 as reported in a recent study (29).
Figure 4. Chromatin Immunoprecipitation (ChIP) analysis of MeCP2 binding to ID1, ID2 and ID3 promoters.
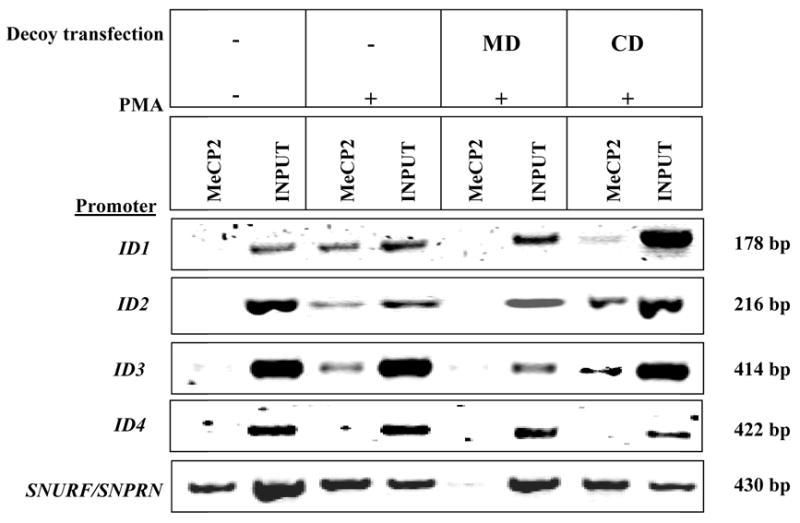
Representative gel of PCR products using primers specific to each of the ID gene promoter following ChIP with C-terminal MeCP2 reactive antibody. The total DNA isolated from chromatin prior to IP (Input) was used as a positive control. The ChIP results show binding of MeCP2 near or within the promoter of ID1, ID2 and ID3 in differentiated SH-SY5Y cells (D-UT) and control decoy transfected cells (D-CD). For ID4, binding of MeCP2 was not observed near or within the promoter region assayed. Additionally, the binding of MeCP2 to SNURF/SNRPN promoter is shown as a positive control for ChIP assay conditions.
DISCUSSION
MeCP2 protein levels are significantly higher in central nervous system (CNS) tissues compared to non-CNS tissues, addressing a major paradox in the pathogenesis of RTT regarding how mutations in ubiquitously transcribed MECP2 result in a phenotype specific to the CNS (19, 45). Several studies have shown that elevated MeCP2 expression is acquired during postnatal brain development, suggesting that MeCP2 might play an important role during neuronal maturation and synaptogenesis (16, 17, 19). In order to identify primary MeCP2 targets in the CNS and understand the cascade of events that follows their dysregulation, studying the right tissue at the right stage of differentiation is critical.
In this study we performed expression microarray analysis to identify novel gene targets of MeCP2 during neuronal maturational differentiation in a human SH-SY5Y neuronal cell culture system. MeCP2 binding to endogenous targets at a precise stage of neuronal maturational differentiation was specifically inhibited by transfection with a MeCP2 decoy. Both transcriptionally up and down regulated MeCP2 target genes have been observed by microarray analysis, consistent with previous studies (24–26), and arguing against a single role for MeCP2 as a transcriptional repressor. Using microarray analysis, we have identified all four known members of the ID subfamily of helix loop helix (HLH) proteins as novel neuronal targets of MeCP2. The microarray results reported in this study were consistent with published data showing that ID1, ID2 and ID3 transcript levels decreased upon SH-SY5Y neuronal differentiation (47) and ID4 expression level increased following neuronal differentiation (48). Our findings are novel in demonstrating that MeCP2 regulates ID gene transcriptional changes during SH-SY5Y neuronal differentiation. However, we also showed significantly increased ID transcripts in postnatal 28 day Mecp2−/y mouse brain tissue using qRT-PCR, demonstrating that ID gene dysregulation by MeCP2 deficiency is not limited to the SH-SY5Y neuronal system. The higher level of transcription of all four ID genes in MeCP2 decoy transfected SH-SY5Y neuronal cells and Mecp2−/y mice is consistent with the predicted role of MeCP2 as a repressor of genes regulating neuronal maturational differentiation (19). The transient developmental stage specific transcriptional changes of ID genes in Mecp2−/y brain observed at P28 (Figure 1B) may explain why ID genes did not appear significant in prior microarray analyses of Mecp2−/y brain samples (24).
In addition to changes in the transcripts we also report an increased expression of ID proteins in Mecp2−/y mice and human RTT cerebral tissues determined by performing immunofluorescence followed by quantitation by LSC. Interestingly, significantly increased ID protein expression in Mecp2−/y was observed at some postnatal time points without a corresponding increase in transcript compared to the controls. Perhaps the developmental stage specific dysregulation of ID transcripts results in long-lived effects on ID protein expression in the brain. We also observed decreased mRNA expression of Neurod1, a neurodevelopmentally essential gene target of ID proteins in Mecp2−/y mice compared to Mecp2+/y controls in all postnatal time points tested. The transient upregulation of ID proteins results in disrupting the auto-regulatory loop and leading to more lasting declines in proteins such as NEUROD1. These results suggest that the Mecp2-deficient neurons in the mouse model and in RTT patients might become arrested in an immature stage of maturational differentiation and inappropriately high ID protein levels could either cause or reflect improper neuronal maturation.
Several tissue-specific bHLH transcription factors such as ASCL1, NEUROD1 and NEUROG1 control the determination and differentiation of neurons in the central and peripheral nervous system (52, 53). bHLH proteins dimerize, bind to E-box sequences containing CANNTG and activate transcription of genes (53). The function of bHLH proteins is blocked by ID proteins that are structurally similar to bHLH except that they lack the basic DNA binding domain (38, 39, 54, 55). Four members, ID1 through ID4, have been identified in mammals (38, 48, 56) as inhibitors of differentiation or inhibitors of DNA binding.
In the absence of ID proteins, bHLH factors function prematurely and terminal differentiation is improperly induced (57). Targeted deletions of all four ID genes have been performed in mouse. Deletion of either Id1 or Id3 individually does not lead to an observable phenotype, but when both are inactivated, telencephalic neurogenesis and angiogenesis are greatly perturbed (58). Mice lacking Id2 are viable, although they have an abnormal immune system and a lactation defect (59, 60). Mice lacking Id4 have a small brain and compromised proliferation of stem cells in the ventricular zone (61).
The role of ID proteins during neuronal differentiation and the overall effect of MeCP2 deficiency on ID gene expression are illustrated in the model as shown in Figure 5. In this model, the two-fold increase in MeCP2 expression during neuronal maturation would serve to down-regulate expression of all four ID genes, resulting in the release of more bHLH transcription factors for binding to the E-box of genes involved in neuronal maturational differentiation. Mutation of MECP2 would therefore increase the levels of ID proteins, thus tipping the balance away from cellular differentiation.
Figure 5. Model for role of ID proteins in regulating neuronal maturational differentiation.
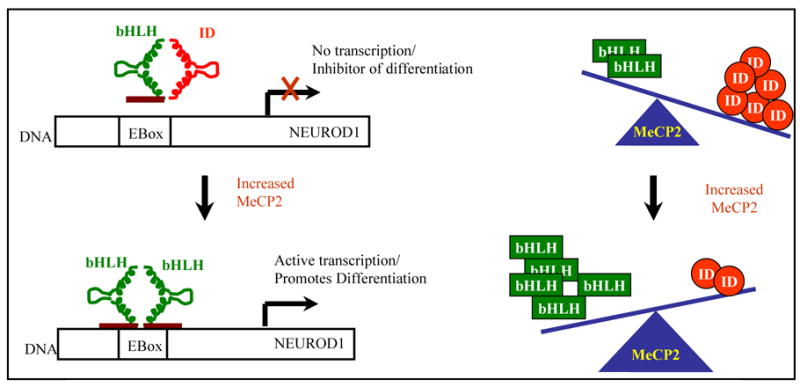
In immature neurons with high expression of ID proteins, heterodimers of bHLH-ID prevent DNA binding and expression of differentiation associated genes like NEUROD1. Upon neuronal maturation, elevated levels of MeCP2 repress the expression of ID genes, resulting in functional bHLH-bHLH dimers that can bind to E-box DNA sequences and activate transcription of differentiation associated genes such as NEUROD1, ASCL1 and NEUROG1.
In a recent study, the levels of BDNF protein were reduced in Mecp2-deficient mouse brain and deletion of Bdnf in Mecp2-deficient mice caused an earlier onset of RTT-like symptoms. In contrast, over-expression of conditional Bdnf transgene in the Mecp2-deficient mice extended lifespan, rescued a locomotor defect, and reversed an electrophysiological deficit (49). Interestingly, within rat BDNF promoter III, a Ca2+-responsive E-box element (CaRE2) was identified that binds to two bHLH transcription factors: upstream stimulatory factors 1 and 2 (USF1/2) (50). In addition, a previous study showed that neurotrophins such as BDNF stimulate neuronal differentiation by altering the balance of expression of various bHLH transcription factors (51). Reduced levels of BDNF and increased levels of ID proteins in Mecp2-deficent mice might result in lower expression of bHLH proteins, resulting in improper neuronal differentiation.
ID proteins are central to pathways regulating proliferation, differentiation, angiogenesis, migration, invasion and cell-cell interaction. Therefore, by understanding the role of ID proteins during neuronal maturation, they might be therapeutically targeted to reverse the arrest in neuronal maturation seen in RTT and other neurodevelopmental disorders.
MATERIALS AND METHODS
Cell culture and MeCP2 decoy Transfections
SH-SY5Y human neuroblastoma cells (ATCC) were grown in complete minimal essential media with 15 % fetal calf serum in T 175 cm2 large tissue culture flasks until 70–80 % confluency. The cells were transfected with decoy mixture prepared in serum free MEM containing 3 μl FuGENE 6 (Roche) transfection reagent and 1 μM of either methylated MD or CD, incubated at room temperature for 30 m before addition to the flasks. MeCP2 decoy (MD) and control decoy (CD) were obtained commercially (GeneDetect.com). Both the control (5' AATCTAGTCTAGACTAGATTA 3') and MeCP2 decoy (5' AATCCGGTCTAGACCGGATTA 3') double-stranded phosphorothioate oligodeoxynucleotides were treated with HpaII methylase (New England Biolabs) (1 U per 1 μg decoy DNA) overnight to methylate the CpG sites. The methylase-treated decoys, MD and CD decoy were digested with HpaII (New England Biolabs) and analyzed by PAGE to confirm methylation. Twelve hours after transfection, cells were treated with 16 nM phorbol 12-myristate 13-acetate (PMA) (Sigma). Total RNA was isolated 48 h after PMA was added using TRIzol reagent (Invitrogen Life technologies) from four different experimental conditions: undifferentiated (UD), differentiated and untransfected (D-UT), differentiated and MeCP2 decoy transfected (D-MD), and differentiated and control decoy transfected (D-CD).
Microarray
Total RNA yield was quantified by spectrophotometric analysis and quality was verified on a gel. A clean up of RNA was performed using RNeasy Mini kit (QIAGEN) before proceeding to cDNA synthesis. 1 μg of RNA under each condition was first reverse transcribed using T7-oligo (dT) promoter primer followed by RNase-H mediated second-stranded cDNA synthesis using One-Cycle cDNA Synthesis kit (Affymetrix). The double stranded cDNA was purified using sample cleanup modules and served as a template in the subsequent in vitro transcription (IVT) reaction for complementary RNA (cRNA) amplification and biotin labeling. The biotinylated cRNA targets were cleaned, fragmented and hybridized to Affymetrix HG-U133 plus 2.0 arrays. Detailed procedure of all the steps mentioned above are described in the GeneChip expression analysis technical manual available on the Affymetrix website (http://www.affymetrix.com). A total of 12 array hybridizations were performed consisting of three biological replicates of four experimental conditions each. The hybridizations were performed in a GeneChip hybridization oven 640 and Gene chip fluidics station 450 at the UC Davis Microarray core facility. The image files were obtained using Affymetrix 3000 GeneChip scanner. The raw CEL and DAT files were analyzed using dChip microarray analysis software available at http://biosun1.harvard.edu/complab/dchip/.
Quantitative Real Time RT-PCR (qRT-PCR)
qRT-PCR was performed using a LightCycler rapid thermal cycler system (Roche, Indianapolis, USA) according to the manufacturer's instructions. Primer sequences specific to all four human ID genes and the control house keeping gene, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) are listed in supplementary table 6 (S6). The primer sequences for mouse ID genes were obtained from PCR primer bank (62). PCR reactions consisted of 1x DNA master SYBR Green I reaction buffer (Roche), 1.5 mM-2 mM MgCl2, 0.5 μM of primers and 10–20 ng of cDNA. First stranded cDNA was synthesized using reverse transcriptase (Invitrogen) according to the manufacture’s protocol from total RNA of SH-SY5Y cells or brain tissue from Mecp2+/y and Mecp2−/y mice. Detection of the PCR product on LightCycler is carried out at the end of the 72°C extension period. To confirm amplification specificity, the PCR products of each primer pair were subjected to a melting curve analysis. The qRT-PCR data was analyzed using the LightCycler analysis software version 2.0. Final quantification was performed using the comparative CT method (Applied Biosystems) and is reported as n-fold difference relative to an undifferentiated SH-SY5Y control cDNA (UD) or Mecp2+/y cDNA of corresponding littermate following normalization to GAPDH control gene.
Chromatin immunoprecipitation
Chromatin from human SH-SY5Y neuroblastoma cells from all four conditions (UD, D-UT, D-MD and D-CD) used for microarray experiment was isolated using a procedure previously described (37). ChIP DNA was amplified using primers designed to span 5' CpG islands near or overlapping the promoter of each ID gene and promoter of SNURF/SNRPN as a positive control for ChIP assay conditions. Primer sequences are listed in supplementary table 7 (S7).
Mecp2-deficient Mice
Mecp2tm1.1Bird/+ (Jackson Laboratories) heterozygous female mice were mated with wild type C57BL/6J mice (Jackson Laboratories) to produce the four possible genotypes. Genotyping was performed using protocols provided by the commercial vendors. Mice were euthanized at different developmental time points (P1 corresponds to day of birth). Whole brain specimens were cut sagitally at the midline using a sterile scalpel and one half was placed in 10 % neutral buffered formalin for 2–3 days and then embedded in paraffin blocks; the other half was placed in TRIzol and stored at −80°C for protein and nucleic acid extractions. Sections of 1 mm were removed from each block followed by sectioning at 5 μm for experimental slides. Hematoxylin and eosin (H & E) slides were prepared for each brain specimen to aid in identification of brain regions. Epitope exposure was carried out as described previously (45).
Mouse Tissue Microarray
Wild type C57BL/6J and Mecp2tm1Bird/y tissue for multiple age time points was obtained, fixed and embedded in paraffin and sampled as described above. Mouse tissue array included triplicate 600 μm cores of grey matter from cerebral cortex of six age-matched male mice time points for Mecp2+/y and Mecp2tm1Bird/y (E 15, P1, P28, P49, P56, and P70).
Human Tissue Microarray
Tissues were received frozen and were fixed, embedded and arrayed as described earlier (46). The human tissue array used contained triplicate 600 μm cores of grey matter from frontal cortex, Broadman area 9, ≤ 30 h PMI. Tissues included on the human array were received from Maryland brain bank and the Harvard brain bank. The array consisted of three cores each of RTT brain samples: RTT 1238 (2y), RTT 1420 (21y), RTT 4312 (24y) and three cores each of three age-matched controls.
Immunofluorescence
Tissue microarrays or whole brain sections were cut into 5 μm sections, placed on glass slides and baked overnight at 56°C. Slides underwent 4 x 5m xylene washes, 2 x 5 m 100 % ethanol washes, dried on a slide warmer (Hybaid) at 50°C, then placed in a 1:10 dilution of DAKO antigen retrieval solution at 95°C for 1 h, cooled to room temperature, and washed for 5 m in 0.2 X SSC. Primary antibodies were diluted in immunofluorescence buffer (IF buffer) (1 X PBS, 0.5 % Tween, 0.01 % Fetal calf serum) added to slides, coversliped and incubated at 37°C for 2 h, washed three times for 5 m in 1 X PBS / 0.5 % Tween. Secondary antibodies diluted in the same IF buffer along with 250 μg/mL RNAse and 3 μg/mL propidium iodide (PI) were then added to slides, coversliped and incubated at 37°C for 1 h followed by 3 x 5 m 1 X PBS / 0.5% Tween washes. Slides were mounted with nuclear counter stain PI (7 μg/ml PI in 50 % glycerol /50% 1X PBS) for analyzing the slides on LSC. Primary antibodies used: anti-ID1 (Santa Cruz, rabbit polyclonal) and anti-ID1 (rabbit monoclonal, kind gift from Dr. Robert Benezra) 1:100, anti-ID2 (Santa Cruz, rabbit polyclonal), anti-ID3 (Santa Cruz, rabbit polyclonal) 1:100, anti-ID4 (Santa Cruz, rabbit polyclonal) 1:100, anti-MeCP2 (Aves, C-terminal, chicken polyclonal) 1:10,000, anti-Histone H1 (Upstate, mouse polyclonal) 1:100. Secondary antibodies used: goat anti-rabbit IgG-Oregon Green (Molecular Probes) 1:100, donkey anti-rabbit IgG-CY5 (Molecular Probes) 1:100, donkey anti-chicken IgY-CY5 (Jackson) 1:100 and goat anti-mouse IgG-Cascade Blue (Molecular Probes) 1:100. Rabbit IgG (Upstate), Chicken IgY (Aves), and Mouse IgG (Upstate) were used at equivalent or higher concentrations than the primary antibodies with the same secondary to test for background levels of staining.
Laser Scanning Cytometry
Slides stained with the above mentioned antibodies were scanned using a Compucyte Laser Scanning Cytometer, as described previously (15, 45, 46). Settings for voltage, PMT, and threshold were identical between negative control and experimental samples. Nuclei, stained red with PI, were contoured within each core and fluorescence was measured for each nucleus for all channels (red, green, blue and long red). Data was analyzed using Wincyte software from Compucyte.
Supplementary Material
Acknowledgments
The authors would like to thank Dr. Michael George, Maggie Chiu and Dan Braunschweig for technical assistance. Dr. Edward Jones, Dr. Robert Benezra, Amber Hogart, Karen N. Thatcher, Ravi Nagarajan, Susan Swanberg for valuable comments on the manuscript. Dr. Robert Benezra (Sloan-Kettering, Cancer Biology and Genetics, NY, USA) kindly provided the ID1 antibody. Human tissue samples were generously provided by the Autism Tissue Program, the University of Maryland Brain and Tissue Bank for Developmental Disorders (supported by NIH N01-HD-1-3138), Harvard Brain Tissue Resource Center (supported in part by PHS MH/NS 31862). This work was supported in part by the Rett Syndrome Research Foundation and the NIH (1R01HD/NS41462). S.P was supported by a predoctoral fellowship from the National Alliance for Autism Research.
Abbreviations
- BDNF
brain derived neurotropic factor
- CD
control decoy (unmethylated oligonucleotide)
- ChIP
chromatin immunoprecipitation
- CpG
Cytosine-phosphate-Guanine (5′-CG-3′)
- ID
inhibitors of differentiation or inhibitors of DNA binding
- LSC
laser scanning cytometer (or cytometry)
- MD
MeCP2 methylated oliconucleotide decoy
- MECP2
methyl-CpG-binding protein 2
- PMA
2-O-tetradecanoylphorbol-13-acetate
- RTT
Rett syndrome
- SNRPN
small nuclear riboprotein N
References
- 1.Sirianni N, Naidu S, Pereira J, Pillotto RF, Hoffman EP. Rett syndrome: confirmation of X-linked dominant inheritance, and localization of the gene to Xq28. Am J Hum Genet. 1998;63:1552–1558. doi: 10.1086/302105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet. 1999;23:185–188. doi: 10.1038/13810. [DOI] [PubMed] [Google Scholar]
- 3.Zoghbi HY, Percy AK, Schultz RJ, Fill C. Patterns of X chromosome inactivation in the Rett syndrome. Brain Dev. 1990;12:131–135. doi: 10.1016/s0387-7604(12)80194-x. [DOI] [PubMed] [Google Scholar]
- 4.Armstrong DD. Review of Rett syndrome. J Neuropathol Exp Neurol. 1997;56:843–849. doi: 10.1097/00005072-199708000-00001. [DOI] [PubMed] [Google Scholar]
- 5.Ellaway C, Christodoulou J. Rett syndrome: clinical update and review of recent genetic advances. J Paediat Child Health. 1999;35:419–426. doi: 10.1046/j.1440-1754.1999.355403.x. [DOI] [PubMed] [Google Scholar]
- 6.Van den Veyver IB, Zoghbi HY. Methyl-CpG-binding protein 2 mutations in Rett syndrome. Cur Opi Genet Dev. 2000;10:275–279. doi: 10.1016/s0959-437x(00)00083-6. [DOI] [PubMed] [Google Scholar]
- 7.Bird AP, Wolffe AP. Methylation-induced repression--belts, braces, and chromatin. Cell. 1999;99:451–454. doi: 10.1016/s0092-8674(00)81532-9. [DOI] [PubMed] [Google Scholar]
- 8.Bird A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002;16:6–21. doi: 10.1101/gad.947102. [DOI] [PubMed] [Google Scholar]
- 9.Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K, Lein ES, Eadie BD, Willhoite AR, Muotri AR, et al. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci U S A. 2003;100:6777–6782. doi: 10.1073/pnas.1131928100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710–723. doi: 10.1101/gad.194101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403–405. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- 12.Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322–326. doi: 10.1038/85899. [DOI] [PubMed] [Google Scholar]
- 13.Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327–331. doi: 10.1038/85906. [DOI] [PubMed] [Google Scholar]
- 14.Shahbazian M, Young J, Yuva-Paylor L, Spencer C, Antalffy B, Noebels J, Armstrong D, Paylor R, Zoghbi H. Mice with truncated MeCP2 recapitulate many Rett syndrome features and display hyperacetylation of histone H3. Neuron. 2002;35:243–254. doi: 10.1016/s0896-6273(02)00768-7. [DOI] [PubMed] [Google Scholar]
- 15.Braunschweig D, Simcox T, Samaco RC, LaSalle JM. X-Chromosome inactivation ratios affect wild-type MeCP2 expression within mosaic Rett syndrome and Mecp2−/+ mouse brain. Hum Mol Genet. 2004;13:1275–1286. doi: 10.1093/hmg/ddh142. [DOI] [PubMed] [Google Scholar]
- 16.Balmer D, Goldstine J, Rao YM, LaSalle JM. Elevated methyl-CpG-binding protein 2 expression is acquired during postnatal human brain development and is correlated with alternative polyadenylation. J Mol Med. 2003;81:61–68. doi: 10.1007/s00109-002-0396-5. [DOI] [PubMed] [Google Scholar]
- 17.Shahbazian MD, Antalffy B, Armstrong DL, Zoghbi HY. Insight into Rett syndrome: MeCP2 levels display tissue- and cell-specific differences and correlate with neuronal maturation. Hum Mol Genet. 2002;11:115–124. doi: 10.1093/hmg/11.2.115. [DOI] [PubMed] [Google Scholar]
- 18.Akbarian S, Chen RZ, Gribnau J, Rasmussen TP, Fong H, Jaenisch R, Jones EG. Expression pattern of the Rett syndrome gene MeCP2 in primate prefrontal cortex. Neurobiol Dis. 2001;8:784–791. doi: 10.1006/nbdi.2001.0420. [DOI] [PubMed] [Google Scholar]
- 19.Zoghbi HY. Postnatal neurodevelopmental disorders: meeting at the synapse? Science. 2003;302:826–830. doi: 10.1126/science.1089071. [DOI] [PubMed] [Google Scholar]
- 20.Nan X, Campoy FJ, Bird A. MeCP2 is a transcriptional repressor with abundant binding sites in genomic chromatin. Cell. 1997;88:471–481. doi: 10.1016/s0092-8674(00)81887-5. [DOI] [PubMed] [Google Scholar]
- 21.Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N, Strouboulis J, Wolffe AP. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187–191. doi: 10.1038/561. [DOI] [PubMed] [Google Scholar]
- 22.Hendrich B, Bird A. Identification and characterization of a family of mammalian methyl-CpG binding proteins. Mol Cell Biol. 1998;18:6538–6547. doi: 10.1128/mcb.18.11.6538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nan X, Tate P, Li E, Bird A. DNA methylation specifies chromosomal localization of MeCP2. Mol Cell Biol. 1996;16:414–421. doi: 10.1128/mcb.16.1.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tudor M, Akbarian S, Chen RZ, Jaenisch R. Transcriptional profiling of a mouse model for Rett syndrome reveals subtle transcriptional changes in the brain. Proc Natl Acad Sci U S A. 2002;99:15536–15541. doi: 10.1073/pnas.242566899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Traynor J, Agarwal P, Lazzeroni L, Francke U. Gene expression patterns vary in clonal cell cultures from Rett syndrome females with eight different MECP2 mutations. BMC Med Genet. 2002;3:12. doi: 10.1186/1471-2350-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colantuoni C, Jeon OH, Hyder K, Chenchik A, Khimani AH, Narayanan V, Hoffman EP, Kaufmann WE, Naidu S, Pevsner J. Gene expression profiling in postmortem Rett Syndrome brain: differential gene expression and patient classification. Neurobiol Dis. 2001;8:847–865. doi: 10.1006/nbdi.2001.0428. [DOI] [PubMed] [Google Scholar]
- 27.Chen WG, Chang Q, Lin Y, Meissner A, West AE, Griffith EC, Jaenisch R, Greenberg ME. Derepression of BDNF transcription involves calcium-dependent phosphorylation of MeCP2. Science. 2003;302:885–889. doi: 10.1126/science.1086446. [DOI] [PubMed] [Google Scholar]
- 28.Martinowich K, Hattori D, Wu H, Fouse S, He F, Hu Y, Fan G, Sun YE. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science. 2003;302:890–893. doi: 10.1126/science.1090842. [DOI] [PubMed] [Google Scholar]
- 29.Horike S, Cai S, Miyano M, Cheng JF, Kohwi-Shigematsu T. Loss of silent-chromatin looping and impaired imprinting of DLX5 in Rett syndrome. Nat Genet. 2005;37:31–40. doi: 10.1038/ng1491. [DOI] [PubMed] [Google Scholar]
- 30.Stancheva I, Collins AL, Van den Veyver IB, Zoghbi H, Meehan RR. A mutant form of MeCP2 protein associated with human Rett syndrome cannot be displaced from methylated DNA by notch in Xenopus embryos. Mol Cell. 2003;12:425–435. doi: 10.1016/s1097-2765(03)00276-4. [DOI] [PubMed] [Google Scholar]
- 31.Ballestar E, Ropero S, Alaminos M, Armstrong J, Setien F, Agrelo R, Fraga MF, Herranz M, Avila S, Pineda M, et al. The impact of MECP2 mutations in the expression patterns of Rett syndrome patients. Hum Genet. 2005;116:91–104. doi: 10.1007/s00439-004-1200-0. [DOI] [PubMed] [Google Scholar]
- 32.Samaco RC, Hogart A, LaSalle JM. Epigenetic overlap in autism-spectrum neurodevelopmental disorders: MECP2 deficiency causes reduced expression of UBE3A and GABRB3. Hum Mol Genet. 2005;14:483–492. doi: 10.1093/hmg/ddi045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nuber UA, Kriaucionis S, Roloff TC, Guy J, Selfridge J, Steinhoff C, Schulz R, Lipkowitz B, Ropers HH, Holmes MC, et al. Up-regulation of glucocorticoid-regulated genes in a mouse model of Rett syndrome. Hum Mol Genet. 2005;14:2247–2256. doi: 10.1093/hmg/ddi229. [DOI] [PubMed] [Google Scholar]
- 34.Pahlman S, Hoehner JC, Nanberg E, Hedborg F, Fagerstrom S, Gestblom C, Johansson I, Larsson U, Lavenius E, Ortoft E, et al. Differentiation and survival influences of growth factors in human neuroblastoma. Eur J Cancer. 1995;31A:453–458. doi: 10.1016/0959-8049(95)00033-f. [DOI] [PubMed] [Google Scholar]
- 35.Jung BP, Jugloff DG, Zhang G, Logan R, Brown S, Eubanks JH. The expression of methyl CpG binding factor MeCP2 correlates with cellular differentiation in the developing rat brain and in cultured cells. J Neurobiol. 2003;55:86–96. doi: 10.1002/neu.10201. [DOI] [PubMed] [Google Scholar]
- 36.Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 2005;19:667–678. doi: 10.1016/j.molcel.2005.07.021. [DOI] [PubMed] [Google Scholar]
- 37.Thatcher KN, Peddada S, Yasui DH, LaSalle JM. Homologous pairing of 15q11-13 imprinted domains in brain is developmentally regulated but deficient in Rett and autism samples. Hum Mol Genet. 2005;14:785–797. doi: 10.1093/hmg/ddi073. [DOI] [PubMed] [Google Scholar]
- 38.Benezra R, Davis RL, Lockshon D, Turner DL, Weintraub H. The protein Id: a negative regulator of helix-loop-helix DNA binding proteins. Cell. 1990;61:49–59. doi: 10.1016/0092-8674(90)90214-y. [DOI] [PubMed] [Google Scholar]
- 39.Norton JD, Deed RW, Craggs G, Sablitzky F. Id helix-loop-helix proteins in cell growth and differentiation. Trends Cell Biol. 1998;8:58–65. [PubMed] [Google Scholar]
- 40.Jen Y, Manova K, Benezra R. Expression patterns of Id1, Id2, and Id3 are highly related but distinct from that of Id4 during mouse embryogenesis. Dev Dyn. 1996;207:235–252. doi: 10.1002/(SICI)1097-0177(199611)207:3<235::AID-AJA1>3.0.CO;2-I. [DOI] [PubMed] [Google Scholar]
- 41.Andres-Barquin PJ, Hernandez MC, Israel MA. Id genes in nervous system development. Histol Histopathol. 2000;15:603–618. doi: 10.14670/HH-15.603. [DOI] [PubMed] [Google Scholar]
- 42.Chae JH, Stein GH, Lee JE. NeuroD: the predicted and the surprising. Mol Cell. 2004;18:271–288. [PubMed] [Google Scholar]
- 43.Liu KJ, Harland RM. Cloning and characterization of Xenopus Id4 reveals differing roles for Id genes. Dev Biol. 2003;264:339–351. doi: 10.1016/j.ydbio.2003.08.017. [DOI] [PubMed] [Google Scholar]
- 44.Ghil SH, Jeon YJ, Suh-Kim H. Inhibition of BETA2/NeuroD by Id2. Exp Mol Med. 2002;34:367–373. doi: 10.1038/emm.2002.52. [DOI] [PubMed] [Google Scholar]
- 45.LaSalle JM, Goldstine J, Balmer D, Greco CM. Quantitative localization of heterogeneous methyl-CpG-binding protein 2 (MeCP2) expression phenotypes in normal and Rett syndrome brain by laser scanning cytometry. Hum Mol Genet. 2001;10:1729–1740. doi: 10.1093/hmg/10.17.1729. [DOI] [PubMed] [Google Scholar]
- 46.Samaco RC, Nagarajan RP, Braunschweig D, LaSalle JM. Multiple pathways regulate MeCP2 expression in normal brain development and exhibit defects in autism-spectrum disorders. Hum Mol Genet. 2004;13:629–639. doi: 10.1093/hmg/ddh063. [DOI] [PubMed] [Google Scholar]
- 47.Lopez-Carballo G, Moreno L, Masia S, Perez P, Barettino D. Activation of the phosphatidylinositol 3-kinase/Akt signaling pathway by retinoic acid is required for neural differentiation of SH-SY5Y human neuroblastoma cells. J Biol Chem. 2002;277:25297–25304. doi: 10.1074/jbc.M201869200. [DOI] [PubMed] [Google Scholar]
- 48.Riechmann V, van Cruchten I, Sablitzky F. The expression pattern of Id4, a novel dominant negative helix-loop-helix protein, is distinct from Id1, Id2 and Id3. Nucleic Acids Res. 1994;22:749–755. doi: 10.1093/nar/22.5.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron. 2006;49:341–348. doi: 10.1016/j.neuron.2005.12.027. [DOI] [PubMed] [Google Scholar]
- 50.Chen WG, West AE, Tao X, Corfas G, Szentirmay MN, Sawadogo M, Vinson C, Greenberg ME. Upstream stimulatory factors are mediators of Ca2+-responsive transcription in neurons. J Neurosci. 2003;23:2572–2581. doi: 10.1523/JNEUROSCI.23-07-02572.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ito H, Nakajima A, Nomoto H, Furukawa S. Neurotrophins facilitate neuronal differentiation of cultured neural stem cells via induction of mRNA expression of basic helix-loop-helix transcription factors Mash1 and Math1. J Neurosci Res. 2003;71:648–658. doi: 10.1002/jnr.10532. [DOI] [PubMed] [Google Scholar]
- 52.Cai L, Morrow EM, Cepko CL. Misexpression of basic helix-loop-helix genes in the murine cerebral cortex affects cell fate choices and neuronal survival. Development. 2000;127:3021–3030. doi: 10.1242/dev.127.14.3021. [DOI] [PubMed] [Google Scholar]
- 53.Lee JE. Basic helix-loop-helix genes in neural development. Curr Opin Neurobiol. 1997;7:13–20. doi: 10.1016/s0959-4388(97)80115-8. [DOI] [PubMed] [Google Scholar]
- 54.Norton JD. ID helix-loop-helix proteins in cell growth, differentiation and tumorigenesis. J Cell Sci. 2000;113 ( Pt 22):3897–3905. doi: 10.1242/jcs.113.22.3897. [DOI] [PubMed] [Google Scholar]
- 55.Ruzinova MB, Benezra R. Id proteins in development, cell cycle and cancer. Trends Cell Biol. 2003;13:410–418. doi: 10.1016/s0962-8924(03)00147-8. [DOI] [PubMed] [Google Scholar]
- 56.Sun XH, Copeland NG, Jenkins NA, Baltimore D. Id proteins Id1 and Id2 selectively inhibit DNA binding by one class of helix-loop-helix proteins. Mol Cell Biol. 1991;11:5603–5611. doi: 10.1128/mcb.11.11.5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yokota Y. Id and development. Oncogene. 2001;20:8290–8298. doi: 10.1038/sj.onc.1205090. [DOI] [PubMed] [Google Scholar]
- 58.Lyden D, Young AZ, Zagzag D, Yan W, Gerald W, O'Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K, et al. Id1 and Id3 are required for neurogenesis, angiogenesis and vascularization of tumour xenografts. Nature. 1999;401:670–677. doi: 10.1038/44334. [DOI] [PubMed] [Google Scholar]
- 59.Yokota Y, Mansouri A, Mori S, Sugawara S, Adachi S, Nishikawa S, Gruss P. Development of peripheral lymphoid organs and natural killer cells depends on the helix-loop-helix inhibitor Id2. Nature. 1999;397:702–706. doi: 10.1038/17812. [DOI] [PubMed] [Google Scholar]
- 60.Mori S, Nishikawa SI, Yokota Y. Lactation defect in mice lacking the helix-loop-helix inhibitor Id2. EMBO J. 2000;19:5772–5781. doi: 10.1093/emboj/19.21.5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yun K, Mantani A, Garel S, Rubenstein J, Israel MA. Id4 regulates neural progenitor proliferation and differentiation in vivo. Development. 2004;131:5441–5448. doi: 10.1242/dev.01430. [DOI] [PubMed] [Google Scholar]
- 62.Wang X, Seed B. A PCR primer bank for quantitative gene expression analysis. Nucleic Acids Res. 2003;31:e154. doi: 10.1093/nar/gng154. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.