

Abstract
Children with acute hypoxic-ischaemic events (e.g. stroke) and chronic neurological conditions associated with hypoxia frequently present to paediatric neurologists. Failure to adapt to hypoxia may be a common pathophysiological pathway linking a number of other conditions of childhood with cognitive deficit. There is evidence that congenital cardiac disease, asthma and sleep disordered breathing, for example, are associated with cognitive deficit, but little is known about the mechanism and whether there is any structural change. This review describes what is known about how the brain reacts and adapts to hypoxia, focusing on epilepsy and sickle cell disease (SCD). We prospectively recorded overnight oxyhaemoglobin saturation (SpO2) in 18 children with intractable epilepsy, six of whom were currently or recently in minor status (MS). Children with MS were more likely to have an abnormal sleep study defined as either mean baseline SpO2 <94% or >4 dips of >4% in SpO2/hour (p = .04). In our series of prospectively followed patients with SCD who subsequently developed acute neurological symptoms and signs, mean overnight SpO2 was lower in those with cerebrovascular disease on magnetic resonance angiography (Mann-Whitney, p = .01). Acute, intermittent and chronic hypoxia may have detrimental effects on the brain, the clinical manifestations perhaps depending on rapidity of presentation and prior exposure.
Introduction
Paediatric neurologists manage children with a range of conditions associated with hypoxia. Survivors of neonatal asphyxia often have long-term motor and cognitive impairments (de Haan, Wyatt, Roth, Vargha-Khadem, Gadian & Mishkin, 2006, this issue), challenging assumptions about the resistance of the immature brain to hypoxia and the degree to which the otherwise expected plasticity may compensate for early brain damage. Other common conditions of childhood are associated with chronic-intermittent hypoxia of varying severity and an increased prevalence of cognitive and behavioural deficits (Kheirandish & Gozal, 2006; Wray, 2006, this issue). Progressive cerebrovascular disease (CVD) (Hogan, Kirkham, Isaacs, Wade & Vargha-Khadem, 2005) or persisting epilepsy may account for some of the poor outcomes in children initially predicted to do well. Variations in presentation, response to treatment and cognitive outcome may also be related to the nature of the injury (focal or global), the stage of brain development at which injury occurs and any acute or chronic systemic disturbance, such as hypotension or hypoxia. The severity and persistence of neurocognitive deficit may be determined not only by the extent to which hypoxia depletes the brain’s energy reserves, but also by the manner in which the brain responds to this challenge.
The aim of this review is to describe what is known about how the brain adapts to hypoxia. Evidence is drawn from animal models and diseases affecting older adults, as well as from studies of populations living at altitude. These mechanisms are subsequently explored in the context of epilepsy and of sickle cell disease, an anaemia associated with oxyhaemoglobin desaturation (Setty, Stuart, Dampier, Brodecki & Allen, 2003), subtle white matter abnormality in the absence of brain infarct and intellectual deficit (Baldeweg, Hogan, Saunders, Telfer, Gadian, Vargha-Khadem & Kirkham, 2006; Schatz & Buzan, 2006).
Mechanisms of adaptation to hypoxia
Cognitive effects of acute hypoxia have been likened to alcohol intoxication (Barcroft, 1920), with headache, mental confusion, drowsiness, muscular weakness, inco-ordination and visual disturbance (McFarland & Evans, 1939). Long-term adaptation to intermittent and sustained hypoxia (see Figure 1) may play a role in modifying the effect of acute hypoxia on neurones, reducing the clinically detectable effects (Miyamoto & Auer, 2000). Altitude studies have identified alternative, parallel or serial adaptive mechanisms (Figure 1), including increased erythropoiesis (Dill, 1964), changes in chemosensitive drive to respiration (Fatemian, Gamboa, Leon-Velarde, Rivera-Ch, Palacios & Robbins, 2003), remodelling of the vasculature (Ng, Tan, Ng & Lim, 2005), upregulation of sympathetic mechanisms (Calbet, 2003) and increased exhaled nitric oxide (NO) protecting against hypoxic pulmonary vasoconstriction and improving oxygen transfer in the lung (Beall, Laskowski, Strohl, Soria, Villena, Vargas, Alarcon, Gonzales & Erzurum, 2001). Some genes upregulated during severe ischaemia, are downregulated by a less severe ‘preconditioning’ insult associated with amelioration of the effects of hypoxia (Figure 1; Stenzel-Poore, Stevens, Xiong, Lessov, Harrington, Mori, Meller, Rosenzweig, Tobar, Shaw, Chu & Simon, 2003).
Figure 1.
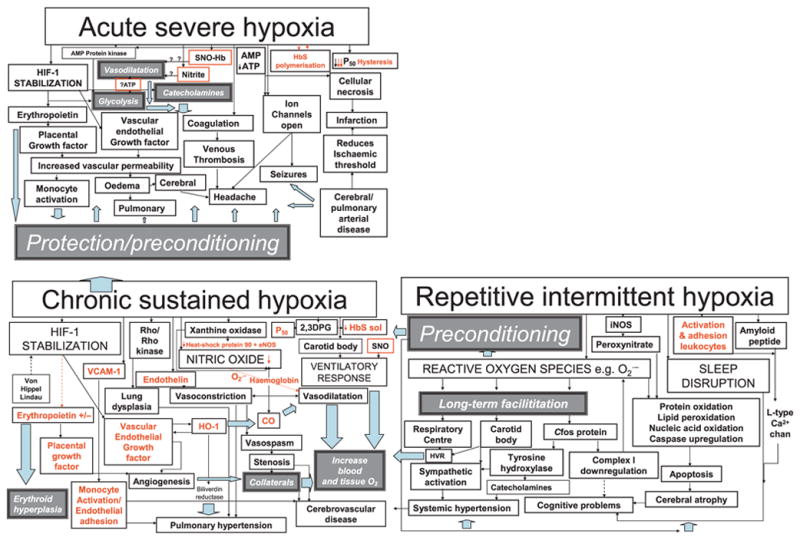
Clinical features are represented by pale grey boxes while white boxes/black arrows refer to mechanisms likely to lead to pathology (red for those for which there is evidence in sickle cell disease, SCD) and dark grey boxes/pale blue arrows refer to potentially protective mechanisms. Red boxes denote mechanisms involving the red cell postulated to be involved in vasodilatation in response to local hypoxia. In chronic sustained hypoxia, hypoxia-inducible factor stabilization leads to upregulation of erythropoietin and growth factors. In addition, there is increased nitric oxide synthesis, although in SCD free haemoglobin secondary to chronic haemolysis and the superoxide generated by increased xanthine oxidase activity may reduce bioavailability and tip the delicate balance between vasodilatation and vasoconstriction in favour of the latter. Vasconstriction is also favoured by the release of endothelin in response to hypoxia. Increased 2,3 dihydophosphoglycerate (2,3 DPG) further increases P50 and decreases Haemoglobin S solubility. Adhesion molecules, such as VCAM-1, are upregulated and favour monocyte adhesion to the endothelium. Upregulation of haem-oxygenase-1 (HO-1) may have protective effects, including vasodilatation and reduction of P50 secondary to the generation of carboxyhaemoglobin, and the antioxidant effects of bilirubin reductase, but also further upregulates vascular endothelial growth factor and may increase carbon monoxide levels locally. The ventilatory response may be increased, probably facilitated by repetitive intermittent hypoxia. Cerebral infarction, atrophy and cognitive problems may be related to a number of mechanisms related to chronic sustained and intermittent hypoxia, including perhaps demyelination secondary to chronically high carbon monoxide levels, the adverse effects of reactive oxygen species generated by repetitive intermittent hypoxia on proteins, nucleic acids and lipids, downregulation of the mitochondrial respiratory chain enzymes and upregulation of amyloid β peptide, as well as the effect of sleep disruption. Chronically hypoxic patients with SCD may be preconditioned and therefore relatively protected from the effects of acute hypoxia, e.g. vasogenic oedema secondary to increased vascular endothelial growth factor, opening of ion channels and venous thrombosis. However, the very rapid polymerization of HbS may cause such severe hypoxia that immediately available compensatory mechanisms, such as increased glycolysis, relative hypertension secondary to catecholamine release, and vasodilatation secondary to release of nitric oxide by S-nitrosylated haemoglobin (SNO-Hb), are overwhelmed and acute neurological complications, such as headache, seizures, cerebral oedema, as well as stroke, are inevitable.
Recent evidence suggests a key role in acclimatization to chronic hypoxaemia for Hypoxia-Inducible Factor (HIF) stabilization which upregulates endothelin, erythropoietin and growth factors such as vascular endothelial growth factor (VEGF) and placental growth factor (PGF) (Figure 1) (Semenza, 2001). Erythropoietin is neuroprotective (Chang, Mu, Wendland, Sheldon, Vexler, McQuillen & Ferriero, 2005), as well as increasing red cell mass and oxygen carrying capacity. HIF stabilization also improves host defence in response to infection (Peyssonnaux, Datta, Cramer, Doedens, Theodorakis, Gallo, Hurtado-Ziola, Nizet & Johnson, 2005) and heat acclimatization (Shein, Horowitz, Alexandrovich, Tsenter & Shohami, 2005), providing a basis for interactions between adaptations (Datta & Tipton, 2006) of relevance to children.
Upregulation of gene products by hypoxia may be age-dependent. As well as inducing angiogenesis, VEGF may induce vascular leakage leading to cerebral oedema (Schoch, Fischer & Marti, 2002) at altitude (Tissot van Patot, Leadbetter, Keyes, Bendrick-Peart, Beckey, Christians & Hackett, 2005) and after status epilepticus (Croll, Goodman & Scharfman, 2004). There are age differences in VEGF expression of potential physiological importance; in the systemic circulation (Rivard, Berthou-Soulie & Principe, 2001) and carotid body (Di Giulio, Bianchi, Cacchio, Artese, Rapino, Macri & Di Ilio, 2005) it is greater in the young with evidence for neuroprotection. HIF stabilization appears to be associated with more apoptotic cell death in older animals (Rapino, Bianchi, Di Giulio, Centurione, Cacchio, Antonucci & Cataldi, 2005); in this age group, lack of expression is neuroprotective (Helton, Cui, Scheel, Ellison, Ames, Gibson, Blouw, Ouyang, Dragatsis, Zeitlin, Johnson, Lipton & Barlow, 2005). Certain polymorphisms increase susceptibility to Alzheimer’s, again suggesting an important role for VEGF in neuroprotection (Del Bo, Scarlato, Ghezzi, Martinelli Boneschi, Fenoglio, Galbiati, Virgilio, Galimberti, Galimberti, Crimi, Ferrarese, Scarpini, Bresolin & Comi, 2005).
Hypoxic induction of NO synthesis is important because NO has a role in HIF stabilization (Hagen, Taylor, Lam & Moncada, 2003) and is a powerful vasodilator increasing tissue oxygen delivery (Bertuglia & Giusti, 2005), although there is ongoing controversy about the precise mechanism. One group has provided evidence that haemoglobin reduces nitrite (Figure 1), releasing NO during haem deoxygenation, with maximal activity observed at 50% haemoglobin oxygenation (P50), stimulating vasodilatation (Crawford, Isbell, Huang, Shiva, Chacko, Schechter, Darley-Usmar, Kerby, Lang, Kraus, Ho, Gladwin & Patel, 2005). Another has shown that during oxygen binding to haem as blood is oxygenated in the lungs, NO binding to a cysteine on the haemoglobin molecule forming S-nitrosylated haemoglobin (SNO) (Figure 1), which changes from the vasoconstricting R state to the vasodilating T state upon deoxygenation, releasing the NO locally (Stamler, Jia, Eu, McMahon, Demchenko, Bonaventura, Gernert & Piantadosi, 1997). Haemoglobin therefore appears to play a key role in matching tissue perfusion to oxygen demand by reacting with nitrite (Gladwin, Crawford & Patel, 2004) or SNO (Singel & Stamler, 2005) (Figure 1) in a manner allosterically regulated by oxygen tension. Central neural transduction of the hypoxic ventilatory response appears to depend on signalling in the brainstem by reactive S-nitrosylated (SNO) molecules, such as S-nitrosoglutathione, formed locally when the NO released from deoxygenated SNO-haemoglobin binds (Lipton, Johnson, Macdonald, Lieberman, Gozal & Gaston, 2001).
The incidence and extent of neurocognitive deficit in children living at altitude is unknown, but these children provide an important naturalistic model for hypoxic-adaptation of relevance to other populations (Virués-Ortega, Garrido, Javierre & Kloezeman, 2006, this issue). For example, the hyperventilatory response (Figure 1), well recognized at altitude, may be particularly important in anaemia, where oxygen delivery may be maintained by increased cardiac output or tissue oxygen extraction (Macarlupu, Buvry, Morel, Leon-Velarde, Richalet & Favret, 2006). In populations native to high altitude, there are at least three distinct phenotypes: Andeans, Ethiopians and Tibetans (Beall, 2000a; Beall et al., 2001), which might represent differences in distribution of genetic polymorphisms, although this awaits confirmation (Mejia, Prchal, Leon-Velarde, Hurtado & Stockton, 2005). Mechanisms of adaptation include: erythropoiesis, enlarged chests and a blunted ventilatory response to hypoxia reducing the work of breathing (Andeans); increased NO production (Tibetans); and hyper-ventilation (Ethiopians) (Beall, 2003). Data in those of African origin are limited but oxyhaemoglobin saturation (SpO2) is relatively normal in Ethiopians living at high altitude (Beall, Decker, Brittenham, Kushner, Gebremedhin & Strohl, 2002) while at lower altitude, tidal volume is reduced, respiratory rate increased and SpO2 lower in the Xhosa than in Caucasians (Terblanche, Tolley, Fahlman, Myburgh & Jackson, 2005). In young adult African Americans, peripheral chemosensitivity is increased during sleep, although baroreceptor responses are reduced compared with Caucasians (Crisostomo, Zayyad, Carley, Abubaker, Onal, Stepanski, Lopata & Basner, 1998), perhaps providing a basis for variation in prevalence of chronic hypertension. These differences may reflect subtle variations in earlier exposure to hypoxia rather than, or as well as, natural selection for genetic polymorphisms (Terblanche et al., 2005).
Adaptations prior to conception, during foetal life and in early childhood (Beall, 2000b) may play an important role. Age-dependent chronic adaptations include attenuation of hypoxic hyperventilatory response after exposure to intermittent hypoxia (Gozal & Gozal, 2001), the degree of alveolar branching (van Tuyl, Liu, Wang, Kuliszewski, Tibboel & Post, 2005), increases in cerebral capillary density (LaManna, Chavez & Pichiule, 2004), size and shape of cerebral smooth muscle and endothelial cells (Williams & Pearce, 2005) and response to sympathetic stimulation and calcium-dependent and -independent mechanisms for smooth muscle contraction (Longo & Pearce, 2005). If a hypoxic challenge occurs later in life, any of these prior adaptations might determine the mechanisms available to respond, and the nature of any sequelae if the response is inadequate.
Adaptation to intermittent or sustained hypoxia might particularly determine the type of neurological complications for which patients are at risk when exposed to acute hypoxia. For example, persistently low levels of tissue oxygen in anaemia may lead to neuroprotective erythropoietin production, but release of younger, more adhesive red cells and any associated haemolysis (Rice & Alfrey, 2005) may adversely affect endothelial function (Gladwin & Kato, 2005), eventually leading to irreversible vascular disease. Erythropoietic changes in skull thickness and facial bone morphology potentially reduce airway size and lead to intermittent hypoxaemia. In obstructive sleep apnoea, upregulation of inflammatory proteins adversely affects endothelial function, leading to reduced arterial diameter (Minoguchi, Yokoe, Tazaki, Minoguchi, Tanaka, Oda, Okada, Ohta, Naito & Adachi, 2005); high middle cerebral artery velocities have also been demonstrated in children with primary snoring (Hill, Hogan, Onugha, Harrison, Cooper, McGrigor, Datta & Kirkham, 2006). Interestingly, patients with Parkinson’s disease (Onodera, Okabe, Kikuchi, Tsuda & Itoyama, 2000) have impaired chemosensitivity to hypoxia. In the elderly, oxyhaemoglobin desaturation is associated with periventricular white matter lesions on MRI (van Dijk, Vermeer, de Groot, van de Minkelis, Prins, Oudkerk, Hofman, Koudstaal & Breteler, 2004).
In summary, there are a number of adaptations to hypoxia, which may depend on genetic factors and on the timing, degree and duration of exposure. The effect of these variables in individuals and the specific consequences in terms of cerebrovascular pathology and neurological disease remain to be determined now that population-based normal data are available (Urschitz, Wolff, Von Einem, Urschitz-Duprat, Schlaud & Poets, 2003).
Exposure to hypoxia in a common neurological disease of childhood: epilepsy
Some childhood epilepsies are recognized by paediatric neurologists as ‘malignant’ in that seizures are frequent and resistant to medication and the child’s development arrests (Dulac & Chiron, 1996; Drury, 2002). Aetiology is obscure in many cases, although some patients have a gene coding for an abnormal component of an ion channel and others may have focal or generalized structural abnormality; children with Sturge-Weber syndrome (see Figure 2) may follow this course (Maria, Neufeld, Rosainz, Drane, Quisling, Ben-David & Hamed, 1998). Many children spend prolonged periods of time in ‘minor status’ (MS), a state of clouded consciousness with frequent absences and sometimes drop attacks, accompanied by continuous discharges on the EEG (Brett, 1966; Drury, 2002).
Figure 2.
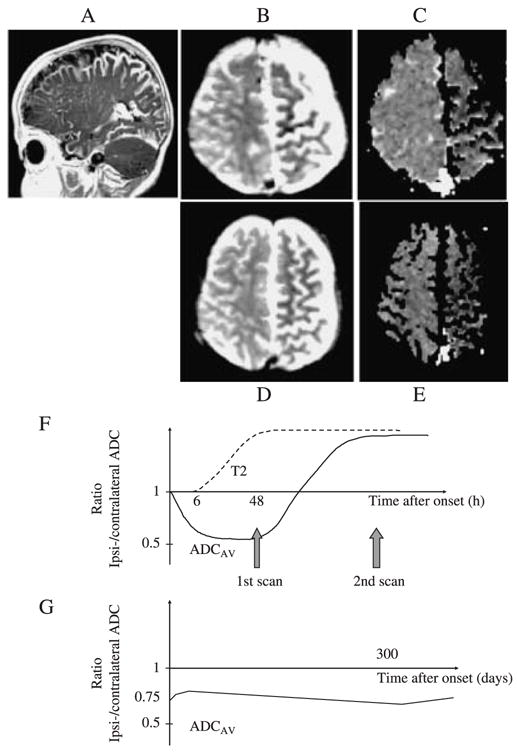
This patient was born with a left capillary haemangioma in the V1–2 distribution and demonstration of a pial angioma confirmed the diagnosis of Sturge-Weber syndrome. She presented at age 4 months with 6 days of frequent treatment-resistant seizures lasting up to 2 hours. T2-weighted imaging (B) and diffusion imaging (C) were performed 6, 12 and 25 days after initial presentation. The apparent diffusion coefficient (ADC) maps obtained at 6 days after initial onset of the seizures showed evidence of ongoing restricted diffusion throughout a substantial part of the abnormal left hemisphere (C). Representative regions showed an ipsi/contralateral ADC ratio of 0.79 (shown graphically in G, compared with values of the order of 0.5 in the first 48 hours in a child with a stroke and 1.0 in controls, shown graphically in F). The ADC remained low in the affected areas at 12 and 25 days after presentation (shown graphically in G, ipsi/contralateral ADC ratio 0.81 and 0.84 respectively). Throughout this period, the T2-weighted images showed hypointensity in corresponding regions. The patient developed a right hemiparesis and chronic epilepsy. A sleep study showed mean and minimum oxyhaemoglobin saturation (SpO2) of 95.8% and 91% respectively (97% is the 5th percentile in normal children; Urschitz et al., 2003). A follow-up scan was performed at the age of 14 months (D, E); the ADC remained low with an ipsi/contralateral ADC ratio of 0.79 (shown graphically in G). At the age of 15 months, her development was delayed to the 10-month level. Persistently low ADC, suggesting ongoing ischaemia, was again demonstrated on MR scans at 16 (G) and 30 months. The patient underwent a hemispherectomy for intractable epilepsy at the age of 33 months and has remained seizure-free post-operatively. It is possible that chronic exposure to mild overnight hypoxaemia had a deleterious effect on brain tissue compromised by chronic venous hypertension and ischaemia.
Hypoxia appears to exacerbate seizures in 10-day-old, but not older or younger rats (Jensen, Holmes, Lombroso, Blume & Firkusny, 1992), suggesting an age-dependent effect. There are chronic abnormalities of cerebral perfusion and metabolism detectable with diffusion (Figure 2) and perfusion MRI or positron emission tomography (PET) (Ferrie, Maisey, Cox, Polkey, Barrington, Panayiotopoulos & Robinson, 1996), which might interact with chronic hypoxaemia in the maintenance of epileptogenicity. Vasculopathy may play a role in some cases (Pascual-Castroviejo, Lopez Martin, Martinez Bermejo & Perez Higueras, 1994). Seizures are usually accompanied by an increase in CBF to meet increased metabolic demand (Brodersen, Paulson, Bolwig, Rogon, Rafaelsen & Lassen, 1973) but not in patients with Sturge-Weber syndrome, rendering them vulnerable to ictal ischaemia (Aylett, Neville, Cross, Boyd, Chong & Kirkham, 1999). Sleep disordered breathing (SDB) and intermittent or sustained oxyhaemoglobin desaturation might contribute to intractability in epilepsy. Abnormal polysomnography has been documented (Malow, Levy, Maturen & Bowes, 2000; Koh, Ward, Lin & Chen, 2000), and appropriate treatment, e.g. adenotonsillectomy or continuous positive airway pressure (CPAP), has been associated with reduction in seizure frequency (Cohen, Lefaivre, Burstein, Simms, Kattos, Scott, Montgomery & Graham, 1997; Holland & Yan, 1997).
We prospectively recorded overnight SpO2 in 18 children with intractable epilepsy, six of whom were currently or recently in MS. An abnormal study was defined as either: (a) mean baseline SpO2 <94%; or, (b) >4 dips of >4% in SpO2/hour. Children with MS were more likely to have an abnormal sleep study (p = .04) and had more dips/hour (p = .04). There was a trend for minimum SpO2 to be lower in those with MS (p = .06) but there was no difference in mean SpO2 between those with and without MS (p = .6). Although it is possible that abnormal sleep studies were directly related to frequent seizures in children with MS, there may be a self-feeding loop such that SDB and nocturnal hypoxaemia in turn contribute to intractability and to cognitive deterioration in epileptic children. The possibility that such a vicious cycle contributes to progressive brain damage could be explored in populations such as those with Sturge-Weber syndrome (Figure 2). Prospective research is needed to understand mechanisms and test therapies.
Exposure and adaptation to hypoxia in a common haemoglobinopathy of childhood: sickle cell disease
Sickle oxyhaemoglobin has a reduced affinity, i.e. the partial pressure of oxygen (PaO2) at which haemoglobin is 50% saturated (P50) is increased, although there is considerable variation between patients (Rackoff, Kunkel, Silber, Asakura & Ohene-Frempong, 1993). Thus arterial blood has lower oxygen saturation for any given arterial PaO2; this has advantages and disadvantages, which may depend on degree of hypoxic exposure (Samaja, Crespi, Guazzi & Vandegriff, 2003). Unloading of oxygen from blood to tissues is facilitated (lessening the drive for chronic adaptative gene upregulation), but oxygen loading at the lungs is reduced. As foetal haemoglobin levels fall during childhood, oxyhaemoglobin affinity must drop, although there are few data on time course. This may influence the hypoxic response and alter cerebral oxygen extraction, as has been suggested for altitude sickness in the general population (Curran-Everett, 2003).
Daytime SpO2 measured using pulse oximetry is low in many SCD patients, in association with degree of anaemia and increasing age (Rackoff et al., 1993), although because of reduced oxygen affinity, arterial hypoxaemia is rarely demonstrated. Overnight oxyhaemoglobin desaturation (Kirkham, Hewes, Prengler, Wade, Lane & Evans, 2001a; Setty et al., 2003) and SDB, usually accompanied by snoring, are common (Samuels, Stebbens, Davies, Picton-Jones & Southall, 1992). Acute chest syndrome (ACS), defined as acute respiratory symptoms accompanied by new lung abnormalities on chest X-ray (Vichinsky, Neumayr, Earles, Williams, Lennette, Dean, Nicherson, Orringer, McKie, Bellevue, Daeschner & Manci, 2000) and acute exposure to altitude (Green, Huntsman & Serjeant, 1971) may be associated with severe acute hypoxaemia. Both low (Fowler, Smith & Greenfield, 1957) and high blood carbon dioxide (Maddern, Reed, Ohene-Frempong & Beckerman, 1989) have been documented, the former probably secondary to an increased hypoxic hyperventilatory response and the latter suggestive of nocturnal hypoventilation, potentially protective as cerebral blood flow (CBF) and oxygen delivery may increase.
Hypoxia-reoxygenation in SCD is characterized by inflammation with upregulation of xanthine oxidase, superoxide generation, oxidative stress and increased monocyte tissue factor (Kaul & Hebbel, 2000) (Figure 1). Although endothelial NO synthase (eNOS) is upregulated, NO is scavenged by superoxide as well as free haemoglobin, released by chronic haemolysis (Reiter, Wang, Tanus-Santos, Hogg, Cannon, Schechter & Gladwin, 2002); bioavailablity is less during acute complications, including chest crisis. Potentially protective interactions, e.g. heat shock protein 90 with eNOS, may be limited, in association with oxidative stress. Circulating endothelin-1, a peptide with vasoconstrictor and bronchoconstrictor effects, is inversely correlated with daytime SpO2 in SCD (Werdehoff, Moore, Hoff, Fillingim & Hackman, 1998); vascular tone may be delicately balanced depending on NO availability (Pluta, Dejam, Grimes, Gladwin & Oldfield, 2005) and endothelin levels (Figure 1). Rapid polymerization of HbS and relative deficiency of SNO (Pawloski, Hess & Stamler, 2005) during acute hypoxia also appears to interfere with red cell oxygen sensing and hypoxic vasodilatation. Hypoxia also increases adhesion of sickle red cells to endothelial wall via mechanisms that include vascular cell adhesion molecule (VCAM-1) and P-selectin (Setty et al., 2003); levels are increased when NO bioavailability is reduced. The challenge is to translate these scientific advances to tackle problems facing clinicians caring for SCD patients with acute or chronic intermittent or sustained hypoxia.
Neurologically asymptomatic SCD patients usually have globally high CBF secondary to anaemia (Herold, Brozovic, Gibbs, Lammertsma, Leenders, Carr, Fleming & Jones, 1986; Prohovnik, Pavlakis, Piomelli, Bello, Mohr, Hilal & De Vivo, 1989), which means that the capacity to respond to other vasodilatory stimuli, e.g. blood carbon dioxide, may be reduced. However, in symptomatic patients diffusely decreased cerebral perfusion is typical (Huttenlocher, Moohr, Johns & Brown, 1984). Studies of regional CBF, using xenon-CT or PET have shown more extensive regional perfusion abnormalities than those shown on anatomical CT or MRI (Numaguchi, Haller, Humbert, Robinson, Lindstrom, Gruenauer & Carey, 1990; Powars, Conti, Wong, Groncy, Hyman, Smith, Ewing, Keenan, Zee, Harold, Hiti, Teng & Chan, 1999). Symptomatic patients with vessel occlusion have reduced CBF distally in the acute phase (Huttenlocher et al., 1984) and chronically (Powars et al., 1999; Kirkham, Calamante, Bynevelt, Gadian, Cox, Evans & Connelly, 2001b; Prengler, Pavlakis, Boyd, Connelly, Calamante, Chong, Saunders, Cox, Bynevelt, Lane, Laverty & Kirkham, 2005). Some patients with normal MRA have CBF reduction focally, particularly posteriorly (Kirkham et al., 2001b).
CBF in SCD appears to depend on bioavailability of endothelial NO (French, Kenny, Scott, Hoffmann, Wood, Hudetz & Hillery, 1997), which may be close to a critically low threshold because it is scavenged (Morris, Kato, Poljakovic, Wang, Blackwelder, Sachdev, Hazen, Vichinsky, Morris & Gladwin, 2005). Dietary supplementation of arginine, an amino acid precursor of NO, may improve endogenous NO bioavailability and performance of motor coordination tasks such as the rotorod in animals (Fasipe, Ubawike, Eva & Fabry, 2004) as well as improving cerebral perfusion and reducing red cell density and mean cell haemoglobin concentration (Romero, Suzuka, Nagel & Fabry, 2002; Kennan, Suzuka, Nagel & Fabry, 2003; Fabry, Etzion, Bookchin, Suzuka & Nagel, 2004).
There are differences in cerebral haemodynamics in response to vasodilatory stimuli in SCD: acetazolamide increases CBF in normal adults but the response was reduced in two-thirds of SCD patients (Kedar, Drane, Shaeffer, Nicole & Adams, 2006). Although there are few formal data on carbon dioxide reactivity, hyper-ventilation may reduce CBF and precipitate posterior circulation infarction (Protass, 1973; Allen, Imbus, Powars & Haywood, 1976; Arnow, Panwalker, Garvin & Rodriguez-Erdmann, 1978). Cerebral oxygen saturation is reduced in patients with SCD compared with controls and falls further during sleep (Nahavandi, Tavakkoli, Hasan, Wyche & Castro, 2004; Raj, Bertolone, Mangold & Edmonds, 2004; Raj, O’Brien, Edmonds, Bertolone & Gozal, 2005). Whereas high oxygen tension reduces CBF in control animals, baseline CBF is decreased in transgenic sickle mice compared with controls and increases with hyperoxia in mice (Kennan, Suzuka, Nagel & Fabry, 2004) and patients (Kennan, Suzuka, Nagel & Fabry, 2002) with SCD. CBF velocity and cerebral oxygenation increase rapidly after blood transfusion (Venketasubramanian, Prohovnik, Hurlet, Mohr & Piomelli, 1994; Nahavandi et al., 2004; Raj et al., 2004). In one patient transfused after stroke, CBF increased immediately after Nifedipine (Ashwal, Bedros & Thompson, 1994) while both increases and decreases have been documented in humans and animals without SCD, perhaps dependent on the resting blood pressure (Grabowski & Johansson, 1985). Blood oxygen level-dependent (BOLD) MRI or transcranial Doppler (TCD) techniques could be employed to explore the relationship between hypoxia, hypercapnia and CBF regulation and to test novel therapies in humans in parallel with carefully designed neuropsychological paradigms and neurological examination (Kennan et al., 2004; Rostrup, Larsson, Born, Knudsen & Paulson, 2005; Hill et al., 2006).
Erythropoetin levels are raised in patients with SCD (Figure 1), but relatively less than in other patients with alternative causes for a comparable anaemia (Sherwood, Goldwasser, Chilcote, Carmichael & Nagel, 1986), probably related to oxyhaemoglobin affinity. VEGF and PGF are raised in SCD (Solovey, Gui, Ramakrishnan, Steinberg & Hebbel, 1999; Gurkan, Tanriverdi & Baslamisli, 2004; Perelman, Selvaraj, Batra, Luck, Erdreich-Epstein, Coates, Kalra & Malik, 2003). PGF levels are higher in those with frequent pain, associated with monocyte activation (Perelman et al., 2003), which is inversely related to overnight hypoxia (Inwald, Kirkham, Peters, Lane, Wade, Evans & Klein, 2000). Haemoxygenase is upregulated and may be protective via antioxidant effects of bilirubin and vasodilatory effects of carbon monoxide (Jison, Munson, Barb, Suffredini, Talwar, Logun, Raghavachari, Beigel, Shelhamer, Danner & Gladwin, 2004), demonstrable by increased plasma carboxyhaemoglobin and exhaled carbon monoxide (Cunnington, Kendrick, Wamola, Lowe & Newton, 2004; Sylvester, Patey, Rafferty, Rees, Thein & Greenough, 2005) and reduced P50 (Figure 1). However, haemoxygenase also upregulates VEGF, leading to a potentially deleterious cycle of angiogenesis (Bussolati, Ahmed, Pemberton, Landis, Di Carlo, Haskard & Mason, 2004).
Angiographic evidence of large vessel occlusion in the majority of sickle-associated strokes (Stockman, Nigro, Mishkin & Oski, 1972) has led to non-invasive population screening with TCD to detect CVD (Adams, McKie, Carl, Nichols, Perry, Brock, McKie, Figueroa, Litaker, Weiner & Brambilla, 1997), an approach founded on significant impact from transfusion programmes on stroke incidence and recurrence (Fullerton, Adams, Zhao & Johnston, 2004). Vasculopathy starts young: high TCD velocities occur in infancy (Hogan, Kirkham, Prengler, Telfer, Lane, Vargha-Khadem & de Haan, 2005) and abnormal MRAs have been demonstrated by the second year (Wang, Langston, Steen, Wynn, Mulhern, Wilimas, Kim & Figueroa, 1998).
As well as anaemia-related increased CBF, high CBFV may reflect vasoconstriction, perhaps secondary to imbalance between endothelin production and NO availability, which might protect against development of cerebral oedema on exposure to acute hypoxia at the cost of increasing ischaemic risk in the territory of the affected arteries. There may be an important interaction between exposure to hypoxia, haemolysis, infection and inflammation in determining whether a vessel previously in (adaptive) spasm (Hill et al., 2006) becomes irreversibly stenosed or occluded. Mechanisms related to hypoxic exposure potentially favouring the development of irreversible CVD include unregulated angiogenesis, thrombosis and endothelial adhesion. There is some evidence for this: for example, intermittent hypoxia and infection increases haemolysis, platelet and white cell activation and endothelial adhesion in SCD (Inwald et al., 2000; Setty et al., 2003). If this is important clinically, an association between SpO2, haemolysis, and severity of vasculopathy might be expected.
In our series of prospectively followed patients with SCD, screened at baseline with TCD, overnight pulse oximetry (Table 2) and MRI and MRA if they were >7 years old, who subsequently developed acute neurological symptoms and signs (Kirkham et al., 2001a, Tables 2 and 3), mean overnight SpO2 was lower and mean reticulocytes higher in those with CVD on MRA (Mann-Whitney, p < .01 for both). All seven with an abnormal sleep study had an abnormal MRA (Table 2) while five children in this series who had normal MRA all had a normal sleep study (prospectively defined as mean SpO2 >92% and no dips). None of the latter group presented with hemiparesis or had infarction in an arterial distribution (Table 3) but three had episodes of dizziness with or without paraesthesiae or confusion compatible with posterior transient ischaemic attacks, one of whom subsequently presented with an organic psychosis (Table 3). Another had a venous sinus thrombosis (Sébire, Tabarki, Saunders, Leroy, Liesner, Saint-Martin, Husson, Williams & Kirkham, 2005). The case history of the remaining patient, who had bilateral borderzone infarction, is given in the legend to Figure 3. These presentations are similar to those documented at altitude (Basnyat, Wu & Gertsch, 2004) and are compatible with the effects of acute hypoxia without prior preconditioning exposure or perhaps with acutely low blood carbon dioxide levels and vasoconstriction. The majority of patients had homozygous sickle cell anaemia (Table 2) but there was no obvious threshold of haemoglobin or haemoglobin F (Powars, Weiss, Chan & Schroeder, 1984) predictive of neurological manifestations and only two patients had ICA/MCA velocities >200 cm/sec (Adams et al., 1997) at any stage. Thirteen patients had recurrent neurological events, which occurred in those who had had a prodromal illness at the time of the index event and in those who had not (Table 3).
Table 2.
Clinical, Transcranial Doppler (internal carotid (ICA)/middle cerebral artery (MCA), haematology, magnetic resonance angiography (MRA) and sleep study data from 19 patients from the East London cohort who had a central nervous system event during a prospective study (Kirkham et al., 2001)
| Overnight oxyhaemoglobin saturation
|
||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Patient | Sex | Haemoglo binopathy | Age | Cerebro-vascular disease on MRA | Initial ICA/MCA Velocity | Maximum ICA/MCA Velocity during follow-up | Hb | Mean reticulocytes | Ts & As | Mean | Min | % <80% | % <90% | Sleep study result |
| 1 | M | SS | 4.98 | N | 126 | 144 | 7.8 | 11.9 | N | 93.34 | 71 | .96 | 11.6 | Normal |
| 2 | M | SS | 7.04 | N | 100 | 138 | 10.7 | 5.8 | N | 97.89 | 67 | .28 | 2.18 | Normal |
| 3 | M | SS | 6.74 | N | 108 | 139 | 9.1 | 9.5 | Y | 98.23 | 86 | .00 | .35 | Normal |
| 4 | F | SS | 3.46 | N | 140 | 152 | 11.4 | 3.2 | N | 98.64 | 82 | .00 | 1.03 | Normal |
| 5 | M | SC | 6.53 | N | 137 | 137 | 13.3 | 2.7 | N | 95.37 | 85 | .00 | 1.29 | Normal |
| 6 | M | SB | 8.88 | Y | 137 | 137 | 12.6 | 13.5 | N | 95.77 | 85 | .00 | 1.68 | Normal |
| 7 | F | SS | 9.63 | Y | 258 | 258 | 7.3 | 10.6 | N | 92.96 | 81 | .00 | 13.5 | Dips |
| 8 | F | SS | 2.82 | Y | 143 | 144 | 10.9 | 11.5 | N | 97.70 | 90 | .00 | .13 | Normal |
| 9 | M | SS | 5.47 | Y | 116 | 141 | 6.9 | 17.1 | N | 90.21 | 76 | .92 | 42.5 | Normal |
| 10 | F | SS | 9.43 | Y | 228 | 267 | 6.5 | 8.6 | N | 95.22 | 85 | .00 | 1.32 | Dips |
| 11 | F | SS | 9.63 | Y | 134 | 134 | 8.1 | 11.6 | N | 95.68 | 41 | .00 | 4.09 | Normal |
| 12 | M | SS | 4.74 | Y | 167 | 167 | 7.6 | 17.1 | Y | 90.58 | 75 | .00 | 38.1 | Dips |
| 13 | M | SS | 4.69 | Y | 198 | 198 | 8.1 | 12.3 | Y | 87.00 | 76 | .50 | 50.0 | Mean <92% |
| 14 | M | SS | 13.29 | Y | 131 | 131 | 8.1 | 18.4 | N | 87.26 | 77 | 1.05 | 97.5 | Dips |
| 15 | F | SS | 10.23 | Y | 72 | 88 | 6.8 | – | N | 91.93 | 81 | .00 | 6.51 | Mean <92% |
| 16 | F | SS | 10.16 | Y | 109 | 124 | 11.1 | 16.9 | Y | 94.70 | 75 | .29 | 3.37 | Normal |
| 17 | F | SS | 2.74 | Y | 125 | 125 | 8.5 | 8.5 | N | 97.05 | 84 | .00 | 2.60 | Normal |
| 18 | M | SS | 11.01 | Y | 98 | 98 | 6.5 | 6.2 | Y | 85.00 | 75 | Dips | ||
| 19 | M | SS | 4.33 | Y | 134 | 134 | 10.2 | 16.3 | N | 92.41 | 89 | .00 | .00 | Normal |
MRA Magnetic resonance angiography, ICA internal carotid artery, MCA middle cerebral artery, Hb Haemoglobin, Dips desaturations, Ts & As Adenotonsillectomy.
Table 3.
Clinical and neuroimaging data from the index and any recurrent central nervous system event in 19 patients from the East London cohort who had a central nervous system event during a prospective study (Table 2, Kirkham et al., 2001a)
| First CNS event after screening (TCD and sleep study)
|
Second CNS event
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Patient | Age | Context | Clinical | Type of event | Age | Context | Clinical | Neuroimaging |
| 1 | 8 | Facial infection | R partial seizure prolonged, coma | Stroke | – | Bilateral borderzone infarcts (Figure 3) | ||
| 2 | 9 | Pain | Headache, dizziness, confusion | Stroke | 12 | Transient parathesia | Normal | |
| 3 | 11 | Pain | Headache, diplopla, dizziness, VIth nerve palsy | TIA | 12 | Pain | Organic psychosis with hallucinations | Normal |
| 4 | 9 | Pain | Dizzy | TIA | . | Normal | ||
| 5 | 0.1 | Twitching right arm, blood stained CSF, ventricular haemorhage, hydrocephalus, ventriculoperitoneal shunt, seizures | TIA | 9 | Collapse, drooling, unable to see, unresponsive, seizures, incontinent, brain death | Venous sinus thrombosis, cerebral oedema (Sébire et al. 2005) | ||
| 6 | 9 | Severe OSA | Left hemiparesis | Stroke | 11 | R facial palsy | Atrophy left parietooccipital | |
| 7 | 10 | Pain | Dizzy, headache, mild diplegia | Stroke | 11 | R hemiparesis | Increase in white matter abnormality | |
| 8 | 2 | URTI | Floppy, unable to hold head, aphasic few hours, looked vacant, staggering to left | Stroke | 7 | R hemiparesis | New infarct middle cerebral artery territory | |
| 9 | 12 | Pain | Severe headache, vomiting, neck stiffness, hypertension | Stroke | . | Haemorrhagic infarct | ||
| 10 | 16 | Aplastic | Dizzy, left paraesthesiae, left weakness, collapsed, shaking upper limbs, fluctuating conscious level | Stroke | . | New infarct basal ganglia (Figure 4) | ||
| 11 | 15 | Pain | Diplopia; divergent squint | TIA | . | ‘Covert’ infarct, no change | ||
| 12 | 3 | Headaches, right sided weakness | TIA | 6 | Drags right foot | Increase in white matter abnormality | ||
| 13 | 1.5 | Not using right arm | TIA | 6 | R weakness, severe behaviour problems | ‘Covert’ infarct, no change | ||
| 14 | 15 | Intermittent difficulty writing right hand | TIA | . | ‘Covert’ infarct, no change | |||
| 15 | 10 | Squint | TIA | 10 | Intermittent blurred visison, 2*episodes of falling, unsteady afterwards, headache | Normal | ||
| 16 | 10 | Headache, blurring vision L eye | Seizure | 13 | Dizzy, not moving arm, headache, EEG-bursts slow both temporal R>L, discharges R posterior temporal | Normal | ||
| 17 | 1.5 | Diarrhoea, dehydration, femoral thrombosis | Extensor posturing, GCS 4 | Seizure | 3 | Scratches table, wall or just blank, sleeps after | New ‘covert’ infarct | |
| 18 | 13 | Pain | Headache, Unconscious, twitching arms, eyes deviated L, brief absences, visual disturbance | Seizure | 13 | Seizure lasting 7 minutes | New ‘covert’ infarct | |
| 19 | 4 | Pain | Headache, uprolling eyes lasted few secs, flickering eyelids, afebrile. Talking to nurse-bizarre facial movements, uprolling eyes, twitching fingers, EEG normal | Seizure | 4 | Episodes shaking down right side*4 at night-wakes him | New ‘covert’ infarct | |
OSA, Obstructive sleep apnoea; TIA, Transient ischaemic attack; EEG, Electroencephalogram; R, Right; L, Left; GCS, Glasgow Coma Score.
Figure 3.
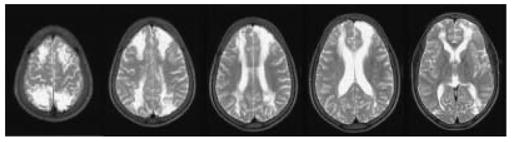
This 8-year-old boy (Patient 1, Tables 2 and 3), one of twins, had uncomplicated sickle cell disease (SCD) but at the age of 6 years, had overnight pulse oximetry classified prospectively as normal (Kirkham et al., 2001) although the mean was less than the 5th percentile for age (Urschitz et al., 2003). His twin developed conditionally high internal carotid/middle cerebral artery velocities on transcranial Doppler (TCD) (maximum 192 cm/sec at the age of 8) but has not had a neurological event over follow-up of 13 years. This patient’s TCD remained normal (maximum 144 cm/sec at the age of 8) but 2 months after this recording, he developed seizures and coma after surgery to drain a painful swelling of his left cheek, associated with fever, after a fall. Preoperative preparation had included hydration and blood transfusion to achieve a haemoglobin of 11.9 g/dl and haemoglobin S of 36% and blood pressure was above 110/50 mmHg (mean arterial blood pressure, MAP 70 mmHg) pre- and post-operatively even after the onset of coma. Intracranial pressure (ICP) was not measured. Initial T2-weighted MRI showed acute infarction with increased signal and swelling in both anterior arterial borderzones and the right posterior arterial borderzone, with mature infarction at follow-up as shown in the figure. TCD, Magnetic Resonance, including arteriography and venography, and four vessel cerebral angiography were normal. Motor function recovered completely. Psychometry showed significant cognitive impairment; compared to premorbid testing full scale IQ was reduced by 30 and performance IQ by 50 points (WISCIII). Covert infarction in the borderzones between the anterior and middle and posterior and middle cerebral arteries are common in SCD, probably because these areas are vulnerable to acute reductions in cerebral blood flow secondary to reduced cerebral perfusion pressure (CPP = MAP − ICP); this patient appears to have had an acute reduction in CPP despite maintenance of blood pressure, perhaps in relation to the cerebral oedema and undiagnosed raised ICP in a child with inadequate preconditioning for the insult experienced and normal cerebral vessels.
ACS is associated with acute hypoxia and relative anaemia (haemoglobin falling at least 1 g/dl from baseline; Henderson, Noetzel, McKinstry, White, Armstrong & DeBaun, 2003). In one series, 3% of ACS patients had neurological symptoms at presentation and 7–10% as a complication (Figure 5; Vichinsky et al., 2000). Brain imaging findings include posterior leukoencephalopathy, bilateral focal cortical oedema, haemorrhage and acute demyelination (Henderson et al., 2003; Lee, McKie, Sekul, Adams & Nichols, 2002). Asthma may be a risk factor for both (Nordness, Lynn, Zacharisen, Scott & Kelly, 2005). Reversibility of the majority of imaging abnormalities and white matter involvement is reminiscent of high altitude cerebral oedema (HACE), venous sinus thrombosis and other neurological syndromes seen in unacclimatized adults who climb mountains quickly and are therefore exposed to acute hypoxia without adequate preconditioning (Kobayashi, Koyama, Kubo, Fukushima & Kusama, 1987; Hackett, Yarnell, Hill, Reynard, Heit & McCormick, 1998; Saito & Tanaka, 2003; Basnyat et al., 2004). Lesion distribution may be different although there are very few imaging data in children with HACE for comparison. Age, rapidity and severity of hypoxia, nature and degree of any adaptive preconditioning or associated pre-existing CVD might influence outcome (Figure 1). The preconditioning effects of chronic hypoxia might lead to relatively small areas of infarction after acute hypoxic exposure compared with the degree of vasculopathy (Figure 4). Oedema might be expected in territories distal to normal (rather than stenosed) vessels in patients who are exposed to acute hypoxia from a normal baseline, i.e. without preconditioning (Figures 3 and 5).
Figure 5.
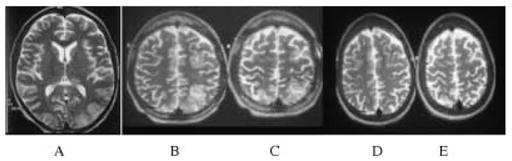
This young woman with haemoglobin SS was born in Africa and immigrated to the United Kingdom aged 13 yrs where she remained well, with no painful or chest crises, until age 18 when she presented acutely with pain and dyspnoea. Blood pressure was 120/70 mmHg and oxyhaemoglobin saturation was 81% on admission. Haemoglobin was 3.8 with a white cell count of 66. Chest X-ray showed bilateral pulmonary infiltrates, reported as compatible with pulmonary oedema. Echocardiography showed moderate left atrial enlargement with left ventricular size at upper limit of normal; systolic function was good and tricuspid regurgitation trivial. She was confused with a Glasgow coma score (GCS) of 13/15 and appeared to have seizures with lip smacking and repetitive jerking of the right leg. When she regained full consciousness (GCS 15/15), she had a right hemianopia. Tone and power were normal in all limbs but reflexes were absent. TCD and MRA were normal. Axial T2 weighted MRI in the acute phase showed high signal changes in the left posterior occipital and bilateral high parietal regions (A, B, C) with hippocampal involvement (A). Repeat MRI 4 weeks later (D, E) showed resolution of all changes but the patient continued to have seizures with visual aura, staring, twitching of right hand, a sharp sensation on the right, hallucinations and memory loss. This is an example of reversible posterior leukencephalopathy (a poor term as there is obviously grey matter involvement and other regions are often affected) secondary to very acute and severe hypoxia in a patient with inadequate preconditioning and normal cerebral vessels. The involvement of the hippocampus in the acute process links this hypoxic insult to the subsequent intractable epilepsy.
Figure 4.
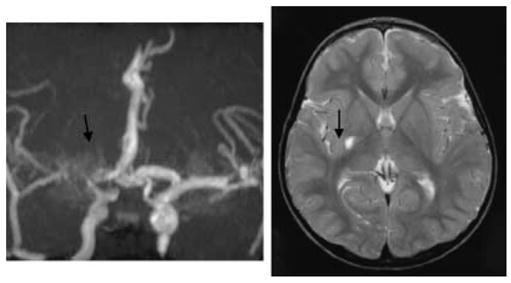
Magnetic resonance angiogram (MRA) and imaging (MRI) from a girl (Patient 10, Tables 2 and 3) with sickle cell anaemia who had a sleep study showing dips and high transcranial velocities (up to 267 cm/second) for 8 years without symptoms but presented with a hemiparesis in the context of acute aplastic anaemia secondary to Parvovirus. Although the MRA shows severe turbulence with some ‘moyamoya’ collaterals (arrow), the MRI shows only a small infarct (arrow) and the patient made a complete recovery. This might be an example of the preconditioning effect of prior exposure to hypoxia in limiting the extent of tissue injury at the time of acute stroke but at the cost of irreversible vascular change.
Although few patients have chronic epilepsy, partial or generalized seizures affect 13% of SCD patients, herald stroke in 10–33% (Liu, Gzesh & Ballas, 1994) and are a risk factor for silent infarction (Kinney, Sleeper, Wang, Zimmerman, Pegelow, Ohene-Frempong, Wethers, Bello, Vichinsky, Moser, Gallagher, DeBaun, Platt & Miller, 1999). Abnormalities of TCD and MR perfusion are commoner in patients with active seizures (Prengler et al., 2005). Rapid change in tissue oxygenation might lead to changes in ion channel function leading to isolated seizures, which might not recur once hypoxic adaptation has occurred, or occasionally to structural damage to the hippocampus and chronic epilepsy (Figure 5). Headache, common in SCD (Palermo, Platt-Houston, Kiska & Berman, 2005) is also seen at altitude (Jaillard, Mazetti & Kala, 1997), apparently in association with oxyhaemoglobin desaturation despite relative polycythaemia (Arregui, Leon-Velarde, Cabrera, Paredes, Vizcarra & Umeres, 1994). Pseudotumour cerebri has been reported in SCD (Henry, Driscoll, Miller, Chang & Minniti, 2004) as well as OSA in adults (Wolin & Brannon, 1995).
Parenchymal and cerebrovascular changes in asymptomatic patients
In addition to infarction visible on CT or MRI (Adams, Nichols, McKie, McKie, Milner & Gammal, 1988; Pavlakis, Bello, Prohovnik, Sutton, Ince, Mohr, Piomelli, Hilal & De Vivo, 1988), there is evidence for subtle abnormality on quantitative T1-weighted MRI, particularly thalamic, in young SCD children (Steen, Langston, Reddick, Ogg, Chen & Wang, 1996). Both increased tortuosity (ectasia) and quantitative T1-weighted MRI changes are related to haematocrit (Steen, Xiong, Mulhern, Langston & Wang, 1999; Steen, Reddick, Glass & Wang, 1998), suggesting that chronic anaemic hypoxia might be an important drive to vascular adaptation with failure to compensate leading to brain pathology.
Possible mechanisms for covert infarction include residua of acute hypoxic posterior leukencephalopathy, cerebral oedema and basal ganglia infarction (Henderson et al., 2003; Usui, Inoue, Kimura, Kirino, Nagaoka, Abe, Nagata & Arai, 2004; Jeong, Kwon, Chin, Yoon & Na, 2002) and venous sinus thrombosis (Sébire et al., 2005) as well as transient ischaemic attack secondary to arterial disease. Patent foramen ovale (PFO) is a well-recognized cause of sustained and intermittent hypoxia common in patients with right ventricular dysfunction, obstructive airways disease and OSA (Shnaider, Shiran & Lorber, 2004; Soliman, Shanoudy, Liu, Russell & Jarmukli, 1999; Shanoudy, Soliman, Raggi, Liu, Russell & Jarmukli, 1998), and is a potentially treatable cause of stroke and migraine in young adults (Finsterer, Sommer, Stiskal, Stollberger & Baumgartner, 2005). However, the possibility that PFO is a risk factor for overt or covert infarction in SCD has received little attention (Dowling, 2005). White matter changes (Baldeweg et al., 2006; Schatz & Buzan, 2006) might also reflect acute reduction in CBF secondary to low blood carbon dioxide levels (Murase & Ishida, 2005) or even local carbon monoxide toxicity (Durak, Coskun, Yikilmaz, Erdogan, Mavili & Guven, 2005) secondary to upregulation of haemoxygenase (Figure 1). Other molecular mechanisms linking chronic sustained and intermittent hypoxia to the neurological manifestations of SCD include the adverse effects of reactive oxygen species generated by repetitive intermittent hypoxia on proteins, nucleic acids and lipids, downregulation of the mitochondrial respiratory chain enzymes and upregulation of amyloid β peptide as well as the effect of sleep disruption (Figure 1).
Implications for future management: strategies to prevent hypoxia-related morbidity
Management strategies to prevent adverse effects of hypoxia on neurocognitive function might include oxygen supplementation for those who are chronically hypoxic, CPAP or surgical approaches, e.g. adenotonsillectomy for OSA. Nutritional supplements, vitamins, e.g. C and E, trace elements, e.g. zinc, and drugs, e.g. aspirin, which are anti-inflammatory, antioxidant and/or increase oxyhaemoglobin affinity might also ameliorate conditions associated with chronic hypoxia, particularly if generation of reactive oxygen species has overwhelmed capacity for compensatory scavenging. To ensure that interventions are appropriate and risk-free, it is important to determine the child’s mechanism of hypoxaemic adaptation, using measurement of overnight oximetry, red cell indices, oxyhaemoglobin affinity, carboxyhaemoglobin, blood pressure, peripheral constriction, respiratory function, exhaled NO and hypoxic ventilatory response (Figure 1). Controlled trials of strategies to prevent morbidity and neurocognitive deficits associated with hypoxia are justified, but should be based on detailed understanding of pathophysiology.
Table 1.
Key terms defined for the non-specialist
| Apoptotic cell death – cell death programmed to occur over hours or days usually by chemical signals from its neighbours |
| Baroreceptor responses – response to input sensing pressure, usually in blood vessels such as the carotid sinus |
| Chemosensitivity – sensing of changes in respiratory gases (carbon dioxide and oxygen) in the brain stem and carotid bodies |
| Erythropoiesis – red blood cell production |
| Haemolysis – lysis of red blood cells with release of haemoglobin |
| Hypoxaemia – low blood oxygen |
| Hypoxia-Inducible Factor (HIF) – protein which is destroyed continuously when there is adequate oxygen but is stabilized during hypoxia and upregulates: |
| Erythropoietin – hormone which increases red cell production |
| Vascular endothelial growth factor (VEGF) – induces vessel growth (angiogenesis) |
| Endothelin – powerful constrictor of blood vessels |
| Nitric Oxide – powerful dilator of blood vessels |
| S-nitrosothiols (SNO) – molecules formed after the reaction of nitric oxide with a critical cysteine residue on proteins |
| Tidal volume – volume of air inhaled and exhaled at each breath |
Acknowledgments
This work was funded by Action Research, the Wellcome Trust, the National Heart, Blood and Lung Institute of the National Institutes of Health (5-R01-HL079937) and the Stroke Association (PROG4). The work was undertaken at NHS Trusts which receive a proportion of their funding from the NHS Executive.
References
- Adams RJ, McKie VC, Carl EM, Nichols FT, Perry R, Brock K, McKie K, Figueroa R, Litaker M, Weiner S, Brambilla D. Long-term stroke risk in children with sickle cell disease screened with transcranial Doppler. Annals of Neurology. 1997;42:699–704. doi: 10.1002/ana.410420505. [DOI] [PubMed] [Google Scholar]
- Adams RJ, Nichols FT, McKie V, McKie K, Milner P, Gammal TE. Cerebral infarction in sickle cell anemia: mechanism based on CT and MRI. Neurology. 1988;38:1012–1017. doi: 10.1212/wnl.38.7.1012. [DOI] [PubMed] [Google Scholar]
- Allen JP, Imbus CE, Powars DR, Haywood LJ. Neurological impairment induced by hyperventilation in children with sickle cell anemia. Pediatrics. 1976;58:124–126. [PubMed] [Google Scholar]
- Annobil SH, Omojola MF, Adzaku FK, Addae SK, Mohammed S. Cerebrovascular accidents (strokes) in children with sickle cell disease residing at high and low altitudes of Saudi Arabia. Annals of Tropical Paediatrics. 1990;10:191–198. doi: 10.1080/02724936.1990.11747429. [DOI] [PubMed] [Google Scholar]
- Arnow PM, Panwalker A, Garvin JS, Rodriguez-Erdmann F. Aspirin, hyperventilation, and cerebellar infarction in sickle cell disease. Archives of Internal Medicine. 1978;138:148–149. [PubMed] [Google Scholar]
- Arregui A, Leon-Velarde F, Cabrera J, Paredes S, Vizcarra D, Umeres H. Migraine, polycythemia and chronic mountain sickness. Cephalalgia. 1994;14:339–341. doi: 10.1046/j.1468-2982.1994.1405339.x. [DOI] [PubMed] [Google Scholar]
- Ashwal S, Bedros A, Thompson J. Nifedipine increases cerebral blood flow in sickle cell disease: a case study. Journal of Child Neurology. 1994;9:337–338. doi: 10.1177/088307389400900326. [DOI] [PubMed] [Google Scholar]
- Aylett SE, Neville BG, Cross JH, Boyd S, Chong WK, Kirkham FJ. Sturge-Weber syndrome: cerebral haemodynamics during seizure activity. Developmental Medicine and Child Neurology. 1999;41:480–485. [PubMed] [Google Scholar]
- Baldeweg T, Hogan AM, Saunders DE, Telfer P, Gadian DG, Vargha-Khadem F, Kirkham FJ. Detecting white matter injury in sickle cell disease using voxel-based morphometry. Annals of Neurology. 2006;59:662–672. doi: 10.1002/ana.20790. [DOI] [PubMed] [Google Scholar]
- Barcroft J. Presidential address on anoxaemia (abridged) Lancet. 1920;199:485. [Google Scholar]
- Basnyat B, Wu T, Gertsch JH. Neurological conditions at altitude that fall outside the usual definition of altitude sickness. High Altitude Medicine and Biology. 2004;5:171–179. doi: 10.1089/1527029041352126. [DOI] [PubMed] [Google Scholar]
- Beall CM. Tibetan and Andean patterns of adaptation to high-altitude hypoxia. Human Biology. 2000a;72:201–228. [PubMed] [Google Scholar]
- Beall CM. Oxygen saturation increases during childhood and decreases during adulthood among high altitude native Tibetians residing at 3800–4200 m. High Altitude Medicine and Biology. 2000b;1:25–32. doi: 10.1089/152702900320658. [DOI] [PubMed] [Google Scholar]
- Beall CM. High-altitude adaptations. Lancet. 2003;362(Suppl):14–15. doi: 10.1016/s0140-6736(03)15058-1. [DOI] [PubMed] [Google Scholar]
- Beall CM, Decker MJ, Brittenham GM, Kushner I, Gebremedhin A, Strohl KP. An Ethiopian pattern of human adaptation to high-altitude hypoxia. Proceedings of the National Academy of Science, USA. 2002;99:17215–17218. doi: 10.1073/pnas.252649199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beall CM, Laskowski D, Strohl KP, Soria R, Villena M, Vargas E, Alarcon AM, Gonzales C, Erzurum SC. Pulmonary nitric oxide in mountain dwellers. Nature. 2001;414:411–412. doi: 10.1038/35106641. [DOI] [PubMed] [Google Scholar]
- Beran RG, Holland GJ, Yan KY. The use of CPAP in patients with refractory epilepsy. Seizure. 1997;6:323–325. doi: 10.1016/s1059-1311(97)80081-7. [DOI] [PubMed] [Google Scholar]
- Bertuglia S, Giusti A. Role of nitric oxide in capillary perfusion and oxygen delivery regulation during systemic hypoxia. American Journal of Physiology and Heart Circulation Physiology. 2005;288:H525–H531. doi: 10.1152/ajpheart.00426.2004. [DOI] [PubMed] [Google Scholar]
- Brett EM. Minor epileptic status. Journal of Neurological Science. 1966;3:52–75. doi: 10.1016/0022-510x(66)90039-6. [DOI] [PubMed] [Google Scholar]
- Brodersen P, Paulson OB, Bolwig TG, Rogon ZE, Rafaelsen OJ, Lassen NA. Cerebral hyperemia in electrically induced epileptic seizures. Archives of Neurology. 1973;28:334–338. doi: 10.1001/archneur.1973.00490230070010. [DOI] [PubMed] [Google Scholar]
- Bussolati B, Ahmed A, Pemberton H, Landis RC, Di Carlo F, Haskard DO, Mason JC. Bifunctional role for VEGF-induced heme oxygenase-1 in vivo: induction of angiogenesis and inhibition of leukocytic infiltration. Blood. 2004;103:761–766. doi: 10.1182/blood-2003-06-1974. [DOI] [PubMed] [Google Scholar]
- Calbet JA. Chronic hypoxia increases blood pressure and noradrenaline spillover in healthy humans. Journal of Physiology. 2003;551:379–386. doi: 10.1113/jphysiol.2003.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YS, Mu D, Wendland M, Sheldon RA, Vexler ZS, Maquillen PS, Ferriero DM. Erythropoietin improves functional and histological outcome in neonatal stroke. Pediatric Research. 2005;58:106–111. doi: 10.1203/01.PDR.0000163616.89767.69. [DOI] [PubMed] [Google Scholar]
- Cohen SR, Lefaivre JF, Burstein FD, Simms C, Kattos AV, Scott PH, Montgomery GL, Graham L. Surgical treatment of obstructive sleep apnea in neurologically compromised patients. Plastic and Reconstructive Surgery. 1997;99:638–646. doi: 10.1097/00006534-199703000-00005. [DOI] [PubMed] [Google Scholar]
- Crawford JH, Isbell TS, Huang Z, Shiva S, Chacko BK, Schechter AN, Darley-Usmar VM, Kerby JD, Lang JD, Jr, Kraus D, Ho C, Gladwin MT, Patel RP. Hypoxia, red blood cells and nitrite regulate NO-dependent hypoxic vasodilation. Blood. 2005;107:566–574. doi: 10.1182/blood-2005-07-2668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisostomo I, Zayyad A, Carley DW, Abubaker J, Onal E, Stepanski EJ, Lopata M, Basner RC. Chemo- and baroresponses differ in African-Americans and Caucasians in sleep. Journal of Applied Physiology. 1998;85:1413–1420. doi: 10.1152/jappl.1998.85.4.1413. [DOI] [PubMed] [Google Scholar]
- Croll SD, Goodman JH, Scharfman HE. Vascular endothelial growth factor (VEGF) in seizures: a double-edged sword. Advances in Experimental and Medical Biology. 2004;548:57–68. doi: 10.1007/978-1-4757-6376-8_4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunnington AJ, Kendrick SF, Wamola B, Lowe B, Newton CR. Carboxyhemoglobin levels in Kenyan children with Plasmodium falciparum malaria. American Journal of Tropical Medicine and Hygiene. 2004;71:43–47. [PubMed] [Google Scholar]
- Curran-Everett D. Oxygen-hemoglobin affinity at sea level may predict acute illness at altitude: theory and simulation. Medical Hypotheses. 2003;60:767–774. doi: 10.1016/s0306-9877(03)00065-3. [DOI] [PubMed] [Google Scholar]
- Datta A, Tipton M. Respiratory responses to cold water immersion: neural pathways, interactions and clinical consequences. Journal of Applied Physiology. 2006 doi: 10.1152/japplphysiol.01201.2005. in press. [DOI] [PubMed] [Google Scholar]
- Davies SC, Stebbens VA, Samuels MP, Southall DP. Upper airways obstruction and cerebrovascular accident in children with sickle cell anaemia. Lancet. 1989;2:283–284. doi: 10.1016/s0140-6736(89)90477-7. [DOI] [PubMed] [Google Scholar]
- Del Bo R, Scarlato M, Ghezzi S, Martinelli Boneschi F, Fenoglio C, Galbiati S, Virgilio R, Galimberti D, Galimberti G, Crimi M, Ferrarese C, Scarpini E, Bresolin N, Comi GP. Vascular endothelial growth factor gene variability is associated with increased risk for AD. Annals of Neurology. 2005;57:373–380. doi: 10.1002/ana.20390. [DOI] [PubMed] [Google Scholar]
- Di Giulio C, Bianchi G, Cacchio M, Artese L, Rapino C, Macri MA, Di Ilio C. Oxygen and life span: chronic hypoxia as a model for studying HIF-1alpha, VEGF and NOS during aging. Respiratory and Physiological Neurobiology. 2005;147:31–38. doi: 10.1016/j.resp.2005.01.006. [DOI] [PubMed] [Google Scholar]
- Dill DB, editor. Handbook of physiology, Section 4. Washington, DC: American Physiological Society; 1964. [Google Scholar]
- Dowling MM. Clinical experience in 24 months of a dedicated sickle cell neurology clinic. Proceedings of the 28th Annual Meeting of the National Sickle Cell Disease Program. 2005;111 [Google Scholar]
- Drury I. EEG in benign and malignant epileptic syndromes of childhood. Epilepsia. 2002;43 (Suppl 3):17–26. doi: 10.1046/j.1528-1157.43.s.3.8.x. [DOI] [PubMed] [Google Scholar]
- Dulac OJ, Chiron C. Malignant epileptic encephalopathies in children. Baillieres Clinical Neurology. 1996;5:765–781. [PubMed] [Google Scholar]
- Durak AC, Coskun A, Yikilmaz A, Erdogan F, Mavili E, Guven M. Magnetic resonance imaging findings in chronic carbon monoxide intoxication. Acta Radiologica. 2005;46:322–327. doi: 10.1080/02841850510021085. [DOI] [PubMed] [Google Scholar]
- Fabry ME, Etzion Z, Bookchin RM, Suzuka SM, Nagel RL. Arginine supplementation of sickle transgenic mice leads to a rapid decrease in MCHC and percent dense cells and a slow increase when supplementation is discontinued. Blood. 2004;104:973. [Google Scholar]
- Fasipe FR, Ubawike AE, Eva R, Fabry ME. Arginine supplementation improves rotorod performance in sickle transgenic mice. Hematology. 2004;9:301–305. doi: 10.1080/10245330410001714185. [DOI] [PubMed] [Google Scholar]
- Fatemian M, Gamboa A, Leon-Velarde F, Rivera-Ch M, Palacios JA, Robbins PA. Selected contribution: ventilatory response to CO2 in high-altitude natives and patients with chronic mountain sickness. Journal of Applied Physiology. 2003;94:1279–1287. doi: 10.1152/japplphysiol.00859.2002. discussion 1253–1254. [DOI] [PubMed] [Google Scholar]
- Ferrie CD, Maisey M, Cox T, Polkey C, Barrington SF, Panayiotopoulos CP, Robinson RO. Focal abnormalities detected by 18FDG PET in epileptic encephalopathies. Archives of Diseases in Childhood. 1996;75:102–107. doi: 10.1136/adc.75.2.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finsterer J, Sommer O, Stiskal M, Stollberger C, Baumgartner H. Closure of a patent foramen ovale: effective therapy of migraine and occipital stroke. International Journal of Neuroscience. 2005;115:119–127. doi: 10.1080/00207450490512687. [DOI] [PubMed] [Google Scholar]
- Fowler NO, Smith O, Greenfield JC. Arterial blood oxygenation in sickle cell anemia. American Journal of Medical Science. 1957;234:449–458. doi: 10.1097/00000441-195710000-00010. [DOI] [PubMed] [Google Scholar]
- French JA, Kenny D, Scott JP, Hoffmann RG, Wood JD, Hudetz AG, Hillery CA. Mechanisms of stroke in sickle cell disease: sickle erythrocytes decrease cerebral blood flow in rats after nitric oxide synthase inhibition. Blood. 1997;89:4591–4599. [PubMed] [Google Scholar]
- Fullerton HJ, Adams RJ, Zhao S, Johnston SC. Declining stroke rates in Californian children with sickle cell disease. Blood. 2004;104:336–339. doi: 10.1182/blood-2004-02-0636. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radical Biological Medicine. 2004;36:707–717. doi: 10.1016/j.freeradbiomed.2003.11.032. [DOI] [PubMed] [Google Scholar]
- Gladwin MT, Kato GJ. Cardiopulmonary complications of sickle cell disease: role of nitric oxide and haemolytic anemia. Hematology (Am Soc Hematol Educ Program) 2005:51–57. doi: 10.1182/asheducation-2005.1.51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gozal E, Gozal D. Respiratory plasticity following intermittent hypoxia: developmental interactions. Journal of Applied Physiology. 2001;90:1995–1999. doi: 10.1152/jappl.2001.90.5.1995. [DOI] [PubMed] [Google Scholar]
- Grabowski M, Johansson BB. Nifedipine and nimodipine: effect on blood pressure and regional cerebral blood flow in conscious normotensive and hypertensive rats. Journal of Cardiovascular Pharmacology. 1985;7:1127–1133. doi: 10.1097/00005344-198511000-00018. [DOI] [PubMed] [Google Scholar]
- Graham DI, Adams JH, Doyle D. Ischaemic brain damage in fatal non-missile head injuries. Journal of Neurological Science. 1978;39:213–234. doi: 10.1016/0022-510x(78)90124-7. [DOI] [PubMed] [Google Scholar]
- Green RL, Huntsman RG, Serjeant GR. The sickle-cell and altitude. British Medical Journal. 1971;4:593–595. doi: 10.1136/bmj.4.5787.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurkan E, Tanriverdi K, Baslamisli F. Clinical relevance of vascular endothelial growth factor levels in sickle cell disease. Annals of Hematology. 2004;84:71–75. doi: 10.1007/s00277-004-0935-y. [DOI] [PubMed] [Google Scholar]
- Hackett PH, Yarnell PR, Hill R, Reynard K, Heit J, McCormick J. High-altitude cerebral edema evaluated with magnetic resonance imaging: clinical correlation and pathophysiology. Journal of the American Medical Association. 1998;280:1920–1925. doi: 10.1001/jama.280.22.1920. [DOI] [PubMed] [Google Scholar]
- Hagen T, Taylor CT, Lam F, Moncada S. Redistribution of intracellular oxygen in hypoxia by nitric oxide: effect on HIF1alpha. Science. 2003;302:1975–1978. doi: 10.1126/science.1088805. [DOI] [PubMed] [Google Scholar]
- Helton R, Cui J, Scheel JR, Ellison JA, Ames C, Gibson C, Blouw B, Ouyang L, Dragatsis I, Zeitlin S, Johnson RS, Lipton SA, Barlow C. Brain-specific knock-out of hypoxia-inducible factor-1alpha reduces rather than increases hypoxicischemic damage. Journal of Neuroscience. 2005;25:4099–4107. doi: 10.1523/JNEUROSCI.4555-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson JN, Noetzel MJ, McKinstry RC, White DA, Armstrong M, DeBaun MR. Reversible posterior leukoencephalopathy syndrome and silent cerebral infarcts are associated with severe acute chest syndrome in children with sickle cell disease. Blood. 2003;101:415–419. doi: 10.1182/blood-2002-04-1183. [DOI] [PubMed] [Google Scholar]
- Henry M, Driscoll MC, Miller M, Chang T, Minniti CP. Pseudotumor cerebri in children with sickle cell disease: a case series. Pediatrics. 2004;113:e265–e269. doi: 10.1542/peds.113.3.e265. [DOI] [PubMed] [Google Scholar]
- Herold S, Brozovic M, Gibbs J, Lammertsma AA, Leenders KL, Carr D, Fleming JS, Jones T. Measurement of regional cerebral blood flow, blood volume and oxygen metabolism in patients with sickle cell disease using positron emission tomography. Stroke. 1986;17:692–698. doi: 10.1161/01.str.17.4.692. [DOI] [PubMed] [Google Scholar]
- Hill CM, Hogan AM, Onugha NN, Harrison D, Cooper S, McGrigor VJ, Datta A, Kirkham FJ. Increased cerebral blood flow velocity in children with primary snoring: associations with cognitive function. Pedriatics. 2006 doi: 10.1542/peds.2006-0092. (joint first authors) (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan AM, Kirkham FJ, Isaacs E. Intelligence after stroke in childhood: a review of the literature and suggestions for future research. Journal of Child Neurology. 2000;15:325–332. doi: 10.1177/088307380001500509. [DOI] [PubMed] [Google Scholar]
- Hogan AM, Kirkham FJ, Isaacs EB, Wade AM, Vargha-Khadem F. Intellectual decline in children with moyamoya and sickle cell anaemia. Developmental Medicine & Child Neurology. 2005;47:824–829. doi: 10.1017/S001216220500174X. [DOI] [PubMed] [Google Scholar]
- Hogan AM, Kirkham FJ, Prengler M, Telfer P, Lane R, Vargha-Khadem F, de Haan M. Physiological correlates of neurodevelopmental delay in infants with sickle cell anaemia: an exploratory study. British Journal of Haematology. 2005;132:99–107. doi: 10.1111/j.1365-2141.2005.05828.x. [DOI] [PubMed] [Google Scholar]
- Huttenlocher PR, Moohr JW, Johns L, Brown FD. Cerebral blood flow in sickle cell cerebrovascular disease. Pediatrics. 1984;73:615–621. [PubMed] [Google Scholar]
- Inwald DP, Kirkham FJ, Peters MJ, Lane R, Wade A, Evans JP, Klein NJ. Platelet and leukocyte activation in childhood sickle cell disease: association with nocturnal hypoxaemia. British Journal of Haematology. 2000;111:474–481. doi: 10.1046/j.1365-2141.2000.02353.x. [DOI] [PubMed] [Google Scholar]
- Jaillard AS, Mazetti P, Kala E. Prevalence of migraine and headache in a high-altitude town of Peru: a population-based study. Headache. 1997;37:95–101. doi: 10.1046/j.1526-4610.1997.3702095.x. [DOI] [PubMed] [Google Scholar]
- Jensen FE, Holmes GL, Lombroso CT, Blume HK, Firkusny IR. Age-dependent changes in long-term seizure susceptibility and behavior after hypoxia in rats. Epilepsia. 1992;33:971–980. doi: 10.1111/j.1528-1157.1992.tb01746.x. [DOI] [PubMed] [Google Scholar]
- Jeong JH, Kwon JC, Chin J, Yoon SJ, Na DL. Globus pallidus lesions associated with high mountain climbing. Journal of Korean Medical Science. 2002;17:861–863. doi: 10.3346/jkms.2002.17.6.861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jison ML, Munson PJ, Barb JJ, Suffredini AF, Talwar S, Logun C, Raghavachari N, Beigel JH, Shelhamer JH, Danner RL, Gladwin MT. Blood mononuclear cell gene expression profiles characterize the oxidant, hemolytic, and inflammatory stress of sickle cell disease. Blood. 2004;104:270–280. doi: 10.1182/blood-2003-08-2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaul DK, Hebbel RP. Hypoxia/reoxygenation causes inflammatory response in transgenic sickle mice but not in normal mice. Journal of Clinical Investigation. 2000;106:411–420. doi: 10.1172/JCI9225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kedar A, Drane WE, Shaeffer D, Nicole M, Adams C. Measurement of cerebrovascular flow reserve in pediatric patients with sickle cell disease. Pediatric Blood Cancer. 2006;46 (2):234–238. doi: 10.1002/pbc.20475. [DOI] [PubMed] [Google Scholar]
- Kennan RP, Suzuka SM, Nagel RL, Fabry ME. Detection of increased cerebral response to hyperoxia in sickle cell patients by transcranial near infrared spectroscopy. Blood. 2002;100:2611. [Google Scholar]
- Kennan RP, Suzuka SM, Nagel RL, Fabry ME. Arginine supplementation ameliorates elevated deoxyhemoglobin and poor perfusion in sickle knockout mice as detected by MRI. Blood. 2003;102:128. [Google Scholar]
- Kennan RP, Suzuka SM, Nagel RL, Fabry ME. Decreased cerebral perfusion correlates with increased BOLD hyperoxia response in transgenic mouse models of sickle cell disease. Magnetic Resonance Medicine. 2004;51:525–532. doi: 10.1002/mrm.20014. [DOI] [PubMed] [Google Scholar]
- Kinney TR, Sleeper LA, Wang WC, Zimmerman RA, Pegelow CH, Ohene-Frempong K, Wethers DL, Bello JA, Vichinsky EP, Moser FG, Gallagher DM, DeBaun MR, Platt OS, Miller ST. Silent cerebral infarcts in sickle cell anemia: a risk factor analysis. The Cooperative Study of Sickle Cell Disease. Pediatrics. 1999;103:640–645. doi: 10.1542/peds.103.3.640. [DOI] [PubMed] [Google Scholar]
- Kirkham FJ, Calamante F, Bynevelt M, Gadian DG, Cox TC, Evans JPM, Connelly A. Perfusion MR abnormalities in patients with sickle cell disease: relation to symptoms, infarction and cerebrovascular disease. Annals of Neurology. 2001b;49:477–485. [PubMed] [Google Scholar]
- Kirkham FJ, DeBaun MR. Stroke in children with sickle cell disease. Current Treatment Options in Neurology. 2004;6:357–375. doi: 10.1007/s11940-996-0028-4. [DOI] [PubMed] [Google Scholar]
- Kirkham FJ, Hewes DK, Prengler M, Wade A, Lane R, Evans JP. Nocturnal hypoxaemia and central-nervous-system events in sickle-cell disease. Lancet. 2001a;357:1656–1659. doi: 10.1016/s0140-6736(00)04821-2. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Koyama S, Kubo K, Fukushima M, Kusama S. Clinical features of patients with high-altitude pulmonary edema in Japan. Chest. 1987;92:814–821. doi: 10.1378/chest.92.5.814. [DOI] [PubMed] [Google Scholar]
- Koh S, Ward SL, Lin M, Chen LS. Sleep apnea treatment improves seizure control in children with neurodevelopmental disorders. Pediatric Neurology. 2000;22:36–39. doi: 10.1016/s0887-8994(99)00114-9. [DOI] [PubMed] [Google Scholar]
- LaManna JC, Chavez JC, Pichiule P. Structural and functional adaptation to hypoxia in the rat brain. Journal of Experimental Biology. 2004;207:3163–3169. doi: 10.1242/jeb.00976. [DOI] [PubMed] [Google Scholar]
- Lee KH, McKie VC, Sekul EA, Adams RJ, Nichols FT. Unusual encephalopathy after acute chest syndrome in sickle cell disease: acute necrotizing encephalitis. Journal of Pediatric Hematology and Oncology. 2002;24:585–588. doi: 10.1097/00043426-200210000-00021. [DOI] [PubMed] [Google Scholar]
- Lipton AJ, Johnson MA, Macdonald T, Lieberman MW, Gozal D, Gaston B. S-nitrosothiols signal the ventilatory response to hypoxia. Nature. 2001;413:171–174. doi: 10.1038/35093117. [DOI] [PubMed] [Google Scholar]
- Liu JE, Gzesh DJ, Ballas SK. The spectrum of epilepsy in sickle cell anemia. Journal of Neurological Science. 1994;123:6–10. doi: 10.1016/0022-510x(94)90196-1. [DOI] [PubMed] [Google Scholar]
- Longo LD, Pearce WJ. Fetal cerebrovascular acclimatization responses to high-altitude, long-term hypoxia: a model for prenatal programming of adult disease? American Journal of Physiology. 2005;288:R16–R24. doi: 10.1152/ajpregu.00462.2004. [DOI] [PubMed] [Google Scholar]
- Macarlupu JL, Buvry A, Morel OE, Leon-Velarde F, Richalet JP, Favret F. Characterisation of the ventilatory response to hypoxia in a model of transgenic anemic mice. Respiratory and Physiological Neurobiology. 2006;150 (1):19–26. doi: 10.1016/j.resp.2005.03.011. [DOI] [PubMed] [Google Scholar]
- McFarland RA, Evans JN. Alterations in dark adaptation under reduced oxygen tensions. Journal of Physiology. 1939;127:37. [Google Scholar]
- Maddern BR, Reed HT, Ohene-Frempong K, Beckerman RC. Obstructive sleep apnea syndrome in sickle cell disease. Annals of Otolaryngology, Rhinology and Laryngology. 1989;98:174–178. doi: 10.1177/000348948909800302. [DOI] [PubMed] [Google Scholar]
- Malow BA, Levy K, Maturen K, Bowes R. Obstructive sleep apnea is common in medically refractory epilepsy patients. Neurology. 2000;55:1002–1007. doi: 10.1212/wnl.55.7.1002. [DOI] [PubMed] [Google Scholar]
- Maria BL, Neufeld JA, Rosainz LC, Drane WE, Quisling RG, Ben-David K, Hamed LM. Central nervous system structure and function in Sturge-Weber syndrome: evidence of neurologic and radiologic progression. Journal of Child Neurology. 1998;13:606–618. doi: 10.1177/088307389801301204. [DOI] [PubMed] [Google Scholar]
- Mejia OM, Prchal JT, Leon-Velarde F, Hurtado A, Stockton DW. Genetic association analysis of chronic mountain sickness in an Andean high-altitude population. Haematologica. 2005;90:13–19. [PubMed] [Google Scholar]
- Minoguchi K, Yokoe T, Tazaki T, Minoguchi H, Tanaka A, Oda N, Okada S, Ohta S, Naito H, Adachi M. Increased carotid intimamedia thickness and serum inflammatory markers in obstructive sleep apnea. American Journal of Respiratory and Critical Care Medicine. 2005;172:625–630. doi: 10.1164/rccm.200412-1652OC. [DOI] [PubMed] [Google Scholar]
- Miyamoto O, Auer RN. Hypoxia, hyperoxia, ischemia, and brain necrosis. Neurology. 2000;54:362–371. doi: 10.1212/wnl.54.2.362. [DOI] [PubMed] [Google Scholar]
- Morris CR, Kato GJ, Poljakovic M, Wang X, Blackwelder WC, Sachdev V, Hazen SL, Vichinsky EP, Morris SM, Jr, Gladwin MT. Dysregulated arginine metabolism, hemolysis-associated pulmonary hypertension, and mortality in sickle cell disease. Journal of the American Medical Association. 2005;294:81–90. doi: 10.1001/jama.294.1.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murase M, Ishida A. Early hypocarbia of preterm infants: its relationship to periventricular leukomalacia and cerebral palsy, and its perinatal risk factors. Acta Paediatrica. 2005;94:85–91. doi: 10.1111/j.1651-2227.2005.tb01793.x. [DOI] [PubMed] [Google Scholar]
- Nahavandi M, Tavakkoli F, Hasan SP, Wyche MQ, Castro O. Cerebral oximetry in patients with sickle cell disease. European Journal of Clinical Investigation. 2004;34:143–148. doi: 10.1111/j.1365-2362.2004.01307.x. [DOI] [PubMed] [Google Scholar]
- Ng I, Tan WL, Ng PY, Lim J. Hypoxia inducible factor-1alpha and expression of vascular endothelial growth factor and its receptors in cerebral arteriovenous malformations. Journal of Clinical Neuroscience. 2005;12:794–799. doi: 10.1016/j.jocn.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Nordness ME, Lynn J, Zacharisen MC, Scott PJ, Kelly KJ. Asthma is a risk factor for acute chest syndrome and cerebral vascular accidents in children with sickle cell disease. Clinical and Molecular Allergy. 2005;3:2. doi: 10.1186/1476-7961-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numaguchi Y, Haller JS, Humbert JR, Robinson AE, Lindstrom WW, Gruenauer LM, Carey JE. Cerebral blood flow mapping using stable xenon-enhanced CT in sickle cell cerebrovascular disease. Neuroradiology. 1990;32:289–295. doi: 10.1007/BF00593047. [DOI] [PubMed] [Google Scholar]
- Onodera H, Okabe S, Kikuchi Y, Tsuda T, Itoyama Y. Impaired chemosensitivity and perception of dyspnoea in Parkinson’s disease. Lancet. 2000;356:739–740. doi: 10.1016/S0140-6736(00)02638-6. [DOI] [PubMed] [Google Scholar]
- Palermo TM, Platt-Houston C, Kiska RE, Berman B. Headache symptoms in pediatric sickle cell patients. Journal of Pediatric Hematology and Oncology. 2005;27:420–424. doi: 10.1097/01.mph.0000175408.27180.8e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascual-Castroviejo I, Lopez Martin V, Martinez Bermejo A, Perez Higueras A. Is cerebral arteritis the cause of the Landau-Kleffner syndrome? Four cases in childhood with angiographic study. Canadian Journal of Neurological Science. 1994;19:46–52. [PubMed] [Google Scholar]
- Pavlakis SG, Bello J, Prohovnik I, Sutton M, Ince C, Mohr JP, Piomelli S, Hilal S, De Vivo DC. Brain infarction in sickle cell anemia: magnetic resonance imaging correlates. Annals of Neurology. 1988;23:125–130. doi: 10.1002/ana.410230204. [DOI] [PubMed] [Google Scholar]
- Pawloski JR, Hess DT, Stamler JS. Impaired vasodilation by red blood cells in sickle cell disease. Proceedings of the National Academy of Science, USA. 2005;102:2531–2536. doi: 10.1073/pnas.0409876102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perelman N, Selvaraj SK, Batra S, Luck LR, Erdreich-Epstein A, Coates TD, Kalra VK, Malik P. Placenta growth factor activates monocytes and correlates with sickle cell disease severity. Blood. 2003;102:1506–1514. doi: 10.1182/blood-2002-11-3422. [DOI] [PubMed] [Google Scholar]
- Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1alpha expression regulates the bactericidal capacity of phagocytes. Journal of Clinical Investigation. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pluta RM, Dejam A, Grimes G, Gladwin MT, Oldfield EH. Nitrite infusions to prevent delayed cerebral vasospasm in a primate model of subarachnoid hemorrhage. Journal of the American Medical Association. 2005;293:1477–1484. doi: 10.1001/jama.293.12.1477. [DOI] [PubMed] [Google Scholar]
- Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921–926. [PubMed] [Google Scholar]
- Powars DR, Conti PS, Wong WY, Groncy P, Hyman C, Smith E, Ewing N, Keenan RN, Zee CS, Harold Y, Hiti AL, Teng EL, Chan LS. Cerebral vasculopathy in sickle cell anemia: diagnostic contribution of positron emission tomography. Blood. 1999;93:71–79. [PubMed] [Google Scholar]
- Prengler M, Pavlakis SG, Boyd S, Connelly A, Calamante F, Chong WK, Saunders D, Cox T, Bynevelt M, Lane R, Laverty A, Kirkham FJ. Sickle cell disease: ischemia and seizures. Annals of Neurology. 2005;58:290–302. doi: 10.1002/ana.20556. [DOI] [PubMed] [Google Scholar]
- Prohovnik I, Pavlakis SG, Piomelli S, Bello J, Mohr JP, Hilal S, De Vivo DC. Cerebral hyperemia, stroke, and transfusion in sickle cell disease. Neurology. 1989;39:344–348. doi: 10.1212/wnl.39.3.344. [DOI] [PubMed] [Google Scholar]
- Protass LM. Possible precipitation of cerebral thrombosis in sickle-cell anemia by hyperventilation. Annals of Internal Medicine. 1973;79:451. doi: 10.7326/0003-4819-79-3-451_1. [DOI] [PubMed] [Google Scholar]
- Rackoff WR, Kunkel N, Silber JH, Asakura T, Ohene-Frempong K. Pulse oximetry and factors associated with hemoglobin oxygen desaturation in children with sickle cell disease. Blood. 1993;81:3422–3427. [PubMed] [Google Scholar]
- Raj A, Bertolone SJ, Mangold S, Edmonds HL., Jr Assessment of cerebral tissue oxygenation in patients with sickle cell disease: effect of transfusion therapy. Journal of Pediatric Hematologly and Oncology. 2004;26:279–283. doi: 10.1097/00043426-200405000-00004. [DOI] [PubMed] [Google Scholar]
- Raj AB, O’Brien LM, Edmonds HL, Bertolone SJ, Gozal D. Use of transcranial near infrared spectroscopy to identify sickle cell patients at risk for nocturnal cerebral hypoxia. Proceedings of the 28th Annual Meeting of the National Sickle Cell Disease Program. 2005;111 [Google Scholar]
- Rapino C, Bianchi G, Di Giulio C, Centurione L, Cacchio M, Antonucci A, Cataldi A. HIF-1alpha cytoplasmic accumulation is associated with cell death in old rat cerebral cortex exposed to intermittent hypoxia. Aging Cell. 2005;4:177–185. doi: 10.1111/j.1474-9726.2005.00161.x. [DOI] [PubMed] [Google Scholar]
- Reiter CD, Wang X, Tanus-Santos JE, Hogg N, Cannon RO, 3rd, Schechter AN, Gladwin MT. Cell-free hemoglobin limits nitric oxide bioavailability in sickle-cell disease. Nature Medicine. 2002;8:1383–1389. doi: 10.1038/nm1202-799. [DOI] [PubMed] [Google Scholar]
- Rice L, Alfrey CP. The negative regulation of red cell mass by neocytolysis: physiologic and pathophysiologic manifestations. Cellular Physiology and Biochemistry. 2005;15:245–250. doi: 10.1159/000087234. [DOI] [PubMed] [Google Scholar]
- Rivard A, Berthou-Soulie L, Principe N. Age dependent defect in vascular endothelial growth factor expression is associated with reduced hypoxia-inducible factor 1 activity. Journal of Biological Chemistry. 2000;275:29643–29647. doi: 10.1074/jbc.M001029200. [DOI] [PubMed] [Google Scholar]
- Romero JR, Suzuka SM, Nagel RL, Fabry ME. Arginine supplementation of sickle transgenic mice reduces red cell density and Gardos channel activity. Blood. 2002;99:1103–1108. doi: 10.1182/blood.v99.4.1103. [DOI] [PubMed] [Google Scholar]
- Rostrup E, Larsson HB, Born AP, Knudsen GM, Paulson OB. Changes in BOLD and ADC weighted imaging in acute hypoxia during sea-level and altitude adapted states. Neuroimage. 2005;28:947–955. doi: 10.1016/j.neuroimage.2005.06.032. [DOI] [PubMed] [Google Scholar]
- Saito S, Tanaka SK. A case of cerebral sinus thrombosis developed during a high-altitude expedition to Gasherbrum I. Wilderness Environmental Medicine. 2003;14:226–230. doi: 10.1580/1080-6032(2003)14[226:acocst]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Samaja M, Crespi T, Guazzi M, Vandegriff KD. Oxygen transport in blood at high altitude: role of the hemoglobin-oxygen affinity and impact of the phenomena related to hemoglobin allosterism and red cell function. European Journal of Applied Physiology. 2003;90:351–359. doi: 10.1007/s00421-003-0954-8. [DOI] [PubMed] [Google Scholar]
- Samuels MP, Stebbens VA, Davies SC, Picton-Jones E, Southall DP. Sleep related upper airway obstruction and hypoxaemia in sickle cell disease. Archives of Disease in Childhood. 1992;67:925–929. doi: 10.1136/adc.67.7.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schatz J, Buzan R. Decreased corpus callosum size in sickle cell disease: relationship with cerebral infarcts and cognitive functioning. Journal of the International Neuropsychological Society. 2006;12:24–33. doi: 10.1017/S1355617706060085. [DOI] [PubMed] [Google Scholar]
- Schoch HJ, Fischer S, Marti HH. Hypoxia-induced vascular endothelial growth factor expression causes vascular leakage in the brain. Brain. 2002;125:2549–2557. doi: 10.1093/brain/awf257. [DOI] [PubMed] [Google Scholar]
- Sébire G, Tabarki B, Saunders DE, Leroy I, Liesner R, Saint-Martin C, Husson B, Williams AN, Kirkham FJ. Venous sinus thrombosis in children. Brain. 2005;128:477–489. doi: 10.1093/brain/awh412. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Hypoxia-inducible factor 1: control of oxygen homeostasis in health and disease. Pediatric Research. 2001;49:614–617. doi: 10.1203/00006450-200105000-00002. [DOI] [PubMed] [Google Scholar]
- Setty BY, Stuart MJ, Dampier C, Brodecki D, Allen JL. Hypoxemia in sickle cell disease: biomarker modulation and relevance to disease pathophysiology. Lancet. 2003;362:1450–1455. doi: 10.1016/S0140-6736(03)14689-2. [DOI] [PubMed] [Google Scholar]
- Shanoudy H, Soliman A, Raggi P, Liu JW, Russell DC, Jarmukli NF. Prevalence of patent foramen ovale and its contribution to hypoxemia in patients with obstructive sleep apnea. Chest. 1998;113:91–96. doi: 10.1378/chest.113.1.91. [DOI] [PubMed] [Google Scholar]
- Shein NA, Horowitz M, Alexandrovich AG, Tsenter J, Shohami E. Heat acclimation increases hypoxia-inducible factor 1alpha and erythropoietin receptor expression: implication for neuroprotection after closed head injury in mice. Journal of Cerebral Blood Flow and Metabolism. 2005;25:1456–1465. doi: 10.1038/sj.jcbfm.9600142. [DOI] [PubMed] [Google Scholar]
- Sherwood JB, Goldwasser E, Chilcote R, Carmichael LD, Nagel RL. Sickle cell anemia patients have low erythropoietin levels for their degree of anemia. Blood. 1986;67:46–49. [PubMed] [Google Scholar]
- Shnaider H, Shiran A, Lorber A. Right ventricular diastolic dysfunction and patent foramen ovale causing profound cyanosis. Heart. 2004;90:e31. doi: 10.1136/hrt.2003.026468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annual Review of Physiology. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- Soliman A, Shanoudy H, Liu J, Russell DC, Jarmukli NF. Increased prevalence of patent foramen ovale in patients with severe chronic obstructive pulmonary disease. Journal of the American Society of Echocardiographers. 1999;12:99–105. doi: 10.1016/s0894-7317(99)70121-5. [DOI] [PubMed] [Google Scholar]
- Solovey A, Gui L, Ramakrishnan S, Steinberg MH, Hebbel RP. Sickle cell anemia as a possible state of enhanced anti-apoptotic tone: survival effect of vascular endothelial growth factor on circulating and unanchored endothelial cells. Blood. 1999;93:3824–3830. [PubMed] [Google Scholar]
- Stamler JS, Jia L, Eu JP, McMahon TJ, Demchenko IT, Bonaventura J, Gernert K, Piantadosi CA. Blood flow regulation by S-nitrosohemoglobin in the physiological oxygen gradient. Science. 1997;276:2034–2037. doi: 10.1126/science.276.5321.2034. [DOI] [PubMed] [Google Scholar]
- Steen RG, Langston JW, Reddick WE, Ogg R, Chen G, Wang WC. Quantitative MR imaging of children with sickle cell disease: striking T1 elevation in the thalamus. Journal of Magnetic Resonance Imaging. 1996;6:226–234. doi: 10.1002/jmri.1880060140. [DOI] [PubMed] [Google Scholar]
- Steen RG, Reddick WE, Glass JO, Wang WC. Evidence of cranial artery ectasia in sickle cell disease patients with ectasia of the basilar artery. Journal of Stroke and Cerebrovascular Disease. 1998;7:330–338. doi: 10.1016/s1052-3057(98)80051-7. [DOI] [PubMed] [Google Scholar]
- Steen RG, Xiong X, Mulhern RK, Langston JW, Wang WC. Subtle brain abnormalities in children with sickle cell disease: relationship to blood hematocrit. Annals of Neurology. 1999;45:279–286. doi: 10.1002/1531-8249(199903)45:3<279::aid-ana2>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Stenzel-Poore MP, Stevens SL, Xiong Z, Lessov NS, Harrington CA, Mori M, Meller R, Rosenzweig HL, Tobar E, Shaw TE, Chu X, Simon RP. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet. 2003;362:1028–1037. doi: 10.1016/S0140-6736(03)14412-1. [DOI] [PubMed] [Google Scholar]
- Stockman JA, Nigro MA, Mishkin MM, Oski FA. Occlusion of large cerebral vessels in sickle-cell anemia. New England Journal of Medicine. 1972;287:846–849. doi: 10.1056/NEJM197210262871703. [DOI] [PubMed] [Google Scholar]
- Sylvester KP, Patey RA, Rafferty GF, Rees D, Thein SL, Greenough A. Exhaled carbon monoxide levels in children with sickle cell disease. European Journal of Pediatrics. 2005;164:162–165. doi: 10.1007/s00431-004-1605-8. [DOI] [PubMed] [Google Scholar]
- Terblanche JS, Tolley KA, Fahlman A, Myburgh KH, Jackson S. The acute hypoxic ventilatory response: testing the adaptive significance in human populations. Comparative Biochemistry and Physiology A – Molecular and Integrative Physiology. 2005;140:349–362. doi: 10.1016/j.cbpb.2005.01.016. [DOI] [PubMed] [Google Scholar]
- Tissot van Patot MC, Leadbetter G, Keyes LE, Bendrick-Peart J, Beckey VE, Christians U, Hackett P. Greater free plasma VEGF and lower soluble VEGF receptor-1 in acute mountain sickness. Journal of Applied Physiology. 2005;98:1626–1629. doi: 10.1152/japplphysiol.00589.2004. [DOI] [PubMed] [Google Scholar]
- Urschitz MS, Wolff J, Von Einem V, Urschitz-Duprat PM, Schlaud M, Poets CF. Reference values for nocturnal home pulse oximetry during sleep in primary school children. Chest. 2003;123:96–101. doi: 10.1378/chest.123.1.96. [DOI] [PubMed] [Google Scholar]
- Usui C, Inoue Y, Kimura M, Kirino E, Nagaoka S, Abe M, Nagata T, Arai H. Irreversible subcortical dementia following high altitude illness. High Altitude Medicine and Biology. 2004;5:77–81. doi: 10.1089/152702904322963717. [DOI] [PubMed] [Google Scholar]
- van Dijk EJ, Vermeer SE, de Groot JC, van de Minkelis J, Prins ND, Oudkerk M, Hofman A, Koudstaal PJ, Breteler MM. Arterial oxygen saturation, COPD, and cerebral small vessel disease. Journal of Neurology, Neurosurgery and Psychiatry. 2004;75:733–736. doi: 10.1136/jnnp.2003.022012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tuyl M, Liu J, Wang J, Kuliszewski M, Tibboel D, Post M. Role of oxygen and vascular development in epithelial branching morphogenesis of the developing mouse lung. American Journal of Physiology and Lung Cell Molecular Physiology. 2005;288:L167–L178. doi: 10.1152/ajplung.00185.2004. [DOI] [PubMed] [Google Scholar]
- Venketasubramanian N, Prohovnik I, Hurlet A, Mohr JP, Piomelli S. Middle cerebral artery velocity changes during transfusion in sickle cell anemia. Stroke. 1994;25:2153–2158. doi: 10.1161/01.str.25.11.2153. [DOI] [PubMed] [Google Scholar]
- Vichinsky EP, Neumayr LD, Earles AN, Williams R, Lennette ET, Dean D, Nickerson B, Orringer E, McKie V, Bellevue R, Daeschner C, Manci EA. Causes and outcomes of the acute chest syndrome in sickle cell disease. National Acute Chest Syndrome Study Group. New England Journal of Medicine. 2000;342:1855–1865. doi: 10.1056/NEJM200006223422502. [DOI] [PubMed] [Google Scholar]
- Wang WC, Langston JW, Steen RG, Wynn LW, Mulhern RK, Wilimas JA, Kim FM, Figueroa RE. Abnormalities of the central nervous system in very young children with sickle cell anemia. Journal of Pediatrics. 1998;132:994–998. doi: 10.1016/s0022-3476(98)70397-x. [DOI] [PubMed] [Google Scholar]
- Werdehoff SG, Moore RB, Hoff CJ, Fillingim E, Hackman AM. Elevated plasma endothelin-1 levels in sickle cell anemia: relationships to oxygen saturation and left ventricular hypertrophy. American Journal of Hematology. 1998;58:195–199. doi: 10.1002/(sici)1096-8652(199807)58:3<195::aid-ajh6>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- Williams JM, Pearce WJ. Age-dependent modulation of endothelium-dependent vasodilatation by chronic hypoxia in ovine cranial arteries. Journal of Applied Physiology. 2005;100:225–232. doi: 10.1152/japplphysiol.00221.2005. [DOI] [PubMed] [Google Scholar]
- Wolin MJ, Brannon WL. Disk edema in an overweight woman. Survey of Ophthalmology. 1995;39:307–314. doi: 10.1016/s0039-6257(05)80108-0. [DOI] [PubMed] [Google Scholar]