

Abstract
Xenobiotic and dietary compounds with hormone-like activity can disrupt endocrine signaling pathways that play important roles during perinatal differentiation and result in alterations that are not apparent until later in life. Evidence implicates developmental exposure to environmental hormone-mimics with a growing list of health problems. Obesity is currently receiving needed attention since it has potential to overwhelm health systems worldwide with associated illnesses such as diabetes and cardiovascular disease. Here, we review the literature that proposes an association of exposure to environmental endocrine disrupting chemicals with the development of obesity. We describe an animal model of developmental exposure to diethylstilbestrol (DES), a potent perinatal endocrine disruptor with estrogenic activity, to study mechanisms involved in programming an organism for obesity. This experimental animal model provides an example of the growing scientific field termed “the developmental origins of adult disease” and suggests new targets of abnormal programming by endocrine disrupting chemicals.
Keywords: fetal exposure, neonatal exposure, overweight, obese, adipocytes, obesogens, diabetes, cardiovascular disease, epigenetic, DOHaD
Introduction
Over the last decade, mounting evidence from wildlife, epidemiological, laboratory animals, and in vitro studies has shown that numerous environmental and dietary chemicals can interfere with an organism’s complex endocrine signaling mechanisms and result in adverse consequences (1–7). Initial concern focused on exposure to chemicals with estrogenic activity and their contributions to reproductive tract disease and dysfunction; however, it has become increasingly evident that estrogenicity is not the only important endocrine mode of action, and that the reproductive tract is not the only organ system affected. Interest has broadened to include chemicals that mimic or interfere with the normal actions of all endocrine hormones including estrogens, androgens, progestins, thyroid, hypothalamic and pituitary hormones; these chemicals are now collectively referred to as “endocrine disruptors”. Organ systems known to be involved include reproductive tract tissues but have been expanded to include other tissues such as those of the respiratory, cardiovascular, and neuroendocrine systems. Most recently, conference proceedings and reports have focused on the involvement of environmental chemicals in the growing obesity problem (8–10). A newly published study points out the effects of environmental chemicals and their disruption of normal development and homeostatic controls over adipogenesis and energy balance (11). While the full extent of the health consequences of endocrine disrupting chemicals is unknown, we are only beginning to understand how chemicals act as endocrine disruptors, and to fully appreciate the complexities and interactions of endocrine signaling mechanisms.
The Developmental Basis of Adult Disease
Adult exposure to endocrine disrupting chemicals is certainly an important factor, however focus on the fetus and/or neonate is of primary concern since developing organisms are extremely sensitive to perturbation by chemicals with hormone-like activity. Adverse effects may be most pronounced in the developing organism and occur at concentrations of the chemical that are far below levels that would be considered harmful in the adult. The exquisite sensitivity of the developing fetus and neonate has been previously described by Howard Bern in a chapter titled “The Fragile Fetus” (12). The protective mechanisms that are available to the adult such as DNA repair mechanisms, a competent immune system, detoxifying enzymes, liver metabolism, and the blood/brain barrier are not fully functional in the fetus or newborn. In addition, the developing organism has an increased metabolic rate as compared to an adult which, in some cases, may result in increased toxicity.
In addition to increased sensitivity during perinatal life, unique problems may be encountered when studying chemical exposures of the fetus and neonate undergoing critical developmental windows of differentiation.
Extrapolation of risks may be difficult since effects may not follow a monotonic dose response curve typically seen in toxicity studies; for example, high concentrations of chemicals may exhibit no or different effects compared with that seen at low concentrations.
Test chemicals may have different effects in the embryo, fetus, or perinatal organism compared to effects seen in adults.
Effects may be manifested in offspring but not in their exposed parent.
Timing of exposure in the developing organism is critical in determining adult outcomes.
Although critical windows of exposure occur during perinatal development, manifestation of the effects may not be apparent until much later in life (the developmental basis of adult disease).
Numerous examples now document that developmental exposure to certain chemicals can lead to adverse effects in adults; many of these studies describe adverse consequences which include tumors in endocrine target tissues, and adverse reproductive effects in males and females.
The scientific hypothesis that adult health and disease have an etiology arising in fetal or early neonatal development is not unique to the field of endocrine disruption. In the late 1980s, studies on maternal nutrition gained prominence by suggesting that the fetal environment, as a reflection of low birth size and poor nutrition, was related to increased risk of non-communicable diseases later in adult life; associations with coronary heart disease quickly extended to included type 2 diabetes, osteoporosis, and metabolic dysfunction (13). These findings led to the development of the “developmental origins of health and disease” (DOHaD) paradigm in which a substantial research effort focused on the perinatal influences on chronic disease (14, 15). Perinatal effects are no longer viewed in terms of just teratogenic changes or acute birth injury such as thalidomide-induced limb malformations, but whether changes induced in early development (pre-implantation through pre-pubertal stages) may lead to life long anomalies. Certainly unique problems exist in studying chemical exposures during development and their relationship to adult disease, but in spite of these difficulties, research findings continue to support the idea that environmental chemicals can have endocrine disrupting effects that result in long-term health consequences.
As an example, the profound effects of estrogens on the developing reproductive tract have been demonstrated by prenatal exposure to DES [for review, (16); NIH 1999;(17)]. Although DES effects were well recognized and firmly documented long before the proposed “DOHaD” paradigm was proposed, DES clearly demonstrates that chemical exposure, in addition to nutrition and other perinatal factors, can significantly alter the developing organism and cause long term effects in the adult.
The Obesity Epidemic
Obesity, defined as excessive body fat (>25% men; >30% women), is fast becoming a significant human health crisis that is receiving worldwide attention (18). The prevalence of obesity has risen dramatically in wealthy developed countries over the last 2 to 3 decades but it is also on the rise in poor nations. The World Health Organization (WHO) has declared excessive weight as one of the top 10 health risks in the world, and has estimated that the number of overweight people in the world is now greater than the number undernourished. In the United States, obesity has reached epidemic proportions with more than 20% of adults defined as clinically obese and an additional 30% defined as overweight. Obesity and overweight are known to seriously affect human health, and to impact the risks and prognosis of a number of diseases including type 2 diabetes, hyperinsulinemia, insulin resistance, coronary heart disease, high blood pressure, stroke, gout, liver disease, asthma and pulmonary problems, gall bladder disease, kidney disease, reproductive problems, osteoarthritis, and some forms of cancer (19). Further, more immediate consequences of being overweight/obese have been linked to psychological problems including social discrimination, poor self-esteem, and depression (20). Obesity is a significant health risk for adults but it is a far more serious problem for children since the incidence of type 2 diabetes is dramatically increasing in children and adolescents along with the rise in obesity. In addition, overweight children have increased risk of becoming overweight adults, thus their prospect for a healthy future is questionable.
Obesity, like many other chronic health problems, is caused by a complex interaction between genetic, behavioral and environmental factors. Commonly held causes of obesity are overeating and a sedentary lifestyle imposed on a background of genetic predisposition for the disease. Although much research has focused on these factors, the exact etiology of obesity remains uncertain. It is clear, however, that obesity is notoriously difficult to treat, so prevention is critical.
An emerging hypothesis proposes that in utero and early developmental exposures to environmental chemicals may play a role in the development of obesity later in life. This hypothesis parallels the models for the action of environmental endocrine disrupting chemicals that affect aspects of reproductive endocrinology and health (10). An increasing number of studies report that exposure to chemicals during critical periods of differentiation, at low environmentally-relevant doses, alters developmental programming resulting in obesity.
In 2002, a novel idea postulated a role for chemical toxins in the etiology of obesity and reviewed data showing that the current epidemic of obesity coincides with the marked increase in use of industrial chemicals in the environment over the past 40 years (19). This study cited numerous studies where chemicals such as pesticides, organophosphates, carbamates, polychlorinated biphenyls, polybrominated biphenyls, phthalates, bisphenol A, heavy metals, and solvents caused weight gain possibly by interfering with weight homeostasis such as alterations in weight–controlling hormones, altered sensitivity to neurotransmitters, or altered activity of the sympathetic nervous system (19). Interestingly, some of the chemicals cited in the review were actually intended to increase weight especially some of those chemicals with “hormone-like” activity like DES (23). Another study showed increased body weight in adulthood following prenatal exposure to several endocrine disrupting compounds including phytoestrogens, bisphenol A and DES supporting the idea that early developmental exposure to some chemicals are related to obesity (24).
Additional studies predict the existence of chemical “obesogens”, molecules that inappropriately regulate lipid metabolism and adipogenesis to promote obesity (11, 25). Identification of these “obesogens” is an exciting area of future research in addition to elucidating their molecular targets, and potential cellular mechanisms through which they might act.
A recent study using genetically altered mice that develop obesity showed that very high levels of the phytoestrogen genistein decreased the occurrence of obesity, resulting in a normal phenotype. These mice are heterozygous for the Avy allele resulting in a genetically invariant background. This gene has been shown to be associated with coat color, obesity and tumorigenesis. Exposure to genistein during development alters the methylation pattern of the promoter region of this gene such that the gene is silenced with increased methylation demonstrating that genistein alters methylation patterns during development. The silencing of this gene reduces its expression and subsequently changes the phenotype to wild type. While this is an interesting model to study epigenetic modification, the authors point out that the implications for obesity remain unclear. This study and others in our laboratory clearly show that genes can be epigenetically modified following developmental exposure to endocrine disrupting chemicals (21, 22) and that perhaps genes involved in the etiology of obesity may be altered.
The Developmental Exposed DES Animal Model to Study Obesity
For over 20 years, research in our laboratory has focused on the effects of estrogenic compounds on development and differentiation. Our working premise has been that the developing organism is extremely sensitive to perturbation by chemicals with estrogenic or endocrine disrupting activity, and that exposure to these chemicals during critical stages of differentiation may have permanent long lasting consequences, some of which may not be expressed or detected until later in life. Diethylstilbestrol (DES) is a well-known example of such a chemical; thus, we have used DES as a model chemical to study environmental estrogens. DES, a synthetic estrogen, was widely prescribed from the 1940s through the 1970s for the prevention of threatened miscarriage. A range of 2–8 million treated pregnancies worldwide has been estimated. Today it is well recognized that prenatal DES treatment results in a low incidence of neoplasia in the female offspring, and high incidence of benign abnormalities in both the male and female offspring (17). To study the mechanisms involved in the toxicity of DES, we developed an animal model using outbred CD-1 mice treated with DES by subcutaneous injections on days 9–16 of gestation (the period of major organogenesis in the mouse) or days 1–5 of neonatal life (a period of cellular differentiation of the reproductive tract, and a critical period of immune, behavioral, and adipocyte differentiation). The prenatal DES animal model has successfully duplicated, and in some cases, predicted, many of the alterations (structural, function, cellular and molecular) observed in similarly DES-exposed humans (26, 27). Although our major focus has been on reproductive tract abnormalities, we also examined the effects of DES on body weight over a wide dose range of exposure. Prenatal DES doses (10 –100 μg/kg maternal body weight) often caused a decrease in the offspring’s body weight at birth and subsequently throughout life; neonatal DES at a dose of 1 μg/kg/day did not affect body weight during treatment but was associated with a significant increase in body weight as adults as we reported in an earlier publication (8). A representative photograph of control and DES treated mice at 4 months of age is shown in Figure 1.
Figure 1. Representative Photograph of Control and DES-treated Mice.
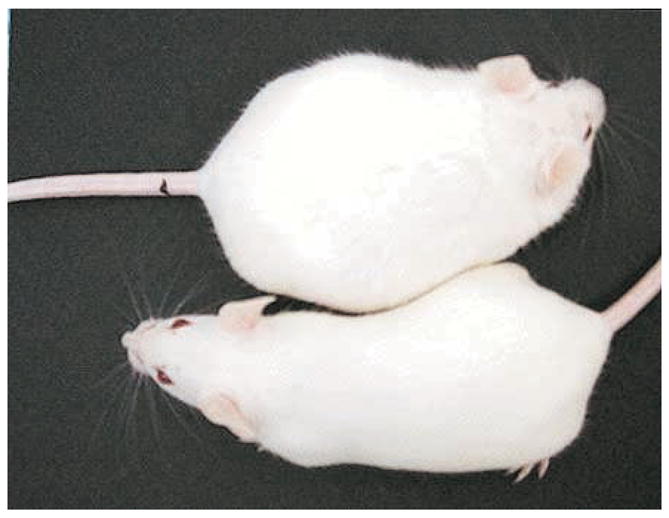
Photograph of 4 month old mice showing the difference in body weight of a control mouse (left panel) and a neonatally DES-treated mouse (right panel).
Unlike the lower dose of DES (1 μg/kg/day), the high DES dose tested (1000 μg/kg/day=1 mg/kg/day) caused a significant decrease in body weight during treatment which was followed by a “catch up” period around puberty and then finally resulting in a significant increase in body weight of DES treated mice as compared to controls after 2 months of age; Figure 2 shows the body weight comparison of control and DES treated mice during the time of treatment and continuing into adulthood. The body weight was significantly lower in DES exposed mice compared to controls during the time of treatment and shortly thereafter as denoted by the * in the graph. However, DES treated mice gradually caught up to controls by 30–45 days, surpassed them by 2 months of age and remained heavier throughout the experiment. Although this experiment was terminated shortly after 4 months of age, earlier data showed this enhanced weight difference was present in DES mice throughout adulthood (8).
Figure 2. Body Weights of Mice Following Neonatal DES Exposure.
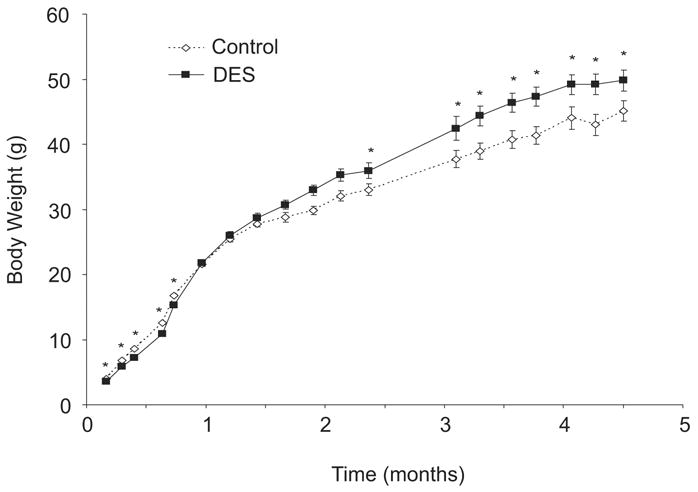
Body weights were measured at various ages. Mice treated with DES had significantly lower body weights during the time of treatment as compared to controls. By 1 month of age the DES treated mice caught up to the controls and by 2 months of age, DES treated mice had surpassed the controls with a significantly higher body weight (n= 16 mice per group); numbers plotted are the mean ± s.e.m.; * denotes significance using ANOVA fallowed by Dunnett’s test (p<0.05).
Further, examination of these animals indicated that the increase in body weight in DES-exposed mice was associated with an increase in the percentage of body fat. Using PIXImus™ mouse densitometry, the percent fat in controls and neonatal DES treated mice were measured at 2 and 6 months of age. A representative image generated from the mouse densitometry at 6 months of age is shown in Figure 3; mice treated with DES are markedly larger than controls. Measurements obtained from the densitometry analysis showed an increase in the estimated body weight, estimated fat weight, and percent fat in DES treated mice compared to controls (Table 1); these differences reached statistical significance at 6 months of age. The higher body weight in DES treated mice was maintained throughout adulthood, however, by 18 months age, statistical differences in body weight between treated and controls were difficult to determine due to increased individual animal variability within groups as they aged (data not shown).
Figure 3. Images of Control and DES-treated Mice as Generated by Piximus Densitometry.
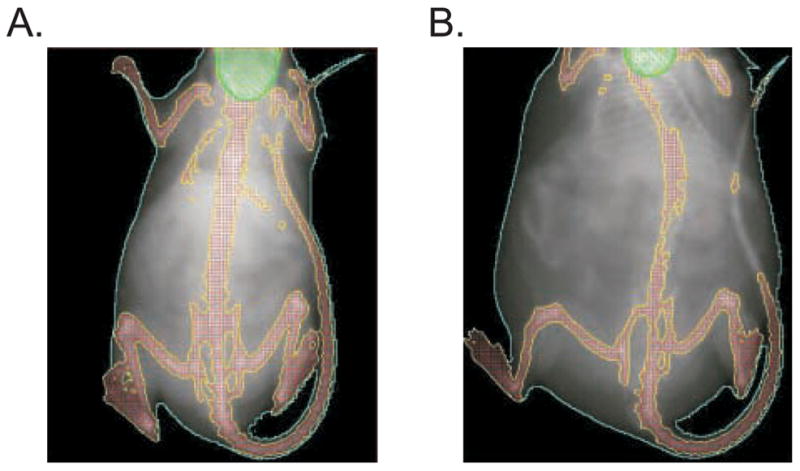
Image captured with PIXImus™ mouse densitometry at 6 months of age. Images are representative of control (left) and DES (right) treated mice. Note that DES mice are significantly larger.
Table 1.
Increased body fat in mice treated neonatally with DES as determined by PIXImus™ Mouse Densitometry.
| 2 months | ||||
|---|---|---|---|---|
| Treatmenta | Estimated Body Weightb | Estimated Fat Weight (g) | % Fat | % Lean |
| Control (n = 8) | 26.8 ± 0.7 | 5.7 ± 0.3 | 21.2 ± 0.8 | 78.8 ± 0.8 |
| DES (n = 8) | 29.3 ± 0.9 * | 6.3 ± 0.3 | 21.4 ± 0.7 | 78.6 ± 0.7 |
| 6 months | ||||
|
| ||||
| Treatmenta | Estimated Body Weightb | Estimated Fat Weight (g) | % Fat | % Lean |
|
| ||||
| Control (n= 4) | 37.9 ± 2.5 | 11.5 ± 1.3 | 29.9 ± 1.8 | 70.1 ± 1.8 |
| DES (n= 4) | 45.0 ± 2.5 | 18.4 ± 1.8 * | 40.8 ± 1.4 * | 59.2 ± 1.8 * |
Mice were treated on days 1–5 with DES 1,000 μg/kg=1 mg/kg (n=4–8 mice per group).
Using the PIXImus™ mouse densitometer, an estimate of the body weight was obtained at 2 and 6 months of age; this measurement excluded the head since the large size of the mice prevented scanning the full body at one time.
p<0.05 using ANOVA followed by Dunnett’s test.
As seen in Figure 3, DES treated mice apparently have excess abdominal fat which has been reported to be associated with cardiovascular disease and diabetes in humans (28). To determine if specific fat depots were affected by DES treatment or whether it was a generalized effect throughout the mouse, the weight of fat depots from control and DES treated mice at 6–8 months of age were measured. In general, fat depots weighed more in the DES treated mice compared to controls and the inguinal and retroperitoneal fat pads showed statistical significance (Table 2). Interestingly, brown fat depot weights were not different between DES and controls (data not shown). Since a recent study found evidence for a role for developmentally expressed genes in the origins of obesity and body fat distribution (29), it is indeed possible that early exposure to environmental chemicals with hormonal activity may be altering the genetic programming of adipocytes and their distribution.
Table 2.
Fat Pad Weights of Mice Treated Neonatally with DES
| Treatment a | Inguinal Fat (g) | Parametral Fat (g) | Gonadal Fat (g) | Retroperitoneal Fat (g) |
|---|---|---|---|---|
| Control | 0.108 ± 0.015 | 1.744 ± 0.211 | 0.608 ± 0.129 | 0.291 ± 0.029 |
| DES | 0.207 ± 0.041 * | 2.084 ± 0.254 | 0.667 ± 0.071 | 0.555 ± 0.080 * |
Mice were treated on days 1–5 with DES 1,000 μg/kg=1 mg/kg and sacrificed at 6–8 months of age (n=8 mice per group).
p<0.05 using ANOVA followed by Dunnett’s test.
Serum profiles of 2 and 6 month old mice revealed an interesting finding as shown in Table 3. Although DES treated mice were similar in weight to controls at 2 months, they had elevated levels of leptin, adiponectin, IL-6 and triglycerides prior to development of overweight and obesity suggesting these may be important early markers of subsequent adult disease such as metabolic syndrome. While most markers were elevated, insulin was decreased in all DES treated mice compared to controls at 2 months of ago. All serum markers were significantly elevated at 6 months of age except triglycerides; the importance of a reduction in this marker is being followed. A similar finding of increased fat depot weight along with decreased serum triglycerides was reported following genistein exposure of pre-pubertal mice (30).
Table 3.
Serum Profiles of Mice Treated Neonatally with DES
| Treatmenta | ||
|---|---|---|
| 2 months | Control | DES |
| Leptin (ng/ml) | 4.8 ± 0.5 | 25.0 ± 1.4 * |
| Adiponectin (μg/ml) | 6.6 ± 0.6 | 38.1 ± 3.6 * |
| IL-6 (pg/ml) | 6.3 ± 0.9 | 60.4 ± 5.0 * |
| Insulin (μU/ml) | 7.4 ± 0.7 | 1.3 ± 0.3 * |
| Triglycerides (mg/ml) | 97.6 ± 3.2 | 122.9 ± 3.5 * |
| 6 months | Control | DES |
|
| ||
| Leptin (ng/ml) | 8.1 ± 0.4 | 60.7 ± 2.3 * |
| Adiponectin (μg/ml) | 9.3 ± 0.6 | 39.2 ± 1.6 * |
| IL-6 (pg/ml) | 10.1 ± 0.4 | 93.8 ± 1.9 * |
| Insulin (μU/ml) | 8.6 ± 0.3 | 10.8 ± 0.3 * |
| Triglycerides (mg/ml) | 116.9 ± 1.7 | 106.8 ± 1.5 |
Mice were treated on days 1–5 with DES 1,000 μg/kg=1 mg/kg (n= 8 per group at 2 months and 16 per group at 6 months of age).
p<0.05 using ANOVA followed by Dunnett’s test.
Since, the balance of activity levels and food intake are known contributors to obesity, we measured a parameter of activity in the DES treated and control mice at 2 months of age before a difference in body weight could be detected. Individual mice were placed in an Opto-Max motor activity chamber (Columbus Instruments, Columbus, OH) and their ambulatory activity measured at 1 minute intervals for a total of 20 minutes each. Data is summarized at five minute intervals in Figure 4. In both DES and control groups, activity levels dropped from the first 5 to 15 minute intervals as the mice acclimated to their environment. Overall, there was no statistical difference in this parameter between the two groups although the DES group showed less movement at 5 minutes than controls. This difference was most likely not enough to explain the increased weight gain in the DES mice as they aged. More comprehensive activity studies may reveal larger differences.
Figure 4. Ambulatory Activity of Adult Mice Following Neonatal DES Exposure.
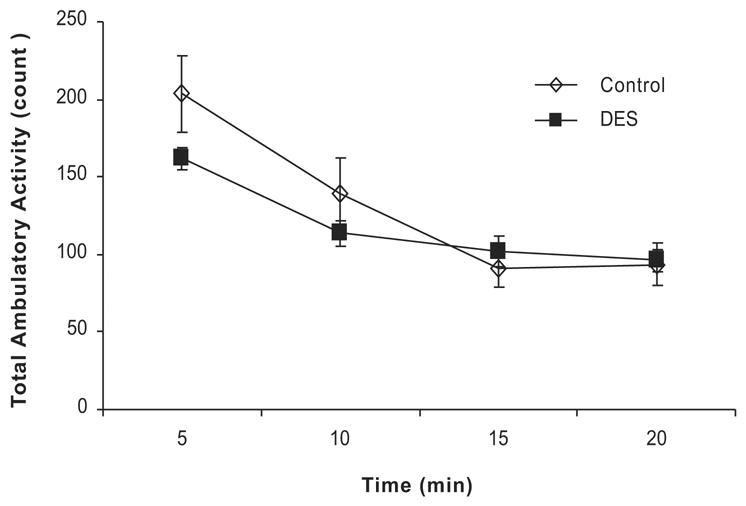
Graph represents total ambulatory activity including movement across the same plane and rearing movement in control and DES treated mice at 2 months of age (n= 8 mice per group). Data is plotted in 5 min intervals over a 20 minute session. The DES mice had less movement as compared to controls at the start of the session although this difference is not significant. DES treated mice exhibited similar activity as controls by the end of the session.
An important consideration for these studies is the diet the mice are fed. We have previously addressed the role of the diet since NIH-31 lab chow contains low levels of phytoestrogens. This diet contains approximately 98 μg/g of genistein and daidzein which is about 16.7 mg/kg/day for a 30 g mouse (31); these levels do not cause overt estrogenic activity. Further, control and DES treated mice were all fed the same diet. Therefore, any contribution of estrogenic activity from phytoestrogens in the diet was minimal. Feed consumption was measured over a 2 week period as shown in Figure 5. Control and DES-treated mice were individually housed and provided a pre-weighed amount of NIH-31 lab chow. At the end of each week, the remaining chow was measured and the total amount was subtracted from the starting amount to determine total feed consumed for each mouse per week. Although DES-treated mice ate more than controls over the course of the experiment (~3 grams more), the amounts were not statistically different. The slight decrease in activity and slight increase in food intake in DES treated mice compared to controls is unlikely to be the sole explanation in the development of obesity in DES treated mice.
Figure 5. Feed Consumption of Adult Mice Following Neonatal DES Exposure.
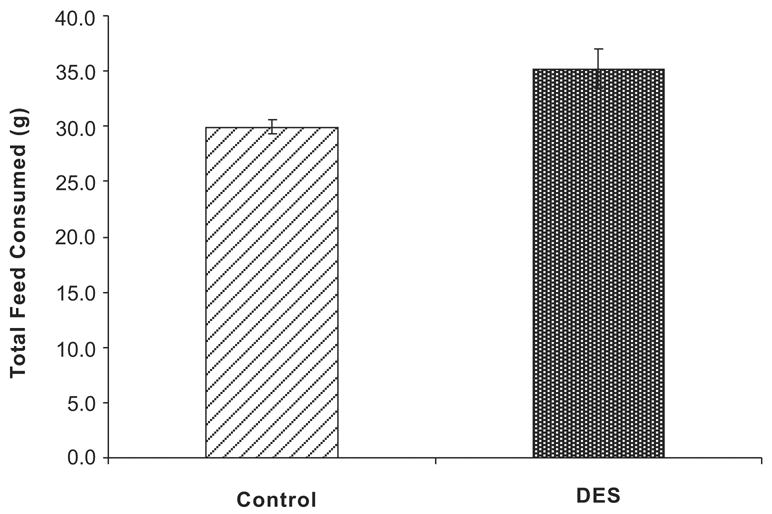
Bars represent the mean total amount of feed consumed in a week for controls and DES treated mice (n=8 per group). Although DES treated mice consumed an average of ~ 3 g more than controls, this increase was not significant.
Finally, glucose levels were measured in DES treated and control mice at 2 mounts of age. Individual mice are plotted in Figure 6 (panel A and B) so that variability across treatment groups can be seen. Following fasting for 18 hours, a drop of blood from the saphenous vein was collected to provide the 0 time point. Blood glucose levels for each mouse were analyzed using Accuchek Advantage glucometer (Roche Diagnostics). Mice were weighed and challenged by intraperitoneal injection of glucose solution (2 g/kg body weight). Blood was analyzed at 20, 40, 60, 80, 100, 120, 140, 160, and 180 minutes following glucose challenge. Two mice in the DES treated group shown in Figure 6 (panel B) had higher glucose levels at 40 minutes following challenge than controls and other DES treated mice; these two mice also showed a slower decrease of glucose levels over the experimental time period although their levels eventually dropped to baseline by 120 minutes. Again, this parameter was altered in some mice before they developed excessive weight. Glucose challenge was also determined in the same group of mice at 6 months of age (Figure 6, panel C and D). While one DES treated mouse showed increased levels of glucose (>600) at 20 and 40 minutes, one control mouse also showed high levels. These data indicate that altered glucose response may occur in aged CD-1 mice under our experimental conditions. The fact that DES treated mice develop this abnormal response to glucose earlier than controls (2 months vs. 6 months) suggests these mice may have earlier onset of obesity associated disease. In support of this, earlier studies from our laboratory have shown a high prevalence of islet cell hyperplasia in the pancreas of DES treated mice supporting the idea that these mice have abnormal glucose metabolism (32).
Figure 6. Glucose Test of Adult Mice Following Neonatal DES Exposure.
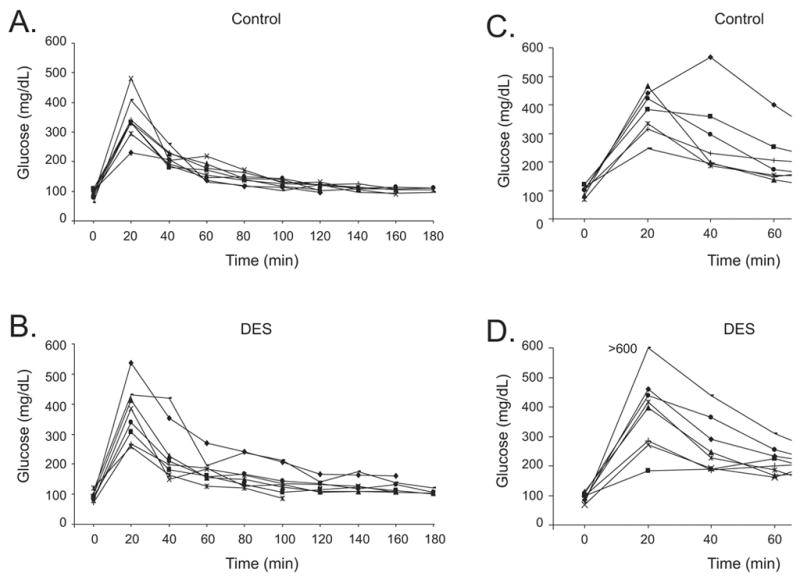
Glucose tolerance was determined at 2 and 6 months of age by measuring morning glucose (mg/dL) after 18 hour fasting period (n=8 per group). Mice were challenged with a glucose solution (2 g/kg) and then glucose levels were measured at 20 min intervals for 180 min. Glucose measurements were plotted individually. Panel A: 2 month control; Panel B: 2 month DES treated; Panel C: 6 month control; Panel D: 6 month DES treated.
Summary and Conclusions
Taken together, our data supports the idea that brief exposure early in life to environmental endocrine disrupting chemicals, especially those with estrogenic activity like DES, increases body weight as the mice age. These data also suggest that these chemicals may contribute to overweight and obesity and other obesity-associated diseases such as type 2 diabetes and cardiovascular disease. Whether our results can be extrapolated to humans as the reproductive abnormalities from the DES mouse model did, remain to be determined but it provides a fruitful area of further research. In addition, the use of this animal model to study “obesogens” and mechanisms involved in altered weight homeostasis (direct and/or endocrine feedback loops, i.e., ghrelin, leptin, etc.) by environmental endocrine disrupting chemicals is an important basic research area that may be addressed by using this model. No longer can we assume than overweight and obesity are simply personal choices, but we have to consider that complex events including environmental chemicals are contributing to this mounting human health problem.
Acknowledgments
The authors would like to thank Page Myers for expert technical assistance in obtaining glucose measurements for this study. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Colborn T, Clement C. Chemically-Induced Alterations in Sexual and Functional Development: The Wildlife/Human Connection. Princeton Scientific; Princeton: 1992. [Google Scholar]
- 2.Colborn T, vom Saal FS, Soto AM. Developmental effects of endocrine-disrupting chemicals in wildlife and humans [see comments] Environ Health Perspect. 1993;101:378–84. doi: 10.1289/ehp.93101378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Colborn T, Dumanski D, Myers JP. Our Stolen Future. Penguin Books, Inc; New York: 1996. [Google Scholar]
- 4.McLachlan JA. Estrogens in the Environment. Elsevier Science Publishing Co; New York: 1985. [Google Scholar]
- 5.McLachlan JA. Estrogens in the Environment II. Elsevier; New York: 1995. [Google Scholar]
- 6.Newbold RR. Diethylstilbestrol (DES) and environmental estrogens influence the developing female reproductive system. CRC Press; Boca Raton: 1999. [Google Scholar]
- 7.Update Endocrine Disruptors: Effects on Male and Female Reproductive Systems. CRC Press; Boca Raton: 2005. [Google Scholar]
- 8.Newbold RR, Padilla-Banks E, Snyder RJ, Jefferson WN. Developmental exposure to estrogenic compounds and obesity. Birth Defects Res A Clin Mol Teratol. 2005;73:478–80. doi: 10.1002/bdra.20147. [DOI] [PubMed] [Google Scholar]
- 9.Heindel JJ, Levin E. Developmental origins and environmental influences--Introduction. NIEHS symposium. Birth Defects Res A Clin Mol Teratol. 2005;73:469. doi: 10.1002/bdra.20141. [DOI] [PubMed] [Google Scholar]
- 10.Heindel JJ. Endocrine disruptors and the obesity epidemic. Toxicol Sci. 2003;76:247–9. doi: 10.1093/toxsci/kfg255. [DOI] [PubMed] [Google Scholar]
- 11.Grun F, Blumberg B. Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology. 2006;147:S50–5. doi: 10.1210/en.2005-1129. [DOI] [PubMed] [Google Scholar]
- 12.Bern H. The fragile fetus. In: Colborn T, Clement C, editors. Chemically-Induced Alterations in Sexual and Functioal Development: The Wildlife/Human Connection. Princeton Scientific Publishing Co; Princeton: 1992. [Google Scholar]
- 13.Barker DJ, Eriksson JG, Forsen T, Osmond C. Fetal origins of adult disease: strength of effects and biological basis. Int J Epidemiol. 2002;31:1235–9. doi: 10.1093/ije/31.6.1235. [DOI] [PubMed] [Google Scholar]
- 14.Hanson M, Gluckman P, Bier D, et al. Report on the 2nd World Congress on Fetal Origins of Adult Disease, Brighton, U.K., June 7–10, 2003. Pediatr Res. 2004;55:894–7. doi: 10.1203/01.PDR.0000115682.23617.03. [DOI] [PubMed] [Google Scholar]
- 15.Gluckman PD, Hanson MA. Developmental origins of disease paradigm: a mechanistic and evolutionary perspective. Pediatr Res. 2004;56:311–7. doi: 10.1203/01.PDR.0000135998.08025.FB. [DOI] [PubMed] [Google Scholar]
- 16.Herbst AL, Bern HA. Developmental Effects of Diethylstilbestrol (DES) in Pregnancy. Thieme-Stratton; New York: 1981. [Google Scholar]
- 17.Giusti RM, Iwamoto K, Hatch EE. Diethylstilbestrol revisited: a review of the long-term health effects. Ann Intern Med. 1995;122:778–88. doi: 10.7326/0003-4819-122-10-199505150-00008. [DOI] [PubMed] [Google Scholar]
- 18.Oken E, Gillman MW. Fetal origins of obesity. Obes Res. 2003;11:496–506. doi: 10.1038/oby.2003.69. [DOI] [PubMed] [Google Scholar]
- 19.Baillie-Hamilton PF. Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med. 2002;8:185–92. doi: 10.1089/107555302317371479. [DOI] [PubMed] [Google Scholar]
- 20.Wardle J, Cooke L. The impact of obesity on psychological well-being. Best Pract Res Clin Endocrinol Metab. 2005;19:421–40. doi: 10.1016/j.beem.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 21.Li S, Hansman R, Newbold R, Davis B, McLachlan JA, Barrett JC. Neonatal diethylstilbestrol exposure induces persistent elevation of c-fos expression and hypomethylation in its exon-4 in mouse uterus. Mol Carcinog. 2003;38:78–84. doi: 10.1002/mc.10147. [DOI] [PubMed] [Google Scholar]
- 22.Li S, Washburn KA, Moore R, et al. Developmental exposure to diethylstilbestrol elicits demethylation of estrogen-responsive lactoferrin gene in mouse uterus. Cancer Res. 1997;57:4356–9. [PubMed] [Google Scholar]
- 23.Newbold RR, McLachlan JA. Transplacental hormonal carcinogenesis: diethylstilbestrol as an example. In: Huff J, Boyd J, Barrett JC, editors. Cellular and Molecular Mechanisms of Hormonal Carcinogenesis: Environmental Influences. Wiley-Liss; New York: 1996. pp. 131–147. [Google Scholar]
- 24.Nikaido Y, Yoshizawa K, Danbara N, et al. Effects of maternal xenoestrogen exposure on development of the reproductive tract and mammary gland in female CD-1 mouse offspring. Reprod Toxicol. 2004;18:803–11. doi: 10.1016/j.reprotox.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 25.Tabb MM, Blumberg B. New modes of action for endocrine-disrupting chemicals. Mol Endocrinol. 2006;20:475–82. doi: 10.1210/me.2004-0513. [DOI] [PubMed] [Google Scholar]
- 26.Newbold R. Cellular and molecular effects of developmental exposure to diethylstilbestrol: implications for other environmental estrogens. Environ Health Perspect 103 Suppl. 1995;7:83–7. doi: 10.1289/ehp.95103s783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newbold RR, Padilla-Banks E, Jefferson WN. Adverse effects of the model environmental estrogen diethylstilbestrol are transmitted to subsequent generations. Endocrinology. 2006;147:S11–7. doi: 10.1210/en.2005-1164. [DOI] [PubMed] [Google Scholar]
- 28.Gillum RF. The association of body fat distribution with hypertension, hypertensive heart disease, coronary heart disease, diabetes and cardiovascular risk factors in men and women aged 18–79 years. J Chronic Dis. 1987;40:421–8. doi: 10.1016/0021-9681(87)90175-5. [DOI] [PubMed] [Google Scholar]
- 29.Gesta S, Bluher M, Yamamoto Y, et al. Evidence for a role of developmental genes in the origin of obesity and body fat distribution. Proc Natl Acad Sci U S A. 2006;103:6676–81. doi: 10.1073/pnas.0601752103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Penza M, Montani C, Romani A, et al. Genistein affects adipose tissue deposition in a dose-dependent and gender-specific manner. Endocrinology. 2006 doi: 10.1210/en.2006-0365. [DOI] [PubMed] [Google Scholar]
- 31.Thigpen JE, Setchell KD, Ahlmark KB, et al. Phytoestrogen content of purified, open- and closed-formula laboratory animal diets. Lab Anim Sci. 1999;49:530–6. [PubMed] [Google Scholar]
- 32.McLachlan JA, Newbold RR, Bullock BC. Long-term effects on the female mouse genital tract associated with prenatal exposure to diethylstilbestrol. Cancer Research. 1980;40:3988–3999. [PubMed] [Google Scholar]