

Abstract
We extend our Bayesian model selection framework for mapping epistatic QTL in experimental crosses to include environmental effects and gene–environment interactions. We propose a new, fast Markov chain Monte Carlo algorithm to explore the posterior distribution of unknowns. In addition, we take advantage of any prior knowledge about genetic architecture to increase posterior probability on more probable models. These enhancements have significant computational advantages in models with many effects. We illustrate the proposed method by detecting new epistatic and gene–sex interactions for obesity-related traits in two real data sets of mice. Our method has been implemented in the freely available package R/qtlbim (http://www.qtlbim.org) to facilitate the general usage of the Bayesian methodology for genomewide interacting QTL analysis.
MAPPING quantitative trait loci (QTL) involves inferring the genetic architecture of complex traits in terms of genomic regions, gene effect, gene action, and possible interactions, given observed phenotype and marker genotype data (Lynch and Walsh 1998). The variation of most complex traits results from interacting networks of multiple QTL and environmental factors (Reifsnyder et al. 2000; Carlborg and Haley 2004; Moore 2005; Stylianou et al. 2006; Valdar et al. 2006; Wang et al. 2006). Inclusion of gene–gene interactions (epistasis) and gene–environment interactions in mapping QTL is expected to aid the discovery of more QTL, improve the accuracy and precision of estimates of their genomic positions and genetic effects, and enhance our ability to understand the genetic basis of complex traits (Jansen 2003; Carlborg and Haley 2004).
Identification of genomewide interacting QTL has been a formidable challenge for geneticists and statisticians, mainly due to numerous possible variables associated with hundreds or thousands of genomic loci (markers and/or loci within marker intervals) that lead to a huge number of possible models (e.g., Yi et al. 2005). The problem is further complicated by the facts that the genomic loci on the same chromosome are highly correlated and the genotypes at many loci are unobservable. Traditional QTL mapping methods utilize prespecified simple statistical models, which fit the effects of only one or two QTL whose putative positions are scanned across the genome (e.g., Lander and Botstein 1989; Haley and Knott 1992; Jansen and Stam 1994; Zeng 1994). Although successful in many applications, such approaches require prohibitive corrections for multiple testing and ignore the nature of complex traits in statistical modeling.
Multiple-QTL mapping has been viewed as a model selection issue (Broman and Speed 2002; Sillanpää and Corander 2002; Yi 2004). Rather than fitting prespecified models to the observed data, model selection approaches proceed by identifying the QTL models from a set of potential QTL models that are best supported by the data. Various model selection methods have been recently proposed for genomewide multiple-QTL mapping from both frequentist and Bayesian perspectives. Frequentist approaches sequentially add or delete QTL using forward and backward or stepwise selection procedures and apply criteria such as P-values or a modified Bayesian information criterion (BIC) to identify the “best multiple-QTL model” (Kao et al. 1999; Carlborg et al. 2000; Reifsnyder et al. 2000; Bogdan et al. 2004; Baierl et al. 2006). Such methods usually pick a single “good” (and maybe useful) model, ignoring the uncertainty about the model itself in the final inference (Raftery et al. 1997; George 2000; Kadane and Lazar 2004).
Several Bayesian model selection approaches for mapping multiple QTL have been developed over the past decade (Satagopan and Yandell 1996; Satagopan et al. 1996; Heath 1997; Sillanpää and Arjas 1998; Stephens and Fisch 1998; Gaffney 2001; Hoeschele 2001; Sen and Churchill 2001; Xu 2003; Wang et al. 2005; Zhang et al. 2005). Bayesian approaches for multiple-QTL mapping build on the likelihood function for the observed phenotypic and marker data, by assigning a prior probability to each model and prior distributions to the unknowns of each model. Inference is then based on the conditional distribution of the unknowns given the observed data, the posterior distribution. The Bayesian approach can simultaneously address both model and parameter uncertainty (Raftery et al. 1997; Chipman et al. 2001). However, its practical implementation entails two major challenges: calculation of the posterior distribution and specification of the prior distributions.
Markov chain Monte Carlo (MCMC) algorithms have been recently developed to map multiple epistatic QTL (Yi and Xu 2002; Yi et al. 2003, 2005; Narita and Sasaki 2004). Yi et al. (2005) described a Bayesian model selection method for identifying epistatic QTL in experimental crosses, based on the composite model space framework of Yi (2004). This approach places an upper bound on the number of detectable QTL and employs latent binary variables to indicate which main and epistatic effects of putative QTL are included in or excluded from the model. The key advantage of the composite model space approach is that it provides a convenient way to reasonably reduce the model space and to construct efficient MCMC algorithms. Yi et al. (2005) developed a full Gibbs sampler to explore the posterior. This Gibbs sampling scheme works well in models with small upper bounds (Yi et al. 2005, 2006). However, it is computationally demanding when the number of possible genetic effects is large.
The contributions of this article are to develop a new, fast sampling scheme to explore the posterior and to propose new prior distributions for two types of key parameters, genetic effects and indicator variables. The new MCMC algorithm has significant computational advantages over the previous algorithms, allowing us to identify interacting QTL fairly quickly even in models with large numbers of possible genetic effects. The new priors can better incorporate our prior knowledge about genetic architecture of complex traits into the model and induce increased posterior probability on more probable models. We extend the composite model space approach to model arbitrary covariates and simultaneously detect gene–gene and gene–environment interactions. While both gene–gene and gene–environment interactions significantly influence many complex traits, simultaneous identification of these interactions has not received significant attention. Benefits of the proposed method are illustrated by analyzing two obesity data sets of mice.
BAYESIAN MODELING OF GENOMEWIDE INTERACTING QTL
Composite model space approach and interacting QTL models:
Here we extend the composite model space approach of Yi et al. (2005) to simultaneously model main and epistatic effects of QTL, environmental effects, and gene–environment interactions. We describe only interactions between main effects of QTL and fixed-effect environments although the proposed method can be extended to more complicated interactions. Most phenotypes under study are affected by both genotype and environment. Accounting for environmental effects can dramatically reduce residual variation. Further, genotypic effects may vary by environment, making it important to consider gene–environment interactions. Here, environment is broadly interpreted as any nongenetic influence that can be measured, including sex, location, and other phenotypic traits under study. We use the term covariate synonymously with environment. Including relevant covariates in QTL mapping can partially address some features of design (e.g., block effects, gradients) and can help identify alternate sets of QTL that may be involved in different pathways (Stylianou et al. 2006).
We approximate positions for all possible QTL using a partition of the entire genome into evenly spaced loci, including all observed markers and additional loci, or pseudomarkers (Sen and Churchill 2001), between flanking markers. Before mapping QTL, we calculate the probabilities of genotypes at these preset loci given the observed marker data as priors of QTL genotypes in our Bayesian framework. We place an upper bound on the number of QTL included in the model. This upper bound is larger than the number of detectable QTL with high probability for a given data set.
We use Cockerham's genetic model to construct main effects, epistasis, and gene– environment interactions, although other genetic models are possible (Kao and Zeng 2002), and we apply conventional methods used in hierarchical linear models to construct environmental effects (e.g., Lynch and Walsh 1998; Gelman et al. 2003). Even with a moderate number of the upper bound, there are many possible genetic effects when considering interactions, but most are negligible and can be excluded. We use an unobserved vector of binary variables γ to indicate which genetic effects (main effects, epistatic effects, and gene–environment interactions) across the possible loci are included in (γj = 1) or excluded from (γj = 0) the model. The indicator vector γ determines the number of included QTL and the activity of the associated genetic effects. We denote the positions of the included QTL by λ. The vector (γ, λ) thus determines the genetic architecture, the number and position of QTL, and their gene action. The goal of our Bayesian approach is to infer the posterior distribution of (γ, λ) and estimate the associated genetic effects.
Suppose all genotypes are known across the genome. We denote the design matrices of selected main, epistatic effects, and gene–environment interactions by XG, XGG, and XGE, respectively, and the design matrix of environmental effects by XE. The design matrices XG, XGG, and XGE are determined from the genotypes of QTL through Cockerham's genetic model. Given (γ, λ) and (XE, XG, XGG, XGE), the phenotype y is expressed as
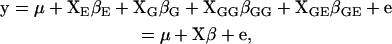 |
(1) |
where μ = (μ, · · · , μ)T is the overall mean; βE are the environmental effects; βG, βGG, and βGE are the selected main effects, epistatic effects, and gene–environment interactions, respectively; and e are independent normal errors with mean zero and variance σ2. We always include the environmental terms XEβE in the model, and hence conventional hierarchical linear models are a special case. To simplify notation, we organize all effects into β and all design matrices into X.
Prior specification:
Bayesian modeling involves explicit priors that state the degree of uncertainty for all unknown aspects of a model. Unknowns for our Bayesian QTL modeling include the indicators γ, positions of QTL λ, effects β, overall mean μ, and residual variance σ2, as well as the QTL genotypes g that determine the design matrix (XG, XGG, XGE). For μ, σ2, λ, and g, we use the priors proposed in Yi et al. (2005). We here suggest new priors on effects β and indicators γ that can restrict their values in a reasonable region of the parameter space and thus induce increased posterior probability on more probable models.
Dependence priors on genetic architecture indicators:
Independence priors for γ work well for many situations (Yi et al. 2005, 2006), but may not be appropriate when either (1) loci with large main effects are more likely to have large interactions or (2) many loci have detectable main effects and thus the probability of detecting additional QTL with weak main effects but strong interactions is low. We here propose dependence priors capturing relations between interaction and main-effect terms (see Chipman 1996, 2004; Chipman et al. 2001). Below we detail dependence priors for epistasis; the same idea extends to gene–environment interactions.
Consider two QTL indexed by j and k, with main-effect and epistasis indicators γj, γk, and γjk. Setting a common inclusion probability for main effects, 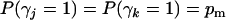 (Yi et al. 2005), we construct conditional inclusion probabilities for epistasis as
(Yi et al. 2005), we construct conditional inclusion probabilities for epistasis as
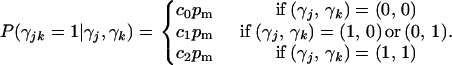 |
Typically, 0 ≤ c0 ≤ c1 ≤ c2 ≤ 1, implying that main effects are more likely to be detected than epistasis and that the importance of an interaction depends on the importance of its “parent” terms. Setting some ch to zero rules out certain interactions: c0 = c1= 0 and c2 > 0 allows interactions only if both main effects are included. These values establish a principle of variable selection, modifying prior mass across possible genetic architecture and greatly reducing the model space.
Hierarchical priors on effects:
We want effect priors that are invariant to the scales of the phenotype and the contrasts and model complexity. This can be accomplished by hierarchical models in which the priors have empirical hyperpriors that depend on the proportion of phenotypic variance explained by the effect. We partition the genetic effects into batches, corresponding to different types of effects, e.g., additive, dominance, additive–additive, additive–environment interactions, etc. Effects in the same batch k follow the same prior, 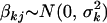 . The prior variance
. The prior variance 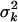 is a random variable with an inverse-χ2 hyperprior,
is a random variable with an inverse-χ2 hyperprior, 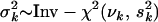 , and has expected value
, and has expected value 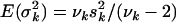 . The degrees of freedom νk controls the skew of the prior for
. The degrees of freedom νk controls the skew of the prior for 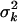 , with larger values recommended (here νk = 6) to tightly center the prior around
, with larger values recommended (here νk = 6) to tightly center the prior around 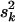 (see Chipman 2004). The scale sk2 is chosen to control the prior confidence region of the proportion of the phenotypic variance explained by βkj (i.e., heritability) (also see Gaffney 2001). The proportion of phenotypic variance explained by βkj is hkj = Vkj
(see Chipman 2004). The scale sk2 is chosen to control the prior confidence region of the proportion of the phenotypic variance explained by βkj (i.e., heritability) (also see Gaffney 2001). The proportion of phenotypic variance explained by βkj is hkj = Vkj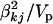 , with Vkj the sample variance for the column of X associated with effect βkj and Vp the total phenotypic variance. Setting
, with Vkj the sample variance for the column of X associated with effect βkj and Vp the total phenotypic variance. Setting 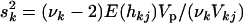 yields E(hkj) = VkjE(
yields E(hkj) = VkjE(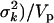 . Expected effect heritabilities, E(hkj), can be set small (say 0.05–0.2) to reflect prior knowledge about genetic architecture. Environmental random effects have normal hierarchical priors similar to the above genetic effects; fixed-effect covariates have uniform empirical priors (see Gelman et al. 2003).
. Expected effect heritabilities, E(hkj), can be set small (say 0.05–0.2) to reflect prior knowledge about genetic architecture. Environmental random effects have normal hierarchical priors similar to the above genetic effects; fixed-effect covariates have uniform empirical priors (see Gelman et al. 2003).
AN EFFICIENT SAMPLING SCHEME
Now that we have established the model, we then describe our MCMC algorithm to explore the posterior. The joint posterior is proportional to the product of the phenotype likelihood function, 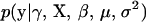 , and the prior distributions of all unknowns,
, and the prior distributions of all unknowns,
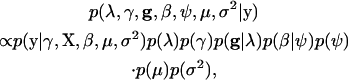 |
(2) |
in which ψ represents all variance parameters for β. For notational convenience, we suppress the dependence on marker data and covariates here and in subsequent notation.
Our algorithm alternately updates unknowns (λ, γ, g, β, ψ, μ, σ2). Given λ, γ, and g, model (1) is a conventional hierarchical linear model, and hence we can update the parameters μ and β given (ψ, σ2) from normal distributions and all elements of (ψ, σ2) from the independent inverse-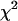 conditional posterior distribution given (μ, β) (Gelman et al. 2003). The conditional posterior distribution of each element of g is multinomial and thus can be sampled directly as well. The conditional posterior of each element of λ has no standard form, but the traditional Metropolis–Hastings algorithm can be used to update the vector λ one at time (Yi et al. 2005).
conditional posterior distribution given (μ, β) (Gelman et al. 2003). The conditional posterior distribution of each element of g is multinomial and thus can be sampled directly as well. The conditional posterior of each element of λ has no standard form, but the traditional Metropolis–Hastings algorithm can be used to update the vector λ one at time (Yi et al. 2005).
We improve our MCMC algorithm efficiently in sampling the indicators γ when there are many effects. We first modify the Gibbs sampler of Yi et al. (2005) to incorporate the new priors proposed herein and describe its drawback in models with very large numbers of effects where many of effects are negligible in size. We then develop a new, fast Metropolis–Hastings algorithm and discuss why the new algorithm is more efficient than the Gibbs sampler.
At each iteration of the MCMC simulation, the full Gibbs sampler proceeds to generate all the indicator variables, γj, from its conditional posterior distribution,
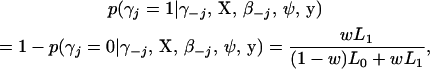 |
(3) |
in which “−j” means all elements except the jth, w = p(γj = 1 | γ−j) is the prior inclusion probability of the jth element, and Lm = p(y | γj = m, γ−j, X, β−j, ψ) for m = 0, 1. Note that βj is integrated out from L1. A convenient way to calculate L1 is to use the following identity:
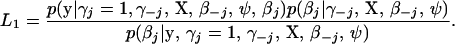 |
(4) |
Since L1 is independent of βj, we can compute it by inserting any value of βj into this expression. A convenient and stable choice for βj is the conditional posterior mean (Gelman et al. 2003).
This Gibbs sampling scheme works reliably (Yi et al. 2005, 2006). However, it is computationally demanding when the number of possible genetic effects (i.e., the number of indicator variables) is large. To understand this, we note that:
Even with a moderate L, the number of possible effects is large. For example, for a F2 population, taking L = 20 leads to 40 (= 2L) possible main effects, 760 (= 2L(L − 1)) possible epistatic effects, and many possible gene–environment interactions. Using the Gibbs sampler it is necessary to compute the conditional posterior probability (3) for each γj. Therefore, the Gibbs sampler is usually computationally demanding.
Most of the genetic effects are near zero and thus γj is zero for most j. If the current value of γj is 0, γj is likely to be regenerated as zero because the prior probability w = p(γj = 1 | γ−j) in (4) is very small. In the Gibbs sampler, it is always necessary to calculate the conditional posterior probability (4) when γj is currently 0. Such computation may be wasteful.
We here propose a new Metropolis–Hastings scheme to update γ that offers significant computational advantages over the Gibbs sampler without sacrifice of statistical efficiency when the number of possible effects is large. Our Metropolis–Hastings scheme extends the Bayesian variable selection method of Kohn et al. (2001) to genomewide interacting QTL analysis. As the full Gibbs sampler, at each iteration of the MCMC simulation, the new algorithm proceeds to update all indicator variables. Denote the current value of γj by C (= 0 or 1). Our new algorithm first proposes a new value P (= 0 or 1) for γj from the conditional prior probability p(γj = C | γ−j). If P = C, the Metropolis–Hastings acceptance probability is 1, and thus γj remains at C and there is no need to compute any values, otherwise, we update γj from the current value C to the proposal 1 − C with acceptance probability
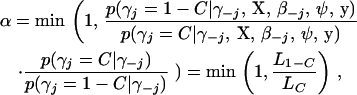 |
(5) |
in which all terms are defined in (3). If γj is currently 1 (i.e., βj is currently included in the model), we can calculate the two values L0 and L1 using the prior variance of βj and the column of X corresponding to the effect βj. If γj is currently 0 (i.e., βj is currently excluded in the model) and the involved QTL(s) is (are) not currently in the model, we first expand X, sampling one or two new QTL position(s) as needed, new genotypes for all individuals, and the prior variance of βj if this parameter is currently out of the model, from the corresponding priors, and then calculate the acceptance probability to update γj. This procedure is also needed for the full Gibbs sampler (Yi et al. 2005).
In this Metropolis–Hastings algorithm, the proposal probability to generate γj = 1 when it is currently 0 is p(γj = 1 | γ−j), which is very small when the number of possible genetic effects is large and most of them are near 0, and thus γj is likely to be proposed as 0. Therefore, it is unnecessary to compute any values for most γj, and hence this new algorithm is much faster than the full Gibbs sampler.
We illustrate the relative advantages of the Gibbs sampler to our new Metropolis–Hastings algorithm in terms of statistical efficiency. The transition probability for γj from C to P, Q(C → P), is
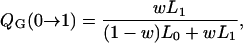 |
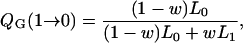 |
and
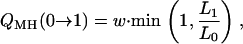 |
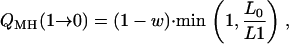 |
for the Gibbs sampler and the Metropolis–Hastings algorithm, respectively, with w = p(γj = 1 | γ−j). Following Kohn et al. (2001), QG(C → 1 − C) > QMH(C → 1 − C). Thus, the Gibbs sampler is statistically more efficient per scan than the Metropolis–Hastings algorithm in terms of transition probabilities. When the upper bound of QTL is large and w is small, the new faster algorithm does not sacrifice much statistical efficiency, since it can be easily shown that QMH(C → 1 − C) ≈ QG(C → 1 − C).
SUMMARIZING AND INTERPRETING THE POSTERIOR SAMPLES
The mixing behavior and convergence rates of MCMC algorithms become a critical issue for a high-dimensional model space problem. Various methods to assess mixing and convergence have been developed and implemented in the package R/coda (Plummer et al. 2004). These diagnostic tools help monitor scalar estimates of interest, such as the numbers of QTL and epistatic effects.
The posterior samples can be used to estimate the posterior distribution and search for models with high posterior probability. Larger effects should appear more often, making them easier to identify. We use all the saved iterations of the Markov chain, corresponding to model averaging, which assesses characteristics of the genetic architecture by averaging over possible models weighted by their posterior probability. Model averaging accounts for model uncertainty and hence provides more robust inference compared to a single “best” model approach (Raftery et al. 1997; Ball 2001; Sillanpää and Corander 2002).
We can use various methods to graphically and numerically summarize and interpret the posterior samples. The posterior inclusion probability for each locus is estimated as its frequency in the posterior samples. Each locus may be included in the model through its main effects and/or interactions with other loci (epistasis) or environmental effects. The larger the effect size is for a locus, the more frequently the locus is sampled. Taking the prior probability into consideration, we use Bayes factors (BF) to show evidence for inclusion against exclusion of a locus. The Bayes factor for a locus is defined as the ratio of the posterior odds to the prior odds for inclusion against exclusion of the locus (Kass and Raftery 1995). Traditionally, a BF threshold of 3, or 2 loge(BF) = 2.1, supports a claim of significance (Kass and Raftery 1995). We can separately estimate the posterior inclusion probability and corresponding Bayes factors of main effects, epistasis, and gene–environment interactions per locus or pair of loci. The proportions of phenotypic variance explained by the different effects (heritabilities) can also be estimated.
IMPLEMENTATION IN R/QTLBIM
We have implemented the method proposed herein and the Gibbs sampler of Yi et al. (2005) in the freely available package R/qtlbim (Yandell et al. 2007). R/qtlbim is an extensible, interactive environment for Bayesian analysis of multiple interacting QTL in experimental crosses. It is built on the widely used R/qtl package (Broman et al. 2003) and includes all its advantages for extensibility. In R/qtlbim, the computationally intensive MCMC algorithms are written in C, with data manipulation and graphics in R.
R/qtlbim provides tools to monitor mixing behavior and convergence of the simulated Markov chain, either by examining trace plots of the sample values of scalar quantities of interest, such as the numbers of QTL and epistatic effects, or by using formal diagnostic methods provided in the package R/coda. R/qtlbim provides extensive informative graphical and numerical summaries of the MCMC output to infer and interpret the genetic architecture of complex traits (Yandell et al. 2007).
REAL DATA EXAMPLES
We illustrate the application of our proposed method by reanalyses of two real obesity data sets. The first data set is a large F2 mouse intercross described in Rocha et al. (2004), where a large number of main-effects QTL were detected using traditional interval mapping for body weight at 6 weeks of age (WK6). We used this data set to show that the use of the new dependence prior on indicator variables can detect stronger evidence for epistatic interactions and the new algorithm has huge computational advantage over the previous algorithm. The second data set is a mouse backcross described in Yi et al. (2005), where three main-effects QTL were found to influence the trait Fat, a sum of right gonadal and hindlimb subcutaneous fat pads. Reanalysis of these backcross data shows that even for models with relatively small numbers of possible genetic effects our new algorithm still gives substantial computational improvement.
For all analyses, the MCMC algorithm ran for 2 × 105 iterations after discarding the first 1000 iterations as burn-in. To reduce serial correlation in the stored samples, the chain was thinned by one in k = 40, yielding 5 × 103 samples for posterior analysis. To assess convergence and mixing behavior, we ran three parallel MCMC sequences with starting points randomly generated from the priors and used the potential scale reduction factor 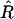 that compares the between- and within-sequence variances for any scalar estimands (Gelman et al. 2003; Plummer et al. 2004). For several scalar estimands (e.g., the numbers of QTL and epistatic effects and the total genetic variance),
that compares the between- and within-sequence variances for any scalar estimands (Gelman et al. 2003; Plummer et al. 2004). For several scalar estimands (e.g., the numbers of QTL and epistatic effects and the total genetic variance), 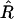 fell below 1.1 quickly, indicating that the chains mixed well and converged rapidly.
fell below 1.1 quickly, indicating that the chains mixed well and converged rapidly.
In the study of the large F2 intercross (Rocha et al. 2004) where there was evidence for many QTL, MCMC sampling time was reduced from 8 hr on a P4 personal computer with the Gibbs sampler of Yi et al. (2005) to ∼1 hr with our new algorithm. For the backcross data analyzed below, the Gibbs sampler took 80 min while the new Metropolis–Hastings algorithm took 15 min. For the two data sets, the two algorithms gave essentially identical results.
Real data I:
A total of 993 mice (554 males and 439 females) were bred from two lines of mice selected for increased 3- to 6-week weight gain (M16i) and low 6-week weight (L6); L6 males were mated to M16i females, with the resulting F1's inter se mated (no full-sib pairings) in two consecutive replicates encompassing a total of 64 full-sib F2 families (Rocha et al. 2004). The two replicates consisted of 490 and 503 mice, respectively, and the numbers of mice in 64 families ranged from 4 to 21. Although many traits were measured in this intercross, for the purpose of illustration, we analyzed only body weight at 6 weeks of age (WK6). A total of 63 fully informative microsatellite markers spanning the 19 autosomes were genotyped. The marker linkage map covered 1200 cM (Kosambi) with an average spacing of 28 cM.
This large F2 data set was analyzed in Rocha et al. (2004), using standard composite-interval mapping (Zeng 1994). For WK6, 11 chromosomes were detected to have evidence of QTL activity with main effects, and most of the detected QTL had only significant additive effects. Marker-based linear models were used to test epistatic interactions among markers. An interaction between two markers on chromosomes 6 and 17 was detected (P = 0.0014). Rocha et al. (2004) performed separate analyses for each gender and found no evidence of gene–sex interactions.
We partitioned each chromosome into a 1-cM grid, resulting in 1200 possible loci across the genome. The factors sex and replicate were treated as fixed binary covariates and family as a categorical random covariate in the model. These three covariates were always included in the model. We considered gene–gene and gene–sex interactions. Two types of priors on the indicator variables γ, independence and dependence priors, were used and compared.
In the analysis with independence priors on γ, the prior number of main-effect QTL was set at lm = 12 and the prior expected number of all QTL (l0) was taken to be lm + 3, allowing for some additional epistatic QTL with weak main effects. The upper bound of the number of QTL, L, was then 26 (= 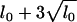 , see Yi et al. 2005). To check prior sensitivity, we reran the algorithm with several other values of lm and l0 and obtained essentially identical results (data not shown). Using the above upper bound and the independence priors on the indicator variables, the total number of genetic effects was 1404, including 52 main effects, 52 gene–sex interactions, and 1300 epistatic effects.
, see Yi et al. 2005). To check prior sensitivity, we reran the algorithm with several other values of lm and l0 and obtained essentially identical results (data not shown). Using the above upper bound and the independence priors on the indicator variables, the total number of genetic effects was 1404, including 52 main effects, 52 gene–sex interactions, and 1300 epistatic effects.
For the analysis with independence prior, the genomewide profile of Bayes factors comparing the model with and without the locus showed evidence of QTL activity on 13 chromosomes (2 logeBF > 2.1) (Figure 1). Most of the loci were included mainly through their additive effects, similar to the results of Rocha et al. (2004). However, our Bayesian analysis found that QTL on chromosomes 3, 4, 6, 11, 12, and 17 interacted with sex, and QTL on chromosomes 3, 6, 12, and 17 had additive–additive interactions. The values of 2 logeBF for additive–additive interactions were ∼2.1 on chromosomes 3, 6, and 12 and 6 on chromosome 17. A QTL on chromosome 3 interacted with a QTL on chromosome 12 and a QTL on chromosome 6 interacted with a QTL on chromosome 17. The proportion of the phenotypic variance explained by each locus (i.e., heritability) was <6%, indicating that WK6 is a typical complex polygenic trait controlled by many loci, each with relatively small effect. Although the proportion of the phenotypic variance explained by epistasis was low, these epistatic effects were detectable using our multiple-QTL approach.
Figure 1.—

F2 data analysis with independence priors on the indicator variables γ and the new Metropolis–Hastings algorithm profiles of Bayes factors (rescaled as 2 logeBF and negative values are truncated as zero): (a) for all combined effects (additive, dominance, epistatic, and gene–sex effects); (b) for main effects, solid and dashed lines represent additive and dominance effects, respectively; (c) for gene–sex interactions, solid and dashed lines represent additive–sex and dominance–sex interactions, respectively; (d) for epistatic interactions, solid lines represent additive–additive interactions and other epistatic effects were not detected. On the x-axis, outer tick marks represent chromosomes and inner tick marks represent markers.
The above analysis with independence priors on the indicator variables detected a large number of main-effect QTL and two epistatic effects whose main effects were detectable. These results indicated that the probability of detecting additional QTL with weak main effects but strong epistasis was low and thus motivated us to use dependence priors on the indicator variables γ. Our second analysis used dependence priors, with c0 = c1 = 0 and c2 = 0.1, thus allowing an interaction to enter the model only if both corresponding main effects were included in the model. This dependence prior ruled out many “unrealistic” models from consideration and thus greatly reduced model space.
Figure 2 displays the genomewide profile of Bayes factors, comparing the model with and without the locus for the analysis with dependence priors. This analysis detected the same chromosomal regions as those in the first analysis. As expected, this second analysis detected the same main effects and gene–sex interactions as in the first analysis. However, the second analysis detected not only much stronger evidence of epistatic effects for chromosomes 3, 6, 12, and 17, but also an additional epistatic effect for chromosome 10 (see the bottom of Figure 2). This may have resulted from the fact that we used dependence priors to focus on promising models. Each main effect explained 3–5% of phenotypic variation while each interaction explained 1–3% when present. As expected, this analysis uncovered the same interaction pattern of chromosomes 3, 6, 12, and 17 as in the first analysis and an additional epistatic interaction between chromosomes 10 and 17, although this interaction was weaker (Figure 3).
Figure 2.—

F2 data analysis with dependence priors on the indicator variables γ and the new Metropolis–Hastings algorithm [one-dimensional profiles of Bayes factors (rescaled as 2 logeBF and negative values are truncated as zero)]: (a) for all combined effects (additive, dominance, epistatic, and gene–sex effects); (b) for main effects, solid and dashed lines represent additive and dominance effects, respectively; (c) for gene–sex interactions, solid and dashed lines represent additive–sex and dominance–sex interactions, respectively; (d) for epistatic interactions, solid lines represent additive–additive interactions and other epistatic effects were not detected. On the x-axis, outer tick marks represent chromosomes and inner tick marks represent markers.
Figure 3.—

F2 data analysis with the new Metropolis–Hastings algorithm: two-dimensional profiles of Bayes factors (rescaled as 2 logeBF and negative values are truncated as zero) and percentage of proportions of variance explained by epistatic effects (heritability) on selected chromosomes. The Bayes factor or heritability of epistasis only is shown above the diagonal; the Bayes factor or heritability comparing the full model with epistasis to no QTL is shown below the diagonal.
Real data II:
A mouse cross was produced from two highly divergent strains: M16i, consisting of large and moderately obese mice, and CAST/Ei, a wild strain of small mice with lean bodies (Yi et al. 2005). CAST/Ei males were mated to M16i females, and F1 males were backcrossed to M16i females, resulting in 54 families and 421 mice (213 males, 208 females) reaching 12 weeks of age. The numbers of mice in 54 families ranged from 4 to 11. All mice were genotyped for 92 microsatellite markers located on 19 autosomal chromosomes. The marker linkage map covered 1214 cM (Haldane). Chromosomes 2, 13, and 15 had 20, 9, and 10 markers, respectively, with an average spacing of 5.5 cM; all other chromosomes had 3–4 markers with average spacing of 28 cM.
In this study, we analyzed Fat, the sum of right gonadal and hindlimb subcutaneous fat pads. In Yi et al. (2005), the phenotypic data were linearly adjusted by sex and family, and the obtained residuals were used as a new phenotype. We here used two models to reanalyze the data. Our first analysis included sex and weight at the age of 12 weeks as binary and continuous fixed covariates, respectively, and family as a categorical random covariate. We permitted the inclusion of gene–gene interactions and gene–sex interactions in the model. In the second analysis, we analyzed log2(Fat) adjusting for sex, log2(weight at 12 weeks), and their interaction as three fixed covariates and include family as a categorical random covariate. Fat and weight distributions were both skewed, corrected by log transformation. We permitted the inclusion of gene–gene interactions and all three types of gene by fixed-covariate interactions in the model.
Each chromosome was partitioned with a 1-cM grid, resulting in 1214 possible loci across the genome. The prior number of main-effect QTL was set at lm = 3, the number of QTL detected in the nonepistatic analyses (Yi et al. 2005), and the prior expected number of all QTL (l0) was taken to be lm + 3. The upper bound of the number of QTL, L, was then 14 (see Yi et al. 2005). The total number of possible genetic effects in this analysis is much smaller than in the first data analysis above. The two algorithms, the Gibbs sampler and the new Metropolis–Hastings algorithm, were used to analyze the data, with independence prior on the indicator variables.
Analysis I:
As shown in Figures 4 and 5, the two algorithms produced essentially identical results. The genomewide profile of Bayes factors comparing the model with and without the locus showed evidence of QTL activity on nine chromosomes. Most of these QTL (i.e., on chromosomes 1, 2, 13, 15, 18, and 19) were detected by Yi et al. (2005), but this reanalysis found new QTL on chromosomes 6, 8, and 14. This supports the fact that including relevant covariates is more appropriate in QTL analysis than using residuals as new phenotypic values. QTL on chromosome 2 had strong main effect and were also found to interact with sex. Except for QTL on chromosome 4, all the detected QTL were found to have strong evidence of epistatic effects; QTL on chromosomes 2, 8, 13, 15, and 19 had not only main effects, but also epistatic effects, while QTL on chromosomes 1, 6, and 18 had only epistatic effects. The two-dimensional profile of Bayes factors comparing the model with and without epistasis is displayed in Figure 6, showing five pairs of strong epistatic interactions, i.e., chromosomes 1 and 18, 2 and 13, 6 and 8, 13 and 15, and 15 and 19.
Figure 4.—

Backcross data analysis with the Gibbs sampler [one-dimensional profiles of Bayes factors as 2 logeBF (negative values are truncated as zero)]: (top) for all combined effects (additive, epistatic, and gene–sex effects); (bottom) for individual effects, solid, dashed, and dotted lines represent additive effects, gene–sex effects, and epistatic interactions, respectively. On the x-axis, outer tick marks represent chromosomes and inner tick marks represent markers.
Figure 5.—

Backcross data analysis with the new Metropolis–Hastings algorithm [one-dimensional profiles of Bayes factors as 2 logeBF (negative values are truncated as zero)]: (top) for all combined effects (additive, epistatic, and gene–sex effects); (bottom) for individual effects, solid, dashed, and dotted lines represent additive effects, gene–sex effects, and epistatic interactions, respectively. On the x-axis, outer tick marks represent chromosomes and inner tick marks represent markers.
Figure 6.—

Backcross data analysis with the new Metropolis–Hastings algorithm: two-dimensional profiles of Bayes factors [rescaled as 2 loge(BF) and negative values are truncated as zero] on selected chromosomes. The Bayes factor of epistasis only is shown above the diagonal; the Bayes factor comparing the full model with epistasis to no QTL is shown below the diagonal.
Analysis II:
The genomewide profiles for main effects, epistasis, and G × E interactions are similar to those in analysis I. We here focus interpretation on the three chromosomes (2, 13, and 15) with denser marker coverage. Figure 7 shows the one-dimensional marginal scan of 2 loge(BF) for (Figure 7a) the complete data set. The combined analysis is dominated by chromosome 2, which shows evidence for main effect, epistasis, and G × E. Chromosomes 13 and 15 show evidence for main effects and possible epistasis and/or G × E. The presence of G × E suggests value in separate analysis for (Figure 7b) females and (Figure 7c) males. Here, log2(weight at 12 weeks) and family were retained as fixed and random covariates, with G × E examined for weight. Figure 7, a–c, shows that there may actually be two distinct QTL on chromosome 2 and that males show evidence for genotype-by-weight interaction on chromosome 13, while females do not.
Figure 7.—

One-dimensional marginal scan of 2 loge(BF) on chromosomes 2, 13, and 15 for (a) both sexes, (b) females, and (c) males. Lines indicate contributions for main effects (solid lines), epistasis (dotted lines), G × E (dashed lines), and the combined sum (dotted-dashed lines). The QTL on chromosome 2 dominates, showing evidence for a main effect and some sort of genotype-by-covariate effect. b and c suggest there may be different QTL for males and females on chromosome 2; thus sex seems to be the primary covariate on this chromosome. While overall evidence of G × E is slight for chromosomes 13 and 15 (a), separate analyses by sex show substantial genotype-by-weight interaction for males (c) but not females (b). Note in males the evidence for some epistasis on chromosomes 2 and 13. Chromosome 15 shows only a modest main-effect QTL for males only.
Figure 8 examines the relationship between Fat and weight at 12 weeks, separating by sex and adjusting within plot by genotype for (Figure 8a) the chromosome 2 QTL or (Figure 8b) the chromosome 13 QTL, using the closest marker to the peaks from Figure 7c. There is a strong QTL effect, but no apparent G × E for chromosome 2. However, the G × E interaction is evident for males when adjusting for chromosome 13 in Figure 8b. A two-dimensional profile of 2 loge(BF) for epistasis found strong evidence between chromosomes 2 and 13, with peak 2 loge(BF) of 5.5 for epistasis and 10.8 for the full model including main effects and epistasis. This evidence for epistasis suggests a more careful look at the G × E interaction with chromosomes 2 and 13, shown in Figure 9. Here we see that the genotype-by-weight interaction is apparent only when the chromosome 2 genotype is H.
Figure 8.—

Lattice plots of log2(Fat2) vs. log2(weight at 12 weeks) by sex, grouped within plot by (a) chromosome 2 QTL genotype or (b) chromosome 13 QTL genotype (A, circles, solid lines; H, triangles, dotted lines). Note the significant difference in slopes only for males grouped by QTL 13.
Figure 9.—

Lattice plots of log2(Fat) vs. log2(weight at 12 weeks) by sex and chromosome 2 QTL genotype (A, H), grouped within plot by chromosome 13 QTL genotype (A, circles, solid lines; H, triangles, dotted lines). Note significant difference in slopes only for males with QTL 2 genotype of H.
DISCUSSION
We have extended the composite model space method for mapping epistatic QTL of Yi et al. (2005) to simultaneously model and detect main effects of multiple QTL, gene–gene interactions, arbitrary covariates, and gene–environment interactions. Our methods are developed in the Bayesian model selection framework, which treats the dimension of models as an unknown and which models uncertainty better than frequentist approaches. We have developed a new sampling for exploring the posterior distribution that can give substantial improvement over the sampling scheme of Yi et al. (2005) in problems with large numbers of possible effects. We have developed new priors on indicator variables and genetic effects that can incorporate our prior knowledge about genetic architecture of complex traits and thereby focus searching on biologically more realistic models. These new priors and the computationally efficient MCMC algorithm greatly improve the ability of the Bayesian model selection methods to rapidly detect complex interactions. We demonstrate the utility of the algorithm and new priors in the analysis of two mouse obesity data sets, in which we report stronger evidence for epistatic interactions than if they were not used and substantial improvement on computational intensity.
We developed our new algorithm using the conventional Metropolis–Hastings technique based on the composite model space. The proposed algorithm is similar to a reversible jump MCMC algorithm, which goes through each indicator variable and uses the prior probability as the proposal and which proceeds to generate one or two new QTL position(s), new genotypes for all individuals, and the prior variance of βj, from the corresponding priors and the associated effect βj from the full conditional posterior. However, this reversible jump MCMC algorithm can be derived only by using our composite model space approach. For nonepistatic models, Yi (2004) showed that the composite model space approach includes many reversible jump MCMC algorithms as special cases.
The methods described herein have been implemented in a software package called R/qtlbim for the open-source R environment (Yandell et al. 2007). The MCMC algorithm is written in compiled C code and wrapped with R code, making the software available for Windows, UNIX, and MacOS operating systems. R/qtlbim is fully compatible with and complementary to R/qtl, an extensive and interactive package of frequentist approaches to QTL mapping in experimental crosses (Broman et al. 2003). A key advantage of the Bayesian approach, as implemented by simulation, is the flexibility with which posterior inferences can be informatively summarized. We have developed various methods to graphically (and numerically) summarize and interpret posterior samples and to diagnose convergence of the Markov chain. These methods have been implemented within R/qtlbim. A detailed description of these graphical methods will be published elsewhere.
The basic framework of our composite model space approach provides flexible ways to reduce the model space. We have incorporated two global constraints on models into our algorithms and software R/qtlbim as options. These constraints dramatically reduce the model space and may be useful for efficiently detecting interacting QTL. The first constraint restricts the spacing among multiple linked QTL. On chromosome c, forcing QTL to be at least dc cM apart excludes the possibility of fitting closely linked QTL if dc is large. The distance dc should depend on the density of markers on chromosome c and on the sample size n. We suggest setting it to the average length of marker intervals on chromosome c. Our second constraint restricts the number of detectable QTL on each chromosome to Lc with L ≤ 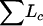 and Lc ≤ Dc/dc, where Dc is the length of chromosome c. End users can use these global constraints to rule out many unrealistic models from consideration.
and Lc ≤ Dc/dc, where Dc is the length of chromosome c. End users can use these global constraints to rule out many unrealistic models from consideration.
The process of Bayesian analysis can be idealized by consisting of four steps: (1) setting up a joint probability distribution for all observable and unobservable quantities, (2) calculating (sampling from) the appropriate posterior distribution, (3) interpreting the posterior sample, and (4) evaluating the fit of the model and the implications of the resulting posterior distribution (Gelman et al. 2003). Despite our best efforts to include as much information in modeling as possible and to search model space as comprehensively as possible, all resulting models are approximate. Hence, checking the fit of a model to data and prior assumptions is always important. Model checking and assessment have been largely ignored in QTL studies. We are also investigating ways to check the fit of inferred QTL models to data and prior assumptions. Our future plans also include extensions to joint analysis of multiple traits, experimental crosses derived from multiple inbred lines, and outbred populations. Computationally efficient algorithms are an essential feature for the practical analysis of complex genetic architectures in these more complicated cases.
Acknowledgments
This work was supported by National Institutes of Health (NIH) grant R01 GM069430 (N.Y.). D.S. was supported in addition by NIH grant T32 DK062710. B.S.Y. was supported in addition by American Diabetes Association grant 7-03-IG-01 and NIH/National Institute of Diabetes and Digestive and Kidney Diseases grants 5803701, 66369, and HL56593.
References
- Baierl, A., M. Bogdan, F. Frommlet and A. Futschik, 2006. On locating multiple interacting quantitative trait loci in intercross designs. Genetics 173: 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball, R. D., 2001. Bayesian methods for quantitative trait loci mapping based on model selection: approximate analysis using the Bayesian information criterion. Genetics 159: 1351–1364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdan, M., J. K. Ghosh and R. W. Doerge, 2004. Modifying the Schwarz Bayesian information criterion to locate multiple interacting quantitative trait loci. Genetics 167: 989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., and T. P. Speed, 2002. A model selection approach for identification of quantitative trait loci in experimental crosses. J. R. Stat. Soc. B 64: 641–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., H. Wu, Ś. Sen and G. A. Churchill, 2003. R/qtl: QTL mapping in experimental crosses. Bioinformatics 19: 889–890. [DOI] [PubMed] [Google Scholar]
- Carlborg, Ö., and C. Haley, 2004. Epistasis: Too often neglected in complex trait studies? Nat. Rev. Genet. 5: 618–625. [DOI] [PubMed] [Google Scholar]
- Carlborg, Ö., L. Andersson and B. Kinghorn, 2000. The use of a genetic algorithm for simultaneous mapping of multiple interacting quantitative trait loci. Genetics 155: 2003–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chipman, H., 1996. Bayesian variable selection with related predictions. Can. J. Stat. 24: 17–36. [Google Scholar]
- Chipman, H., 2004. Prior distributions for Bayesian analysis of screening experiments, pp. 235–267 in Screening: Methods for Experimentation in Industry, Drug Discovery, and Genetics, edited by A. Dean and S. M. Lewis. Springer, New York.
- Chipman, H., E. I. Edwards and R. E. McCulloch, 2001. The practical implementation of Bayesian model selection, pp. 65–116 in Model Selection, edited by P. Lahiri. Institute of Mathematical Statistics, Beachwood, OH.
- Gaffney, P. J., 2001. An efficient reversible jump Markov chain Monte Carlo approach to detect multiple loci and their effects in inbred crosses. Ph.D. Dissertation, University of Wisconsin, Madison, WI.
- Gelman, A., J. Carlin, H. Stern and D. Rubin, 2003. Bayesian Data Analysis. Chapman & Hall, London.
- George, E. I., 2000. The variable selection problem. J. Am. Stat. Assoc. 95: 1304–1308. [Google Scholar]
- Haley, C. S., and S. A. Knott, 1992. A simple regression method for mapping quantitative trait loci in line crosses using flanking markers. Heredity 69: 315–324. [DOI] [PubMed] [Google Scholar]
- Heath, S. C., 1997. Markov chain Monte Carlo segregation and linkage analysis for oligogenic models. Am. J. Hum. Genet. 61: 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeschele, I., 2001. Mapping quantitative trait loci in outbred pedigrees, pp. 599–644 in Handbook of Statistical Genetics, edited by D. J. Balding, M. Bishop and C. Cannings. Wiley, New York.
- Jansen, R. C., 2003. Studying complex biological systems using multifactorial perturbation. Nat. Rev. Genet. 4: 145–151. [DOI] [PubMed] [Google Scholar]
- Jansen, R. C., and P. Stam, 1994. High resolution of quantitative traits into multiple loci via interval mapping. Genetics 136: 1447–1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadane, J. B., and N. A. Lazar, 2004. Methods and criteria for model selection. J. Am. Stat. Assoc. 99: 279–290. [Google Scholar]
- Kao, C. H., and Z.-B. Zeng, 2002. Modeling epistasis of quantitative trait loci using Cockerham's model. Genetics 160: 1243–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao, C. H., Z.-B. Zeng and R. D. Teasdale, 1999. Multiple interval mapping for quantitative trait loci. Genetics 152: 1203–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kass, R. E., and A. E. Raftery, 1995. Bayes factors. J. Am. Stat. Assoc. 90: 773–795. [Google Scholar]
- Kohn, R., M. Smith and D. Chen, 2001. Nonparametric regression using linear combinations of basis functions. Stat. Comput. 11: 313–322. [Google Scholar]
- Lander, E. S., and D. Botstein, 1989. Mapping Mendelian factors underlying quantitative traits using RFLP linkage maps. Genetics 121: 185–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer Associates, Sunderland, MA.
- Moore, J. H., 2005. A global view of epistasis. Nat. Genet. 37: 13–14. [DOI] [PubMed] [Google Scholar]
- Narita, A., and Y. Sasaki, 2004. Detection of multiple QTL with epistatic effects under a mixed inheritance model in an outbred population. Genet. Sel. Evol. 36: 415–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plummer, M., N. Best, K. Cowles and K. Vines, 2004. CODA: output analysis and diagnostics for MCMC, v. 0.9–5. Institute of Mathematical Statistics, Beachwood, OH (http://www-fis.iarc.fr/coda).
- Raftery, A. E., D. Madigan and J. A. Hoeting, 1997. Bayesian model averaging for linear regression models. J. Am. Stat. Assoc. 92: 179–191. [Google Scholar]
- Reifsnyder, P. R., G. Churchill and E. H. Leiter, 2000. Maternal environment and genotype interact to establish diabesity in mice. Genome Res. 10: 1568–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha, J. L., E. J. Eisen, D. L. Van Vleck and D. Pomp, 2004. A large sample QTL study in mice. I: Growth. Mamm. Genome 15: 83–99. [DOI] [PubMed] [Google Scholar]
- Satagopan, J. M., and B. S. Yandell, 1996. Estimating the number of quantitative trait loci via Bayesian model determination. Special Contributed Paper Session on Genetic Analysis of Quantitative Traits and Complex Disease, Biometric Section, Joint Statistical Meetings, Chicago.
- Satagopan, J. M., B. S. Yandell, M. A. Newton and T. C. Osborn, 1996. Markov chain Monte Carlo approach to detect polygene loci for complex traits. Genetics 144: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen, Ś., and G. Churchill, 2001. A statistical framework for quantitative trait mapping. Genetics 159: 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and E. Arjas, 1998. Bayesian mapping of multiple quantitative trait loci from incomplete inbred line cross data. Genetics 148: 1373–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillanpää, M. J., and J. Corander, 2002. Model choice in gene mapping: what and why. Trends Genet. 18: 301–307. [DOI] [PubMed] [Google Scholar]
- Stephens, D. A., and R. D. Fisch, 1998. Bayesian analysis of quantitative trait locus data using reversible jump Markov chain Monte Carlo. Biometrics 54: 1334–1347. [Google Scholar]
- Stylianou, I. M., R. Korstanje, R. Li, S. Sheehan, B. Paigen et al., 2006. Quantitative trait locus analysis for obesity reveals multiple networks of interacting loci. Mamm. Genome 17: 22–36. [DOI] [PubMed] [Google Scholar]
- Valdar, W., L. C. Solberg, D. Gauguier, W. O. Cookson, J. N. P. Rawlins et al., 2006. Genetic and environmental effects on complex traits in mice. Genetics 174: 959–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, H., Y. M. Zhang, X. Li, G. L. Masinde, S. Mohan et al., 2005. Bayesian shrinkage estimation of quantitative trait loci parameters. Genetics 170: 465–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, S., N. Yehya, E. E. Schadt, H. Wang, T. A. Drake et al., 2006. Genetic and genomic analysis of a fat mass trait with complex inheritance reveals marked sex specificity. PLoS Genet. 2: 0148–0159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, S., 2003. Estimating polygenic effects using markers of the entire genome. Genetics 163: 789–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yandell, B. S., T. Mehta, S. Banerjee, D. Shriner, R. Venkataraman et al., 2007. R/qtlbim: QTL with Bayesian interval mapping in experimental crosses. Bioinformatics 23: 641–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., 2004. A unified Markov chain Monte Carlo framework for mapping multiple quantitative trait loci. Genetics 167: 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., and S. Xu, 2002. Mapping quantitative trait loci with epistatic effects. Genet. Res. 79: 185–198. [DOI] [PubMed] [Google Scholar]
- Yi, N., D. B. Allison and S. Xu, 2003. Bayesian model choice and search strategies for mapping multiple epistatic quantitative trait loci. Genetics 165: 867–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., B. S. Yandell, G. A. Churchill, D. B. Allison, E. J. Eisen et al., 2005. Bayesian model selection for genome-wide QTL analysis. Genetics 170: 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, N., D. K. Zinniel, K. Kim, E. J. Eisen, A. Bartolucci et al., 2006. Bayesian analysis of multiple epistatic QTL models for body weight and body composition in Mice. Genet. Res. 87: 45–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, Z.-B., 1994. Precision mapping of quantitative trait loci. Genetics 136: 1457–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, M., K. L. Montooth, M. T. Wells, A. G. Clark and D. Zhang, 2005. Mapping multiple quantitative trait loci by Bayesian classification. Genetics 169: 2305–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]