

Abstract
Nonrandom mating induces correlations in allelic states within and among loci that can be exploited to understand the genetic structure of natural populations (Wright 1965). For many species, it is of considerable interest to quantify the contribution of two forms of nonrandom mating to patterns of standing genetic variation: inbreeding (mating among relatives) and population substructure (limited dispersal of gametes). Here, we extend the popular Bayesian clustering approach STRUCTURE (Pritchard et al. 2000) for simultaneous inference of inbreeding or selfing rates and population-of-origin classification using multilocus genetic markers. This is accomplished by eliminating the assumption of Hardy–Weinberg equilibrium within clusters and, instead, calculating expected genotype frequencies on the basis of inbreeding or selfing rates. We demonstrate the need for such an extension by showing that selfing leads to spurious signals of population substructure using the standard STRUCTURE algorithm with a bias toward spurious signals of admixture. We gauge the performance of our method using extensive coalescent simulations and demonstrate that our approach can correct for this bias. We also apply our approach to understanding the population structure of the wild relative of domesticated rice, Oryza rufipogon, an important partially selfing grass species. Using a sample of n = 16 individuals sequenced at 111 random loci, we find strong evidence for existence of two subpopulations, which correlates well with geographic location of sampling, and estimate selfing rates for both groups that are consistent with estimates from experimental data (s ≈ 0.48–0.70).
UNDERSTANDING the mating structure of natural populations is a major goal of population biology. Here we consider the problem of using genotype data from a sample of individuals to distinguish between two forms of nonrandom mating: inbreeding or mating among relatives and population subdivision or limited dispersal of gametes. As Sewall Wright demonstrated, both of these evolutionary forces induce a correlation in allelic state among uniting gametes (i.e., autozygosity) (Wright 1931, 1965). Specifically, writing {Ai, Aj} to denote the outcome of inheriting alleles i and j at a particular locus of interest, Wright thought about the problem in terms of the correlation in state:
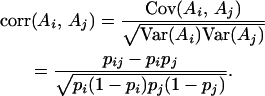 |
In a randomly mating population, the probability of inheriting a combination of alleles {Ai, Aj} is, by definition, given by the product of their marginal probabilities (i.e., pij = pi pj). Therefore, under random mating there is no correlation in allelic state among the genes inherited from the two parents.
In a subdivided population with inbreeding, however, the correlation in allelic state, FIT, may be nonzero and is given by Wright's famous equation
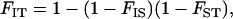 |
(1) |
where FIS is equivalent to the correlation in state conditional on subpopulation of origin, and FST is the correlation in state among randomly sampled alleles within subpopulations. The first is a measure of inbreeding and the second is a measure of population substructure. This equation demonstrates that the relative contribution of the two forces to deviations from random mating are of comparable magnitude and depend critically on the particular values of the parameters.
Although this phenomenon is appreciated by many population geneticists, many modern statistical approaches for analyzing genotype data ignore one of these two components. For example, methods for identifying population structure among a sample of individuals assume random mating within subpopulations (Pritchard et al. 2000; Dawson and Belkhir 2001; Corander et al. 2003; Falush et al. 2003). Likewise, methods for estimating self-fertilization rates from genotype data assume individuals are sampled from a single population (Ayres and Balding 1998; Enjalbert and David 2000) or require labor-intensive approaches such as progeny arrays (direct genotyping of offspring–mother pairs) (Ritland 2002). Therefore, considerable interest exists in the development of an approach that can reliably estimate the degree of population subdivision and inbreeding rates from a sample of genotyped individuals of unknown relatedness.
Our starting point in this study is the widely used program STRUCTURE (Pritchard et al. 2000; Falush et al. 2003), which implements a Bayesian clustering algorithm that simultaneously estimates locus allele frequencies and probabilistically assigns individuals to one of K subpopulations. STRUCTURE works by exploiting a key concept in population genetics: undetected population substructure leads to a genomewide deficit of heterozygotes in a sample as compared to the predictions of the Hardy–Weinberg equilibria (HWE) (Wahlund 1928; Hartl and Clark 1997). Informally, by assigning individuals probabilistically across a fixed number of K subpopulations, the algorithm minimizes deviations from HWE across the whole sample by maximizing within-subpopulation HWE as well as linkage equilibrium among unlinked loci. It is important to note, however, that various genetic and evolutionary forces can also lead to a genomewide deficiency of heterozygotes in a sample. In hermaphroditic populations, for example, partial self-fertilization reduces heterozygosity by a factor 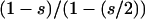 , where s is the proportion of progeny produced by self-fertilization (Haldane 1924). Since STRUCTURE assumes that individuals in the sample are either fully outcrossing or haploid, application of the algorithm to partially selfing populations may result in spurious inference of population structure and/or admixture as pointed out in Falush et al. (2003). (It is important to note that under the extreme case of complete self-fertilization, one can sidestep this issue by treating each diploid individual as haploid.)
, where s is the proportion of progeny produced by self-fertilization (Haldane 1924). Since STRUCTURE assumes that individuals in the sample are either fully outcrossing or haploid, application of the algorithm to partially selfing populations may result in spurious inference of population structure and/or admixture as pointed out in Falush et al. (2003). (It is important to note that under the extreme case of complete self-fertilization, one can sidestep this issue by treating each diploid individual as haploid.)
To investigate spurious evidence for admixture in the presence of partial self-fertilizaton, we modified Hudson's implementation of the standard coalescent algorithm (Hudson 1997) to accommodate partial selfing (Nordborg and Donnelly 1997) and generated a sample of 100 individuals drawn from a population with selfing rate s = 0.5 genotyped at 100 loci. We then ran the standard STRUCTURE 1.0 algorithm assuming two clusters (K = 2) on this data set (see the Simulations section for details). We expect STRUCTURE to assign all individuals to one of the two clusters shown in Figure 1c, since we have simulated data from a single unstructured population. Figure 1, a and b, generated by the Distruct program (Rosenberg et al. 2002), summarizes the posterior assignment probabilities. For this data set drawn from a single population, STRUCTURE classified all individuals as “admixed” with 50% of their genome coming from cluster 1 (green) and 50% coming from cluster 2 (purple). This result holds regardless of whether one considers the correlated (i.e., F model) or uncorrelated allele frequency models and suggests that application of STRUCTURE to data from a partially selfing population may lead to spurious signals of population substructure as initially suggested by Falush et al. (2003).
Figure 1.—





Population assignments for a single data set of 100 individuals simulated under partial selfing (s = 50%) and no population substructure and analyzed assuming K = 2. (a and b) The Distruct graph from STRUCTURE using (a) the correlated alleles model and (b) the uncorrelated alleles model. (c) The Distruct graph from InStruct of the same data set. (d) Distribution of log-likelihood difference between the K = 2 and the K = 1 model under six levels of population selfing rates as estimated by STRUCTURE using the F model (A)/InStruct (B). Each colored line represents the density of average log-likelihood difference with 100 replicate data sets simulated without population structure and under a specific selfing rate, indicated in the inset.
To quantify this effect further, we repeated the procedure above for 100 data sets simulated for each of six levels of selfing and ran STRUCTURE under both  and
and 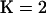 . To gauge the improvement in fit between the
. To gauge the improvement in fit between the 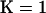 and
and 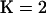 models, we compared the difference in average log-likelihood score across retained draws from Markov chain Monte Carlo (MCMC):
models, we compared the difference in average log-likelihood score across retained draws from Markov chain Monte Carlo (MCMC):
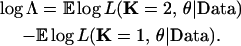 |
(2) |
The distribution of log Λ for different values of s is plotted in Figure 1d(A). We note that when s = 0.0, the population is completely outcrossing and the distribution of log Λ provides the null distribution of the test statistic under the hypothesis of no selfing and no population structure. Figure 1d(A) shows that as selfing rate increases so does the distribution of log-likelihood difference between 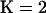 and
and 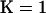 leading to increased rejection of the null hypothesis. When the selfing rate is >0.5, the whole of the distribution of log Λ exceeds the critical value, resulting in a 100% false positive rate.
leading to increased rejection of the null hypothesis. When the selfing rate is >0.5, the whole of the distribution of log Λ exceeds the critical value, resulting in a 100% false positive rate.
Therefore, we concluded that a modification to the basic model of STRUCTURE is essential when wanting to infer population structure for partially selfing species or those with a recurrent pattern of inbreeding. This article presents and validates such an approach, which we term “InStruct.” When InStruct is applied to the data sets above, it both reduces the false positive rate dramatically (see Figure 1d) and corrects for spurious admixture completely (see Figure 1c).
The new algorithm we present here extends the STRUCTURE 1.0 framework by incorporating the possibility of inbreeding among individuals in the sample. Much of this article is focused on self-fertilization, but the program has been written generally so as to estimate inbreeding coefficients as well. We consider two general scenarios: a population-specific process by which all individuals within one subpopulation share the same selfing potential (which may reflect a shared environment, for example) as well as a model where selfing probabilities vary among individuals in the whole sample. This model is particularly useful for modeling population substructure when some samples have been artificially propagated in the lab (or the field) through enforced selfing. For this scenario, we use a Bayesian density estimation algorithm called the Dirichlet process mixture model (DPMM), which offers great flexibility in estimating the distribution of latent (or unobserved) variables in the probabilistic model. It has recently been used to estimate the distribution of ω = dN/dS along a protein-coding sequence (Huelsenbeck et al. 2006). We quantify the power, robustness, and accuracy of the approach using data simulated under a myriad of scenarios, varying both the degree of selfing and population substructure.
A major motivation for our research was the desire to understand population structure in the wild ancestor of domesticated Asian rice (Oryza rufipogon), in an effort to identify wild germplasm for improvement of this important crop species. Therefore, to illustrate the application of our method and to investigate the role of inbreeding and population substructure in O. rufipogon, we apply InStruct to multilocus data from a sample of 16 individuals collected from various localities across Southeast Asia. We find strong evidence of population subdivision in O. rufipogon, as well as evidence for geographic variation in the rates of self-fertilization. Potentially the most important feature of InStruct is that it allows the identification of variation in mating system in either structured or unstructured populations, which in turn opens the door to using molecular population genetic approaches to investigate the evolution of mating systems.
THEORY
A myriad of factors influence selfing rates in natural populations, including genetic and developmental factors (such as presence/absence of self-incompatibility loci, flower shape, deleterious mutation rate, etc.) as well as abiotic and biotic environmental factors (such as availability of animal pollinators, local population density, rainfall variation, etc.). Furthermore, plants obtained from intensively managed populations (such as seed centers that propagate varieties of food crops) are often the result of artificial selfing (i.e., purification) and different lines may have been propagated for different numbers of generations via self-fertilization.
Our model is not explicit as to which of these factors (if any) is influencing selfing rate, but rather, we start from the premise that each individual in the sample has a constant but unknown selfing potential that we wish to estimate from the available genetic data. The selfing potential of an individual is defined as the probability that the individual reproduces via self-fertilization (see below). We consider two models for how selfing varies among individuals in the sample: a “population-specific” model and an “individual” model.
Under the population-specific model, the selfing potentials are equal for individuals assigned to the same population and equivalent to the proportion of offspring produced via self-fertilization each generation. This is a reasonable model if local environmental factors are the chief determinants of selfing rate. Under the individual model, we use a form of Bayesian probability density estimation to estimate the selfing rate for each individual in the sample, potentially combining individuals with statistically similar rates and splitting up individuals with statistically different rates. This is a particularly useful model for analyzing genetic material from seed centers where different lines may have been the result of propagation by self-fertilization and the number of generations of propagation differs among lines (and is often unknown).
Parameter notation:
We borrow much of our notation from Pritchard et al. (2000). Probability densities are denoted by calligraphy fonts: 𝒰 represents the uniform distribution, 𝒢 the geometric distribution, and 𝒟 the Dirichlet distribution. Uppercase italic letters (e.g., P, G, X) are vectors or matrices of random variables and lowercase italic letters (e.g., p, g, x) represent instantiations of the random variables. Letters in boldface type represent constants (e.g.,  ,
,  ) and every effort is made to retain the same notation as in the original STRUCTURE articles.
) and every effort is made to retain the same notation as in the original STRUCTURE articles.
Assume a sample of 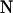 individuals genotyped at
individuals genotyped at 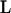 loci are to be classified into
loci are to be classified into 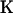 populations with ploidy
populations with ploidy 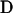 . (Throughout this article we consider the diploid case
. (Throughout this article we consider the diploid case 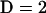 ). We incorporate the possibility of admixture into the model by allowing an individual's genotype at a locus to be composed of alleles from distinct populations. This is true even for selfing individuals since their genomes can be mosaics of haplotypes recently derived from selfing of an admixed parent.
). We incorporate the possibility of admixture into the model by allowing an individual's genotype at a locus to be composed of alleles from distinct populations. This is true even for selfing individuals since their genomes can be mosaics of haplotypes recently derived from selfing of an admixed parent.
As in Pritchard et al. (2000), denote marker allele frequencies by 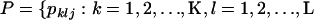 , and
, and 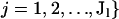 such that pklj is the allele frequency of the jth allele type at the lth locus in the kth population, where
such that pklj is the allele frequency of the jth allele type at the lth locus in the kth population, where 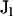 is the number of distinct alleles at the lth locus. For each individual i, let
is the number of distinct alleles at the lth locus. For each individual i, let 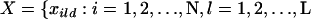 , and
, and 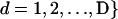 , where xild is the allele carried at locus l for the dth copy. In accordance with Pritchard et al. (2000), let
, where xild is the allele carried at locus l for the dth copy. In accordance with Pritchard et al. (2000), let 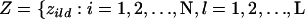 , and
, and 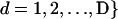 represent the matrix of zild, the population of origin of the dth allele copy at the lth locus in the ith individual and let
represent the matrix of zild, the population of origin of the dth allele copy at the lth locus in the ith individual and let 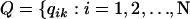 and
and 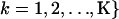 be the matrix of qik, the proportion of the ith individual's genome originating from population k.
be the matrix of qik, the proportion of the ith individual's genome originating from population k.
Write 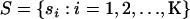 to denote the selfing rates for the
to denote the selfing rates for the 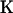 subpopulations and
subpopulations and 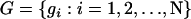 to denote the vector containing the number of generations until each individual experiences an outcrossing event in the past. Furthermore, let
to denote the vector containing the number of generations until each individual experiences an outcrossing event in the past. Furthermore, let 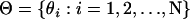 be the vector of individual selfing potentials, where θi is the probability that individual i reproduces via self-fertilization in a given generation. We assume that this parameter is constant in time for a given individual. Under the population-specific model, we further assume that all individuals from a given population have the same value of θi and that this quantity is equivalent to sk, the percentage of offspring produced via selfing in subpopulation k. To estimate selfing rates for individuals of admixed ancestry, we need to make some mathematical assumptions as to how to combine selfing potentials. The model we employ in InStruct is a weighted average of population-specific selfing rates. In particular, if an individual cannot be classified unambiguously into one of K subpopulations, we model the individual's selfing potential as the weighted average of the K population selfing rates with weighting constants equal to the qik, the proportion of individual i's genome that we estimate to originate from population k (see Equation 7 below).
be the vector of individual selfing potentials, where θi is the probability that individual i reproduces via self-fertilization in a given generation. We assume that this parameter is constant in time for a given individual. Under the population-specific model, we further assume that all individuals from a given population have the same value of θi and that this quantity is equivalent to sk, the percentage of offspring produced via selfing in subpopulation k. To estimate selfing rates for individuals of admixed ancestry, we need to make some mathematical assumptions as to how to combine selfing potentials. The model we employ in InStruct is a weighted average of population-specific selfing rates. In particular, if an individual cannot be classified unambiguously into one of K subpopulations, we model the individual's selfing potential as the weighted average of the K population selfing rates with weighting constants equal to the qik, the proportion of individual i's genome that we estimate to originate from population k (see Equation 7 below).
We use a superscript to track parameters within MCMC iterations such that 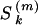 is the value of the selfing rate for population k at iteration m of an MCMC chain. When available, we use conjugate priors since these make the MCMC much more efficient by often enabling Gibbs sampling. These priors can also easily accommodate previous information about population structure and self-fertilization rates.
is the value of the selfing rate for population k at iteration m of an MCMC chain. When available, we use conjugate priors since these make the MCMC much more efficient by often enabling Gibbs sampling. These priors can also easily accommodate previous information about population structure and self-fertilization rates.
Modeling selfing:
We model the number of generations gi until an outcrossing event for the ith individual as a geometric random variable with probability of success 1 − θi, where θi is the selfing rate for individual i:
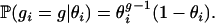 |
(3) |
This amounts to assuming that whether an individual selfs or not is independent from generation to generation and constant in time. Thus gi(m) = 1 indicates that at step m in our MCMC, the ith individual is generated by an outcrossing event in the previous generation, whereas gi(m) > 1 implies individual i was produced via selfing that extends gi(m) − 1 generations into the past.
The reason for conditioning on G is that the likelihood of the data given parameters P, G, and Z does not depend on S or Q, greatly simplifying our calculations (see Equations 5 and 6). Specifically, we write the likelihood of the genotype data given allele frequencies, population assignments, and number of generations back until an outcrossing event as
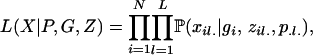 |
(4) |
where 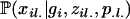 is the genotype frequency of individual i at locus l. If the two alleles for this genotype are from different subpopulations (i.e., zil1 ≠ zil2), we assume the genotype frequency is the product of the population allele frequencies (amounting to random mating among populations). If the population assignment is the same, our probabilities follow directly from basic population genetic theories. If individual i is the result of gi − 1 generations of selfing, then the probability of homozygosity for the A allele is
is the genotype frequency of individual i at locus l. If the two alleles for this genotype are from different subpopulations (i.e., zil1 ≠ zil2), we assume the genotype frequency is the product of the population allele frequencies (amounting to random mating among populations). If the population assignment is the same, our probabilities follow directly from basic population genetic theories. If individual i is the result of gi − 1 generations of selfing, then the probability of homozygosity for the A allele is
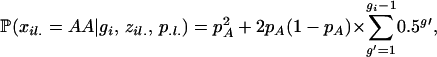 |
(5) |
where pA is the allele frequency of A in its assigned subpopulation. If individual i is heterozygous at locus l (suppose the genotype is Aa at that locus), the genotype probability is
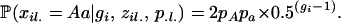 |
(6) |
In modeling inbreeding more generally, we can replace the above equations by their usual analogs in Wright's formulation conditional on the inbreeding coefficient F (see appendix). For simplicity, we remain for the rest of this article focused on selfing, but note that InStruct has an option for modeling inbreeding as well. Next we turn to models for how selfing rates vary among individuals and populations.
Population-specific model:
For the population-specific model, we define the selfing potential θi conditional on the population assignments of individual i as
 |
If we assume that the probability that individual i comes from population k equals the proportion of individual i's genome that originates from population k that has selfing rate sk, we obtain
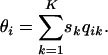 |
(7) |
Individual variation in selfing model:
A clear limitation of the population-specific model is that it does not allow for selfing rate variation among individuals within subpopulations, which may be an important feature of the data. To relax this assumption, we employ the DPMM. The rationale behind this approach is not biological, but statistical. Instead of assuming a distribution for selfing rates among individuals and estimating parameters of the model (e.g., beta distribution, logit, probit, etc.), we use a Bayesian version of nonparametric density estimation to “learn” the selfing rates from the data. Informally, it is equivalent to smoothing a histogram of individually estimated selfing rates and taking our uncertainty in the smoothing function into account. Smoothing occurs via collapsing and expanding sets of individuals that have been assigned the same identical selfing rate (a class) and updating the selfing rate assigned to each class. The parameter governing the smoothing function, α, works mathematically by influencing the prior distribution on the number of classes.
In essence, the DPMM model generates partitions of selfing rates where within a partition all individuals have the same selfing rate. Formally, we think of the Dirichlet process mixture model as a finite mixture model where the number of mixture components is a random variable. We treat each individual's selfing rate as arising from the same distribution family with different parameters for each component. The joint prior distribution of all selfing rates in the DPMM model corresponds to a generalized Polya urn scheme. The hierarchical structure of the Dirichlet process mixture model is
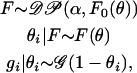 |
where 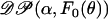 is the Dirichlet process with base distribution F0 and scaling parameter α > 0, and F is a random distribution drawn from the 𝒟𝒫, with the graphical model representation shown in supplemental Figure 1 at http://www.genetics.org/supplemental/. In words, the above is saying that the distribution F from which the selfing rate for individual i is drawn follows a Dirichlet process. Conditional on the parameters governing F, the selfing rate θi is drawn. Conditional on the selfing rate θi, the number of generations until outcrossing gi is geometrically distributed. The Bayesian framework treats the probability distribution F as an infinite-dimensional parameter, whose prior distribution is Dirichlet process and posterior is a mixture of Dirichlet processes (MacEachern and Muller 1998 and McAuliffe et al. 2004). In our case F0 is assumed to be the uniform distribution on [0, 1]. In practice, this amounts to modeling the selfing rate for individual i as either sampled from the uniform distribution or identical to one of existing selfing rates according to the following probabilities:
is the Dirichlet process with base distribution F0 and scaling parameter α > 0, and F is a random distribution drawn from the 𝒟𝒫, with the graphical model representation shown in supplemental Figure 1 at http://www.genetics.org/supplemental/. In words, the above is saying that the distribution F from which the selfing rate for individual i is drawn follows a Dirichlet process. Conditional on the parameters governing F, the selfing rate θi is drawn. Conditional on the selfing rate θi, the number of generations until outcrossing gi is geometrically distributed. The Bayesian framework treats the probability distribution F as an infinite-dimensional parameter, whose prior distribution is Dirichlet process and posterior is a mixture of Dirichlet processes (MacEachern and Muller 1998 and McAuliffe et al. 2004). In our case F0 is assumed to be the uniform distribution on [0, 1]. In practice, this amounts to modeling the selfing rate for individual i as either sampled from the uniform distribution or identical to one of existing selfing rates according to the following probabilities:
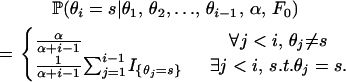 |
(8) |
To update θi under the individual selfing rate model, we use iterative Gibbs sampling. That is, we sample θi from its posterior distribution conditional on all other selfing rates in the sample θ(−i) and G,
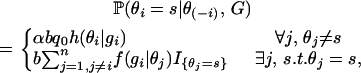 |
(9) |
where f(gi | θj) is the density function for the geometric distribution and b is a normalizing constant: 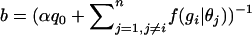 . Here, q0 is the probability of the number of generations until outcrossing gi,
. Here, q0 is the probability of the number of generations until outcrossing gi, 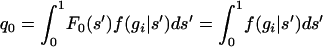 , since F0(s) = 1 for s ∈ [0, 1]. And h(θi | gi) is the posterior distribution on θi (the selfing rate for individual i), given gi; i.e.,
, since F0(s) = 1 for s ∈ [0, 1]. And h(θi | gi) is the posterior distribution on θi (the selfing rate for individual i), given gi; i.e., 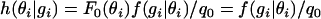 . In words, the equation above states: assign individual i a unique selfing rate drawn from the posterior distribution h(θi | gi) with probability αbq0; otherwise, assign individual i to an existing selfing rate s with probability proportional to the sum of likelihood of generations of individuals that already carry selfing rate s multiplied by the normalizing term b. The number of classes of selfing rates is randomly determined by the Polya urn model, which is governed by the scaling parameter α. It is interesting to note that the prior distribution on the number of classes is identical to the Ewens sampling distribution for a panmictic neutrally evolving Wright–Fisher population as has been pointed out by several authors (e.g., Tavare and Ewens 1998).
. In words, the equation above states: assign individual i a unique selfing rate drawn from the posterior distribution h(θi | gi) with probability αbq0; otherwise, assign individual i to an existing selfing rate s with probability proportional to the sum of likelihood of generations of individuals that already carry selfing rate s multiplied by the normalizing term b. The number of classes of selfing rates is randomly determined by the Polya urn model, which is governed by the scaling parameter α. It is interesting to note that the prior distribution on the number of classes is identical to the Ewens sampling distribution for a panmictic neutrally evolving Wright–Fisher population as has been pointed out by several authors (e.g., Tavare and Ewens 1998).
Markov chain Monte Carlo procedure:
To sample from the posterior distribution of all parameters in our model, we use a single-component Metropolis algorithm with blockwise updating. The sampling scheme consists of five updating steps. For the mth iteration, the sequence of parameter updating is
Update allele frequencies P(m) via the Gibbs sampler.
Update selfing rates S(m) at either population or individual levels. Under the population-specific model, selfing rates are updated using the back-reflection sampler (BRS) or the “adaptive independence sampler” (AIS) (see appendix for more information). Selfing rates under the individual model are produced from the Dirichlet process mixture model.
Update the number of generations until outcrossing events G(m) via an independent Metropolis–Hastings step.
Update the population assignments Z(m) via the Gibbs sampler.
Update the proportion of genome assignments Q(m) via the Gibbs sampler.
The mathematical details are provided in the appendix. The above algorithm has been implemented in an ANSI C computer program, InStruct (Inbreeding and Substructure) available from bustamantelab.cb.bscb.cornell.edu/software.shtml. A web interface for InStruct is also available through cbsuapps.tc.cornell.edu/InStruct.aspx.
Inference:
The selfing rate of each population (or individual) is estimated as the sample average over M retained MCMC draws:
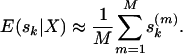 |
Posterior credibility intervals are constructed using the symmetric percentage method [i.e., 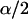 and (
and (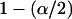 ) empirical quantiles of the MCMC draws for an α-level credibility interval] since we have found that the posterior mean is often very close to the posterior median, implying symmetric posterior distribution of population selfing rates. We also consider the posterior median as a point estimator of individual selfing rates since the posterior distribution of selfing rates is often quite skewed. Inference for the rest of the parameters is done in a similar manner as in Pritchard et al. (2000).
) empirical quantiles of the MCMC draws for an α-level credibility interval] since we have found that the posterior mean is often very close to the posterior median, implying symmetric posterior distribution of population selfing rates. We also consider the posterior median as a point estimator of individual selfing rates since the posterior distribution of selfing rates is often quite skewed. Inference for the rest of the parameters is done in a similar manner as in Pritchard et al. (2000).
Assessing convergence:
To assess convergence of our MCMC scheme, we use the Gelman–Rubin statistics that are based on the one-way analysis of variance (ANOVA) and compare the within-chain variance to the between-chain variance (Gelman and Rubin 1992). At stationarity, these should be equal. We use the Gelman–Rubin statistics to check the convergence of log-likelihood and selfing rates across different chains after applying the following identifiability constraint to the retained MCMC draws:
As in other Bayesian mixture settings, we are faced with the label-switching problem across chains [i.e., for different chains the algorithm may switch the labels of which population is 1, 2, etc., without affecting the likelihood (Jasra et al. 2005)]. We apply a simple identifiability constraint on the parameter space to break the symmetry in the likelihood; namely, the posterior mean selfing rate of each population along the MCMC is calculated and sorted in ascending order and the population with lowest average selfing rate is labeled 1; thus only one permutation of population labeling is obtained. This constraint is obviously effective only when the selfing rates differ substantially among subpopulations.
Simulations:
To assess the power and robustness of this approach under different selfing scenarios, we simulate data using standard coalescent theory with selfing and population structure. We treat each diploid individual as a deme of two chromosomes and use a separation-of-timescales approach to draw samples under selfing (Nordberg and Donnelly 1997; Nordborg 2000; Wakeley 2000). The simulation was a two-step process:
- Step 1. Calculate for each locus the number of lineages
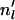 that make it through the scattering phase:
that make it through the scattering phase:- Sample the number of generations G = {gi: i = 1, 2, … , N} until an outcrossing event in the past for each individual from the geometric distribution
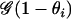 . (This random variable is a constant across all the loci for a given individual and will strongly influence whether lineages for a given individual coalesce due to selfing or scatter through outcrossing.)
. (This random variable is a constant across all the loci for a given individual and will strongly influence whether lineages for a given individual coalesce due to selfing or scatter through outcrossing.) - If an individual is the product of outcrossing in the previous generation (i.e., gi = 1), then for all loci the pair of chromosomes do not coalesce within individual i. Therefore, the probability that the two chromosomes coalesce in the past, denoted as ρi, is 0. If an individual is a product of selfing in the previous generation (gi = 2), then ρi is simply
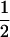 and if an individual is generated via multiple generations of selfing (i.e., gi > 2), then ρi is
and if an individual is generated via multiple generations of selfing (i.e., gi > 2), then ρi is 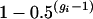 .
. - For each locus l, draw Uil an independent uniform(0, 1) random variable for i = 1, … , N. If Uil < 1 − ρi, set the number of lineages
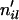 that make it out of the scattering phase to 2 for individual i; otherwise, set it to 1.
that make it out of the scattering phase to 2 for individual i; otherwise, set it to 1. - Sum up among individuals to obtain the number of lineages at locus l that make it out of the scattering phase:
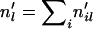 .
.
Step 2. Given
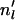 , simulate allelic history at locus l via the standard coalescent software “ms” (Hudson 2002). For all loci where individual i has
, simulate allelic history at locus l via the standard coalescent software “ms” (Hudson 2002). For all loci where individual i has 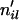 = 1, store the individual as homozygous due to selfing.
= 1, store the individual as homozygous due to selfing.
Using this procedure, we consider several substructure and selfing models assuming equal and constant subpopulation sizes, no migration among subpopulations, and a divergence time τ of 0.5 measured in standard units of 2N generations. We use “model k” to identify the simulated population models, where k represents the number of subpopulations in the sample, in our cases, k = {1, 2, 3, 6}.
We also consider several “individual”-based models for how selfing varies among individuals in the sample:
Model Ident: A single population with identical selfing rates across individuals.
Model Norm: A single population with variable selfing rates across individuals and the logit-transformed selfing rates follow the normal distribution with mean 0 and standard deviation σ; i.e.,
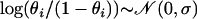 .
.Model Beta: A single population with variable selfing rates across individuals, which follow the beta distribution with different combinations of scale and shape parameters α and β; i.e.,
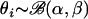 .
.
RESULTS
Application to simulated data:
Using the simulation scheme outlined above, we generated 100 data sets per parameter combination per population model and one representative data set per parameter combination per individual model. Detailed information regarding choice of parameters is provided in Table 1. For each data set, InStruct was run for five independent chains, each chain with 1,000,000 iterations in total, 500,000 burn-in iterations, and a thinning interval of 10 iterations between retained draws. For all the simulated runs, the reported diagnostic Gelman–Rubin statistic is <1.10, indicating good convergence in both log-likelihood and selfing rates. We also used the direct plotting method to show the convergence of five MCMC chains with distinct initial starting conditions. Diagnostic graphs of convergence of selfing rates are provided in supplemental Figure 2 at http://www.genetics.org/supplemental/, showing the first 2000 iterations of two randomly chosen data sets under model 1 with selfing rates 0.3 and 0.7. The values of the selfing rates converge quickly, normally entering the stationary distribution within a few hundred iterations. The convergence of population structure is slower than that of selfing rates, but it is usually on the same order as STRUCTURE. We observed that as the complexity of population structure increased (i.e., as k increased), so did the number of iterations of the MCMC algorithm required to ensure convergence (data not shown).
TABLE 1.
Parameters used for data simulated under each model
| Model | Data set no. | Subpop. no. | Subpop. size | Sample size | Loci no. | Combinations or distributions of selfing rates |
|---|---|---|---|---|---|---|
| 1 | 100 | 1 | 100 | 100 | 100 | 0, 0.1, 0.3, 0.5, 0.7, 0.9 |
| (0, 0.3), (0, 0.9) | ||||||
| 2 | 100 | 2 | 50 | 100 | 100 | (0.3, 0.3), (0.3, 0.6) |
| (0.3, 0.9), (0.9, 0.9) | ||||||
| (0.1, 0.1, 0.1), (0.9, 0.9, 0.9) | ||||||
| 3 | 100 | 3 | 50 | 150 | 100 | (0.4, 0.5, 0.6), (0.1, 0.5, 0.9) |
| (0.25, 0.6, 0.85), (0.05, 0.45, 0.75) | ||||||
| 6 | 50 | 6 | 50 | 300 | 100 | (0.05, 0.3, 0.45, 0.55, 0.75, 0.95) |
| Ident | 1 | 1 | 100 | 100 | 100 | s = 0.3 or s = 0.7 |
| Norm | 1 | 1 | 100 | 100 | 100 | 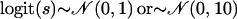 |
| Beta | 1 | 1 | 100 | 100 | 100 |
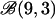 or or 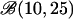
|
Data set number indicates the number of replications to be simulated under a specific model. Subpop. number indicates the number of subpopulations assumed in the simulation. Subpop. size is the number of individuals belonging to each subpopulation. Sample size means the total number of individuals. Loci number is the number of unlinked loci genotyped in each individual. Combinations of selfing rates are the different selfing levels used in the simulation; e.g., (0.3, 0.6) means two subpopulations with selfing rates 0.3 and 0.6, respectively.
Inference of selfing rates for population-specific models:
Our inference goals are twofold. First, we are concerned with the accuracy of selfing rates estimation under each of the simulation scenarios described above. Second, we wish to assess the accuracy of population assignments once selfing rates have been estimated.
Under model 1, each sample contains partially selfing individuals and no population substructure. In Figure 2, we report the distribution of estimated posterior mean selfing rates among replicate data sets for varying levels of s. With partial self-fertilization (i.e., s > 0), we see that the distribution of the posterior mean estimates of selfing rates falls mostly within the range containing the true selfing rates ± 0.1. For example, for data simulated under s = 0.5 the vast majority of the estimated rates across the 100 replicate data sets lie within [0.4, 0.6]. It is also interesting to note that the modes of the distributions of posterior mean estimates are the true selfing rates (Figure 2, dashed lines).
Figure 2.—

The posterior distribution of selfing rates estimated from simulations without population structure under six levels of population selfing rates. Each colored line represents the density of the posterior mean of selfing rates of 100 simulation runs under a specific selfing rate in the key.
Model 2 assumes two subpopulations with equal or distinct selfing rates split from a common ancestral population in the recent past (τ = 0.5 in units of 2Ne generations). In Figure 3, we report the distribution of the posterior estimates of the selfing rates for the two subpopulations under varying levels of outcrossing. In comparison to model 1, the variance in estimated selfing rates among replicate data sets increased (Figure 3). Population assignment worked extremely well for this model with nearly 100% correct assignment probabilities for all individuals in all replicate data sets.
Figure 3.—

The posterior distribution of selfing rates estimated from simulations under model 2 with six combinations of selfing rates: (A) s = {0.0, 0.3}, (B) s = {0.0, 0.9}, (C) s = {0.3, 0.3}, (D) s = {0.3, 0.6}, (E) s = {0.3, 0.9}, and (F) s = {0.9, 0.9}. Each colored line represents the density of the posterior mean of a subpopulation selfing rate from 100 simulation runs under a specific combination of selfing rates in the key.
Figures 4 and 5 illustrate the accuracy of our selfing rate estimation under a more sophisticated population structure model. By comparing Figure 4 (model 3, where the sample is drawn from three populations) vs. Figure 2 (model 1) and Figure 3 (model 2) we can assess how population structure affects our inference regarding selfing. We note that the width of the distribution of the posterior mean of population selfing rates increases, implying that the variance of the estimator becomes larger and estimation becomes slightly upwardly biased, potentially due to population misidentification for some individuals, especially when K = 6 subpopulations are simulated (Figure 5). It is also important to note that for the case of a large variance among populations in selfing rates, a small fraction of replicate data sets converged to a point with high selfing and low population structure (i.e., high “bump” near 0.90 in Figure 4D). In summary, InStruct has high accuracy in estimating selfing rates under a myriad of selfing rate combinations for K = 1, 2, 3, and 6 populations.
Figure 4.—

The posterior distribution of selfing rates estimated from simulations under model 3 with six combinations of selfing rates: (A) S = {0.4, 0.5, 0.6}, (B) S = {0.1, 0.5, 0.9}, (C) S = {0.1, 0.1, 0.1}, (D) S = {0.25, 0.6, 0.85}, (E) S = {0.05, 0.45, 0.75}, and (F) S = {0.9, 0.9, 0.9}. Each colored line represents the density of the posterior mean of a subpopulation selfing rate from 100 data sets simulated under a specific selfing rate combination in the key.
Figure 5.—

The posterior distribution of selfing rates estimated from simulations with six subpopulations of unequal selfing rates. Each colored line represents the density of the posterior mean of a subpopulation selfing rate from 50 simulation runs under a specific selfing rate in the key.
Another interesting result from Figures 2–5 is that regardless of K when the selfing rates are near 0 or 1, the estimator has a lower variance than when the selfing rate is near 50%. That is, when a population is nearly completely selfing or completely outcrossing, the mating system strongly affects patterns of genetic variation, which makes it easy to detect and estimate selfing. In contrast, when selfing rates are moderate and the population is substructured, the precision of our estimator decreases as evidenced by the appearance of multimodal or flat posterior distributions for sk.
We expect the accuracy of our selfing rate estimation to be influenced by several facets of the data, including sample size and number of loci. To address this question, we compared the coverage of 90% credibility intervals for sk under different combinations for the total number of individuals sampled and the number of loci genotyped (see Table 2, 100 data sets per combination). Several interesting patterns emerged from this analysis. First, when there is a single population (model 1), the Bayesian credibility intervals are conservative since almost all entries in the table are significantly >90% and none has an observed coverage statistically <90%. Second, when we sampled n = 50 individuals per subpopulation and 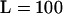 loci (first line of all comparisons in the table), the coverage of the credibility intervals was well behaved across different population structure scenarios except those with extreme differences in sk among subpopulations. That is, model 1, model 2, and many combinations in model 3 had excellent coverage. One exception was model 3 with sk ∈ {0.05, 0.45, 0.75} where the realized coverage is closer to 82% rather than 90%. Likewise, in model 6 the average coverage among the five subpopulations with selfing rates <s = 0.95 was only 84% (for the s = 0.95 subpopulation the coverage was conservative). The third interesting pattern that emerges from Table 2 is that reducing both sample size per subpopulation and number of loci per genotype tended to decrease the coverage of the credibility intervals, but not systematically. That is, in all models investigated, the coverage of both the n = 10 individuals per subpopulation and
loci (first line of all comparisons in the table), the coverage of the credibility intervals was well behaved across different population structure scenarios except those with extreme differences in sk among subpopulations. That is, model 1, model 2, and many combinations in model 3 had excellent coverage. One exception was model 3 with sk ∈ {0.05, 0.45, 0.75} where the realized coverage is closer to 82% rather than 90%. Likewise, in model 6 the average coverage among the five subpopulations with selfing rates <s = 0.95 was only 84% (for the s = 0.95 subpopulation the coverage was conservative). The third interesting pattern that emerges from Table 2 is that reducing both sample size per subpopulation and number of loci per genotype tended to decrease the coverage of the credibility intervals, but not systematically. That is, in all models investigated, the coverage of both the n = 10 individuals per subpopulation and 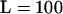 loci sampled as well as the n = 50 individuals per subpopulation and
loci sampled as well as the n = 50 individuals per subpopulation and 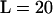 loci sampled tended to have worse coverage than the standard of n = 50 individuals and
loci sampled tended to have worse coverage than the standard of n = 50 individuals and 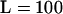 loci. There are exceptions, however, when the coverage for the smaller n treatment had better (or more conservative) coverage than the large n treatment. This is probably due to a larger variance of the selfing rate estimator.
loci. There are exceptions, however, when the coverage for the smaller n treatment had better (or more conservative) coverage than the large n treatment. This is probably due to a larger variance of the selfing rate estimator.
TABLE 2.
Coverage of 90% credible intervals of selfing rates under models 1, 2, 3, and 6 with respect to specific population size and locus number based on 100 data sets per selfing rate combination (50 data sets for model 6)
| Model 1
|
|||||||
|---|---|---|---|---|---|---|---|
| Sample size | Locus no. | 0.0 | 0.1 | 0.3 | 0.5 | 0.7 | 0.9 |
| 100 | 100 | 1.00 | 0.93 | 0.93 | 0.912 | 0.95 | 0.958 |
| 20 | 100 | 0.988 | 0.99 | 0.92 | 0.888 | 0.93 | 0.92 |
| 100 | 20 | 0.99 | 0.958 | 0.932 | 0.94 | 0.924 | 0.96 |
| Model 2
|
|||||||
| Sample size | Locus no. | 0.0 | 0.3 | 0.0 | 0.9 | 0.3 | 0.3 |
| 100 | 100 | 0.976 | 0.878 | 0.96 | 0.94 | 0.882 | 0.914 |
| 20 | 100 | 0.732 | 0.892 | 0.734 | 0.938 | 0.93 | 0.91 |
| 100 | 20 | 0.772 | 0.99 | 0.742 | 0.97 | 0.95 | 0.91 |
| Model 2
|
|||||||
| Sample size | Locus no. | 0.3 | 0.6 | 0.3 | 0.9 | 0.9 | 0.9 |
| 100 | 100 | 0.91 | 0.948 | 0.968 | 0.924 | 0.902 | 0.99 |
| 20 | 100 | 0.948 | 0.94 | 0.88 | 0.926 | 0.88 | 0.98 |
| 100 | 20 | 0.898 | 0.9 | 0.928 | 0.924 | 0.894 | 1.00 |
| Model 3
|
|||||||
| Sample size | Locus no. | 0.4 | 0.5 | 0.6 | 0.1 | 0.5 | 0.9 |
| 150 | 100 | 0.948 | 0.958 | 0.948 | 0.832 | 0.92 | 0.97 |
| 30 | 100 | 0.962 | 0.976 | 0.916 | 0.856 | 0.932 | 0.86 |
| 150 | 20 | 0.964 | 0.97 | 0.964 | 0.792 | 0.868 | 0.954 |
| Model 3
|
|||||||
| Sample size | Locus no. | 0.25 | 0.6 | 0.85 | 0.05 | 0.45 | 0.75 |
| 150 | 100 | 0.89 | 0.924 | 0.97 | 0.816 | 0.818 | 0.836 |
| 30 | 100 | 0.852 | 0.884 | 0.896 | 0.788 | 0.91 | 0.892 |
| 150 | 20 | 0.86 | 0.97 | 0.978 | 0.766 | 0.972 | 0.968 |
| Model 6
|
|||||||
| Sample size | Locus no. | 0.05 | 0.30 | 0.45 | 0.55 | 0.75 | 0.95 |
| 300 | 100 | 0.800 | 0.900 | 0.840 | 0.800 | 0.860 | 1.000 |
Each data set was run for five independent MCMCs, with 1,000,000 iterations, 500,000 burn-in iterations, and a thinning interval of 10 iterations (for model 6 one chain per data set). The proposal method for selfing rate here is the AIS.
Inference of selfing rates—individual variation models:
Figure 6 shows the results of the DPMM method on a single typical data set under various models for how θ varies among individuals. We observe that for all the cases considered, DPMM estimation of the distribution of selfing rates across 100 individuals approximates the true distribution well. That is, the mean, the median, and the mode are mostly centered at their true values, especially when selfing rates follow a beta distribution (Figure 6, C and F). It is important to note that the peaky and multimodal shape of posterior distribution is an inherent property of the DPMM model as DPMM generates finite discrete classes within which individuals share the same selfing rate and once a large class is formed, the potential that an individual value belongs to this class is greatly increased.
Figure 6.—

The distributions of posterior medians of selfing rates of 100 individuals drawn from the Dirichlet process mixture model. The magenta dashed lines represent the true distribution of selfing rates in the simulation. The red, green, blue, and yellow solid lines are the estimated densities from the Dirichlet process mixture model with scaling parameters α = 1, α = 5, α = 10, and α = 20, respectively. The individual selfing rates were simulated under three different scenarios in three columns: (1) model ident (A) S = 0.3 and (D) S = 0.7, (2) model norm (B) 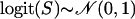 and (E)
and (E) 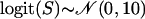 , and (3) model beta (C) S ∼ beta(9, 3) and (F) S ∼ beta(10, 25).
, and (3) model beta (C) S ∼ beta(9, 3) and (F) S ∼ beta(10, 25).
A key part of the DPMM method is a choice for the α-parameter that governs the prior distribution on the number of classes of selfing rates. Figure 6 summarizes simulations with various values of α. According to McAuliffe et al. (2004), for n observations the prior expected number of classes in the data is ∼α log n. We chose values of α within the range 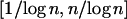 , corresponding to one class for all the observations and one class per observation, respectively. Smaller values of α lead to a “peaky” distribution with many values clustered in one class. When α is large, the proportion of values sampled from the base distribution increases, resulting in smoother density estimation. Intermediate values of α tend to classify a reasonable number of values into each class, generally resulting in a better approximation to the true distribution.
, corresponding to one class for all the observations and one class per observation, respectively. Smaller values of α lead to a “peaky” distribution with many values clustered in one class. When α is large, the proportion of values sampled from the base distribution increases, resulting in smoother density estimation. Intermediate values of α tend to classify a reasonable number of values into each class, generally resulting in a better approximation to the true distribution.
When evaluating the performance of DPMM in estimating the distribution of selfing rates among individuals, a key issue should be considered: each θi parameter is effectively estimated from one single data point. That is, the most amount of information one can have in our model about selfing rate θi is the number of generations until an outcrossing event gi. Even if gi were known without error, there would still be high uncertainty in θi since one has observed only a single geometric random variable. Therefore, allowing selfing rates to vary among individuals in the sample when one has little information about a particular θi may produce density estimation that is wildly different from the true distribution. That is, the inherent uncertainty due to sampling variation coupled with overshrinkage of parameters (see discussion below) may lead to shape estimation quite different from the true density. To address this issue, in supplemental Figure 3 (http://www.genetics.org/supplemental/) we plot the distribution of the difference between the estimated selfing rate and its true value of all the individuals in the simulations of the three individual selfing rate models assuming α = 5. Most of them appear to follow a nearly normal distribution, with mean 0 and standard deviation <0.15 for almost all the parametric simulations conducted. We also report the estimated densities for 20 data sets simulated under a beta distribution for selfing rates, using two parameter combinations in supplemental Figure 4 (http://www.genetics.org/supplemental/). It appears that the distributions of estimated selfing rates are similar in shape to the underlying true beta distribution with considerable among-sample variation.
Inference of population assignment for simulated data:
Our accuracy in classifying individuals into populations is comparable to that of STRUCTURE with the original model when no self-fertilization exists. For the 100-data-set replications under model 2 and model 3 at various levels of selfing, each individual is separated into one of the major groups appropriately with frequency 0.99. The accuracy of classification decreases slightly for model 6 (the assignment proportion is ∼0.95) as might be expected with a more complex demographic scenario. One disadvantage of InStruct is the tendency of merging subpopulations with similar allele frequencies and similar selfing rates when the data do not provide sufficient evidence of differentiation. This phenomenon, which has also been observed in the STRUCTURE-like algorithm BAPS (Corander et al. 2003) and the Bayesian clustering algorithm with hidden Markov random field (Francois et al. 2006), mainly occurs when assuming more subpopulations than are represented in the real data or when sample size per true subpopulation is very small.
Application to rice data:
To gauge the performance of our algorithm on real data, we applied InStruct to 111 single-nucleotide polymorphisms (SNPs) discovered via direct sequencing across 111 unlinked loci of n = 16 individuals of O. rufipogon, a wild ancestor of the cultivated rice species (A. L. Caicedo, S. H. Willamson, A. Fledel-Alon, T. L. York, N. Polato, K. M. Olsen, R. Nielsen, S. McCouch, C. D. Bustamante, and M. D. Purugganan, unpublished results). Each SNP has two alleles and only one SNP per locus was used in our analysis. The individuals in the sample were collected from the wild with 9 sampled from China, 5 from Nepal, 1 from India, and 1 from Laos. We focus on a subset of the data [n = 91 (78.4%) SNPs] that contains no missing data. We ran InStruct and STRUCTURE on these data for five independent chains, each chain with 200,000 iteration steps, 100,000 burn-in, and a thinning interval of 10 steps, assuming different starting points. Graphical representations of population assignments from STRUCTURE and InStruct were produced from the program Distruct (Rosenberg et al. 2002).
When two subpopulations are assumed, the estimation of selfing rates and substructure converged very well among the five independent chains. The classification of individuals is consistent with geographical separation in that all the individuals from China formed one major cluster and the other cluster mainly contains Nepalese individuals. The fact that the Indian individual is clustered with Nepal is quite reasonable as India is nearer to Nepal than China geographically and the Himalayan mountains likely reduce pollen flow to and from China. The Laos individual falls in between the two clusters with a larger part of its alleles (91.14%) as likely of Nepalese origin and ∼8.86% of Chinese origin. This classification is almost the same as that of STRUCTURE, although the proportion of the genome that originates in each population is slightly different for several individuals, which might be due to our accounting for self-fertilization (Figure 7a). One critical difference is the classification of a Chinese individual that STRUCTURE predicts as admixed with nearly equal ancestry in the two clusters. Using InStruct, this same individual is now classified with high posterior probability 0.999 [90% C.I.: (0.996, 1.000)] in the “Chinese” cluster. The lack of overlap in credibility intervals implies there is significant discrepancy in classification of this individual as was observed in the simulated data presented in Figure 1. When we ran InStruct assuming three subpopulations, the convergence rate was poor with some runs converging on all individuals assigned only two clusters, leaving the third cluster empty. This is due to the tendency of the Bayesian clustering algorithm to merge subpopulations with similar allele frequencies. A likely reason for this in our case is the small sample size of just 16 individuals and the optimal classification is to assume K = 2.
Figure 7.—


(a) The Distruct plot of population assignment for n = 16 rice accessions assuming K = 2 from STRUCTURE and InStruct. The two clusters are represented by pink and light blue. For InStruct, the corresponding selfing rates of subpopulations are indicated at the top. (b) Estimated selfing rates under the individual model using the Dirichlet process prior model. The points represent the posterior mean of individual selfing rates and their different shapes indicate the countries where that individual was collected: squares with x's inside represent China, diamonds represent Nepal, circles represent India, and triangles indicate Laos. The x-axis represents the index of 16 individuals collected from the wild. The red lines across the points represent the 90% posterior confidence intervals of individual selfing rates.
The posterior means of selfing rates for the Chinese and Nepalese subpopulations under the population model are 0.697 and 0.484 with 90% confidence intervals (0.553, 0.826) and (0.260, 0.699), respectively. While the confidence intervals overlap, this is suggestive of potential regional differences in selfing rate for O. rufipogon. This result should be interpreted with caution, however, since the Nepalese material was collected recently from the wild while the Chinese individuals come mainly from an existing germplasm collection and may have undergone purification as part of standard germplasm propagation (S. McCouch, personal communication). In Figure 7b, we present the results of running the individual-based model of InStruct that uses DPMM for density estimation. We note that the majority of individuals have posterior means for θ, the selfing rate parameter, between 0.5 and 0.7, which is consistent with previous estimates based on pollen count (Oka 1988). It is important to note that confidence intervals for θ are much wider under the individual-based model as compared to the population-based estimate of selfing rates.
DISCUSSION
In this article, we present a modification of the popular Bayesian clustering program STRUCTURE (Pritchard et al. 2000) for inferring population substructure and self-fertilization simultaneously. Using extensive simulations with four distinct demographic models (K = 1, 2, 3, 6), we demonstrate that our method can accurately estimate selfing rates in the presence of population structure in the data. Additionally it can classify individuals into their appropriate subpopulations without the assumption of Hardy–Weinberg equilibrium within subpopulations.
It is important to note that the accuracy of selfing rate estimation is influenced by multiple factors, including sample size and number of loci, with decreased precision when they are small, as is illustrated in Table 2. Likewise, we find that the complexity of the true demographic history underlying data (e.g., the number of subpopulations derived from a common ancestral population) also influences accuracy. In general, more complicated models lead to decreased precision in selfing rate estimation. For example, when we simulated six subpopulations split from one ancestral population, the coverages of 90% credible intervals of selfing rates are near 85%.
As with other methods for inference of population structure, InStruct explores a complex multimodal likelihood surface using a stochastic search algorithm. This means that the program may “get stuck” in suboptimal parts of the parameter space. We, therefore, encourage users to run several chains and compare the expected log-likelihood as with other MCMC schemes. In practice, we have observed that InStruct infrequently merges subpopulations, especially ones with correlated allele frequencies, which can result in “empty” clusters and poor convergence in population assignments and selfing rate estimation. This phenomenon has been described previously for other STRUCTURE-like algorithms such as BAPS (Corander et al. 2003) and the Bayesian clustering algorithm with hidden Markov random field (Francois et al. 2006). One idea we have explored is to use simulated annealing to “heat and cool chains” so as to allow movement among local maxima. We have also investigated stopping MCMC chains with “empty clusters,” where an empty cluster contains less than one expected individual after sufficient burn-in. While this suggestion is ad hoc and in a sense does not solve the poor convergence problem, we have found that it tends to control against merging populations into an extreme pathological case of K = 1 with high selfing for data simulated under K > 1.
We employ the Dirichlet process mixture model to estimate how individual selfing rates vary among individuals in the sample. Instead of assuming a distribution for selfing rates among individuals and estimating parameters of the model, we use a Bayesian version of nonparametric density estimation to “learn” the selfing rates from the data. We anticipate that the individual specific model will facilitate plant breeding by providing a fairly accurate estimate of individual selfing rates divorced from the consequences of population structure. There are a few statistical caveats, however, that we raise.
In many statistical inference problems, the number of parameters to be estimated is much smaller than the sample size. Therefore, “large-sample” estimators such as maximum likelihood or method-of-moments have good statistical properties (e.g., unbiased, consistent, efficient, etc.). In our case, we wish to estimate a selfing rate parameter for each individual in the sample based on a single (unobserved) data point, namely, G, the number of generations of selfing in the genealogy of the individual until an outcrossing event looking back in time. For this type of inference problem, standard large-sample statistical approaches are not accurate and approaches that “share” information across related parameters (so-called “shrinkage” estimators) often have better performance. That is, when estimating the selfing rate of a given individual i we use information regarding selfing rates for all other individuals in the sample and iterate this procedure. Shrinkage methods reduce (or shrink) the variance of estimated parameters by drawing outliers nearer to the mean value. The drawback to such an approach is that we may sometimes “overshrink” and downwardly or upwardly bias the estimation for some individuals with selfing rates in the tails of the distribution.
We find that the distribution of estimated selfing rates minus the corresponding true values has the shape of normal distribution with mean zero and standard deviation ∼0.15 under various simulated individual models as shown in supplemental Figure 3 (http://www.genetics.org/supplemental/). Estimation is more accurate when no substructure exists or subpopulations have similar selfing rates, compared to subpopulations with very distinct selfing rates as the Dirichlet process mixture model tends to find a local maximum and thus cluster individual data points into big categories of selfing rates. When DPMM is applied to data sets simulated with two subpopulations and two distinct selfing rates, it sometimes peaks at two true selfing rates (supplemental Figure 5D at http://www.genetics.org/supplemental/) or peaks at a value in the middle of the two true selfing rates and clusters all individual values into that class (supplemental Figure 5, A–C). It is important to note that the DPMM model is a nonparametric method of density estimation, which is less efficient than the parametric estimation approach and thus takes longer to reach stationary states.
Due to the structure of the likelihood function under the individual model and the limitation of data available, confidence intervals for individual selfing rates will likely be large unless the posterior mean or median is close to complete selfing (θi = 1). The reason for this is that the most information one can have in our model regarding θi is the true number of generations until outcrossing gi. Depending on the magnitude of gi, many possible values θi may be consistent with the observed data. For example, if there has been only one generation since an outcrossing event (gi = 1), this observation is consistent with nearly the whole of the interval [0, 1) and the posterior mean for θi | gi = 1 is 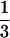 under a uniform prior for θi.
under a uniform prior for θi.
Another practical issue for our approach is how to choose the appropriate scaling parameter and base distribution for inference under the individual selfing rate model (Figure 6). If the scaling parameter is small, then the expected number of selfing rate classes is small, leading to the peaky distribution of selfing rates. If the scaling parameter is large, then one class contains only one data point, which adds much uncertainty to estimation, leading to biased estimation of the underlying distribution. According to McAuliffe et al. (2004), the nonparametric estimation method of the scaling parameter and base distribution can be incorporated into the MCMC scheme, which may facilitate estimation, or a hierarchical uninformative prior distribution can be placed on the scaling parameter and base distribution to integrate out the uncertainty of estimation on these nuisance parameters.
Although the estimation accuracy is dependent on multiple factors, we expect that this model will have wide applications in many aspects of sequence analysis as it has great flexibility for analyzing multilocus marker data. However, several points need to be addressed with respect to improving the basic model presented here.
First, InStruct assumes loci are unlinked and conditionally independent given model parameters. It is known that pairwise linkage disequilibrium increases with selfing and can extend very far in highly selfed organisms (Nordborg 2000). The flip side of this is that selfing may leave a strong linkage disequilibrium (LD) signal that may be exploited for further refinement of our inference of individual selfing rates. Therefore, linkage disequilibrium should be incorporated into this model as in a new version of STRUCTURE (Falush et al. 2003). One approach might be to include a linkage map for the markers explicitly in the model with predictions from population genetic theory regarding how selfing affects LD among loci conditional on known recombination rates. A second limitation of our model is that it is applicable only to diploid individuals. It would be more practical, particularly for inference in plant populations, to extend the model to polyploid individuals. Two complications on this front are that the number of genotypes at a polyploid locus exponentially increases with the ploidy of the genome and two types of polyploid exist, autopolyploid and allopolyploid, which increase the complexity of calculating genotype frequencies for each locus.
The application of InStruct to data from the partially selfing wild relative of domesticated rice O. rufipogon gives results consistent with geographic sampling and with the program STRUCTURE. Our estimates of the selfing rates for each subpopulation overlap, suggesting an outcrossing rate for wild rice near 50%. Partial outcrossing has several potential evolutionary advantages in regard to either complete outcrossing or complete selfing. For example, advantageous mutations can be fixed in the population at a faster rate as compared to outcrossing. Likewise, when mates are rare (e.g., in an adverse environment), selfing ensures the likely survival of the lineage. Last, partial outcrossing can purge the population of deleterious mutations without inducing a high genetic load. We hope the development of InStruct will allow estimation of selfing rates among natural plant populations, enabling the community to test hypotheses regarding the evolutionary and ecological context for selfing rate evolution.
Acknowledgments
We are grateful to Susan McCouch and John Kelly for many thoughtful comments on an early version of the manuscript. Two anonymous reviewers greatly helped the exposition of this work. This work is funded by National Science Foundation award 0319553 to Michael D. Purugganan, Susan McCouch, Carlos D. Bustamante, and Rasmus Nielsen.
APPENDIX: DETAILS OF THE MARKOV CHAIN MONTE CARLO ALGORITHM
Initiation of MCMC:
Under the population-specific model, the initial states of population selfing rate parameters sk are generated from the uniform distribution 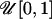 . The initial number of generations until an outcrossing event gi for each individual is drawn independently by sampling from the geometric distribution with unique uniform random probabilities of success. Under the individual selfing model, the θi's are first drawn from the Dirichlet process prior and then the gi's are sampled from the geometric distribution with a probability of success 1 − θi. Initiation of Z and Q is congruent with Pritchard et al. (2000).
. The initial number of generations until an outcrossing event gi for each individual is drawn independently by sampling from the geometric distribution with unique uniform random probabilities of success. Under the individual selfing model, the θi's are first drawn from the Dirichlet process prior and then the gi's are sampled from the geometric distribution with a probability of success 1 − θi. Initiation of Z and Q is congruent with Pritchard et al. (2000).
Updating of MCMC:
In the blockwise updating scheme of MCMC, the update of P, Z, and Q follows Pritchard et al. (2000). The rest of the parameters are updated with the single-component Metropolis–Hastings algorithm as detailed below:
- Update S:
-
At the population level, selfing rates are proposed with either the BRS or the AIS. For the BRS, we update the selfing rate vector S(m) by using Metropolis sampling with a K-dimensional uniform proposal distribution centered on the current vector of population selfing rates. That is, a proposed selfing rate
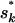 for population k is drawn from
for population k is drawn from 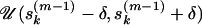 with back reflection in [0, 1], where δ is a tuning parameter.For the AIS, we assume three classes of states for the selfing rate parameter: s0 equivalent to complete outcrossing, s(0,1) that denotes the case of partial outcrossing (s ∈ (0, 1)), and s1 that represents complete selfing. Let p0 represent the probability of proposing a jump to state s0 on the basis of the current value of s, p(0,1) be the probability of proposing a jump to state s(0,1) on the basis of current s, and p1 be the probability of proposing a jump to state s1 on the basis of current s. In our model, we use the probabilities in the table below to calculate the proposal density q(s, s*), where the first column in the table shows three starting states for selfing rates and the first row represents three ending states,
with back reflection in [0, 1], where δ is a tuning parameter.For the AIS, we assume three classes of states for the selfing rate parameter: s0 equivalent to complete outcrossing, s(0,1) that denotes the case of partial outcrossing (s ∈ (0, 1)), and s1 that represents complete selfing. Let p0 represent the probability of proposing a jump to state s0 on the basis of the current value of s, p(0,1) be the probability of proposing a jump to state s(0,1) on the basis of current s, and p1 be the probability of proposing a jump to state s1 on the basis of current s. In our model, we use the probabilities in the table below to calculate the proposal density q(s, s*), where the first column in the table shows three starting states for selfing rates and the first row represents three ending states,s p0 p(0,1) p1 s = 0 0.50 0.50 0.0 s ∈ (0, 1) 0.05 0.90 0.05 s = 1 0.0 0.50 0.50
where δi(j) is a Kronecker delta function defined by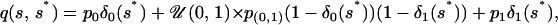
(A1) 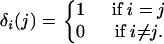 Since the prior on S is uniform and the proposal of the BRS is symmetric, the Metropolis acceptance probability r depends only on the ratio of the likelihood function at the two points proposed,
Since the prior on S is uniform and the proposal of the BRS is symmetric, the Metropolis acceptance probability r depends only on the ratio of the likelihood function at the two points proposed,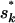 and current sk:
and current sk: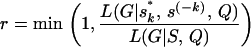 The allele frequencies P or population assignments Z are ignored from the above formula as the relevant likelihood does not depend on them conditional on G and Q.For the AIS, the Metropolis–Hastings ratio needs to multiply a proposal term:
The allele frequencies P or population assignments Z are ignored from the above formula as the relevant likelihood does not depend on them conditional on G and Q.For the AIS, the Metropolis–Hastings ratio needs to multiply a proposal term: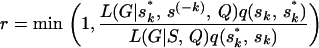 Since we assume individuals are independently sampled and use the formula (3), the likelihood is
Since we assume individuals are independently sampled and use the formula (3), the likelihood is
where θi is calculated as the expected selfing rate for individual i using Equation 7.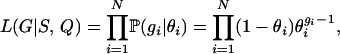 The rationale for needing two samplers is that when the selfing rate value of our MCMC is near the boundaries, one needs to be able to jump in and out of the states for complete selfing (s = 1) or complete outcrossing (s = 0). As we illustrate below, the AIS is not as efficient as the BRS, so when the MCMC chain is not near sk = 0 or sk = 1, the BRS is recommended.
The rationale for needing two samplers is that when the selfing rate value of our MCMC is near the boundaries, one needs to be able to jump in and out of the states for complete selfing (s = 1) or complete outcrossing (s = 0). As we illustrate below, the AIS is not as efficient as the BRS, so when the MCMC chain is not near sk = 0 or sk = 1, the BRS is recommended. - Updating of individual selfing rates is described in the Modeling selfing section.
-
- Update G: We choose an independent sampler to update each component of G. Specifically, the proposed update
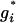 is drawn from a geometric distribution independently for each individual
is drawn from a geometric distribution independently for each individual 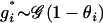 , where
, where 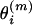 is calculated using formula (7). And an upper bound 50 is placed on
is calculated using formula (7). And an upper bound 50 is placed on 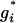 to facilitate the computation as the value of gi > 50 does not affect likelihood calculation much compared to the value of 50. Since the proposal distribution we employ is an independence sampler and the likelihood does not depend on the current values of S or Q, the Metropolis–Hastings ratio is thus
to facilitate the computation as the value of gi > 50 does not affect likelihood calculation much compared to the value of 50. Since the proposal distribution we employ is an independence sampler and the likelihood does not depend on the current values of S or Q, the Metropolis–Hastings ratio is thus
where L(X | G, Z, P) is the likelihood Equation 4.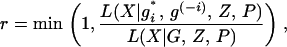
Joint inference of inbreeding coefficients and substructure:
Estimating inbreeding coefficients while accounting for population structure is done in a similar manner to inference of selfing rates, except that there is no “G” component and the likelihood of data is calculated using Wright's formula. This likelihood now depends on the inbreeding coefficients F and allele frequencies P and assignment of alleles Z,
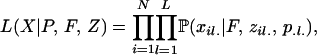 |
(A2) |
where 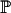 (xil. | F, zil., p.l.) is the genotype frequency of individual i at locus l. If the two alleles for this genotype are from different subpopulations (i.e., zil1 ≠ zil2), we assume the genotype frequency is the product of the population allele frequencies (amounting to random mating among populations). If the population assignment is the same, our probabilities follow directly from basic population genetic theory. The probability of homozygosity for the A allele is a function of the general inbreeding coefficient in the population assigned to individual i at position l
(xil. | F, zil., p.l.) is the genotype frequency of individual i at locus l. If the two alleles for this genotype are from different subpopulations (i.e., zil1 ≠ zil2), we assume the genotype frequency is the product of the population allele frequencies (amounting to random mating among populations). If the population assignment is the same, our probabilities follow directly from basic population genetic theory. The probability of homozygosity for the A allele is a function of the general inbreeding coefficient in the population assigned to individual i at position l 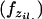 ,
,
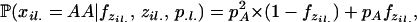 |
(A3) |
where pA is the allele frequency of A in its assigned subpopulation. If individual i is heterozygous at locus l (suppose the genotype is Aa at that locus), the genotype probability is
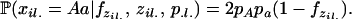 |
(A4) |
We use the BRS and AIS to propose inbreeding coefficients and then accept it with the Metropolis–Hastings algorithm.
We find that the BRS is very efficient and easily tunable, but has the disadvantage that it can never attain the boundary values of complete outcrossing (0.0) or complete selfing (1.0). The AIS can generate proposal draws for any value in the interval [0, 1], but, as implemented, the rejection rate for AIS is high. One can observe from the convergence graphs (see supplemental Figure 2 at http://www.genetics.org/supplemental/) that the patterns of selfing rate updating are remarkably different between the two methods. This is likely because a fraction of new proposed selfing rates by AIS are randomly sampled from the uniform distribution on [0, 1], which have low a priori probability of explaining the data. The AIS sampler can easily get stuck in one value for several iterations while BRS tends to reject new proposed jumps much less often (interestingly the convergence efficiency of AIS is similar to that of BRS). The importance of using AIS near the boundaries is illustrated in supplemental Figure 6 at http://www.genetics.org/supplemental/, where we note that the BRS density for zero selfing rate is strongly right shifted as compared to AIS. In actual application of InStruct to real data, the selfing rate proposal density should be chosen according to context and necessity.
References
- Ayres, K. L., and D. J. Balding, 1998. Measuring departures from Hardy-Weinberg: a Markov chain Monte Carlo method for estimating the inbreeding coefficient. Heredity 80(6): 769–777. [DOI] [PubMed] [Google Scholar]
- Corander, J., P. Waldmann and M. Sillanpaa, 2003. Bayesian analysis of genetic differentiation between populations. Genetics 163: 367–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, K. J., and K. Belkhir, 2001. A Bayesian approach to the identification of panmictic populations and the assignment of individuals. Genet. Res. 78: 59–77. [DOI] [PubMed] [Google Scholar]
- Enjalbert, J., and J. L. David, 2000. Inferring recent outcrossing rates using multilocus individual heterozygosity: application to evolving wheat populations. Genetics 156: 1973–1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francois, O., S. Ancelet and G. Guillot, 2006. Bayesian clustering using hidden Markov random fields in spatial population genetics. Genetics 174: 805–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gelman, A., and D. B. Rubin, 1992. Inference from iterative simulation using multiple sequences (with discussion). Stat. Sci. 7: 457–511.
- Haldane, J. B. S., 1924. A mathematical theory of natural and artificial selection. ii. The influence of partial self-fertilisation, inbreeding, assortative mating, and selective fertilisation on the composition of Mendelian populations, and on natural selection. Proc. Camb. Philos. Soc. Biol. Sci. 1: 158–163. [Google Scholar]
- Hartl, D., and A. Clark, 1997. Principles of Population Genetics. Sinauer Associates, Sunderland, MA.
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18: 337–338. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck, J. P., S. Jain, S. W. D. Frost and S. L. K. Pond, 2006. A Dirichlet process model for detecting positive selection in protein-coding DNA sequences. Proc. Natl. Acad. Sci. USA 103: 6263–6268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasra, A., C. C. Holmes and D. A. Stephens, 2005. Markov chain Monte Carlo methods and the label switching problem in Bayesian Markov chain Monte Carlo methods and the label switching problem in Bayesian mixture modeling. Stat. Sci. 20: 50–67. [Google Scholar]
- MacEachern, S. N., and P. Muller, 1998. Estimating mixture of Dirichlet process models. J. Comput. Graph. Stat. 7: 223–238. [Google Scholar]
- McAuliffe, J. D., D. M. Blei and M. I. Jordan, 2004. Nonparametric empirical Bayes for the Dirichlet process mixture model. Technical Report 675. University of California, Berkeley, CA.
- Nordborg, M., 2000. Linkage disequilibrium, gene trees and selfing: an ancestral recombination graph with partial self-fertilization. Genetics 154: 923–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordborg, M., and P. Donnelly, 1997. The coalescent process with selfing. Genetics 146: 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oka, H. I., 1988. Origin of Cultivated Rice. Japan Scientific Societies Press, Tokyo; Elsevier, Amsterdam/New York.
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritland, K., 2002. Extensions of models for the estimation of mating systems using n independent loci. Heredity 88: 221–228. [DOI] [PubMed] [Google Scholar]
- Rosenberg, N., J. K. Pritchard, J. L. Weber, H. Cann, K. Kidd et al., 2002. Genetic structure of human populations. Science 298: 2381–2385. [DOI] [PubMed] [Google Scholar]
- Tavare, S., and W. J. Ewens, 1998. The Ewens sampling formula, pp. 230–234 in Encyclopedia of Statistical Sciences Update, Vol. 2. Wiley, New York.
- Wahlund, S., 1928. Composition of populations from the perspective of the theory of heredity. Hereditas 11: 65–105. [Google Scholar]
- Wakeley, J., 2000. The effects of subdivision on the genetic divergence of populations and species. Evol. Int. J. Org. Evol. 54: 1092–1101. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16: 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1965. The interpretation of population structure by f-statistics with special regard to systems of mating. Evolution 19: 395–420. [Google Scholar]