

Abstract
We present theoretical explanations and show through simulation that the individual admixture proportion estimates obtained by using ancestry informative markers should be seen as an error-contaminated measurement of the underlying individual ancestry proportion. These estimates can be used in structured association tests as a control variable to limit type I error inflation or reduce loss of power due to population stratification observed in studies of admixed populations. However, the inclusion of such error-containing variables as covariates in regression models can bias parameter estimates and reduce ability to control for the confounding effect of admixture in genetic association tests. Measurement error correction methods offer a way to overcome this problem but require an a priori estimate of the measurement error variance. We show how an upper bound of this variance can be obtained, present four measurement error correction methods that are applicable to this problem, and conduct a simulation study to compare their utility in the case where the admixed population results from the intermating between two ancestral populations. Our results show that the quadratic measurement error correction (QMEC) method performs better than the other methods and maintains the type I error to its nominal level.
IGNORING confounders in genetic association studies can lead to inflated false positive rates and also to inflated false negative rates (Weinberg 1993). Simply stated, confounders are additional variables that are correlated with the risk factor under consideration and can independently cause the outcome of interest (Greenland and Robins 1985). In the presence of a confounder, an association observed between two variables may just reflect their correlation with a third variable (a confounder) that is not included in the model. If all other conditions are appropriate, the type I error of the statistical test for association may be controlled at its nominal level by conditioning upon the confounder.
Population stratification and genetic admixture are the most commonly discussed sources of confounding in genetic association studies (Knowler et al. 1988; Spielman et al. 1993; Devlin and Roeder 1999). Genomic control and structured association testing (SAT) are statistical approaches that have been proposed to control for stratification in association studies. In the presence of population stratification, Devlin and Roeder (1999) demonstrated that the chi-square test statistic of association is inflated by a constant λ (>1). When the confounder and the phenotype can be represented on a categorical or an ordinal scale, genomic control allows for a simple correction by dividing the observed test statistic by 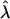 , which is estimated from the data. SAT is more appropriate when the genetic background variable (the confounder) is defined on a continuous scale (Pritchard and Rosenberg 1999; Pritchard et al. 1999). These methods attempt to reduce the false positive rate (type I error) associated with confounding due to population stratification or genetic admixture.
, which is estimated from the data. SAT is more appropriate when the genetic background variable (the confounder) is defined on a continuous scale (Pritchard and Rosenberg 1999; Pritchard et al. 1999). These methods attempt to reduce the false positive rate (type I error) associated with confounding due to population stratification or genetic admixture.
Several researchers have used the SAT methods to control for confounding in association studies. These methods can be divided into two categories: those that estimate the ancestry proportion of each individual in the sample and use this estimate as a covariate in the test for association (Pritchard and Rosenberg 1999; Pritchard and Donnelly 2001; Ziv and Burchard 2003) and those that rely upon a measure of genetic background obtained by performing a principal-component analysis (PCA) on the genotypic data to provide control for population stratification in the test for genetic association (Zhang et al. 2003, 2006; Price et al. 2006). Although the principal components can still be contaminated with measurement error and hence reduce their ability to provide adequate control over the overall type I error in genetic association studies, this article focuses on the first category of SAT methods. It may happen that even after controlling for a measure of genetic ancestry and other appropriate covariates one can still observe statistically significant associations between ancestry informative markers (AIMs) (a marker is said to be ancestry informative when its alleles are differentially distributed among the ancestral populations considered in the study) with extreme allele frequency disparity and various phenotypes. It is unclear whether these observed associations are just false positives due to lack of control or signs of genuine trait-influencing markers. Some SAT approaches (e.g., Pritchard et al. 2000a,b; Pritchard and Donnelly 2001) implicitly assume that the individual ancestry proportions used as a genetic background variable in association testing are measured without error. This assumption, however, is not always valid and may consequently affect the results of an association test. The objectives of this article are to show (1) that the admixture estimates obtained from existing software should be considered as error-contaminated measurements of individual ancestry, (2) that ignoring these errors leads to an inflated false positive rate, (3) how existing measurement error correction methods can be applied to this problem, and (4) results of a simulation study examining the performance of four of the measurement error correction methods described in objective 3. We concede that objectives 1 and 2 are not entirely new to the field. However, we show in the results section that some measurement error accommodation may be required even in cases where the correlation between the estimated ancestry proportion and the true individual ancestry value is as high as 0.95. Once this is established we focus on illustrating how measurement correction methods can be applied to this type of problem and describe the degree of improvement that can be obtained by using them in SATs.
MATERIALS AND METHODS
We focus on individual ancestry instead of individual admixture as a way to control for confounding on the basis of the proof given in Redden et al. (2006), showing that it is interindividual variation in ancestry, not admixture, that causes residual confounding. An individual ancestry proportion (IAP) defined with respect to a specific ancestral population 𝓅 is the proportion of that individual's ancestors who originated from 𝓅 whereas this individual's admixture proportion with respect to 𝓅 is simply the proportion of his/her genome that is derived from 𝓅. From these definitions it is easy to realize that two full siblings have the same ancestry proportion but not necessarily the same admixture proportion due to random variation that occurred during each meiosis process.
Admixture as an error-contaminated measure of ancestry:
From the above definitions, one can conclude that only an estimate of admixture is produced by existing software. Admixture is an imperfect measure of ancestry for several reasons. Only a relatively small subset of markers (with respect to the entire genome) is considered, and therefore variation between the statistic (admixture) and the parameter (ancestry) should be expected. The markers used to compute individual admixture proportions are not completely ancestry informative; that is, the allele frequency difference (at each marker) between two ancestral populations is 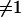 . This difference is referred to as the δ-value and has been used as a measure of the degree of ancestry informativeness of each marker when only two ancestral populations are considered. In some cases the δ-values may be insufficient to adequately describe the best set of markers to use in the estimation of an individual's ancestry, especially when the admixed population is derived from more than two ancestral populations and multiallelic markers are used to estimate the ancestry proportion (Rosenberg et al. 2003; Pfaff et al. 2004). Despite these issues, we chose to use the δ-values because our examples focus on admixture generated by two founding populations and consider simulated single-nucleotide polymorphism data in the analysis (Weir 1996; ROSENBERG et al. 2003). Genotyping error can clearly bias the estimate of ancestry provided by the existing algorithms and software. Poor knowledge regarding the history of the admixed population may cause the investigator to consider the wrong ancestral populations, which affects the estimation of the allele frequencies used to quantify the informativeness of each marker and the starting values in the algorithms that estimate ancestry. As an imperfect measure, admixture can be seen as a manifestation of the unobserved ancestry, the variations (“errors”) due to biological variation (meiosis), and other errors (genotyping errors, incorrect assumptions about ancestral allele frequencies, using AIMs that are less than completely ancestry informative markers, etc.).
. This difference is referred to as the δ-value and has been used as a measure of the degree of ancestry informativeness of each marker when only two ancestral populations are considered. In some cases the δ-values may be insufficient to adequately describe the best set of markers to use in the estimation of an individual's ancestry, especially when the admixed population is derived from more than two ancestral populations and multiallelic markers are used to estimate the ancestry proportion (Rosenberg et al. 2003; Pfaff et al. 2004). Despite these issues, we chose to use the δ-values because our examples focus on admixture generated by two founding populations and consider simulated single-nucleotide polymorphism data in the analysis (Weir 1996; ROSENBERG et al. 2003). Genotyping error can clearly bias the estimate of ancestry provided by the existing algorithms and software. Poor knowledge regarding the history of the admixed population may cause the investigator to consider the wrong ancestral populations, which affects the estimation of the allele frequencies used to quantify the informativeness of each marker and the starting values in the algorithms that estimate ancestry. As an imperfect measure, admixture can be seen as a manifestation of the unobserved ancestry, the variations (“errors”) due to biological variation (meiosis), and other errors (genotyping errors, incorrect assumptions about ancestral allele frequencies, using AIMs that are less than completely ancestry informative markers, etc.).
Sensitivity of the empirical α-level to measurement error:
A simulation study was designed to assess the effect of measurement error in the individual ancestry proportion on the false positive rates observed in SAT. We simulated the underlying individual ancestry distribution (D) by drawing from the mixed distribution described in Tang et al. (2005), where a mixture of uniform and normal distributions is used to mimic the ancestry distributions observed in the African-American population. We generated 1000 markers with different degrees of ancestry informativeness such that the mean δ-value was 0.9 for the first 200 markers, 0.6 for markers 201–400, 0.3 for markers 401–600, and 0.1 for the remaining markers. The allele frequency of each marker in the admixed sample is computed as the weighted average of the two ancestral allele frequencies. That is, if we let 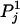 denote the frequency of allele 1 at the jth marker in the first ancestral population and
denote the frequency of allele 1 at the jth marker in the first ancestral population and 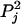 denote the frequency of the same allele in the second ancestral population then the frequency of this allele for the ith admixed individual is given by
denote the frequency of the same allele in the second ancestral population then the frequency of this allele for the ith admixed individual is given by 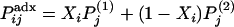 , where
, where 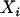 is the simulated ancestry for the ith admixed individual. Finally, we generated a phenotypic variable that is influenced by individual ancestry and markers g280, g690, and g870, using the following equation:
is the simulated ancestry for the ith admixed individual. Finally, we generated a phenotypic variable that is influenced by individual ancestry and markers g280, g690, and g870, using the following equation:
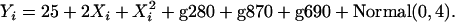 |
(1) |
More detail about the simulation procedure can be found in the appendix. Hence the phenotype is generated such that it is associated with an individual's true ancestry proportion and three markers located in regions with medium-to-low ancestry informativeness. Because the phenotypic value is associated with individual ancestry, a large number of the generated markers are spuriously associated with the phenotypic variable in addition to the three markers g280, g690, and g870 that have a genuine effect. This illustrates the need to control for individual ancestry, which is the only source of confounding in this simulation. We let D be the simulated true individual ancestry proportions from the mixture distribution described above and generated two error-contaminated variables D1 and D2 such that 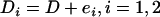 , and
, and 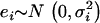 . This is the formulation of the classical measurement error model that is assumed for the remainder of this article. We set the values of Di that fall outside the [0, 1] range to 0 if they are negative and 1 if they are >1. The number of values of Di that falls outside this range is negligible and represents <0.1% of the entire data set. This number is not large enough to affect the overall conclusion of this analysis. We chose
. This is the formulation of the classical measurement error model that is assumed for the remainder of this article. We set the values of Di that fall outside the [0, 1] range to 0 if they are negative and 1 if they are >1. The number of values of Di that falls outside this range is negligible and represents <0.1% of the entire data set. This number is not large enough to affect the overall conclusion of this analysis. We chose 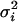 , the variance of the measurement error variable, such that the observed correlations between D and D1 and D and D2 are 0.95 and 0.80, respectively. We chose these values to illustrate the fact that even a measure of ancestry proportion that is highly correlated with the true ancestry proportion can lead to significant type I error inflation. This inflation gets worse as the correlation between true and measured ancestry proportion decreases or in other words as the measurement error variance increases. We then used a sample size of 1000 individuals to test for association between the simulated phenotype and every marker in the data set controlling D1 and D2. As can be seen in Figure 1, the ratio of empirical to nominal type I error increases greatly with the amount of noise in the individual admixture proportion.
, the variance of the measurement error variable, such that the observed correlations between D and D1 and D and D2 are 0.95 and 0.80, respectively. We chose these values to illustrate the fact that even a measure of ancestry proportion that is highly correlated with the true ancestry proportion can lead to significant type I error inflation. This inflation gets worse as the correlation between true and measured ancestry proportion decreases or in other words as the measurement error variance increases. We then used a sample size of 1000 individuals to test for association between the simulated phenotype and every marker in the data set controlling D1 and D2. As can be seen in Figure 1, the ratio of empirical to nominal type I error increases greatly with the amount of noise in the individual admixture proportion.
Figure 1.—

Type I error inflation due to the fact that a surrogate is used instead of the true genetic background variable to control an association study. Nonnegligible type I error inflation still occurs when a surrogate variable is used that is highly correlated with the true ancestry values. Highly ancestry-informative markers (AIMs) are more likely to be falsely associated with the phenotype whenever the genetic background variable used to control for type I error inflation is measured with error. The nominal α-level considered is 0.05.
Measurement errors are ubiquitous to individual ancestry estimates: Recent advances in computing and statistics have made it possible to estimate individual admixture proportions. Software packages such as STRUCTURE, ADMIXMAP, and ANCESTRYMAP, among others, will produce these estimates (Pritchard et al. 2000a,b; Falush et al. 2003; Hoggart et al. 2003; Patterson et al. 2004). Simulation studies showed that other than a few considerations relative to the convergence of the algorithm being used, the quality of the admixture estimates provided by these packages depends on the following set of parameters: (1) the number of AIMs, (2) the degree of ancestry informativeness, (3) the amount of linkage disequilibrium (LD) among markers, (4) the number of generations since admixture, and (5) the number of founders included in the data set (Darvasi and Shifman 2005; McKeigue 2005). In Figure 2, we show how the number of AIMs, the number of founders considered in the analysis, and the degree of ancestry informativeness as measured by (δ) affects the type I error rate of the association test.
Figure 2.—

Observed type I error as a function of the number of AIMs, their degree of ancestry informativeness, and the number of founders considered in the study. These factors determine the level of “noise” in the ancestry estimates and illustrate the need for measurement error correction. Left graph: 250 markers generated on 1000 individuals. The allele frequency (p) of each marker was drawn from a beta (80, 20) for an individual originated from ancestral population 1. At the corresponding marker, an individual from the second ancestral population had an allele frequency of 1 − p. We then used the estimated individual ancestry proportion and its squared value to control for potential confounding and test each marker for association with a simulated phenotype. This graph shows that, all other things being equal, the level of type I error inflation decreases as the number of ancestry informative markers (AIMs) used to estimate the individual ancestry proportion increases. One can also observe that the founder effect is less important than the AIM effect. Right graph: created by testing for an association between a simulated phenotype and each marker present in the data set. The generated sample contained 1000 admixed individuals and 1000 founders (500 from each ancestral population). The individual ancestry estimates used to control for admixture are computed with only the markers that have δ shown in the graph where each group contained 100 AIMs.
The quality of the individual ancestry estimates improves with the number of markers in the data set. This is particularly clear when maximum-likelihood (ML) methods are used to estimate individual admixture because of the consistency property of ML estimators (an estimator is said to be consistent if it converges to the true parameter that it is estimating as the sample size increases). The presence of high-quality AIMs makes it easier to trace the origin of each allele inherited by the sampled individual. A consequence of the admixture process among ancestral populations with differing allele frequencies at many loci is the creation of long stretches of LD in the genome of admixed individuals (Long 1991; McKeigue 1997, 1998, 2005). The longer these blocks are, the easier they are to match to specific founder populations. However, these blocks of LD deteriorate with time; therefore, the precision of admixture estimates decreases with the number of generations since admixture, which results in an increase in the number of markers needed to accurately estimate individual admixture (Shifman et al. 2003; Darvasi and Shifman 2005; McKeigue 2005).
Earlier methods used to estimate individual admixture assumed that the allele frequency of each marker in the ancestral population was known, which represented a serious impediment to their application since this information is rarely available. New algorithms proposed by Pritchard et al. (2000a,b), Pritchard and Przeworski (2001), and Tang et al. (2005) relax this assumption. In practice, it is required that only a few individuals from what is believed to be the founder population be available in the sample to provide a good starting point for the program. The accuracy of this starting point is important to ensure timely convergence to the true values.
Measurement error in admixture estimates:
Following from previous sections, it is evident that the admixture estimates provided by the existing software packages can be seen only as imperfect measurements of an individual's true ancestry. Redden et al. (2006) showed how association testing controlling for ancestry can be anchored in a regression framework so that existing statistical methodology and well-tested statistical packages can be used to conduct this type of test. However, the measurement error problem needs to be addressed before proceeding with the association test. In a simple linear model, using the error-contaminated variable instead of the true variable leads to an underestimation or attenuation of the slope parameter of the linear regression and higher-than-expected residual variance (Fuller 1987; Carroll et al. 1995). The effects of measurement errors on parameter estimates and hypothesis testing are compounded as the regression model considered becomes more complicated. For example, all the parameter estimates in a multiple regression are known to be biased even if only one of the independent variable is measured with error (Carroll et al. 1995). The level of control attained by controlling for a known confounder is severely reduced in the presence of measurement error. This lack of control and residual confounding is illustrated in Figure 1.
The SAT approach described by Redden et al. (2006) includes the individual ancestry proportion and the product of the ancestral ancestries in the association test to completely control for confounding effects due population substructure. This requirement is justified by the fact that the number of alleles that an admixed individual inherits at specific marker is a function of the ancestry of his/her two parents. We have shown in this article that simply controlling for individual ancestry is appropriate only when one is testing for an additive effect. Since investigators seldom consider only additive effects, adding the product of the two parental ancestries guards against type I error inflation. Since submission of Redden et al.'s (2006) article, our simulation (data not shown) has indicated that ancestry squared adequately approximates the product of ancestral ancestries in situations that we are simulating here. Furthermore, these simulations studies have shown that in the type of situation that we have considered the square value of the estimated ancestry proportion correlates more highly with this product than the estimation method proposed by Redden et al. (2006). Let 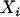 ,
, 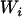 ,
, 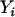 , and
, and 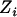 denote, respectively, the ith individual's true ancestry, an error-contaminated measure of true ancestry, the phenotype value, and the observed genotype at a specific marker. We use these letters to denote these variables for the remainder of this article. The objective is to test for association between
denote, respectively, the ith individual's true ancestry, an error-contaminated measure of true ancestry, the phenotype value, and the observed genotype at a specific marker. We use these letters to denote these variables for the remainder of this article. The objective is to test for association between 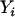 and
and 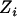 while controlling for true ancestry. In the SAT framework the model is written as
while controlling for true ancestry. In the SAT framework the model is written as
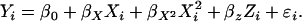 |
(2) |
However, an individual's true ancestry proportion is not directly observable and is therefore considered to be a latent variable. In principle, one should not simply replace 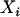 , the true unobserved individual ancestry proportion, by
, the true unobserved individual ancestry proportion, by 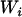 , the observed individual admixture estimate, because doing so will yield only the so-called naive estimates and likely lead to an inflation of the empirical type I error (Carroll et al. 1985; Carroll 1989). In the next sections we describe the relationship between
, the observed individual admixture estimate, because doing so will yield only the so-called naive estimates and likely lead to an inflation of the empirical type I error (Carroll et al. 1985; Carroll 1989). In the next sections we describe the relationship between 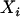 and
and  , show how an estimate of the measurement error variance can be obtained, and present a few simple measurement error correction methods that can be applied to this problem.
, show how an estimate of the measurement error variance can be obtained, and present a few simple measurement error correction methods that can be applied to this problem.
Measurement error models:
We have described above the relationship between individual admixture and individual ancestry. Although the functional form of this relationship is unknown, we use the classical measurement error specification and assume that
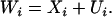 |
(3) |
The classical model seems appropriate in this case because we want to control for the unobserved individual ancestry. In the event that the relationship between 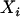 and
and 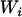 is multiplicative instead of additive, one can always return to the additive specification by taking the logarithm of both sides. A few assumptions underlie this model, the most common being that the errors are independently and normally distributed with mean and constant variance,
is multiplicative instead of additive, one can always return to the additive specification by taking the logarithm of both sides. A few assumptions underlie this model, the most common being that the errors are independently and normally distributed with mean and constant variance, 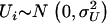 . It is also assumed that the error term
. It is also assumed that the error term  is independent of the latent variable
is independent of the latent variable . We further investigate the effect of violating these assumptions on the error distribution in the discussion section.
. We further investigate the effect of violating these assumptions on the error distribution in the discussion section.
To perform measurement error correction, one needs information regarding the measurement error variance. Replication and validation are the most common methods used to estimate this variance. Replication data are used when several measurements of  are available and there are good reasons to believe that
are available and there are good reasons to believe that 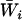 the average of the
the average of the  's is a better estimate of
's is a better estimate of 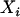 than
than  alone. For example, the average ancestry proportion computed on a set of full siblings would be a more accurate measure of their ancestry proportion than the value observed on a single individual. Validation entails obtaining the true value of individual ancestry on a small subset of people and building a model that relates observed ancestry to true ancestry. Here, we offer a novel approach using an old statistic that provides an estimate of the upper bound of the measurement error variance.
alone. For example, the average ancestry proportion computed on a set of full siblings would be a more accurate measure of their ancestry proportion than the value observed on a single individual. Validation entails obtaining the true value of individual ancestry on a small subset of people and building a model that relates observed ancestry to true ancestry. Here, we offer a novel approach using an old statistic that provides an estimate of the upper bound of the measurement error variance.
Cronbach's α as a measure of reliability:
Redden et al. (2006) showed how Cronbach's α can be used to estimate the reliability of the individual admixture estimates as a surrogate for individual ancestry. Under the assumption of independence between the measurement errors and true ancestry, the reliability of individual admixture as an estimate of individual ancestry is given by
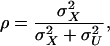 |
(4) |
where 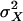 and
and 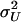 denote, respectively, the variance of the true but unobserved variable X and the variance of the measurement error U. To compute Cronbach's α, let m be the total number of unlinked AIMs selected on each chromosome and let k denote the number of chromosomes for which marker information is available. Therefore, there are km markers available for estimating the individual ancestry proportion. In practice, all the markers typed on a single chromosome are unlikely to be independent, but, conditional on individual ancestry, the marker genotypes across chromosomes are independent. Therefore, one can then use existing software packages to obtain individual ancestry estimates on each chromosome. Let
denote, respectively, the variance of the true but unobserved variable X and the variance of the measurement error U. To compute Cronbach's α, let m be the total number of unlinked AIMs selected on each chromosome and let k denote the number of chromosomes for which marker information is available. Therefore, there are km markers available for estimating the individual ancestry proportion. In practice, all the markers typed on a single chromosome are unlikely to be independent, but, conditional on individual ancestry, the marker genotypes across chromosomes are independent. Therefore, one can then use existing software packages to obtain individual ancestry estimates on each chromosome. Let 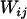 denote the ancestry proportion estimate computed on the jth chromosome for the ith individual. Let
denote the ancestry proportion estimate computed on the jth chromosome for the ith individual. Let  be a squared matrix denoting the variance–covariance matrix calculated from the admixture estimates obtained from each subset. Cronbach (1951) shows that a measure of reliability of the sum or the average (when all chromosome are equally weighted) of the Wij's as an overall measure of the unobserved individual ancestry is given by
be a squared matrix denoting the variance–covariance matrix calculated from the admixture estimates obtained from each subset. Cronbach (1951) shows that a measure of reliability of the sum or the average (when all chromosome are equally weighted) of the Wij's as an overall measure of the unobserved individual ancestry is given by
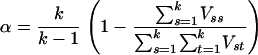 |
(5) |
Once 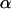 is computed, an upper bound of the measurement error variance is given by
is computed, an upper bound of the measurement error variance is given by
 |
(6) |
where 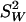 is the estimated sample variance of W, the variable measured with error. The only assumption needed to compute Cronbach's α is that the items—in this case, the chromosome-specific ancestry estimates—are estimating the same underlying latent variable, i.e., the individual true ancestry proportion with a finite variance. See Figure 3 for a comparison between true measurement error variance and the measurement error variance estimated with Cronbach's α for high, medium, and low correlation between true and estimated ancestry proportions.
is the estimated sample variance of W, the variable measured with error. The only assumption needed to compute Cronbach's α is that the items—in this case, the chromosome-specific ancestry estimates—are estimating the same underlying latent variable, i.e., the individual true ancestry proportion with a finite variance. See Figure 3 for a comparison between true measurement error variance and the measurement error variance estimated with Cronbach's α for high, medium, and low correlation between true and estimated ancestry proportions.
Figure 3.—

Comparison between true and estimated measurement variance for high, medium, and low correlation between the true and the estimated individual admixture proportion as estimated by Cronbach's α. Cronbach's α provides an upper bound of the measurement error variance.
Measurement error correction methods:
This article concentrates on measurement error in the frequentist framework. In this section we discuss four measurement correction methods and later conduct simulation studies to judge their performance under different conditions.
The measurement error correction methods considered are (1) the quadratic measurement error correction (QMEC) method, (2) the regression calibration method, (3) the expanded regression calibration, and (4) the simulation extrapolation (SimEx) algorithm. These methods have been extensively studied and have been applied to a wide range of problems.
QMEC:
Several methods were proposed to correct for measurement errors in polynomial regression (Wolter and Fuller 1982; Fuller 1987; Cheng and Schneeweiss 1998; Cheng and Van Ness 1999; CHENG et al. 2000). We focus on the treatment given by Cheng and Schneeweiss (1998; Cheng and Van Ness 1999), which assumes that an estimate of the measurement error variance is available. This assumption is valid as shown in our discussion about how an estimate of the measurement error variance can be obtained using Cronbach's α.
Let
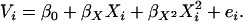 |
(7) |
Equation 1 can then be rewritten as
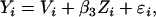 |
(8) |
where 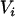 corresponds to the section of Equation 1 that is measured with error. Measurement error correction is then achieved in three steps: (1) Apply methods similar to those of Fuller (1987), Wolter and Fuller (1982), and Cheng and Schneeweiss (1998) and Cheng and Van Ness (1999) to obtain
corresponds to the section of Equation 1 that is measured with error. Measurement error correction is then achieved in three steps: (1) Apply methods similar to those of Fuller (1987), Wolter and Fuller (1982), and Cheng and Schneeweiss (1998) and Cheng and Van Ness (1999) to obtain  ; (2) compute the residual from that regression that we denote by
; (2) compute the residual from that regression that we denote by 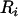 ; and (3) test for association between the residual that can also be seen as a corrected phenotype and
; and (3) test for association between the residual that can also be seen as a corrected phenotype and 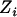 . In general, these measurement correction methods assume that the
. In general, these measurement correction methods assume that the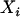 's are unknown constants and seek to replace the error-contaminated variables
's are unknown constants and seek to replace the error-contaminated variables 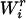 by a new variable
by a new variable 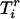 such that
such that 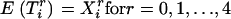 .
.
Assuming 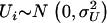 and no specification error, Cheng and Schneeweiss (1998) showed that
and no specification error, Cheng and Schneeweiss (1998) showed that 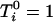 ,
, 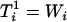 ,
,  ,
, 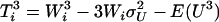 , and
, and 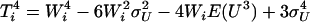 and defined for each individual in the data set matrix
and defined for each individual in the data set matrix  whose elements
whose elements 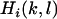 are given by
are given by 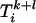 for (l,k) = 0, 1, 2. Corrections on the dependent variable lead to the vector denoted
for (l,k) = 0, 1, 2. Corrections on the dependent variable lead to the vector denoted 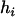 , whose elements are
, whose elements are 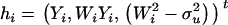 . Since
. Since 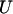 is assumed to follow a normal distribution with mean 0 and constant variance,
is assumed to follow a normal distribution with mean 0 and constant variance, 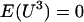 , which allows one to further simplify
, which allows one to further simplify 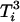 and
and 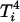 . The adjusted least-squares estimator is obtained by solving
. The adjusted least-squares estimator is obtained by solving
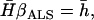 |
(9) |
where 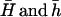 are the averages computed over the entire data set. Once
are the averages computed over the entire data set. Once 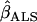 is obtained, this estimate can be plugged into Equation 7 to compute
is obtained, this estimate can be plugged into Equation 7 to compute 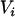 . The residuals or corrected phenotype
. The residuals or corrected phenotype 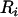 can then be used to test whether the marker
can then be used to test whether the marker  has an effect on the phenotype of interest.
has an effect on the phenotype of interest.
Regression calibration:
Proposed independently by Gleser (1990) and Carroll (Carroll and Stefanski 1990), the regression calibration's objective, given Equation 1, is to condition on the observed values of 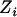 and
and 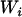 to construct a new variable
to construct a new variable 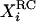 such that
such that 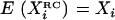 . For a model like that of Equation 1, following Carroll's (D. W. Schafer, unpublished data; Carroll et al. 1995) notation by letting
. For a model like that of Equation 1, following Carroll's (D. W. Schafer, unpublished data; Carroll et al. 1995) notation by letting 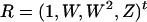 represent the matrix of all the observed variables, the calibrated variable is given by
represent the matrix of all the observed variables, the calibrated variable is given by  , where
, where 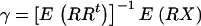 . In practice,
. In practice, 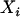 is replaced by
is replaced by 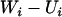 so that
so that  and
and 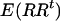 is replaced by
is replaced by 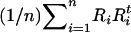 .
.
Expanded regression calibration:
Thus far the measurement error correction methods we have discussed have not made any assumption about the probability distribution of the unobserved individual ancestry proportions. The expanded regression calibration models assume that unobserved true values are random draws from a known underlying distribution. Given that distribution, the mean and the variance of the distribution 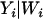 are modeled as functions of the mean and variance of
are modeled as functions of the mean and variance of 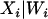 . From Equation 1, conditioning on the observed value
. From Equation 1, conditioning on the observed value 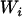 , we have
, we have
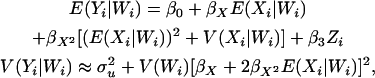 |
(10) |
where the variance follows because the  is considered to be a nonrandom variable. Note that it is not necessary to condition on both the observed ancestry proportion (W) and the marker being tested (Z) since all the information contained in Z regarding the true ancestry (X) has already been used to estimate W. Moreover, given the number of markers required to provide a “good” estimate of W, the inclusion of an additional marker that was not used in the initial estimation of W is not likely to significantly affect the estimation of W. The parameters of these models are estimated in a quasi-likelihood framework. Under the normality assumption, the mean and variance of
is considered to be a nonrandom variable. Note that it is not necessary to condition on both the observed ancestry proportion (W) and the marker being tested (Z) since all the information contained in Z regarding the true ancestry (X) has already been used to estimate W. Moreover, given the number of markers required to provide a “good” estimate of W, the inclusion of an additional marker that was not used in the initial estimation of W is not likely to significantly affect the estimation of W. The parameters of these models are estimated in a quasi-likelihood framework. Under the normality assumption, the mean and variance of 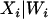 are given, respectively, by
are given, respectively, by
 |
(11) |
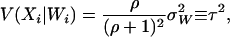 |
(12) |
where 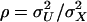 ; in practice,
; in practice,  is estimated by
is estimated by 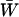 . Using Cronbach's α, an upper bound of
. Using Cronbach's α, an upper bound of 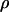 is given by
is given by  . Not including the
. Not including the  term, Kuha and Temple (2003) defined the vector
term, Kuha and Temple (2003) defined the vector 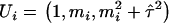 , where
, where 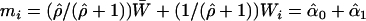 Wi and two functions,
Wi and two functions,  with
with 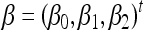 and
and 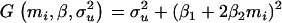 , that they used to write the estimating equations derived from Equations 11 and 12. These estimating equations are
, that they used to write the estimating equations derived from Equations 11 and 12. These estimating equations are
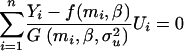 |
(13) |
and
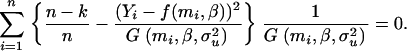 |
(14) |
These equations are solved iteratively by using the naive or regression calibration parameter estimates as starting values. Given the starting values, a solution of Equation 13 is given by weighted least squares. For parameter estimates, the Newton–Raphson algorithm can be used to solve Equation 14. (For a more complete presentation of these methods see Schneeweiss and Nitter 2001 and Kuha and Temple 2003.) Once convergence is reached, one again can compute the residuals 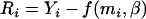 and test for association between the corrected phenotype
and test for association between the corrected phenotype 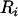 and the observed genotype
and the observed genotype 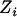 .
.
SimEx:
SimEx, a more computationally intensive method of measurement error correction, relies on simulation to either estimate or reduce the bias due to measurement error (Cook and Stefanski 1994). The simulation steps work by considering additional data sets with increasing measurement error variance. Assuming that the variance of the measurement error 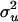 is known, one can simulate new data sets where the measurement error variance is an increasing function of a parameter
is known, one can simulate new data sets where the measurement error variance is an increasing function of a parameter 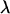 . That is, one simulates
. That is, one simulates 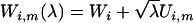 , where
, where 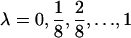 and
and 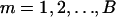 . For a fixed
. For a fixed 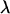 , the variables
, the variables 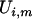 are assumed to be mutually independent and independent of all the other variables present in Equation 1. For each value of
are assumed to be mutually independent and independent of all the other variables present in Equation 1. For each value of 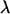 , the parameters
, the parameters 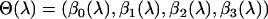 of Equation 1 and their corresponding standard errors are estimated B times using a chosen estimation method (ordinary least squares, weighted least squares, ML, etc.). The average value of these parameters estimates
of Equation 1 and their corresponding standard errors are estimated B times using a chosen estimation method (ordinary least squares, weighted least squares, ML, etc.). The average value of these parameters estimates 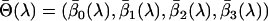 represents the parameter estimate corresponding to the fixed value of
represents the parameter estimate corresponding to the fixed value of 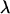 . The standard error of these parameters can also be obtained similarly by subtracting the sample covariance of
. The standard error of these parameters can also be obtained similarly by subtracting the sample covariance of 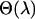 from the average variance of
from the average variance of 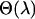 . The extrapolation step is conducted by first modeling each component of
. The extrapolation step is conducted by first modeling each component of 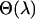 as an increasing linear, quadratic, or hyperbolic function of
as an increasing linear, quadratic, or hyperbolic function of 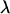 and then setting
and then setting  in the estimated equation to extrapolate back to the parameter estimates that one would observe without measurement error. The drawback of the SimEx algorithm in addition to the extrapolation step is that it is very computationally expensive. (For more details about SimEx see Cook and Stefanski 1994 and Carroll et al. 1995.)
in the estimated equation to extrapolate back to the parameter estimates that one would observe without measurement error. The drawback of the SimEx algorithm in addition to the extrapolation step is that it is very computationally expensive. (For more details about SimEx see Cook and Stefanski 1994 and Carroll et al. 1995.)
Assuming that an estimate of the covariance matrix of the measurement error variable is available, these measurement error correction methods can all be extended to correct for measurement error when estimating ancestry from admixed individuals resulting from the intermating of more than two parental populations. However, to our knowledge little is known regarding their statistical properties in the multivariate setting and more research is warranted in this area. For this reason, we restrict our comparison to the univariate case. Thus, our conclusions apply only to admixed populations that are derived from the intermating among individuals originating from exactly two founding populations.
Degree of measurement error considered in the analysis:
The measurement error correction methods do not appear to be beneficial when the initial ancestry proportions are very poorly estimated. Thus we consider only estimates of the admixture proportions that have reliability coefficients of at least 0.50 (i.e., 50%). This restriction is based only on the fact that one should always strive to start with the best ancestry proportion estimates possible and apply measurement error correction methods on these estimates to minimize type I error rate inflation. Indeed, this inflation worsens as the correlation between the available measure of individual ancestry and the true value decreases.
Data simulation:
We consider three scenarios: The first scenario is designed to mimic situations where the reliability of the estimated ancestry proportion—as defined in Equation 4—is very high and varies around 0.95; this reliability is ∼0.75 in the second scenario and ∼0.55 in the last scenario. We simulate 1000 replications of a data set containing 1000 individuals and 1000 markers. The markers are divided in blocks of 100 markers having δ-values varying from 0 to 0.9. That is, the average δ-value between the first 100 markers is ∼0.9 whereas the last 100 markers have approximately the same allele frequency in the two ancestral populations. Additional details about the simulation procedure can be found in the appendix. The estimated ancestry proportions are obtained by averaging the 22 chromosome-specific individual ancestry proportions. These proportions are obtained using, respectively, 10, 5, and 3 markers for the first, second, and third scenarios. These markers are taken in regions where the δ-values are at least ≥0.3. The ancestral allele frequencies are simulated such that their difference is around the desired δ-value. Each marker is generated using the simulated allele frequency in each ancestral population and conditioning on the individual ancestry proportion. These ancestry proportions were obtained from the same mixture of a uniform and normal distribution presented in Tang et al. (2005). We use a function similar to Equation 1 to generate the phenotypic variable with the only difference being that the phenotype now depends on only one variable that is removed from the list of markers to be tested. This is done to guarantee that every significant association detected between the phenotypic variable and a marker can be classified as a false positive.
RESULTS
It is noteworthy to mention that Cronbach's α provides a reliability coefficient for the average of the chromosome-specific ancestry estimates as a measure of the underlying true ancestry proportion. Consequently, we have used this average as an estimate of the true individual ancestry proportion. Our simulations have shown that for unlinked markers, the correlation between the individual ancestry estimate obtained by averaging over the chromosome-specific estimates and the estimate that one would obtain by considering all the markers together is estimated around 99.7%. This correlation is estimated at 99.5% when we considered a real data set that contained a little over 6000 individuals in which 1312 AIMs based on the marker panel described in Smith et al. (2004) were typed.
We first show how accurately the true measurement error variance is estimated using the upper bound provided by Cronbach's α. Figure 3 presents the average value over the 1000 replications of the true and estimated measurement error variance for each scenario. The estimated variance also seems to follow nicely the variation in the true variance observed from one replicate to another. The correlations between the true and the estimated measurement error variance are 0.97, 0.88, and 0.78 when the reliability coefficients are 0.95, 0.75, and 0.55, respectively. It is important, however, to keep in mind that Cronbach's α provides only the upper bound of the measurement error variance. One should also keep in mind that the bias observed in estimating this variance is directly associated with the quality of ancestry proportions estimates available in the study, which in turns determines the performance of the existing measurement error correction methods.
Figure 4 shows the comparison between each measurement error correction method and the case where measurement errors are ignored when the reliability coefficients of the estimated IAPs are ∼95, 75, and 55%, respectively, at the 5% significance level. The bars represent the average of the observed type I error computed over 1000 replications.
Figure 4.—

Comparison between the four measurement error correction methods and the case where no adjustment for measurement error correction is made for high correlation between the estimated individual ancestry proportions (IAPs) and the true IAPs. The estimated IAPs are computed using 220 markers having a δ-value of ≥0.5. There is apparently no need to apply measurement error correction methods under the ideal situation depicted in case I (the far left graph) where the reliability of the estimated ancestry proportion is 95% reliable as a measure of the true ancestry. However, since the true IAPs are not observed in a real study, one can never be sure that this level of correlation is achieved. Note that applying the QMEC, the ERC, or SimEx would provide adequate type I error control. In caseII, the reliability coefficient is 75%. The estimated IAPs are computed using 110 markers having a δ-value of ≥0.3. Applying measurement error correction in this situation provides better type I error control than naively using the estimated IAPs. In this case, QMEC maintains the desired significance level whereas RC and SimEx show 15% inflation at the significance level of 1% compared to 45% inflation that is observed when no measurement error correction is considered. Case III represents the scenario where the reliability coefficient of the estimated ancestry proportions as a measure of the true ancestry proportion is only 55%. To simulate this situation, the estimated IAPs are computed using 66 markers having a δ-value of ≥0.3. Applying the discussed measurement error correction methods in this case provides better type I error control than the naive method. The IAPs are poorly estimated and using them alone will lead to a 240% inflation rate at the significance level of 1%. The QMEC method still has the correct type I error. Although the other methods provide significantly less type I error inflation than QMEC, they show their limitations, which are mostly due to the fact that the assumption that the measurement errors are normally distributed does not hold.
Case I shows that when the estimated IAPs are highly reliable with the true IAPs, all the measurement error correction methods except the regression calibration approach perform well at the 5 and 1% nominal significance levels. Figure 4 shows that a very small amount of measurement errors in the estimated IAPs does not greatly increase the false positive rate of association tests that control for the right function of individual ancestry. However, since the true IAPs are not available in a real study, one cannot compute the correlation between the true and the observed IAPs and will never know the reliability of the estimated IAPs. However, applying the measurement error correction in this case would still provide the appropriate type I error.
Case II shows the observed type I error produced by each method when the reliability coefficient of the estimated IAPs is ∼75%. One can begin to see the advantage of using these methods to address measurement error in the IAPs. Without adjusting for measurement errors, at the 0.01 significance level (graph not shown), one would observe a 45% type I error inflation compared to the <2% inflation observed with QMEC. One can also start ranking the performance of each method at this level and realize that the quadratic measurement error correction method seems the most accurate of the four methods considered.
Case III allows one to better appreciate the need for measurement error correction. When the reliability of the estimated IAPs is only ∼55%, serious type I error inflation is observed when no measurement correction is considered. In fact, the inflation rate is ∼240% at the nominal rate of 1% whereas the best-performing measurement error correction method shows only a 12% inflation rate. This number also allows one to realize that these measurement error correction methods are not “fix all” methods. That is, one cannot start with bad measures of IAPs and expect adequate type I error control by applying these methods.
Figure 4 also shows that in general, the QMEC method performs better than the other methods presented in this article, maintaining the nominal type I error, which is set at 0.05. However, most genetic association analyses would consider much lower type I error levels. We compare the QMEC method to the case where measurement error in the ancestry proportion is ignored at the 10−4 nominal significance level. We conducted a more thorough simulation analysis based on 10,000 replications. Since we test 1000 hypotheses for each replicate, 10,000 replications is large enough to provide an acceptable amount of Monte Carlo error. The Monte Carlo error associated with this simulation study is given in Table 1. Figure 5 shows the effect of measurement error on the type I error is much more accentuated at lower nominal α-values. Figure 5 also demonstrates the benefit of applying measurement error correction in structured association tests. In the very rare cases where the IAPs are perfectly measured, the QMEC maintains its nominal type I error. In all other cases it will either maintain or suffer very slight type I error inflation compared to double-digit inflation that can be observed when these measurement errors are ignored.
TABLE 1.
Monte Carlo error of the 10,000 replications simulation study comparing the QMEC method to the case where measurement error in the ancestry proportion estimates is ignored
| Reliability coefficient | No correction | QMEC |
|---|---|---|
| High (ρ ∼ 0.95) | 0.00033 | 0.00038 |
| Medium (ρ ∼ 0.75) | 0.00054 | 0.00033 |
| Low (ρ ∼ 0.55) | 0.00196 | 0.00035 |
The comparison was done at the 10−4 significance level.
Figure 5.—

Comparison between QMEC and the case where measurement error in the ancestry proportion estimates is ignored for the reliability coefficients considered. (Nominal α = 10−4) The effect of measurement error on the type I error inflation is much more severe at the lower nominal significance value. Serious type I error inflation is observed for even relatively well-measured estimates of ancestry proportion. When the reliability coefficient ratio is 75%, which corresponds to a correlation coefficient close to 87% between true and estimated ancestry proportions, a type I error inflation close to 170% is observed when measurement error is ignored. This inflation is only 20% after correction for measurement error using the QMEC method. This inflation worsens as the reliability coefficient decreases and measurement error is ignored as shown in the graph when ρ ∼ 0.55. That is when the correlation between estimated and true ancestry proportions is ∼0.75. In this case, the inflation is ∼16 times higher than the nominal level whereas the QMEC method shows only slight deviation from its nominal type I error rate.
Finally, we ran three simulation studies to evaluate the effect of departure from the normality assumption on QMEC's type I error rate. We consider two skewed distributions for the measurement error variable, namely the asymmetric Gaussian (AG) and the lognormal distribution. By definition, the AG has three parameters, one overall mean and two variances: a variance (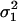 ) that corresponds to values that are less than or equal to the mean and another variance (
) that corresponds to values that are less than or equal to the mean and another variance (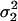 ) that is observed with values that are higher than the mean. The asymmetry or skewness coefficient of the AG depends on these two variances. Specifically, skewness increases when the absolute difference is higher between these two variances. We set the asymmetry coefficient at 0.333 in the first case, which is denoted by AG1, and at 0.5 in the second case, which is denoted by AG2. All three distributions (AG1, AG2, and lognormal) have a mean of zero and a variance that is computed such that the reliability of the error-contaminated variable is 0.95, 0.75, or 0.55, depending on the scenario that we want to mimic. We centered the lognormal distribution around its expected value in each case to obtain a measurement error distribution that has a mean of zero.
) that is observed with values that are higher than the mean. The asymmetry or skewness coefficient of the AG depends on these two variances. Specifically, skewness increases when the absolute difference is higher between these two variances. We set the asymmetry coefficient at 0.333 in the first case, which is denoted by AG1, and at 0.5 in the second case, which is denoted by AG2. All three distributions (AG1, AG2, and lognormal) have a mean of zero and a variance that is computed such that the reliability of the error-contaminated variable is 0.95, 0.75, or 0.55, depending on the scenario that we want to mimic. We centered the lognormal distribution around its expected value in each case to obtain a measurement error distribution that has a mean of zero.
Table 2 shows the ratio of the observed to nominal type I error rate observed when the measurement error variable follows an AG1, an AG2, or a lognormal distribution and the reliability coefficient is 0.95, 0.75, or 0.55. The robustness analysis shows that QMEC can in some cases be slightly conservative when the distribution of the measurement error variable is skewed. It seems that estimated ancestry proportions that are ∼75% reliable for the true ancestry proportions are the most affected. The type I error observed with highly reliable ancestry estimates is slightly inflated at the 10−4 significance level when the measurement error variable follows an asymmetric Gaussian and quite conservative where the errors follow a centered lognormal distribution. The type I error rate of the QMEC for less reliable measures of individual ancestry proportion is slightly conservative independently of the distribution of the measurement error variable. The ratio of the observed to nominal type I error is at least 2.5—at the nominal α-level of 10−4—in each case when no correction for measurement error is applied. Therefore, it would still be beneficial to apply QMEC even in cases where the distribution of the measurement error variable is not normal.
TABLE 2.
Ratio of the observed to nominal type I error when the measurement error variable follows a skewed distribution
| Significance level
|
|||||
|---|---|---|---|---|---|
| Error distribution | Reliability | 0.05 | 0.01 | 0.001 | 0.0001 |
| Asymmetric Gaussian 1 | 0.95 | 1.037 | 1.068 | 1.095 | 1.123 |
| 0.75 | 0.868 | 0.784 | 0.705 | 0.610 | |
| 0.55 | 0.944 | 0.923 | 0.861 | 0.860 | |
| Asymmetric Gaussian 2 | 0.95 | 1.025 | 1.043 | 1.044 | 1.048 |
| 0.75 | 0.869 | 0.797 | 0.715 | 0.680 | |
| 0.55 | 0.944 | 0.923 | 0.861 | 0.860 | |
| Lognormal | 0.95 | 0.867 | 0.784 | 0.645 | 0.510 |
| 0.75 | 0.968 | 0.931 | 0.851 | 0.920 | |
| 0.55 | 0.944 | 0.923 | 0.861 | 0.860 | |
DISCUSSION
We used simulations to demonstrate the importance of incorporating measurement error correction methods in association studies that use an estimate of individual ancestry as a covariate in a regression analysis. We then described four measurement error correction methods and showed how they can be applied to reduce the potential for residual confounding created by measurement errors inherent in the individual ancestry estimate. Finally, we focused on the method that seems to perform the best and ran more simulations to study its behavior at the 10−4 significance level and its robustness to deviation from the normality assumption that is made regarding the distribution of the measurement error variable. All measurement error correction methods require prior knowledge of the measurement variance. Because the classical methods that provide this estimate are either straightforward or cost effective, we showed how a measure of reliability as computed by Cronbach's α can be used to provide an estimate of the upper bound of the measurement error variance.
The expanded regression calibration (ERC) approach seems to have the highest type I error inflation rate. ERC assumes that the IAPs and the measurement error variable are normally distributed with known mean and variance. Our simulated data have shown that neither of these assumptions holds. In fact, the distribution of the ancestry proportions is highly skewed. More research in this area is required to derive the exact distribution of the true ancestry proportions. This distribution is crucial in determining the performance of the measurement error correction methods that we have considered. In fact, all methods that rely on the normality of the true ancestry proportions variable seem to produce higher than expected type I error when the correlation between true and estimated IAP is decreasing.
The regression calibration method as described here requires the inversion of a large matrix that is not guaranteed to be of full rank because the genotypic information was simulated at random. In this case the Penrose generalized inverse is used, which introduces greater variation in the corrected individual ancestry estimates, thus increasing the potential for residual confounding. Moreover, regression calibration, by definition, does not use the available phenotypic data, which consequently makes it less apt to break the confounding triangle.
The SimEx algorithm that we used also relies on the normality assumption. It did not provide adequate control of the type I error rate. This method is also quite computationally expensive. For example, to run 1000 replications of the SimEx algorithm with 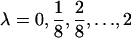 and 100 replicates for each noise level while investigating 1000 markers, one needs to run (1000 × 1000 × 100 × 17) = 17 × 108 regression analyses (Carroll et al. 1995).
and 100 replicates for each noise level while investigating 1000 markers, one needs to run (1000 × 1000 × 100 × 17) = 17 × 108 regression analyses (Carroll et al. 1995).
The QMEC method provides the most reliable type I error control for all three conditions considered in the analysis. It is important to note that this method relies less on the normality assumptions made regarding the distribution of the IAPs. In fact, unlike the expanded regression calibration method and the SimEx algorithm, it treats them as fixed constants instead of random draws from a normal distribution, and, contrary to the regression calibration approach, it uses the phenotypic variables in estimating the parameter of the quadratic regression. This method and the regression calibration take ∼1 min to run for one replicate, which makes them very fast compared to the SimEx algorithm and the expanded regression calibration method.
We showed that the individual admixture estimates obtained from a set of AIMs should be seen only as an error-contaminated measurement of the individual true ancestry proportion, which represents one of the variables that should be controlled for in a test of genetic association in an admixed population. We also demonstrated how small measurement error in these admixture estimates can inflate the type I error committed in this type of test and presented four measurement correction methods developed in the frequentist framework. Because all of these methods require an a priori estimate of the measurement error variance, we showed how Cronbach's α can be used to obtain the upper bound of this variance.
The results of the larger simulation study that compared the QMEC method to the case where measurement error is ignored confirm the initial findings regarding this method's ability to provide adequate the type I error control and highlighted the overall benefit of addressing the measurement error problem inherent to the estimation of individual ancestry proportion. This result is based on the assumption that the errors are normally distributed, with mean 0 and a constant variance, which implies that 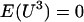 . The violation of this assumption may cause QMECs to become slightly conservative. However, centering the observed ancestry proportion on their observed mean should reduce this bias.
. The violation of this assumption may cause QMECs to become slightly conservative. However, centering the observed ancestry proportion on their observed mean should reduce this bias.
Although there is a consensus in the field regarding the value of controlling for genetic background variables in SAT to control both the false positive and the false negative rates, it is noteworthy to mention that the precision with which these variables are measured will affect the degree of type I error control they provide. When the variables are error contaminated, existing measurement error correction methods can help maintain the specified type I error rate. Caution in the choice of measurement error correction methods is merited, as is evaluation as to whether the assumptions underlying these methods are met and assurance that their a priori estimate of the measurement error variance is reasonably close to the true value.
Acknowledgments
We thank Raymond J. Carroll for his comments on an earlier version of this manuscript. We also thank Amit Patki and Vinodh Srinivasasainagendra for their help in programming these methods and parallelizing the SimEx algorithm. This work was supported in part by National Institutes of Health grants T32HL072757, P30DK056336, K25DK062817, T32AR007450, P01AR049084, and R01DK067426.
APPENDIX: EQUATIONS USED IN OUR SIMULATIONS
True ancestry proportion:
We used the mixture distribution described in Tang et al. (2005) to simulate directly the European ancestry in African-Americans. This distribution can be written as follows: 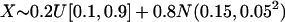 .
.
Allele frequency in the ancestral populations:
For a given δ-value, we use the following procedure to simulate the allele frequency in the ancestral population:
Draw δ from
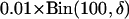 .
.- Let
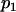 and
and  denote, respectively, the allele frequency in each ancestral population; then
denote, respectively, the allele frequency in each ancestral population; then 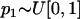 and
and 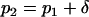 .
.- If (
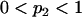 ) the allele frequencies are set at
) the allele frequencies are set at 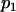 and
and 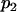 .
. - Else
- Repeat
- Until (
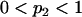 ).
).
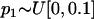
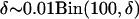
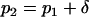
Allele frequency of the admixed individual:
The allele frequency of the admixed individual is given by 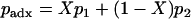 , where X represent the ancestry proportion obtained in step 1.
, where X represent the ancestry proportion obtained in step 1.
The allele at each marker is then generated by drawing from a Bernoulli distribution with frequency 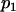 for an individual coming from the first ancestral population,
for an individual coming from the first ancestral population, 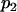 for an individual drawn from the second founding population, and
for an individual drawn from the second founding population, and 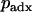 for an admixed individual.
for an admixed individual.
Phenotypic variable:
The phenotypic variable is generated using an equation of the form 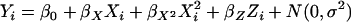 .
.
References
- Carroll, R. J., 1989. Covariance analysis in generalized linear measurement error models. Stat. Med. 8: 1075–1093. [DOI] [PubMed] [Google Scholar]
- Carroll, R. J., and L. Stefanski, 1990. Approximate quasilikelihood estimation in models with surrogate predictors. J. Am. Stat. Assoc. 85: 652–663. [Google Scholar]
- Carroll, R. J., P. P. Gallo and L. J. Gleser, 1985. Comparisons of least squares and errors-in-variables regression, with special reference to randomized analysis of covariance. J. Am. Stat. Assoc. 80: 929–932. [Google Scholar]
- Carroll, R. J., D. Ruppert and L. A. Stefanski, 1995. Measurement Error in Nonlinear Models. Chapman & Hall/CRC, London.
- Cheng, C. L., and H. Schneeweiss, 1998. Polynomial regression with errors in the variables. J. R. Stat. Soc. Ser. B Stat. Methodol. 60: 189–199. [Google Scholar]
- Cheng, C. L., and J. Van Ness, 1999. Statistical Regression With Measurement Error. Oxford University Press, New York.
- Cheng, C. L., H. Schneeweiss and M. Thamerus, 2000. A small sample estimator for a polynomial regression with errors in the variables. J. R. Stat. Soc. Ser. B Stat. Methodol. 62: 699–709. [Google Scholar]
- Cook, J. R., and L. A. Stefanski, 1994. Simulation-extrapolation estimation in parametric measurement error models. J. Am. Stat. Assoc. 89: 1314–1328. [Google Scholar]
- Cronbach, L., 1951. Coefficient alpha and the internal structure of tests. Psychometrika 16: 297–334. [Google Scholar]
- Darvasi, A., and S. Shifman, 2005. The beauty of admixture. Nat. Genet. 37: 118–119. [DOI] [PubMed] [Google Scholar]
- Devlin, B., and K. Roeder, 1999. Genomic control for association studies. Am. J. Hum. Genet. 65: A83. [DOI] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller, W. A., 1987. Measurement Error Models. John Wiley & Sons, New York.
- Gleser, L. J., 1990. Improvements of the naive approach to estimation in nonlinear errors-in-variables regression models, pp. 99–114 in Statistical Analysis of Measurement Error Models and Application. American Mathematical Society, Providence, RI.
- Greenland, S. A. N. D., and J. M. Robins, 1985. Confounding and misclassification. Am. J. Epidemiol. 122: 495–506. [DOI] [PubMed] [Google Scholar]
- Hoggart, C. J., E. J. Parra, M. D. Shriver, C. Bonilla, R. A. Kittles et al., 2003. Control of confounding of genetic associations in stratified populations. Am. J. Hum. Genet. 72: 1492–1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowler, W. C., R. C. Williams, D. J. Petitt and A. G. Steinberg, 1988. Gm3–5,13,14 and type-2 Diabetes-Mellitus - an association in American-Indians with genetic admixture. Am. J. Hum. Genet. 43: 520–526. [PMC free article] [PubMed] [Google Scholar]
- Kuha, J., and J. Temple, 2003. Covariate measurement error in quadratic regression. Int. Stat. Rev. 71: 131–150. [Google Scholar]
- Long, J. C., 1991. The genetic structure of admixed populations. Genetics 127: 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue, P. M., 1997. Mapping genes underlying ethnic differences in disease risk by linkage disequilibrium in recently admixed populations. Am. J. Hum. Genet. 60: 188–196. [PMC free article] [PubMed] [Google Scholar]
- McKeigue, P. M., 1998. Mapping genes that underlie ethnic differences in disease risk: methods for detecting linkage in admixed populations, by conditioning on parental admixture. Am. J. Hum. Genet. 63: 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeigue, P. M., 2005. Prospects for admixture mapping of complex traits. Am. J. Hum. Genet. 76: 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, N., N. Hattangadi, B. Lane, K. E. Lohmueller, D. A. Hafler et al., 2004. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 74: 979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaff, C. L., J. Barnholtz-Sloan, J. K. Wagner and J. C. Long, 2004. Information on ancestry from genetic markers. Genet. Epidemiol. 26: 305–315. [DOI] [PubMed] [Google Scholar]
- Price, A. L., N. J. Patterson, R. M. Plenge, M. E. Weinblatt, N. A. Shadick et al., 2006. Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38: 904–909. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and P. Donnelly, 2001. Case-control studies of association in structured or admixed populations. Theor. Popul. Biol. 60: 227–237. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., and M. Przeworski, 2001. Linkage disequilibrium in humans: models and data. Am. J. Hum. Genet. 69: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., and N. A. Rosenberg, 1999. Use of unlinked genetic markers to detect population stratification in association studies. Am. J. Hum. Genet. 65: 220–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. J. Donnelly, 1999. Correcting for population stratification in linkage disequilibrium mapping studies. Am. J. Hum. Genet. 65: A101. [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. a Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens, N. A. Rosenberg and P. Donnelly, 2000. b Association mapping in structured populations. Am. J. Hum. Genet. 67: 170–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redden, D., J. Divers, L. Vaughan, H. Tiwari, T. Beasley et al., 2006. Regional admixture mapping and structured association testing: conceptual unification and an extensible general linear model. PLoS Genet. 2: 1254–1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg, N. A., L. M. Li, R. Ward and J. K. Pritchard, 2003. Informativeness of genetic markers for inference of ancestry. Am. J. Hum. Genet. 73: 1402–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneeweiss, H., and T. Nitter, 2001. Estimating a polynomial regression with measurement errors in the structural and in the functional case-a comparison, pp. 195–207 in Data Analysis From Statistical Foundations. Nova Science, New York.
- Shifman, S., J. Kuypers, M. Kokoris, B. Yakir and A. Darvasi, 2003. Linkage disequilibrium patterns of the human genome across populations. Hum. Mol. Genet. 12: 771–776. [DOI] [PubMed] [Google Scholar]
- Smith, M. W., N. Patterson, J. A. Lautenberger, A. L. Truelove, G. J. Mcdonald et al., 2004. A high-density admixture map for disease gene discovery in African Americans. Am. J. Hum. Genet. 74: 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spielman, R. S., R. E. Mcginnis and W. J. Ewens, 1993. Transmission test for linkage disequilibrium: the insulin gene region and insulin-dependent diabetes mellitus (IDDM). Am. J. Hum. Genet. 52: 506–516. [PMC free article] [PubMed] [Google Scholar]
- Tang, H., J. Peng, P. Wang and N. J. Risch, 2005. Estimation of individual admixture: analytical and study design considerations. Genet. Epidemiol. 28: 289–301. [DOI] [PubMed] [Google Scholar]
- Weinberg, C. R., 1993. Toward a clearer definition of confounding. Am. J. Epidemiol. 137: 1–8. [DOI] [PubMed] [Google Scholar]
- Weir, B. S., 1996. Genetic Data Analysis II. Sinauer Associates, Sunderland, MA.
- Wolter, K. M., and W. A. Fuller, 1982. Estimation of the quadratic errors-in-variables model. Biometrika 69: 175–182. [Google Scholar]
- Zhang, S., X. Zhu and H. Zhao, 2006. On a semiparametric test to detect associations between quantitative traits and candidate genes using unrelated individuals. Genet. Epidemiol. 24: 44–56. [DOI] [PubMed] [Google Scholar]
- Zhang, S. L., X. F. Zhu and H. Y. Zhao, 2003. On a semiparametric test to detect associations between quantitative traits and candidate genes using unrelated individuals. Genet. Epidemiol. 24: 44–56. [DOI] [PubMed] [Google Scholar]
- Ziv, E., and E. G. Burchard, 2003. Human population structure and genetic association studies. Pharmacogenomics 4: 431–441. [DOI] [PubMed] [Google Scholar]