

Abstract
Crossover interference in meiosis is often modeled via stationary renewal processes. Here we consider a new model to incorporate the known biological feature of “obligate chiasma” whereby in most organisms each bivalent almost always has at least one crossover. The initial crossover is modeled as uniformly distributed along the chromosome, and starting from its position, subsequent crossovers are placed with forward and backward stationary renewal processes using a chi-square distribution of intercrossover distances. We used our model as well as the standard chi-square model to simulate the patterns of crossover densities along bivalents or chromatids for those having zero, one, two, or three or more crossovers; indeed, such patterns depend on the number of crossovers. With both models, simulated patterns compare very well to those found experimentally in mice, both for MLH1 foci on bivalents and for crossovers on genetic maps. However, our model provides a better fit to experimental data as compared to the standard chi-square model, particularly regarding the distribution of numbers of crossovers per chromosome. Finally, our model predicts an enhancement of the recombination rate near the extremities, which, however, explains only a part of the pattern observed in mouse.
MEIOSIS is a key step in the life cycle of organisms that reproduce sexually because it is the process that leads to the halving of chromosome number; ploidy level is restored at the fertilization step. In the meiotic process, crossovers (COs) play a crucial role, ensuring the proper segregation of homologous chromosomes at the first division. Indeed, they make a mechanical link between homologs that is essential to ensure that the cell will send each homolog to opposite poles. A CO may be observed cytologically because it forms a structure called chiasma. In addition, genetic recombination due to COs is an important evolutionary force involved in shaping the genetic diversity of natural and artificial populations. For instance, the frequency of COs may be different between sexes or according to the mode of reproduction (Lenormand and Dutheil 2005; Roze and Lenormand 2005) and seems to play an evolutionary role.
COs lead to reciprocal exchange of large fragments of homologous nonsister chromatids (supplemental Figure 1 at http://www.genetics.org/supplemental/). Thus, the genetic outcome of COs is the reassociation of genetic markers located on both sides of the CO point. COs occur in the context of a bivalent that is a structure associating two homologous chromosomes, each formed by two sister chromatids. Each gamete will inherit one of these chromatids. When a bivalent exhibits one CO, two nonsister chromatids are recombined while the other two remain unchanged. With two COs on one bivalent, three kinds of chromatids are recovered with zero, one, or two COs (supplemental Figure 1). It is generally considered that there is no chromatid interference (Zhao et al. 1995; Copenhaver et al. 1998); i.e., when two COs occur on the bivalent the chromatids involved in each CO are randomly and independently chosen.
CO distribution along chromosomes may be estimated at the bivalent level, by direct cytological observation of chiasmata or by immunofluorescent observation of foci of different proteins associated with CO formation (Lawrie et al. 1995; Froenicke et al. 2002; de Boer et al. 2006). In such cases, one measures the density of chiasmata or foci along the bivalent, and the distance is measured in micrometers under the microscope. Alternatively, CO distributions may be estimated at the chromatid level by analyzing segregation data from high-density linkage-mapping experiments (Broman et al. 2002). In such cases, the CO position is between two adjacent genetic markers that display recombination, and the distance is measured in units of genetic distance (centimorgans). In all cases, CO density is not uniformly distributed along physical chromosomes; the pattern varies greatly among species and even among chromosomes within a species. A very common rule is that COs are strongly suppressed at the centromere regions, even if in some species COs have been found preferentially localized in pericentromeric regions (Jones 1984; Choo 1998; see review by Mezard 2006). Although the causes of such variability are still not understood, it is clear that CO distribution is a highly regulated process. First, in most organisms, each pair of chromosomes, whatever their size, bears at least one CO (Jones and Franklin 2006). Second, COs are not independent of each other: the occurrence of one CO inhibits the occurrence of others in a distance-dependent manner, resulting in COs being spaced more evenly through the chromosomes than would be expected if they occurred independently. This phenomenon is known as positive interference (hereafter referred to as interference; Jones 1984). As a consequence, the number of COs is often limited (typically one to two COs per chromosome in mouse). The existence of interference has been confirmed in most species tested (exceptions include Schizosaccharomyces pombe and Aspergillus nidulans) although its mechanism remains mysterious (Zickler and Kleckner 1999). In addition, data obtained in yeast (Hollingsworth and Brill 2004; Stahl et al. 2004), human (Housworth and Stahl 2003), and Arabidopsis thaliana (wild type, Lam et al. 2005; Copenhaver et al. 2002; mutants, Higgins et al. 2004; Mercier et al. 2005) strongly suggest that while most COs are subject to interference, some of them are not, defining two classes of COs (Mezard et al. 2007). The ratio of interfering COs to noninterfering COs differs between the species in which it has been studied. In yeast, ∼30% of the COs seem to escape the interference mechanism and in A. thaliana it is ∼15%. In mouse, several results (reviewed in de Boer et al. 2006) suggest that noninterfering COs constitute ∼10% of the total CO number. At the two extremes are Caenorhabditis elegans that has only interfering COs and S. pombe with only noninterfering COs. Interestingly, interfering COs can be specifically detected during meiosis, using immunodetection of the MLH1, one of the proteins involved in this pathway of CO formation.
Interference is thus important for the dynamics of diversity and genetic linkage in natural populations or in controlled crosses. In particular, it should be properly modeled when building linkage maps because the map function converting recombination fractions into additive genetic distances depends on the frequency of double COs, and this frequency is sensitive to interference. Moreover, interference reduces the probability of close recombination events on a gamete, thereby strongly affecting the way the genetic material is shuffled at each generation.
Numerous models have been proposed to describe the role of interference in the positioning of COs along chromosomes (Mezard et al. 2007). An important mathematical model of the field is the “chi-square model” (also called “counting model”) that produces a very satisfactory fit of predicted CO distribution to genetic data sets in many cases (Foss et al. 1993). The chi-square model is based on the assumption that a fixed number of failed events separate COs. That was motivated by the fact that biologically, meiotic recombination is known to be initiated by DNA double-strand breaks that are repaired as either COs or non-CO (Bishop and Zickler 2004). Thus, the failed events would be the non-CO events. However, the chi-square model based on a fixed number of failed events between two COs is challenged by data that suggest that the number of failed events varies between sexes and individuals in the same species, between chromosomes in the same nucleus, and even along chromosomes or in mutant context (Lin et al. 2001; Esch 2005; Martini et al. 2006). Furthermore, the biological foundation of the chi-square model remains speculative as no biological process has been described that directly counts DNA double-strand breaks, although some interesting hypotheses of ordered clusters of recombination intermediates have been proposed by Stahl et al. (2004). Nevertheless, this model still provides a very good fit to genetic data and can be used to model CO distribution without assuming any particular biological process (de Boer et al. 2006). Very similar to this chi-square model, the gamma model (McPeek and Speed 1995) is obtained by pulling inter-CO distances from a gamma probability distribution. For integer values of its parameter, the gamma model is strictly equivalent to the chi-square model.
It has been known for some time that in a majority of organisms, successful meiosis requires at least one chiasma to be formed on each bivalent, a constraint referred to as obligate chiasma (OC) (Jones 1984; Jones and Franklin 2006). Surprisingly, although there is a vast literature on mathematical modeling of interference (Zhao and Speed 1996), little has been done to include OC. To take into account the OC, we introduce a model of CO formation with OC. One possible explanation for the OC phenomenon is that in a process sequential in time, at least one CO should arise if enough double-strand break sites attempt to produce COs. Then, once a site is sufficiently advanced in the CO process, interference sets in; i.e., that site will inhibit the sites in its neighborhood. In this work, we modified the chi-square model to take into account this OC constraint and examine the general pattern that follows for the COs along bivalents and along chromatids. We first do this at the bivalent level, comparing simulations obtained with the chi-square and our models to experimental distributions of the subset of COs marked by MLH1 along mouse bivalents. These COs are believed to belong to the interfering pathway. The data we use were published by Froenicke et al. (2002). Then we extend this study to CO patterns at the chromatid level, comparing simulations obtained with our model to experimental distributions of recombination along mouse linkage maps published by Broman et al. (2002). Finally we show that our model naturally leads to an enhanced recombination rate along physical chromosomes near the telomeres and compare this to the mouse data in Froenicke et al. (2002).
MODELS
Distance units and chromosome length:
Throughout this article, positions of loci along a chromosome are expressed either in physical micrometer distance along the synaptonemal complex (SC) or in genetic distance as for linkage maps. In addition, we also introduce the interference-relevant distance (IRD), which is the distance for the coordinate space in which we suppose that interference is produced.
SC distances can be measured from microscope image analysis as described by Froenicke et al. (2002). They reflect a certain degree of chromatin condensation that may be heterogeneous, so this distance is not expected to be linear in the physical distance (measured in base pairs along the DNA molecule).
Genetic distances are expressed in morgans; this unit is defined as the distance over which the mean number of COs arising on a chromatid is one per meiosis. In the coordinate space of genetic distance, the CO density is thus uniformly distributed along the chromosome. In general, genetic distance is proportional neither to SC distance nor to physical distance in base pairs along the DNA molecule.
We define IRD as the coordinate space in which we model interference. This space is most commonly supposed to be the same as the genetic distance space, since most interference models suppose that two COs are less likely to occur when the genetic distance between them becomes low. This means that such models consider that the closer two intervals are on the linkage map, the less likely they are to be both recombinant in the same meiosis. In this article, we introduce a new interference model in which IRD and genetic distances are not proportional to each other.
It is possible to convert positions from one coordinate space to another, provided that the CO density along each space is known. So if 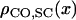 ,
, 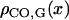 , and
, and 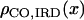 are the CO densities at position x in the SC space, the genetic space, and the IRD space, respectively, and XSC, XG, and XIRD are the positions of a given locus in these three spaces, respectively, correspondences between these positions may be calculated using the relation
are the CO densities at position x in the SC space, the genetic space, and the IRD space, respectively, and XSC, XG, and XIRD are the positions of a given locus in these three spaces, respectively, correspondences between these positions may be calculated using the relation
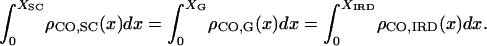 |
At the chromatid level, 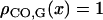 for any x by definition of the genetic distance. At the bivalent level, we see later that
for any x by definition of the genetic distance. At the bivalent level, we see later that 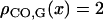 for any x.
for any x.
From bivalents to chromatids:
Interference arises at the bivalent level, which consists of the four aligned chromatids tied together in the SC. For each CO generated, the two (nonsister) chromatids involved are chosen at random with equal probabilities among the four possible pairs, following the hypothesis of no chromatid interference (supplemental Figure 1 at http://www.genetics.org/supplemental/). Thus, once COs have been placed along a bivalent by using any model of interference, it is possible to model the position of COs along a chromatid derived from this bivalent by randomly selecting on average half of the COs and discarding the other ones. This is called “random thinning” (McPeek and Speed 1995). Then, the mean density of COs on the bivalent must be 2.0 per morgan because each CO has a probability of 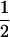 to be passed on to a chromatid and there the density of COs must be 1.0 by definition of the morgan unit for genetic distance.
to be passed on to a chromatid and there the density of COs must be 1.0 by definition of the morgan unit for genetic distance.
Interference models using stationary renewal processes:
The main models of CO interference are based on stationary renewal processes (SRPs) (Zhao and Speed 1996). In a renewal process, one generates a succession of points (positions) along a line, going from say left to right. The distances between successive points are independent and identically distributed random variables. Such a process becomes “stationary” (statistically independent of where one sets the origin of the coordinates on the line) if the first point is laid down far enough to the left; in particular, the expected density of points becomes uniform along the line. For the modeling of COs along bivalents, first, place on the line a segment of length equal to the genetic length of the bivalent under consideration; then, use the SRP to lay down points on the line; finally, identify the positions of the SRP points lying within the segment as COs at the corresponding genetic positions on the bivalent. By stationarity of the SRPs, the density of COs is uniform along the bivalent. Note that in such SRPs, the IRD space is the genetic distance space, so inter-CO distances are expressed in morgans. Interference is naturally associated with the distribution of distances (d) between successive COs: if interference is high, the points will tend to be regularly spaced. If, on the contrary, there is no interference, then d has an exponential distribution, corresponding to the Haldane no interference model (Haldane 1919). SRPs are useful mathematical tools for modeling the interference that one CO has on its surrounding. One of the most commonly used SRPs is the chi-square model (Foss et al. 1993), also called the counting model. We focus on it because of its simplicity and goodness-of-fit to data in previous works. In that model, d is obtained by summing m + 1 random variables (m is an integer parameter corresponding to the number of precursors that do not turn into COs), each of them following an exponential distribution. This is equivalent to pulling d from a chi-square distribution with 2(m + 1) d.f. To ensure an average number of two COs per morgan unit of bivalent length, each value of d has to be divided by 2(m + 1). The biological interpretation of this model is that the current CO prevents the following m putative ones from being realized so they are “skipped.” When m = 0, we recover the noninterfering model (Haldane's model). The chi-square model has been applied to describe interference in a number of different organisms, and adjustments to biological data lead to average values of m = 2 for Neurospora, m = 4 for Drosophila (Foss et al. 1993), and m = 3–9 for Arabidopsis, depending on chromosomes (Copenhaver et al. 2002; Lam et al. 2005).
“Forced initial CO” model for obligate chiasma:
For each bivalent, having at least one CO ensures that meiosis will correctly segregate the chromosomes. In general, SRPs do not lead to OC because there is some chance that the interval generated by the SRP will be larger than the length of the chromosome.
We describe here an interference model in which the OC rule is enforced by construction: we directly generate the position of an initial CO with a uniform distribution along the IRD space. Then we generate further COs successively toward each end of the bivalent via SRPs using for each inter-CO distance d a sum of m + 1 exponentially distributed random variables. The resulting process is referred to as forced initial CO (FIC). Both the FIC and the chi-square models involve SRPs parameterized by the integer m, which specifies the number of putative points to skip. Note that both the chi-square and our FIC model involve a single parameter m that describes the intensity of the interference; no other adjustment to the data can be made because we assume given the genetic length of the chromosome.
The FIC model is not a SRP, and it generates CO distributions that are not uniform if m is different from zero. In other words, the IRD space within the FIC model is different from the genetic distance space. So we have to go from this IRD space to the distance of interest that can be either the SC distance or the genetic distance, depending on the type of data that we want to compare to. For this, we simulate 5 × 105 bivalents to numerically obtain the CO density distribution along IRD space, and, after CO positions have been simulated, we convert their IRD position into either SC or genetic positions as described before (no adjustable parameter here).
To ensure an average number of 2 COs per morgan unit of bivalent length, we do not know of a mathematical solution to calculate the rescaling coefficient that should be applied to d. So we determine this rescaling coefficient numerically by simulating COs on 104 bivalents and iterating this process on the rescaling coefficient until the average simulated number of COs per morgan unit of bivalent length reaches 2.0 ± 10−3.
DATA SETS AND ANALYSIS
Modeling interference between MLH1 foci using data from Froenicke et al. (2002):
The mouse data kindly provided to us by Lorrie Anderson specify, for each chromosome separately, the positions of the MLH1 foci, measured in relative SC distances, decomposed according to whether one focus or two foci are observed on the same SC, as displayed in Figure 3 of Froenicke et al. (2002). That study used male mice of strain C57BL/6J. During meiosis, the MLH1 foci were detected by immunofluorescence, their position (in micrometers) on the SC was measured, and the identification of the corresponding chromosome number was obtained by chromosome “painting.” MLH1 foci are associated with the formation of a subset of COs corresponding to the interfering pathway (Argueso et al. 2004; de Boer et al. 2006). See Froenicke et al. (2002) for further details.
Figure 3.—

Experimental and simulated frequencies of MLH1 foci along individual synaptonemal complexes (SCs) from centromere (left end) to telomere (right end), for bivalents having one (circles) or two (triangles) MLH1 and for all bivalents (diamonds). Experimental data (open symbols and dashed lines) come from cytogenetical observations of MLH1 foci on male mice bivalents by Froenicke et al. (2002). Simulations (solid symbols and lines) are computed using the chi-square model or our FIC model (see text). Chromosome length (in morgans) is calculated as half the average number of MLH1 foci per bivalent in experimental data (see values in Table 1). The interference parameter m is adjusted for each chromosome on the basis of the distribution of inter-CO distances (see text; values in Table 1) and adjusted independently for the chi-square model and the FIC model. x-axis: relative position of the middle of the bin along the synaptonemal complex (SC), expressed in relative SC distance. All bin sizes are 0.1. Frequencies are shown for chromosomes 1 (long) and 16 (short) and for all 19 autosomes pooled (see text). Graphs of other individual chromosomes are given in supplemental Figure 2 at http://www.genetics.org/supplemental/. All simulations are based on 5 × 105 simulated meioses.
We calculated 95% confidence intervals on frequencies of MLH1 foci on bivalents estimated from experimental data as 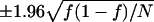 , where f is the estimated frequency of these foci and N is the total number of bivalents observed.
, where f is the estimated frequency of these foci and N is the total number of bivalents observed.
We use both our FIC model and the standard chi-square model to simulate (i) frequencies of SCs with zero, one, two, or three or more COs and (ii) patterns of COs along SCs having one CO and along SCs having two COs. The MLH1 mapping data of Froenicke et al. (2002) give the position in fractions of the total SC length. We convert this fraction into the usual genetic distance as described before, using the density distribution of MLH1 foci along the SC space. This last density distribution is taken from the data of Froenicke et al. (2002). We also use the experimental data set to determine the genetic length of each chromosome, as half the average number of MLH1 foci per bivalent. To stay as close as possible to the data published in Froenicke et al. (2002), we use here only fractions of total SC lengths, so standardized positions on each chromosome go from zero to one. The interference parameter m is the only quantity we can adjust; for this adjustment, we proceed as follows: (1) for every pair of adjacent MLH1 foci in the experimental data set, we calculate the genetic distance d between both foci; (2) we calculate the distribution of d as the frequencies of d-values falling into 10 bins of identical width, spanning the length of the chromosome; (3) for all integer values of m between 0 and 50, we simulate 107 meioses using our models, derive a series of simulated d values, and calculate their distribution as for experimental data; (4) for each value of m, we measure the goodness-of-fit of the model as the sum of squares of differences between experimental and simulated frequencies over the 10 distance bins; and (5) finally we set m to the value for which this sum of squares of differences reaches a minimum.
To pool MLH1 density data over all chromosomes, the numbers of foci observed in a given position bin are summed over all chromosomes, and global densities are then calculated for each bin. For simulations, chromosome lengths are determined from the data for each chromosome separately and then m is adjusted. Similarly, simulated COs are pooled over chromosomes as for experimental MLH1 foci.
Modeling interference between COs using data from Broman et al. (2002):
The data kindly provided to us by Karl Broman specify separately for each chromosome the positions of COs, measured in genetic distance, on a high-density mouse linkage map reflecting female meiosis. Data are decomposed according to whether zero, one, two, or three COs are observed on the same gametic chromosome as displayed in Figure 3 of Broman et al. (2002). That work pooled two data sets from 94 progeny of each of the interspecific BSB and BSS backcross populations, respectively (C57BL/6J × Mus spretus) F1 × C57BL/6J and (C57BL/6J × SPRET/Ei) F1 × SPRET/Ei (Rowe et al. 1994), genotyped with respectively 1372 and 4913 genetic markers, 904 markers being common to both populations. See Broman et al. (2002) for further details.
Here for each chromosome we set m to ν − 1, where ν is the parameter of the gamma model estimated by Broman et al. (2002). Indeed, the gamma model with an integer value of ν is strictly equivalent to a chi-square model with m = ν − 1 (McPeek and Speed 1995). So we apply our model without using any adjusted parameter. For chromosomes 17 and 18, genetic length was estimated by Broman et al. (2002) to values slightly <50 cM. Since such values are not in accordance with the OC constraint and thus are not allowed for the FIC model, we set those chromosome lengths to 50 cM for the simulations. In general, increasing the genetic length will lower the goodness-of-fit to the data, and so this procedure should not skew the analysis in favor of the model.
Density data pooling over chromosomes is performed as for MLH1 data, by pooling COs instead of MLH1 foci.
Modeling the heterogeneity of MLH1 foci distribution along chromosomes:
If there were a unique pathway to produce COs, the distribution of MLH1 foci would give an exact picture of the pattern of recombination rate along the bivalent. Under this simplified hypothesis, we investigate to what extent the observed distribution of MLH1 foci along SCs could be a consequence of both interference and the OC constraint.
To do this, we simulate crossovers on bivalents with our FIC model considering IRD positions to be the same as SC positions, and so no conversion between these positions is necessary. We thus compare directly the simulated pattern of CO density along IRD space with the pattern of MLH1 foci density along SC space, to see to what extent these patterns may be shaped by interference and OC.
As before, we pool data over chromosomes by taking the experimental densities of foci for each chromosome and pooling them. The same procedure is used in the simulations where for each chromosome the value of m is adjusted to the data as seen before.
So far we have never considered centromere effects, one reason being that both the Froenicke et al. (2002) and the Broman et al. (2002) studies are on mice where chromosomes are telocentric. Nevertheless, in most organisms, the centromere suppression effect is the dominant effect patterning recombination rate along chromosomes, so we cannot escape modeling it here. We have thus extended our model to take into account in an all-or-none fashion the effect of the centromere on recombination. In this extended model, we simulate the COs as follows: the first CO must fall outside the centromere; and subsequent ones are generated as before but only those outside the centromere are kept.
RESULTS
Patterning of MLH1 foci along the chromosome:
In Figure 1 we show the histograms of simulated CO frequencies on bivalents (along the genetic map) when one has one, two, and three COs. The pattern appears as a ripple effect, the more COs there are, the more ripples appear. For meioses with just one CO, the distribution is lower at the chromosome ends while it is higher for meioses with two COs. Further ripples appear when the number of COs grows, and the density is higher in the middle for an odd number of COs and lower for an even number of COs. If interference is absent (m = 0), each of the curves is completely flat, while as m grows, the ripples become stronger (Figure 1). Another feature is the dependence on the chromosome length; for short chromosomes (between 50 and 70 cM) and m intermediate to large, the curve associated with having two COs has a deep trough near the middle. For long chromosomes, the patterning becomes weaker (Figure 1), and for very long chromosomes, the ripples disappear, leaving just the uniform distribution. Qualitatively, the chi-square model and our FIC model lead to similar patterns (Figure 1). However, for short chromosomes, the FIC model predicts a lower proportion of bivalents with two and three COs and a greater proportion of bivalents with one CO, as compared to the chi-square model (Figure 1). This difference becomes greater for shorter chromosomes and weaker interference, and, inversely, the differences between the models become negligible for long chromosomes or strong interference.
Figure 1.—

Frequencies of crossovers (COs) along bivalents having one (circles), two (triangles), or three COs (squares) and for all bivalents (diamonds). Positions are based on genetic distances. The chromosome is taken to have a genetic size of 100 or 60 cM. The data were obtained from simulations using the standard chi-square model (open symbols and dashed lines) and our FIC model enforcing the obligate chiasma constraint (solid symbols and lines), using m = 4 and m = 12. x-axis: relative position of the middle of the bin along the synaptonemal complex (SC), expressed in relative genetic distance. All bin sizes are 0.05. CO frequencies are calculated from 5 × 107 simulated meioses. The total curve for all bivalents is flat because of the definition of genetic distance.
Let us now compare our model with the experimental distributions obtained by Froenicke et al. (2002). Table 1 gives the values of interference parameter m in the chi-square model and in our FIC model after adjustment to the experimental data for each chromosome. For short chromosomes 15–19, because of OC there are only a very small number of bivalents with two MLH1 foci in the experimental data, so that sometimes no adjustment was possible for m. In such cases, however, the value of m has almost no effect on the outcome of the models, so that m was arbitrarily set to 50.
TABLE 1.
Estimates of chromosome genetic length L and interference parameter m from female mouse CO mapping along genetic maps (data from Broman et al. 2002) or from male mouse MLH1 foci mapping along bivalents (data from Froenicke et al. 2002)
| Chromosome | LMLH1 (cM) | mMLH1 FIC | mMLH1 χ2 | LCO (cM) | mCO FIC |
|---|---|---|---|---|---|
| 1 | 76.2 | 25 | 20 | 98.4 | 9 |
| 2 | 77.7 | 9 | 8 | 87.2 | 9 |
| 3 | 63.1 | 26 | 30 | 75.5 | 22 |
| 4 | 70.1 | 14 | 12 | 89.4 | 6 |
| 5 | 72.0 | 50 | 45 | 82.4 | 10 |
| 6 | 59.7 | 15 | 17 | 67.0 | 12 |
| 7 | 68.1 | 9 | 10 | 71.8 | 9 |
| 8 | 60.8 | 15 | 18 | 67.6 | 9 |
| 9 | 55.7 | 24 | 34 | 67.6 | 9 |
| 10 | 61.7 | 8 | 10 | 62.2 | 28 |
| 11 | 69.9 | 14 | 12 | 68.1 | 11 |
| 12 | 54.6 | 4 | 8 | 56.4 | 4 |
| 13 | 51.4 | 5 | 9 | 66.0 | 23 |
| 14 | 53.2 | 50 | 50 | 61.7 | 11 |
| 15 | 49.6 | 50a | 22 | 54.3 | 62 |
| 16 | 50.0 | 50a | 9 | 61.2 | 35 |
| 17 | 50.9 | 50a | 16 | 49.5b | 35 |
| 18 | 50.9 | 50a | 10 | 41.5b | 15 |
| 19 | 50.0 | 50a | 50a | 51.6 | NA |
| X | NA | NA | NA | 76.6 | 9 |
For MLH1 foci data, estimates of m are obtained with either the FIC model or the standard chi-square model. For CO data, m was estimated with the FIC model.
For short chromosomes, given the very small number of bivalents with two MLH1 foci, an arbitrary value of m = 50 was used.
For chromosomes 17 and 18, genetic length was estimated from the data of Broman et al. (2002) to values <50 cM. Since such values are not in accordance with the OC constraint and thus are not allowed for the FIC model, we set these chromosome lengths to 50 cM for the simulations.
Figure 2 shows the observed and expected frequencies of bivalents with zero, one, two, and three or more MLH1 foci (experimental data) or simulated COs. The chi-square and the FIC models give a similarly good fit to the data for longer chromosomes (1–11), whereas in shorter chromosomes (12–19) the chi-square model often tends to predict too few bivalents with one CO and too many ones with zero or two COs, so that the FIC model provides a more satisfactory fit to the data. To obtain an objective measure of the goodness-of-fit to the data for each model, we calculated sums of squares of differences between observed and expected frequencies of bivalents with zero, one, two, and three or more MLH1 foci or simulated COs (Table 2). The lower this value the better the model fits the data. Pooling all chromosomes together as explained before, this sum of squares is 5.2 × 10−4 for the FIC model and 7.9 × 10−3 for the chi-square model, a value 15 times higher than with the FIC model.
Figure 2.—

Experimental and simulated frequencies of bivalents having zero, one, two, or three or more (x-axis) experimental MLH1 foci (open bars) or simulated COs (solid and dashed bars). Error bars indicate 95% confidence intervals (see text). Experimental data come from cytogenetical observations of MLH1 foci on male mice synaptonemal complexes (SCs) by Froenicke et al. (2002). Simulations are computed using the standard chi-square model (dashed bars) and our FIC model enforcing the obligate chiasma constraint (solid bars). Chromosome length (in morgans) is calculated as half the average number of MLH1 foci per bivalent in experimental data (see values in Table 1). The interference parameter m is adjusted for each chromosome on the basis of the distribution of inter-CO distances (see text; values in Table 1) and adjusted independently for the chi-square model and the FIC model. Frequencies are calculated for each autosome and in the last graph for all 19 autosomes pooled (see text). Simulated CO frequencies are calculated from 5 × 105 simulated meioses.
TABLE 2.
Sum of squares of differences between simulated and experimental frequencies of (1) MLH1 foci in 10 different bins along bivalents having one, two, and three or more foci (patterns of MLH1 density) and (2) bivalents with zero, one, two, and three or more MLH1 foci (frequency of bivalent classes) for the chi-square and FIC models
| Patterns of MLH1 density (1)
|
Frequency of bivalent classes (2)
|
|||||
|---|---|---|---|---|---|---|
| Chromosome | χ2 | FIC | Ratioa | χ2 | FIC | Ratioa |
| 1 | 2.45 × 10−3 | 1.16 × 10−3 | 2.11 | 8.48 × 10−4 | 2.86 × 10−4 | 2.97 |
| 2 | 5.00 × 10−3 | 3.30 × 10−3 | 1.52 | 6.07 × 10−3 | 2.92 × 10−3 | 2.08 |
| 3 | 8.88 × 10−3 | 6.61 × 10−3 | 1.34 | 1.98 × 10−5 | 1.63 × 10−4 | 0.12 |
| 4 | 3.30 × 10−3 | 2.81 × 10−3 | 1.17 | 1.29 × 10−4 | 2.07 × 10−4 | 0.62 |
| 5 | 2.31 × 10−3 | 3.24 × 10−3 | 0.71 | 6.22 × 10−4 | 2.74 × 10−4 | 2.27 |
| 6 | 4.20 × 10−3 | 2.29 × 10−3 | 1.83 | 5.68 × 10−3 | 1.25 × 10−5 | 454.68 |
| 7 | 2.74 × 10−3 | 2.54 × 10−3 | 1.08 | 2.30 × 10−3 | 4.74 × 10−5 | 48.62 |
| 8 | 1.53 × 10−3 | 1.01 × 10−3 | 1.51 | 1.48 × 10−3 | 1.32 × 10−3 | 1.12 |
| 9 | 2.41 × 10−3 | 1.50 × 10−3 | 1.61 | 1.87 × 10−3 | 3.37 × 10−4 | 5.55 |
| 10 | 4.51 × 10−3 | 3.33 × 10−3 | 1.36 | 6.43 × 10−3 | 1.59 × 10−3 | 4.04 |
| 11 | 1.74 × 10−3 | 1.29 × 10−3 | 1.35 | 7.10 × 10−4 | 2.45 × 10−4 | 2.90 |
| 12 | 1.73 × 10−2 | 3.88 × 10−3 | 4.46 | 4.28 × 10−2 | 7.79 × 10−4 | 54.91 |
| 13 | 1.50 × 10−2 | 1.68 × 10−3 | 8.93 | 5.24 × 10−2 | 3.30 × 10−3 | 15.89 |
| 14 | 1.46 × 10−3 | 7.46 × 10−4 | 1.95 | 9.46 × 10−4 | 3.68 × 10−4 | 2.57 |
| 15 | 1.13 × 10−2 | 3.19 × 10−4 | 35.36 | 2.87 × 10−2 | 1.16 × 10−3 | 24.82 |
| 16 | 1.51 × 10−2 | 1.66 × 10−3 | 9.09 | 5.70 × 10−2 | 4.46 × 10−3 | 12.77 |
| 17 | 9.45 × 10−3 | 6.25 × 10−4 | 15.12 | 3.68 × 10−2 | 1.08 × 10−3 | 34.13 |
| 18 | 1.08 × 10−2 | 1.29 × 10−3 | 8.36 | 5.08 × 10−2 | 3.06 × 10−3 | 16.59 |
| 19 | 4.19 × 10−3 | 3.45 × 10−4 | 12.14 | 8.99 × 10−3 | 5.05 × 10−4 | 17.81 |
| Pooled | 2.27 × 10−3 | 4.42 × 10−4 | 5.15 | 7.93 × 10−3 | 5.17 × 10−4 | 15.34 |
Ratios between sums of squares of differences obtained for chi-square and FIC models: ratio values <1 indicate a better fit to the data for the chi-square model, whereas values >1 indicate a better fit for the FIC model. Data are given for individual chromosomes (1–19) and by pooling MLH1 foci or bivalent counts over all chromosomes (pooled).
Figure 3 shows the expected and observed patterns of MLH1 foci density along the SC for bivalents having exactly 1 or exactly 2 MLH1 foci, as well as for all bivalents. These last curves for all bivalents show a perfect fit between models and data by construction. They are strongly asymmetric, with very low MLH1 densities at the centromeric end and very high ones at the telomeric end. Figure 3 shows graphs for two chromosomes and for all 19 autosomes pooled together, and a similar figure with each of the 19 autosomes is given as supplemental Figure 2 at http://www.genetics.org/supplemental/. Note that a difference between Figure 1 and Figure 3 is that IRD positions were converted into SC positions in Figure 3 and into genetic positions in Figure 1. As observed in Figure 2, simulations reproduce the experimental data equally well for the chi-square and the FIC models in longer chromosomes (1–11), whereas for shorter ones (12–19) the FIC model shows a slightly better fit. The smallest chromosomes are also noisier because they have so few meioses with two MLH1 foci. As previously, we measured goodness-of-fit to the data for the FIC and chi-square models by calculating sums of squares of differences between observed and expected frequencies of MLH1 foci in the different bins along bivalents (Table 2). Lower values again indicate better fits to the data. Pooling all chromosomes together as explained before, this sum of squares is 4.4 × 10−4 for the FIC model and 2.3 × 10−3 for the chi-square model, so that the FIC model gives a value five times lower than the chi-square model.
Interestingly, among 2020 bivalents analyzed in the experimental data set, 20 of them had no CO belonging to the subset marked by MLH1. If such observations are not artifacts, the FIC model alone, by construction, is not able to account for them (see discussion). We also see that essentially no cases of three or more COs are produced within our FIC model; the reason is that interference in the mouse is strong (m is large).
Modeling the pattern of COs along chromatids:
Figure 4 shows the predicted patterns of CO density along chromatids within the FIC and chi-square models. Qualitatively, the patterns are similar to the case of the bivalents (Figure 1), although the variations are less pronounced here, in particular for the case of the single CO curve. Again, the intensity of the pattern depends on chromosome length and m and is stronger for smaller chromosomes and higher values of m. Differences between chi-square and FIC models (Figure 4) are qualitatively similar to those observed at the bivalent level (Figure 1), though less pronounced.
Figure 4.—

Simulated distribution of CO frequency along the genetic map for gametes having one, two, or three COs. Symbols and other elements are the same as described in the Figure 1 legend. Here, the model randomly suppresses on average half of the COs from the bivalents to go to the chromatid level.
In Figure 5 we compare experimental data to simulations obtained with the FIC model, in direct analogy with what was done for Figures 2 and 3 except that the x-axis corresponds to genetic distance in Figure 5, d–f, instead of SC distance in Figure 3. The experimental data are linkage mapping data of Broman et al. (2002), and we also use his values of m for the interference parameter. For chromosome 1 (Figure 5, a and d), the intensity of the patterning is weak and the data are noisy, but this is the chromosome where the patterning effect is weakest because it is long. Most of the chromosomes have a clear patterning effect and resemble that given in Figure 5e: the single CO events arise dominantly near the center, and if a gamete has more than one CO these arise only rarely near the center. This is confirmed in Figure 5f, where counts of COs are pooled over all chromosomes. A figure similar to Figure 5, but for all individual chromosomes is available as supplemental Figure 3 at http://www.genetics.org/supplemental/.
Figure 5.—

Experimental and simulated distributions of COs. Experimental data come from mouse female meiosis analyzed via mapping experiments Broman et al. (2002). Simulations are computed using our FIC model. Values of genetic chromosome length (in morgans) and interference parameter m used for simulations are calculated from Broman et al. (2002) respectively as the average number of COs per gamete in experimental data and the estimated parameter ν of the gamma model − 1 (see values in Table 1). For chromosomes 17 and 18, where Broman found genetic lengths <50 cM, we use a length of 50 cM for simulations (see text). (a and d) Chromosome 1 (long); (b and e) chromosome 16 (short); (c and f) frequencies from pooled numbers of gametes or COs over all 20 chromosomes. Graphs of other individual chromosomes are given in supplemental Figure 3 at http://www.genetics.org/supplemental/. (a–c) Frequency of gametes with zero, one, two, and three or more COs (x-axis) in experimental and simulated data. Open bars indicate experimental data, and solid bars indicate simulated data. Error bars indicate 95% confidence intervals (see text). (d–f) Frequency distribution of COs along the genetic map, from centromere (left end) to telomere (right end) for gametes with exactly one (circles) or two (triangles) COs. x-axis: relative position of the middle of the bin along the genetic map. All bin sizes are 0.1. Open symbols and dashed lines indicate mouse experimental data. Solid symbols and lines indicate simulated data. Simulated CO frequencies are calculated from 5 × 105 simulated meioses using our FIC model.
Obligate chiasma produces a “smile” in the CO density:
Here we point out that the obligate chiasma constraint along with interference leads to a natural enhancement of CO rates at the extremities of a chromosome. To see this, we apply our FIC model of OC with interference and determine the density of COs as a function of IRD position. Then comparing the results to the data of Froenicke et al. (2002) indicates to what extent the density distribution of COs along IRD space may explain this distribution along SC space. Our results show that COs arise more frequently near the extremities of the chromosome and less in the center. We call this a smile effect. The results are presented in Figure 6. Not surprisingly, the strength of the smile depends on the length of the chromosome and on the parameter m. As an example, the enhancement of the CO rate at telomeres compared to the center reaches 24% for m = 8. Note that SRP models without OC, like the chi-square model, do not lead to any such smile effect.
Figure 6.—

Simulated frequencies of COs along gametes. Positions are expressed in the interference-relevant distance (IRD) space (see text). The initial CO is uniformly distributed except for the curves with centromere effect. Genetic size of chromosome: 100 and 60 cM. Interference is modeled using our FIC model with interference parameter m. Triangles, m = 0; circles, m = 4; squares, m = 12, without centromere effect (solid symbols and lines) or with all recombination suppressed between positions 0.4 and 0.6 (centromere effect, open symbols and dashed lines). x-axis: relative position of the middle of the bin along the IRD space. All bin sizes are 0.05. CO frequencies are calculated from 5 × 107 simulated meioses.
We also modeled the case of a metacentric chromosome with complete suppression of recombination within the centromere and none outside its range. Results are shown in Figure 6; the recombination rate appears to be higher both near the centromere and near the telomeres (double smile) for 100-cM chromosomes, whereas this effect is not observed with 60-cM chromosomes.
Finally, we compare our theoretically motivated smile shapes to the actual data of total MLH1 foci densities (see Figure 3), using the SC distances provided in Froenicke et al. (2002) and considering that SC and IRD distances are identical. The results are displayed in Figure 7. The theoretical smile leads to an enhancement at the telomere compared to the center of ∼34%, while that of the experimental data is 179%.
Figure 7.—

Experimental frequencies of mouse MLH1 foci along synaptonemal complexes (dashed line and open symbols) compared to simulated frequencies of crossovers (COs, solid line and symbols) under the hypothesis that IRD distance is the same as SC distance. MLH1 foci and simulated COs are first counted for each chromosome independently and then pooled. Experimental data come from cytologenetical observations of mouse chromosomes by Froenicke et al. (2002). Simulations are computed using our FIC model with obligate chiasma, without any centromere or telomere effect. Genetic sizes of the chromosomes and values of the interference parameter m for each chromosome are the same as in Figures 2 and 3. x-axis: classes of relative SC position along chromosome. Simulated CO frequencies are calculated from 5 × 105 simulated meioses.
DISCUSSION
Multiple distance coordinate spaces and interference:
As explained before, positions along a bivalent or a chromatid may be expressed in four different metrics: (1) the physical distance in base pairs along the DNA molecule, (2) the cytological distance measured in micrometers along a chromosome axis that may be observed under the microscope (e.g., the SC distance), (3) the IRD space that we define here as the relevant metric to study the relation between the strength with which two loci interfere and the distance between these loci, and (4) the genetic distance related to the expected number of COs. The experimental relationship between genetic and physical (SC) distances is illustrated by the curves of total MLH1 foci density in Figure 3. Concerning IRD distances, no experimental data are available to date, but the predicted relationship between genetic and IRD distances under the hypotheses of the FIC model is illustrated in Figure 6.
In studies of interference, a major question is to know more precisely to what the IRD space may be related. For instance, the well-known Kosambi mapping function (Kosambi 1944) assumes that considering two segments of equal size on a linkage map, the probability of having a CO in the second segment, conditional on having a CO in the first segment, is proportional to the genetic distance between both segments. This model thus assumes that the IRD space is the genetic distance space. This holds for all SRP models, in particular for the chi-square (Foss et al. 1993) and gamma (McPeek and Speed 1995) models. On the other hand, in our FIC model, as well as the beam-film model (Kleckner et al. 2004), the polymerization model (King and Mortimer 1990), and particular count-location models (Goldgar and Fain 1988), COs simulated by the models are not uniformly distributed, so the IRD space is different from the genetic distance space. Unfortunately, to our knowledge there are to date no biological data that would explain the mode of action of interference so that one would be able to interpret IRD space in clear biological terms.
FIC model:
Our FIC model was developed to enforce the biological OC constraint (Jones and Franklin 2006) observed in many organisms. Previous work (Broman and Weber 2000) enforced this OC constraint only within the so-called “count-location” models (different from the chi-square model; Sturt 1976; Karlin and Liberman 1978; Goldgar and Fain 1988; Lange et al. 1997); however, there is no biological hypothesis motivating these models. Moreover, except for Goldgar and Fain (1988), they do not incorporate inhibition effects between nearby COs, so such models do not describe distance-dependent interference.
In the FIC model, we have enforced the OC constraint in a biologically motivated way as follows. We consider that in the initial phase there are many attempts to generate a CO; one commonly estimates the number of double-strand breaks potentially initiating a CO to be 10–40 times larger than the final number of COs that succeed in doing so (Moens et al. 2002; Anderson and Stack 2005; Chelysheva et al. 2005; see review in Mezard et al. 2007). When one of those attempts succeeds, we hypothesize that it then inhibits nearby attempts in progress. Such an inhibition could be steric and molecular based, but its mechanism is still unknown. Note that when m = 0, the chi-square model coincides with the Haldane model of no interference, while the FIC coincides with a count location model (Karlin and Liberman 1978) where zero CO events are forbidden.
Within the FIC model, we place the first CO along the chromosome with the uniform distribution as measured in IRD space. However, this first CO may be placed by following another distribution. For instance, one may hypothesize that this density distribution is that observed for early recombination nodules along the bivalent.
Validation of the FIC model:
Our FIC model incorporates both the obligate chiasma and interference effects of the chi-square model type. This FIC model gives a very good fit to mouse experimental data, both for MLH1 foci distribution along the SC and for CO distribution along the genetic map. The mouse data are especially appropriate in this respect because interference is strong there, but in certain other organisms achiasmatic bivalents have been reported so the FIC model will be less appropriate for them. In both simulated and experimental data, when there are two COs, they tend to be away from the middle of the chromosome, whereas single COs are clustered toward the middle. Such a patterning was commonly observed a long time ago in experimental data sets (Jones 1984), for example, from Drosophila (Charles 1938) and from humans (Laurie and Hulten 1985).
The fits on individual chromosomes show some local discrepancies (see supplemental Figures 2 and 3 at http://www.genetics.org/supplemental/ for details) whereas this is not the case when data are pooled for all chromosomes. In the case of MLH1 foci, such local discrepancies may be interpreted as local variations of interference. Indeed, such local variations of interference along chromosomes have already been demonstrated in humans (Lin et al. 2001) and in A. thaliana (Drouaud et al. 2007) although they are difficult to observe. In the case of COs, local discrepancies can be explained either by local variations of interference or by local variations of the intensity of the non-MLH1 pathway, also referred to as the noninterfering pathway (Mezard et al. 2007).
The values of the interference parameter mMLH1 (see Table 1) that we obtained by fitting the distribution of inter-CO distances may be interpreted as a measure of the intensity of interference between MLH1 foci in mouse male meiosis. However, the values of the parameter ν of the gamma function estimated by de Boer et al. (2006) were, respectively, 11.5 and 11.8 for chromosomes 1 and 2, which is not really in accordance with our estimates of m, although the observations were made in both cases with MLH1 foci during mouse male meiosis. In their work, de Boer et al. (2006) estimated ν by fitting the histogram of inter-MLH1 distances (based on SC distances) to a gamma function and applied subsequent corrections for the finite length of the chromosome and for the limited resolution of immunofluorescent observations. These histograms were based on the SC distance between MLH1 foci, so the underlying model corresponded to a modified gamma model in which the SRP would be applied along the SC coordinate space instead of the genetic space. On the other hand, the SRPs used in our approach are based on genetic distance just as the standard gamma and chi-square models (McPeek and Speed 1995). Given the high heterogeneity of MLH1 foci density along SC distances (Figure 7), it is not surprising that the two methods lead to different estimates of m.
As discussed above, local variations of interference along chromosomes have been demonstrated in some organisms. In such cases, the use of a constant value for m in both chi-square and FIC models is questionable and should probably be restricted to chromosome segments where interference may be considered homogeneous. Moreover, the use of a constant value for m has also been seriously challenged in yeast by the fact that the ratio crossovers/noncrossovers increases as the frequency of double-strand breaks decreases, which is referred to as “crossover homeostasis” (Martini et al. 2006). To us this suggests that the interference in IRD space extends over distances that are not sensitive to precursor density; IRD distance itself is then the main factor determining the strength of interference. Further developments toward interference models including possible variations of m would be highly valuable provided that enough high-resolution data are available to estimate the parameters.
Relevance of OC modeling:
Our comparison of FIC and chi-square models for their ability to reproduce the histograms of relative frequencies of mouse bivalents with zero, one, two, and three or more MLH1 foci shows better adjustments with the FIC model for most of the individual chromosomes as well as for all chromosomes pooled (Figure 2). Considering the patterns of MLH1 foci along mouse bivalents with one and with two foci, the FIC model also globally gives better results than the chi-square model without obligate chiasma. Note, however, that the difference is clearly visible in this second case only for some chromosomes, especially the shortest ones, and nearly visible when all chromosomes are pooled (Figure 3; supplemental Figure 2 at http://www.genetics.org/supplemental/). Furthermore, there is no case where the chi-square model gives a clearly better adjustment than the FIC model even though both models involve just one adjustable parameter. In addition, the FIC model leads to better goodness-of-fit values for (1) the frequencies of bivalents with zero, one, two, or three or more MLH1 foci and (2) MLH1 densities in different bins along chromosomes. It is thus relevant, given today's high-quality data, to use models incorporating the OC constraint in mouse and maybe in other organisms with OC too.
In our approach (the FIC model), we forced explicitly the presence of an initial chiasma, so the OC rule was absolutely respected. In other approaches such as the beam-film model (Kleckner et al. 2004) or the polymerization model (King and Mortimer 1990), there are numerous precursor sites from which at least one chiasma later should appear, nearly always enforcing the OC hypothesis in practice. Mixture models including one interfering and one noninterfering pathway (Copenhaver et al. 2002; Stahl et al. 2004) may also lead to few achiasmatic bivalents.
But we also saw that the Froenicke et al. (2002) data set gave rise to 20 bivalents without MLH1. These bivalents cannot be accounted for with our FIC model, even though their frequency is <1%. Two possible explanations may be considered:
MLH1 foci may escape cytological observation, as discussed by Froenicke et al. (2002). These authors explain that the number of MLH1 foci may be underestimated if “some MLH1 protein was lost during the spreading procedure or was not accessible to antibodies” or because of synchronism issues.
MLH1 foci do not mark all COs, so that given the probable existence of ∼10% MLH1-independent noninterfering COs in mouse (de Boer et al. 2006), it may simply be concluded that 20 bivalents had only MLH1-independent COs. This shows the need for more global models incorporating both the OC featured in the FIC model and mixtures of interfering and noninterfering pathways as used by Copenhaver et al. (2002) in Arabidopsis and by Stahl et al. (2004) and Malkova et al. (2004) in yeast.
Heterogeneity in the CO rate along chromosomes:
The correspondence between physical and genetic distance is determined by the recombination rate along each chromosome. In all organisms, these rates vary a lot with in particular a frequent strong suppression effect in and near the centromeres and typically enhancements near the telomeres (Jones 1984; Mezard 2006). Many forces shape these variations: (1) the distribution of chiasma precursors, which may be approached by counting early recombination nodules; (2) chromatin compaction, leading in particular to the centromere effect (Choo 1998); and (3) factors that may determine the way precursors commit or not to turn into COs, including the forces behind interference and obligate chiasma effects. In the case of mouse chromosomes, where the centromere is at one end and the telomere at the other end, this second force may explain the strong asymmetry observed in the experimental curves of Figures 3 and 7. Qualitatively, the theoretical smile shown by our FIC model can be understood as related to the third force and may be explained as follows. In our model of obligate chiasma, the first CO is distributed uniformly, and the next ones are produced by a stationary renewal process. When the interference is positive and acts out to a distance comparable to the length of the chromosome, putting down a second CO will be possible only when the first one is toward one end of the chromosome; this leads to an enhancement of the CO density toward the ends. The simulations proceed in an “interference” coordinate system that is not known ahead of time; there the density of COs shows a smile effect, with an enhancement toward the ends of the chromosome. Similar end effects arise in other models of interference such as the beam-film (Kleckner et al. 2004) and polymerization (King and Mortimer 1990) models. It is not unreasonable to consider that this interference-relevant coordinate system is tied to physical space (in micrometers) and the SC coordinate system is a natural candidate. Using the FIC model with OC then reveals a hitherto underappreciated fact that a combination of effects due to OC, interference, and genetic length of the chromosome may give rise to the enhancement of recombination at the ends.
We saw that this predicted smile effect appeared in the MLH1 data of Froenicke et al. (2002), though with a much larger enhancement effect at the telomere than predicted by our FIC model. However, male and female meioses behave differently in mice (de Boer et al. 2006). de Boer et al.'s Figure 1.E compares the densities of male and female mice along the SC and shows a much steeper rise near the telomere in males compared to females. Our model thus seems more applicable to the case of female meiosis.
In male mice, other forces must be driving the telomere enhancement of recombination. They can be due to heterogeneity in the distribution of recombination precursors, leading to an enhancement of CO near telomeres. This is not specific to the mouse: in maize, the distribution of early recombination nodules marking precursors increases from centromere to telomeres (Stack and Anderson 2002) but this increase is less pronounced than the one of late recombination nodules (chiasmata). So both effects, distribution of precursors and the smile, may shape the final distribution of COs. On the centromere side, it is more difficult to conclude anything: the experimental data do show a local maximum in the density of MLH1 foci, but this density is strongly lowered when one approaches too much the centromere. Clearly it would be necessary to go beyond our all-or-none modeling of centromeric suppression. Nevertheless, the local maximum observed in experimental data suggests that our smile effect might be somewhat at work in mouse meiosis.
Acknowledgments
We thank warmly Lorrie Anderson and Karl Broman who provided us with their remarkable data sets. We also thank Jan Drouaud, Adrienne Ressayre, Christine Dillmann, and Domenica Manicacci for fruitful discussions on interference and helpful comments on the manuscript and four anonymous reviewers for their valuable criticisms.
References
- Anderson, L. K., and S. M. Stack, 2005. Recombination nodules in plants. Cytogenet. Genome Res. 109: 198–204. [DOI] [PubMed] [Google Scholar]
- Argueso, J. L., J. Wanat, Z. Gemici and E. Alani, 2004. Competing crossover pathways act during meiosis in Saccharomyces cerevisiae. Genetics 168: 1805–1816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop, D. K., and D. Zickler, 2004. Early decision; meiotic crossover interference prior to stable strand exchange and synapsis. Cell 117: 9–15. [DOI] [PubMed] [Google Scholar]
- Broman, K. W., and J. L. Weber, 2000. Characterization of human crossover interference. Am. J. Hum. Genet. 66: 1911–1926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broman, K. W., L. B. Rowe, G. A. Churchill and K. Paigen, 2002. Crossover interference in the mouse. Genetics 160: 1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charles, D. R., 1938. The spatial distribution of cross-overs in X-chromosome tetrads of Drosophila melanogaster. J. Genet. 36: 103–126. [Google Scholar]
- Chelysheva, L., S. Diallo, D. Vezon, G. Gendrot, N. Vrielynck et al., 2005. AtREC8 and AtSCC3 are essential to the monopolar orientation of the kinetochores during meiosis. J. Cell Sci. 118: 4621–4632. [DOI] [PubMed] [Google Scholar]
- Choo, K. H., 1998. Why is the centromere so cold? Genome Res. 8: 81–82. [DOI] [PubMed] [Google Scholar]
- Copenhaver, G. P., W. E. Browne and D. Preuss, 1998. Assaying genome-wide recombination and centromere functions with Arabidopsis tetrads. Proc. Natl. Acad. Sci. USA 95: 247–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copenhaver, G. P., E. A. Housworth and F. W. Stahl, 2002. Crossover interference in Arabidopsis. Genetics 160: 1631–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer, E., P. Stam, A. J. Dietrich, A. Pastink and C. Heyting, 2006. Two levels of interference in mouse meiotic recombination. Proc. Natl. Acad. Sci. USA 103: 9607–9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drouaud, J., R. Mercier, L. Chelysheva, A. Bérard, M. Falque et al., 2007. Sex-specific crossover distributions and variations in interference level along Arabidopsis thaliana chromosome 4. PLoS Genet. (in press). [DOI] [PMC free article] [PubMed]
- Esch, E., 2005. Estimation of gametic frequencies from F-2 populations using the EM algorithm and its application in the analysis of crossover interference in rice. Theor. Appl. Genet. 111: 100–109. [DOI] [PubMed] [Google Scholar]
- Foss, E., R. Lande, F. W. Stahl and C. M. Steinberg, 1993. Chiasma interference as a function of genetic distance. Genetics 133: 681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froenicke, L., L. K. Anderson, J. Wienberg and T. Ashley, 2002. Male mouse recombination maps for each autosome identified by chromosome painting. Am. J. Hum. Genet. 71: 1353–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgar, D. E., and P. R. Fain, 1988. Models of multilocus recombination: nonrandomness in chiasma number and crossover positions. Am. J. Hum. Genet. 43: 38–45. [PMC free article] [PubMed] [Google Scholar]
- Haldane, J. B. S., 1919. The combination of linkage values and the calculation of distance between loci of linked factors. J. Genet. 8: 299–309. [Google Scholar]
- Higgins, J. D., S. J. Armstrong, F. C. Franklin and G. H. Jones, 2004. The Arabidopsis MutS homolog AtMSH4 functions at an early step in recombination: evidence for two classes of recombination in Arabidopsis. Genes Dev. 18: 2557–2570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollingsworth, N. M., and S. J. Brill, 2004. The Mus81 solution to resolution: generating meiotic crossovers without Holliday junctions. Genes Dev. 18: 117–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Housworth, E. A., and F. W. Stahl, 2003. Crossover interference in humans. Am. J. Hum. Genet. 73: 188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, G. H., 1984. The control of chiasma distribution. Symp. Soc. Exp. Biol. 38: 293–320. [PubMed] [Google Scholar]
- Jones, G. H., and F. C. Franklin, 2006. Meiotic crossing-over: obligation and interference. Cell 126: 246–248. [DOI] [PubMed] [Google Scholar]
- Karlin, S., and U. Liberman, 1978. Classifications and comparisons of multilocus recombination distributions. Proc. Natl. Acad. Sci. USA 75: 6332–6336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King, J. S., and R. K. Mortimer, 1990. A polymerization model of chiasma interference and corresponding computer simulation. Genetics 126: 1127–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleckner, N., D. Zickler, G. H. Jones, J. Dekker, R. Padmore et al., 2004. A mechanical basis for chromosome function. Proc. Natl. Acad. Sci. USA 101: 12592–12597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosambi, D., 1944. The estimation of map distances from recombination values. Ann. Eugen. 12: 172–175. [Google Scholar]
- Lam, S. Y., S. R. Horn, S. J. Radford, E. A. Housworth, F. W. Stahl et al., 2005. Crossover interference on nucleolus organizing region-bearing chromosomes in Arabidopsis. Genetics 170: 807–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange, K., H. Zhao and T. P. Speed, 1997. The Poisson-skip model of crossing-over. Ann. Appl. Probab. 7: 299–313. [Google Scholar]
- Laurie, D. A., and M. Hulten, 1985. Further studies on chiasma distribution and interference in the human male. Ann. Hum. Genet. 49: 203–214. [DOI] [PubMed] [Google Scholar]
- Lawrie, N. M., C. Tease and M. A. Hultén, 1995. Chiasma frequency, distribution and interference maps of mouse autosomes. Chromosoma 104: 308–314. [DOI] [PubMed] [Google Scholar]
- Lenormand, T., and J. Dutheil, 2005. Recombination difference between sexes: a role for haploid selection. PLoS Biol. 3: 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin, S., R. Cheng and F. A. Wright, 2001. Genetic crossover interference in the human genome. Ann. Hum. Genet. 65: 79–93. [DOI] [PubMed] [Google Scholar]
- Malkova, A., J. Swanson, M. German, J. H. McCusker, E. A. Housworth et al., 2004. Gene conversion and crossing over along the 405-kb left arm of Saccharomyces cerevisiae chromosome VII. Genetics 168: 49–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini, E., R. L. Diaz, N. Hunter and S. Keeney, 2006. Crossover homeostasis in yeast meiosis. Cell 126: 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPeek, M. S., and T. P. Speed, 1995. Modeling interference in genetic recombination. Genetics 139: 1031–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mercier, R., S. Jolivet, D. Vezon, E. Huppe, L. Chelysheva et al., 2005. Two meiotic crossover classes cohabit in Arabidopsis: one is dependent on MER3, whereas the other one is not. Curr. Biol. 15: 692–701. [DOI] [PubMed] [Google Scholar]
- Mezard, C., 2006. Meiotic recombination hotspots in plants. Biochem. Soc. Trans. 34: 531–534. [DOI] [PubMed] [Google Scholar]
- Mezard, C., J. Vignard, J. Drouaud and R. Mercier, 2007. The road to crossovers: plants have their say. Trends Genet. 23: 91–99. [DOI] [PubMed] [Google Scholar]
- Moens, P. B., N. K. Kolas, M. Tarsounas, E. Marcon, P. E. Cohen et al., 2002. The time course and chromosomal localization of recombination-related proteins at meiosis in the mouse are compatible with models that can resolve the early DNA-DNA interactions without reciprocal recombination. J. Cell Sci. 115: 1611–1622. [DOI] [PubMed] [Google Scholar]
- Rowe, L. B., J. H. Nadeau, R. Turner, W. N. Frankel, V. A. Letts et al., 1994. Maps from two interspecific backcross DNA panels available as a community genetic mapping resource. Mamm. Genome 5: 253–274. [DOI] [PubMed] [Google Scholar]
- Roze, D., and T. Lenormand, 2005. Self-fertilization and the evolution of recombination. Genetics 170: 841–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stack, S. M., and L. K. Anderson, 2002. Crossing over as assessed by late recombination nodules is related to the pattern of synapsis and the distribution of early recombination nodules in maize. Chromosome Res. 10: 329–345. [DOI] [PubMed] [Google Scholar]
- Stahl, F. W., H. M. Foss, L. S. Young, R. H. Borts, M. F. F. Abdullah et al., 2004. Does crossover interference count in Saccharomyces cerevisiae? Genetics 168: 35–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturt, E., 1976. A mapping function for human chromosomes. Ann. Hum. Genet. 40: 147–163. [DOI] [PubMed] [Google Scholar]
- Zhao, H., and T. P. Speed, 1996. On genetic map functions. Genetics 142: 1369–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, H., M. S. McPeek and T. P Speed, 1995. Statistical analysis of chromatid interference. Genetics 139: 1057–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zickler, D., and N. Kleckner, 1999. Meiotic chromosomes: integrating structure and function. Annu. Rev. Genet. 33: 603–754. [DOI] [PubMed] [Google Scholar]